
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrophage-targeting oligopeptides from Mortierella alpina†
Jacob M.
Wurlitzer
a,
Aleksa
Stanišić
b,
Sebastian
Ziethe
a,
Paul M.
Jordan
 c,
Kerstin
Günther
c,
Oliver
Werz
c,
Hajo
Kries
b and
Markus
Gressler
*a
c,
Kerstin
Günther
c,
Oliver
Werz
c,
Hajo
Kries
b and
Markus
Gressler
*a
aDepartment Pharmaceutical Microbiology at the Leibniz Institute for Natural Product Research and Infection Biology (Hans-Knöll-Institute), Friedrich-Schiller-University, Winzerlaer Strasse 2, Jena 07745, Germany. E-mail: markus.gressler@leibniz-hki.de
bJunior Group Biosynthetic Design of Natural Products at the Leibniz Institute for Natural Product Research and Infection Biology (Hans-Knöll-Institute), Beutenbergstrasse 11a, Jena 07745, Germany
cDepartment Pharmaceutical/Medicinal Chemistry at the Friedrich-Schiller-University, Philosophenweg 14, Jena 07743, Germany
First published on 15th July 2022
Abstract
The realm of natural products of early diverging fungi such as Mortierella species is largely unexplored. Herein, the nonribosomal peptide synthetase (NRPS) MalA catalysing the biosynthesis of the surface-active biosurfactants, malpinins, has been identified and biochemically characterised. The investigation of the substrate specificity of respective adenylation (A) domains indicated a substrate-tolerant enzyme with an unusual, inactive C-terminal NRPS module. Specificity-based precursor-directed biosynthesis yielded 20 new congeners produced by a single enzyme. Moreover, MalA incorporates artificial, click-functionalised amino acids which allowed postbiosynthetic coupling to a fluorophore. The fluorescent malpinin conjugate penetrates mammalian cell membranes via an phagocytosis-mediated mechanism, suggesting Mortierella oligopeptides as carrier peptides for directed cell targeting. The current study demonstrates substrate-specificity testing as a powerful tool to identify flexible NRPS modules and highlights basal fungi as reservoir for chemically tractable compounds in pharmaceutical applications.
Introduction
Among natural products, non-ribosomal peptides (NRP) are of medicinal importance and include antibiotic, anticancer or immunomodulating drugs. Attempts have been made to produce novel NRPs via classical solid-phase peptide synthesis1 or by enzyme engineering within the emerging field of synthetic biology.2 However, most clinically relevant compounds are still isolated from their microbial producers, i.e. bacteria and higher fungi. In contrast, basal fungi such as Mortierella sp. have traditionally been used as a resource for polyunsaturated fatty acids in the food industry,3 but have not been considered as NRP producers. However, recent investigations of the secondary metabolism of Mortierella alpina revealed an unexpected potential for the production of small oligopeptides of pharmaceutical interest including the surface-active malpinins A–E (1–5) (Fig. 1).4 Additionally, cyclic pentapeptides such as malpibaldins and the antibacterial malpicyclins are produced.5 The biosynthesis of the latter compounds has recently been assigned to two bacterial-like nonribosomal peptide synthetases (NRPSs), malpibaldin synthetase MpbA and malpicyclin synthetase MpcA, respectively.5 Both enzymes are composed of five consecutive modules (C-A-T) each harbouring a condensation (C) domain, an ATP-dependent adenylation (A) domain and a thiolation (T) domain, which act in concert to provide a linear pentapeptide that is subsequently cyclised by a C-terminal thioesterase domain (TE). Whilst L-amino acids are incorporated by the action of canonical C domains, D-amino acid building blocks are introduced by bacterial-like dual epimerization/condensation (E/C) domains.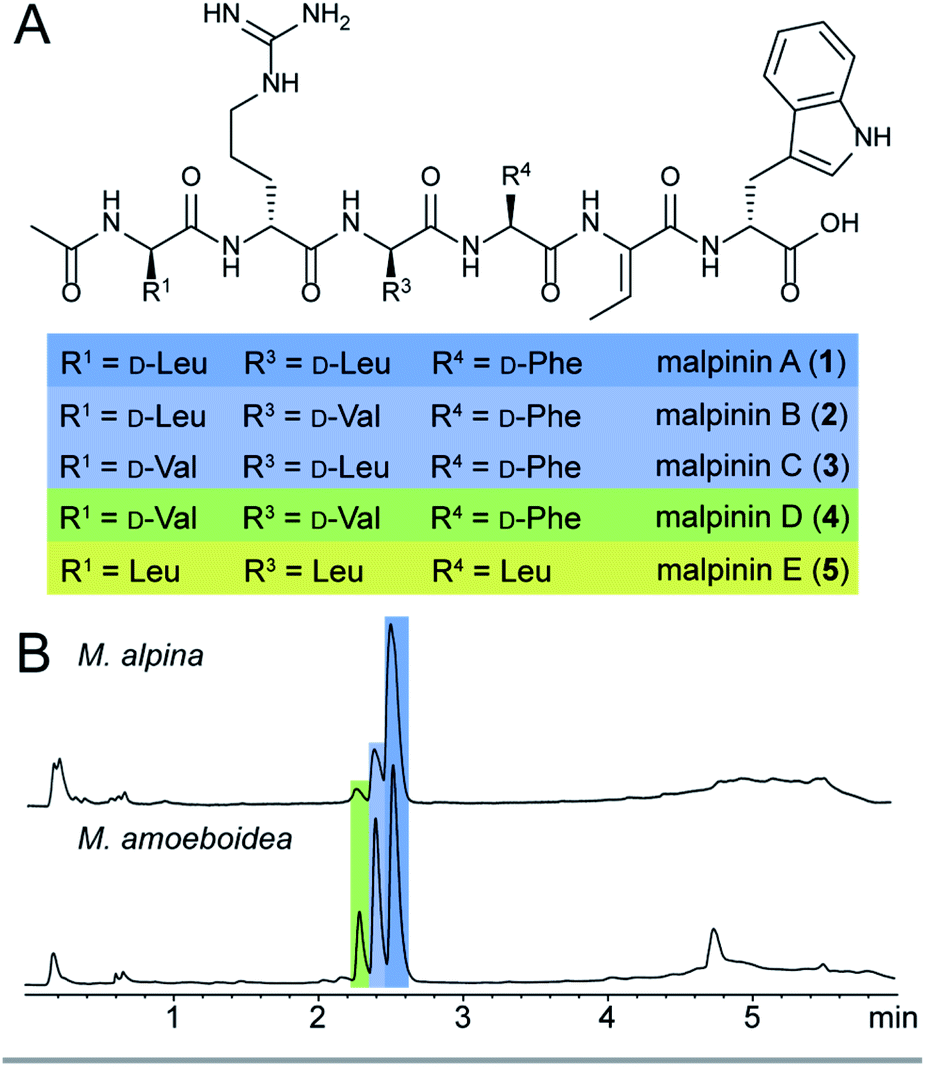 | ||
| Fig. 1 Native malpinins. A. Chemical structures of malpinin A–E (1–5). The stereoconfiguration of 5 has not been determined. B. UV chromatograms (λ = 280 nm) of crude extracts of mycelia from M. alpina ATCC32222 and M. amoeboidea CBS 889.72 grown on YPD. Compound 1 is the predominant metabolite, whereas compound 5 is detectable only in traces (not depicted). | ||
In contrast to Mortierella cyclopentapeptides, the biosynthetic origin of the biosurfactant 1–5 is still enigmatic. Malpinins have a surface tension-lowering activity with a critical micelle concentration (CMC) of 14 μM that is 580-fold lower than that of the commonly used anionic detergent sodium dodecyl sulfate (SDS).4 Their low cytotoxicity makes these biosurfactants valuable candidates for pharmaceutical or medicinal applications. However, information on the malpinin uptake into mammalian cells has been lacking. Malpinins are acetylated hexapeptides (Ac-D-Leu/Val-D-Arg-D-Leu/Val-L-Phe/Leu-Dhb-D-Trp) with two striking structural features (Fig. 1): (i) a non-canonical amino acid, (Z)-dehydrobutyrine (Dhb), at position 5, and (ii) a C-terminal D-amino acid, D-tryptophan, that can be oxidised to kynurenine.4 Moreover, incorporated D-amino acids in positions 1, 3 and 4 were variable resulting in the 1-congeners 2–5. Consequently, a hexamodular NRPS with flexible modules 1, 3 and 4 is expected for the malpinin biosynthesis matching the above-mentioned catalytic features.
Substrate promiscuity is a key feature of many enzymes which enables both natural enzyme evolution6 and enzyme engineering7 in synthetic biology.8 In NRPSs, substrate promiscuity has been occasionally encountered in A domains9 and harnessed for producing non-natural products7 but has not been systematically investigated. Several adenylation assays have been established to measure the activity of A domains.10,11 Recently, the hydroxamate specificity assay (HAMA) was developed which unravels specificity profiles of A domains under competition conditions in a straightforward fashion. HAMA is based on the quenching of aminoacyl adenylates by hydroxylamine and LC-MS/MS detection of respective hydroxamate products allowing parallel testing of multiple competing substrates.12 Hence, we choose HAMA to investigate putatively substrate tolerant A domains5 in the malpinin synthetase.
In this report, we use a combination of state-of-the-art genome sequencing techniques and UHPLC-MS/MS-based metabolite screening to identify the malpinin synthase MalA. A thorough substrate specificity analysis of the complete set of its NRPS modules led to the identification of 20 novel malpinin congeners, among them unusual methionine-containing compounds. Using substrate profiling by HAMA, the enzyme's relaxed substrate tolerance was determined and exploited to incorporate non-natural, “clickable” amino acid substrates. The bioorthogonally labelled fluorescent Mortierella oligopeptide permeates membranes of mammalian cells in a phagocytose-dependent process. Hence, malpinins represent potential carrier peptides for drugs in macrophage-associated diseases.
Results and discussion
Identification of the malpinin synthetase MalA
The published genome of M. alpina13 does not provide appropriate NRPS candidate genes for malpinin biosynthesis. Therefore, a comparative genome analysis of M. alpina ATCC32222 and its close relative Mortierella amoeboidea CBS889.72 (ref. 14) was used to identify the malpinin synthetase gene. Both species produce 1–5 as determined by UHPLC-MS and ESI-MS/MS (Fig. 1, and ESI, S1†) and were subjected to genome sequencing (Table S1†). Subsequent analysis of both genomes using the antiSMASH15 platform lead to the identification of at least 16 potential NRPS and NRPS-like genes in both species (Table S2†). Consistent with the metabolite screening (Fig. S1†), either genome encodes the two cyclopentapeptide NRPSs MpbA (85% amino acid identity between both species) and MpcA (90%), which catalyse the production of malpibaldins and malpicyclins, respectively. In addition, both species share three large genes encoding one hexamodular (Nps5, 89% identity), one heptamodular (Nps3 [MalA], 90% identity) and one octamodular NRPS (Nps2, 73% identity), which are plausible candidates for malpinin biosynthesis (Tables 1, S2 and Fig. S2†). An expression analysis by qRT-PCR revealed that the nps2 gene is hardly expressed under laboratory conditions, whilst transcripts of both nps3 (malA) and nps5 are highly abundant (Fig. S3†). However, solely malA expression levels correlated with titers of 1, i.e. the major metabolite in Mortierella metabolite extracts, according to LC-MS-based metabolite quantification (Fig. S3†). Moreover, the candidate enzyme MalA shows the required distribution of C and E/C domains (Table 1), although the 7th module seems to be obsolete for the production of a hexapeptide.| Gene | Length incl. introns (bp) | Protein size (aa) | Domain architecture of the enzyme |
|---|---|---|---|
| nps2 | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 963 963 |
8125 | A-T-E/C-A-T-C-A-T-E/C-A-T-C-A-T-C-A-T-C-A-T-C-A-T-TE |
| nps3 (malA) | 24471 |
7760 | Cs-A-T-E/C-A-T-E/C-A-T-E/C-A-T-C-A-T-C*-A-T-E/C-A-T-TE |
| nps5 | 20143 |
6489 | Cs-A-T-E/C-A-T-C-A-T-E/C-A-T-E/C-A-T-C-A-T-TE |
MalA modules 1, 3 and 4 are highly promiscuous
The full-length 24.5 kb transcript of malA has been verified by Sanger sequencing and encodes a heptamodular NRPS MalA spanning over 7760 aa (853 kDa). As described for NRPSs of other basal fungi,5,16 the A domains of MalA cluster phylogenetically with bacterial counterparts (Fig. S4†). Since knock-out strategies are hardly applicable for early diverging fungi,17 we used in vitro specificity profiling to assign all A domains of MalA to a particular adenylation step in the malpinin biosynthesis. Heterologous production of all seven C-A-T modules (M1-7) as bi-terminal His6-tagged fusion proteins was accomplished in Escherichia coli (Fig. S2, S5 and Table S3†). The formation of aminoacyl adenylate during the adenylation reactions was tracked by conversion to stable aminoacyl hydroxamates that were quantified using multiplexed LC-MS/MS measurements (HAMA, Fig. 2 and Table S4†).12 HAMA revealed that both M1 and M3 have relaxed specificity towards aliphatic amino acids (L-Leu > L-Met > L/D-Val > L-Cys), explaining Val at position 1 and 3 in 1-congeners 2–4 (Fig. 1). The incorporation of L-Leu, but not N-acetyl-L-Leu by module 1 was also confirmed (Fig. S6†) suggesting the N-terminal acetylation occurs at a later stage of biosynthesis. Similar to M1 and M3, M4 has a broad substrate spectrum showing the highest activity with L-Phe followed by other hydrophobic amino acids (L-Phe > L-Met = L-Trp). In contrast, M2, M5 and M6 are highly specific for L-Arg, L-Thr, and L-Trp, respectively (Fig. 2). Solely, M7 converted its preferred substrate (L-Phe) with a 15000-fold reduced turnover rate compared to the most active module M6, indicating that its A domain cannot contribute to the malpinin biosynthesis due to low activity. The residual activity for L-Phe may indicate that a 7th residue was present in an evolutionary precursor to the malpinin family. An inactive terminal module has been shown for the indanomycin synthase in Streptomyces antibioticus.18 Since the last T domain in MalA is apparently not loaded with an amino acid, either the TE domain must offload the oligopeptide from the preceding T domain or the dual E/C domain of M7 must transfer the final peptide chain to the free acceptor T domain of M7 prior to release. In any case, this E/C domain is required for the stereo inversion of the C-terminal L-Trp to D-Trp in 1–5. A similar mechanism is proposed for the epimerase (E) domain of the β-(L-α-aminoadipyl)-L-Cys-D-Val (LLD-ACV) synthetase from β-lactam producing fungi.19–21 Building on our expression analysis and HAMA-based in vitro activity assays, a biosynthetic pathway for malpinins by MalA is proposed, which includes (i) a canonical successive peptide biosynthesis, (ii) epimerizations of L-amino acids by dual E/C domains, if required, and (iii) an early peptide offloading at module 7 after five condensation steps (Fig. 3).
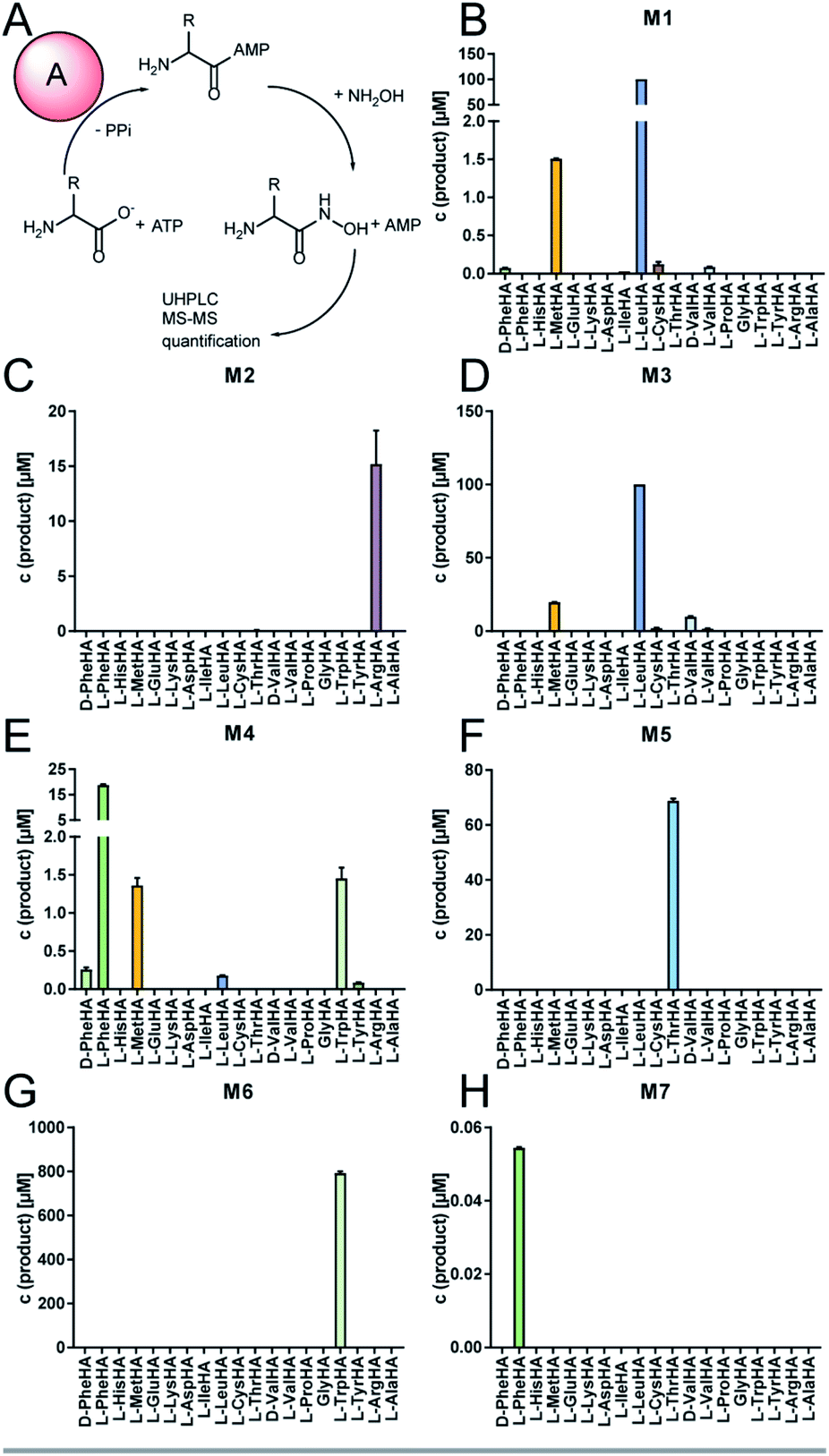 | ||
| Fig. 2 Specificity profiles of the NRPS modules of MalA. (A) Adenylation reaction and hydroxamate formation during HAMA. Modules and substrates were mixed at a final concentration of 1 μM and 1 mM, respectively, and were incubated for 60 min at 37 °C. The resulting aminoacyl hydroxamates (HA) were subsequently analysed by UHPLC-MS. (B–H) Specificity profiles of the complete set of NRPS modules 1–7 (M1–M7) of MalA determined by the HAMA assay. Note that M7 shows lowest activity. | ||
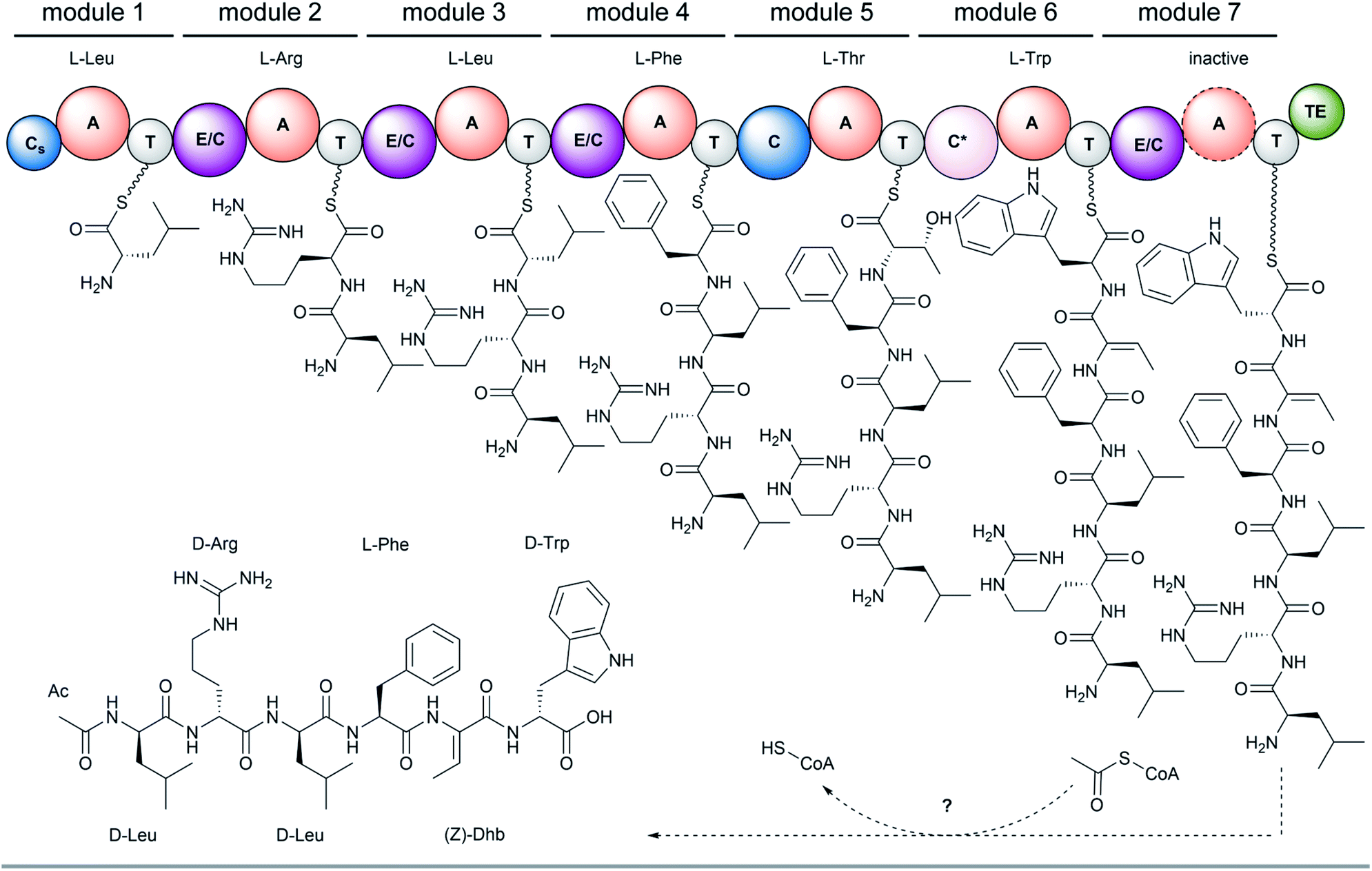 | ||
| Fig. 3 Biosynthesis of malpinins by MalA shown for 1. Involved domains are: A, adenylation domain; C, canonical condensation domain; Cs, starter condensation domain (inactive); C*, dual dehydration/condensation domain; E/C, dual epimerization/condensation domain; T, thiolation domain; TE, thioesterase domain. Note, that the final adenylation domain (in M7) is inactive and offloading occurs either by the final dual E/C domain or the C-terminal T or TE domain. | ||
In bacteria, N-terminal acylation of peptides is catalysed by C-starter (Cs) domains that transfer various acyl chains from acyl-CoA, a standalone acyl carrier protein (ACP) or an acylated C-A-T module selectively to the N-terminus of the nascent NRP.22–24 Recently, a fungal Cs domain has been described to N-terminally acetylate the NRP aspergillicin A from Aspergillus flavus.25 However, the Cs domain of MalA is truncated and lacks the essential C1–C4 core motifs at its N-terminal region including the highly conserved tandem His–His motif in the active site of C3 (Fig. S7 and S8†). An acetylation of L- or D-leucyl-SNAC, that mimics the thiotemplated leucyl moiety,24 could not be determined for MalA-Cs (Fig. S9†) suggesting an inactive Cs domain. The presence of such truncated, non-functional, but structurally required Cs domains have been demonstrated for fungal NRPSs, e.g. in cyclosporine and penicillin biosynthesis.26,27 However, N-acetylation of leucyl-SNAC thioesters was detectable in M. alpina protein crude extracts when incubated with acetyl-CoA (Fig. S9†). These findings and the observation that no acetyl transferase gene is co-expressed in the malpinin A gene cluster (Fig. S10†), point to a co- or post-synthetic acetylation in trans by an acetyltransferase encoded elsewhere in the genome – a phenomenon that has recently been described for the erinacine biosynthesis in the mushroom Hericium erinaceus.28 Indeed, the precursor deacetyl-1 (m/z 817.4716 [M + H]+) is detectable in M. alpina with moderate abundance (Fig. S11 and S12†).
Remarkably, malpinins contain (Z)-Dhb as non-proteinogenic amino acid. Dhb is present in a variety of cyanobacterial NRP.29–32 To incorporate Dhb in ribosomally and post-translationally modified peptides (RiPPs) such as the lantibiotic precursor prenisin or the lacticin-481 propeptide, Thr residues are post-translationally dehydrated by a downstream processing dehydratase (NisB)33 or by a bifunctional dehydratase/cyclase (LctM),34 respectively. However, no homologous genes are encoded in the genomes of M. alpina and M. amoeboidea. During malpinin biosynthesis, Thr is most likely incorporated by M5 (Fig. 2) and is then dehydrated by the subsequent dual E/C domain of M6 to give the α,β-unsaturated amino acid (Z)-Dhb (Fig. 3). In dual E/C domains, the α-C atom is first deprotonated resulting in an enolate intermediate and the epimerisation is achieved through addition of a proton from the opposite side.35 However, the latter step is apparently avoided by M6 and a hydroxyl group is eliminated instead. Recently, the origin of such α,β-unsaturated amino acyl moieties in the NRP albopeptide from Streptomyces albofaciens has been assigned to a novel class of dual β-elimination/condensation domains (C*) in the NRPS AlbB.36 Consistently, the amino acid sequence of the dual E/C domain of M6 shows the same conserved motifs as the C* domains of AlbB, i.e. the catalytic 136HHXXXD141 condensation motif and a E367 residue, which is discussed to be involved in the dehydration reaction.36 Hence, our findings imply a similar mechanism of incorporation of (Z)-Dhb in malpinins. This assumption is further supported by the fact that Thr- and Dhb-specific A domains from bacteria and Mortierella share the same residues in the specificity determining binding pocket (Table S5†).
Enzymatic promiscuity facilitates the production of diverse malpinin congeners
The substrate specificity data confirmed MalA as malpinin synthetase and disclosed substrate tolerance of its modules M1, M3, and M4. In-depth analysis of kinetic parameters of the A domain for M3 by MesG activity assays11 revealed highest specificity for L-Leu (kcat/KM = 61 mM−1 min−1) followed by L-Val (kcat/KM = 4.8 mM−1 min−1) and L-Met (kcat/KM = 0.47 mM−1 min−1) which matches the probability of occurrence of Leu over Val at position 3 in native malpinins 1–5 (Fig. 1 and S13†). However, Met-containing malpinins have never been detected. Surprisingly, the turnover number for the three tested substrates is similarly high (kcat ∼ 2.2 min−1) and seems sufficient to support a typical rate of peptide formation (approx. 1 min−1) with all of them.37 In other words, the adenylation kinetics suggest that the amino acid composition of malpinins can be simply altered by providing elevated concentrations of alternative substrates such as L-Met and L-Cys (for M1, M3, and M4) or L-Trp (for M4). To test this hypothesis, fungal cultures were supplemented with six different canonical amino acids (L-Leu, L-Val, L-Met, L-Trp, L-Cys, L-Phe) in a precursor-directed biosynthesis approach. While L-Leu feeding resulted in production of 1 and 5 as main metabolites, L-Val feeding enlarged the metabolite variety mainly to 2–5, which is consistent with a previous report.4 In accordance to the predictions made by the HAMA assay, L-Met supplementation strongly increased the metabolic diversity by at least 12 additional compounds (malpinins F-Q, 6–17), according to EIC-based quantification by two independent UHPLC-MS methods (Fig. 4A and Tables S6–S8, Fig. S14†). Subsequent ESI-MS/MS-fragmentation confirmed their 1-derived lead structures and suggested a replacement of Leu and/or Phe by one, two or three Met moieties at the expected flexible positions 1, 3, and/or 4 (Fig. S15–S23†).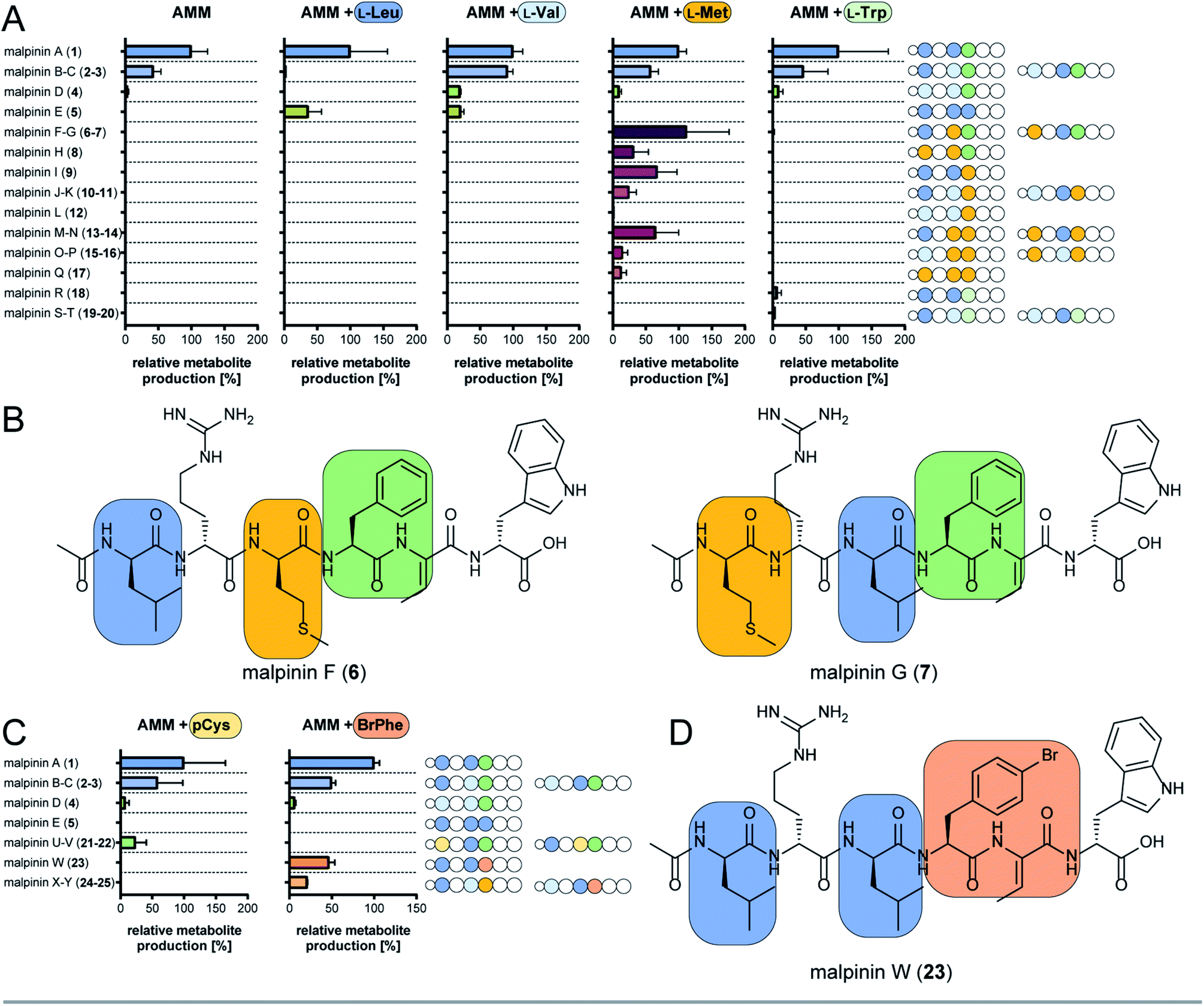 | ||
| Fig. 4 Precursor-directed biosynthesis of novel malpinins. (A) Distribution of native (1–5) and novel malpinins (6–20) by feeding canonical amino acids predicted by HAMA. For detailed HR-ESI-MS/MS analysis see Table S6 (properties of metabolites) and Fig. S14–S23 and S37–S39.† Experiments were carried out in Aspergillus minimal medium without supplementation (AMM) or amended with 5 mM L-Leu, L-Val, L-Met, or L-Trp. Amino acid sequences of 1–20 are shown schematically as stings of beads with variable amino acids highlighted in colour (indigo, Leu; light blue, Val; green, Phe; brown, Met; dark green, Trp). (B) NMR-verified novel malpinins F (6) and G (7). For structure elucidation refer to Tables S7, S9, S10 and Fig. S24–S34.† (C) Distribution of native (1–5) and novel malpinins with non-natural amino acids (21–25) after feeding S-propargyl-L-Cys (pCys) or 4-bromo-L-Phe (BrPhe). Amino acid sequence of 1–5 and 21–25 are shown schematically as a string of beads with variable amino acids highlighted in colour (indigo, Leu; light blue, Val; green, Phe; gold, pCys; ochre, Br-Phe). For detailed HR-ESI-MS/MS analysis see ESI (Table S7 and Fig. S41–S45†). (D) NMR-verified, clickable malpinin W (23). For structure elucidation refer to Tables S10, S11 and Fig. S46–S51.† | ||
Two metabolites (malpinin F and G, 6 and 7) show nearly the same molecular weight (m/z 877.4368 and 877.4374 [M + H]+) and are most likely constitutional isomers (Fig. 4B). They were purified from upscaled fungal cultures by preparative HPLC subjected to extensive 1D and 2D NMR analyses (Table S9 and Fig. S24–S34†). 13C NMR spectra of 6 and 7 revealed seven signals above 160 ppm, accounting for seven carbonyl moieties. Fifteen signals in the aromatic range of the spectrum were identified. Five signals ranging from δC 50 to 55 ppm hinted to five α-carbon atoms, that could be confirmed by HSQC spectra, linking them to their respective α-C protons (δH 4.20–4.70 ppm). As expected, the eight amide protons (δH 7.54–9.18 ppm) did not show any scalar couplings in HSQC spectra. The final peptide backbone was constructed using COSY couplings between amide and α-C protons as well as HMBC correlations between α-C protons and the following carbonyl C atoms (Fig. S34†). Starting from the α-C proton signals, the amino acid side chains were elucidated by COSY, HSQC and HMBC. In 6 and 7, the 13C signal of the terminal methylene group in the aliphatic side chains of Met (δC 29.0 ppm and δC 29.5 ppm, respectively) correlated with a highly abundant 1H singlet signal derived from the lone-standing methyl group (δH 1.92 ppm and 2.00 ppm). With a chemical shift of δC 156.6 ppm as part of the guanidinium group of Arg, δC 128.0 as double signal of Dhb, and 3 and 4 double bond signals each for Phe and Trp, respectively, a total number of eight C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X double bonds and eight CC double bonds were identified. The stereo configuration of 6 and 7 was finally determined by advanced Marfey's analysis38 (Table S10†) and revealed incorporation of D-configured Met in both metabolites.
X double bonds and eight CC double bonds were identified. The stereo configuration of 6 and 7 was finally determined by advanced Marfey's analysis38 (Table S10†) and revealed incorporation of D-configured Met in both metabolites.
In sum, the NMR analysis confirmed the incorporation of D-Met in position 1 and 3 in 6 and 7, respectively, as suggested by the previous ESI-MS/MS experiments. Among the proteogenic amino acids, Met is underrepresented as building block in NRPs.39 Met-containing peptides have been extracted from cyanobacteria40 or marine sponges41–43 and harbour diverse biological activities including anti-cancer and phosphatase–inhibitory properties. However, 6 and 7 do not show any antimicrobial activity (Fig. S35†), but have a similar critical micelle concentration (CMC) as 1 (Fig. S36†).
Aside from the Met-containing metabolites 6–17, but to a lower extent, three Trp-containing 1-congeners (malpinin R-T, 18–20) were produced by L-Trp feeding (Fig. 4A and S37–S39†). In all cases, Trp was incorporated at position 4 in agreement with the relaxed specificity observed in the HAMA profile of M4 (Fig. 2). However, no altered metabolite profiles were obtained by feeding L-Cys or L-Phe (Fig. S40†).
The first A domain of the hexamodular anabaenopeptin synthetase accepts the chemically divergent amino acids Arg and Tyr.7,44 Similarly, substrate flexibility led to the biosynthesis of up to 16 structural variants of microcystins in the cyanobacterium Phormidium.45 Here, promiscuity of A domains 2 and 4 has been assigned to altered residues in positions 236, 239 and 278 in the substrate binding pockets. Similar to cyanobacteria, relaxed substrate specificity is the major driver of metabolic diversity in Mortierellaceae resulting in 20 natural malpinins (1–20). Modules M1, M3, and M4 of MalA incorporate hydrophobic amino acids such as L-Met or L-Trp – in addition to their native substrates L-Leu, L-Val or L-Phe in vitro and in vivo. This raised the question, if non-natural clickable amino acids would be accepted as well.
Clickable amino acids enable synthesis of malpinin conjugates
Malpinins possess surface-active properties, but marginal cytotoxicity, and are hence promising for medicine and material science. Despite the Dhb moiety, the bottleneck in chemical tractability is the availability and accessibility of coupling moieties for “click” chemistry within the molecules. To investigate a potential incorporation of non-proteinogenic, but click-functionalised amino acids, fungal cultures were fed with the Met-congener S-propargyl-L-cysteine (pCys) or the Phe-homolog 4-bromo-L-phenylalanine (BrPhe) and malpinin derivative production was quantified by UHPLC-MS (Fig. 4C).Feeding with pCys led to poor growth of the fungus and moderate incorporation (24%) into malpinins: the Leu moieties in positions 1 and 3 were replaced by pCys in malpinin U (21), m/z 887.4224 [M + H]+ and malpinin V (22), m/z 887.4224 [M + H]+, respectively (Fig. S41, S42 and Tables S6, S7†). In contrast, BrPhe was incorporated in suitable amounts (47%) in place of Phe in position 4: the 1-congener malpinin W (23, m/z 937.3920 [M + H]+), and the two minor 2- and 3-congeners malpinin X (24, m/z 923.3766 [M + H]+) and malpinin Y (25, m/z 923.3767 [M + H]+) were detected (Fig. 4D, S43–S45 and Tables S6, S7†). This relaxed substrate specificity is a remarkable feature of MalA, since acceptance of non-natural amino acids by A domains of other enzymes required time-consuming, systematic mutagenesis of residues in the substrate binding pocket.7 Preferred incorporation of halogenated and other Phe analogs has been previously accomplished by a Trp-to-Ser point mutation in module 1 of the gramicidin S synthetase GrsA and module 4 of the tyrocidine synthetase TycA.46,47 However, MalA M4 contains the respective Trp (W3852) and the intrinsic substrate flexibility must be due to other reasons (Table S5†).
We isolated 12.8 mg of 23 from a 3 L culture and confirmed its structure by NMR (Table S11 and Fig. S46–S51†) and Marfey's analysis (Table S10†). The inspection of the 1H-NMR spectrum revealed the absence of the δH 7.17 signal corresponding to the replacement of the C4 proton by a bromine atom in the Phe moiety at position 4 when compared to the 1H-NMR spectrum of 1 (Fig. S46 and Table S11†). The high field chemical shift of C4 from δC 126.3 ppm (in 1) to δC 119.5 ppm (in 23) in the 13C-NMR spectrum confirmed the successful 4-bromo-L-Phe integration (Fig. S47†).
To test the suitability of malpinins as cell-permeating compounds, 23 was coupled to the fluorescent dye 5-carboxy-fluorescein (5-FAM). To this end, the aryl-halide (23) was substituted by an aryl-azide (26, m/z 900.4836 [M + H]+, Fig. S52†) via an Ullmann-type copper catalysed nucleophilic aromatic substitution48 (Fig. 5A). The clickable 26 was successfully coupled to the 5′-FAM-alkyne by a Cu(I)-catalysed azide-alkyne click reaction (CuAAC) to finally yield 24 mg of 27 (m/z 1313.5734 [M + H]+, Fig. S53†). The structure of the product was confirmed by 1D- and 2D-NMR (Fig. S54–S59 and Table S12†).
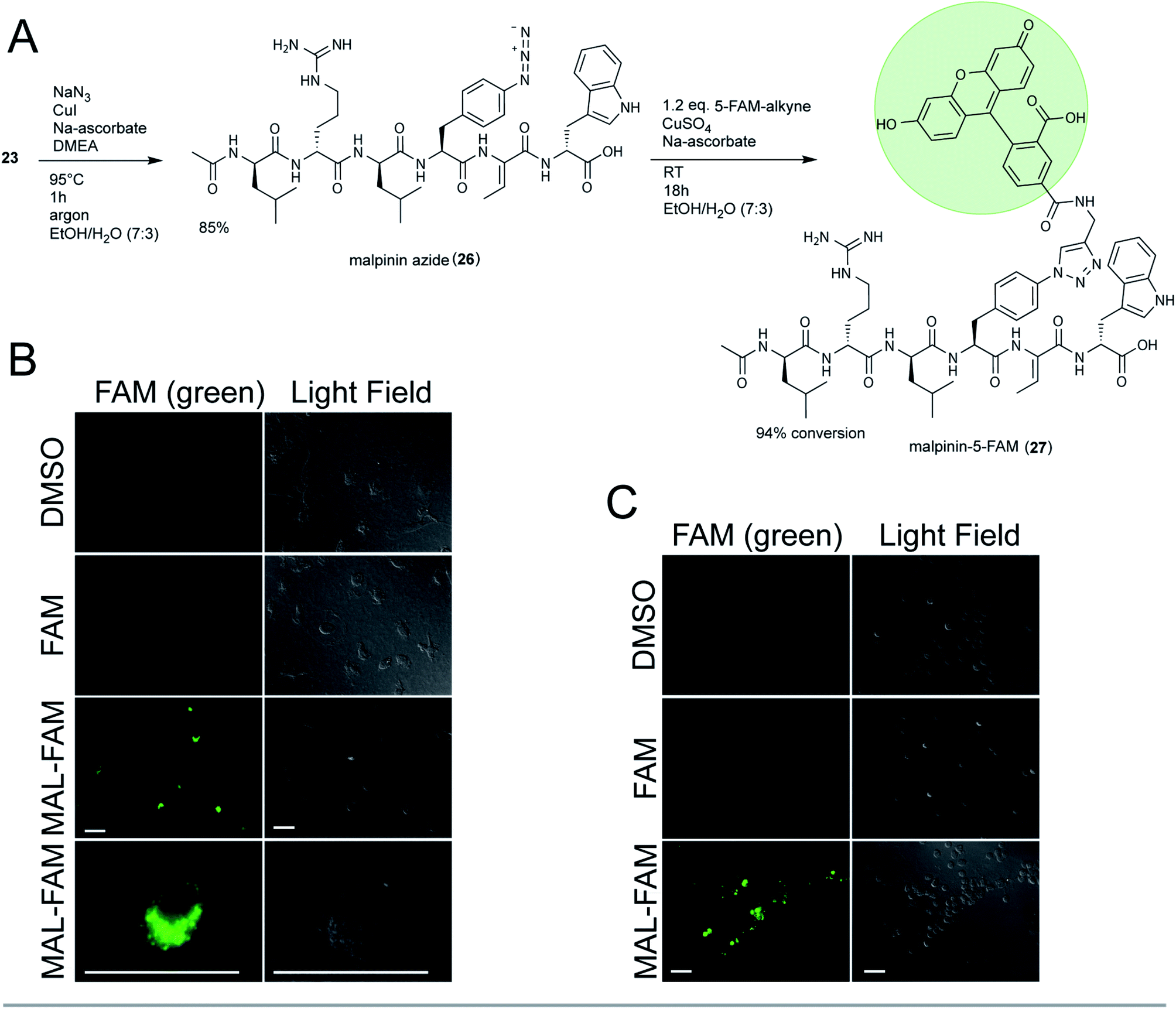 | ||
| Fig. 5 Phagocytosis-triggered uptake of the fluorescent malpinin-conjugate 27. (A) Synthesis of the triazole-linked 5-FAM conjugate 27. (B and C) Microscopy of human macrophages (B) or neutrophils (C) after fluorophore treatment. Cells were incubated with 30 μM 5-FAM, 27 (MAL-FAM) or pure DMSO (negative control) for 3 h (macrophages) or 1 h (neutrophils) prior to microscopy. Pictures were taken using the green fluorescent channel (λex = 492 nm, λem = 518 nm, left panels) or light field (right panels). Scale bar = 10 μm. | ||
Consistently with 13C-NMR data from 1,2,3-triazole ring systems,49 the chemical shifts of C1′ and C2′ (δC 73.1 ppm and δC 80.9 ppm in the alkyne) were altered towards δC 120.9 ppm and δC 145.9 ppm in 27. The bond between malpinin and 5-FAM was established by inspection of the HMBC spectra through a weak coupling between C1′ (δC 120.9 ppm) of 5-FAM and the C21 proton (δH 7.81) of Phe in malpinin. Similarly to 5-FAM, the malpinin-conjugate 27 showed fluorescent properties, i.e. an emission maximum at λ = 526 nm (Fig. S60†).
Malpinin-conjugates penetrate phagocytic cells
Malpinins show surface-active properties concomitant with low cytotoxicity. To test their potential pharmaceutical application, malpinin trafficking into human cells was studied. 5-FAM and 27 were incubated with human monocyte-derived macrophages and fluorophore localization was determined by fluorescence microscopy. Whilst pure 5-FAM was not able to penetrate the cell membrane, the fluorescent 27 was taken up by the cells (Fig. 5B). In contrast to our expectations, 27 did not attach to the cell membrane, but persisted in the cytoplasm. Hence, we concluded that malpinins enable the uptake of coupled compounds of interest, for which FAM serves as a proof of principle. To study the mechanism of uptake more profoundly, also human neutrophils and embryonic kidney cells (HEK293) were tested. Phagocytic neutrophils share a similar intracellular fluorescence pattern when incubated with 27 (Fig. 5C). In contrast, nonphagocytic HEK293 cells50 hardly showed fluorescence signals, when 27 was applied (Fig. S61†). Hence, uptake of malpinin conjugates appears specific for phagocytotic cells. The use of macrophage-targeting agents is a promising prospect in cancer immunotherapy by modulation of the recruitment and survival of macrophages or by inhibition of their tumor-promoting function.51 Moreover, macrophages are used as “trojan horses” to carry drug-loaded nanoparticles to a source of infection or into a tumor environment.52 Hence, malpinin-conjugates may provide a small-molecular alternative with low cytotoxicity. Future studies will reveal the effectiveness of this approach. In this respect, the high production titers of malpinins up to 10% of the fungal dry biomass53 provide an environmentally sustainable alternative to solid-phase peptide biosynthesis.Conclusions
The current study demonstrates the potential of substrate profiling of A domains for biosynthetic diversification of NRPs. Feeding alternative substrates to a flexible NRPS of a basal fungus strongly shifted product profiles and enabled the production of click-functionalised compounds. The final bioorthogonal-labelling of malpinins uncovered its new pharmaceutical application as peptide carriers for phagocyte-specific drugs.In the current study, a complete determination of substrate specificities of an NRPS system was carried out, which in turn led to the identification of a wealth of new metabolites (Fig. S62†). Promiscuity-based broadening of product diversity is a common strategy in nature to switch the biosynthesis from one compound to another with a higher selective advantage (screening hypothesis).54 The synthetic biology can benefit from this evolutionary-driven approach: In the past ten years, tremendous progress has been made to reconstitute and engineer trajectorial NRP biosynthesis in vitro by combining several NRPS modules by either an inter-domain peptide linker or docking domains55–58 or, alternatively, a DNA template as binding platform.59 The substrate tolerant A domains of NRPSs from basal fungi bear excellent prospects for combinatorial enzyme engineering60 in the future.
Besides their use in food industry, basal fungi are a promising resource of antibiotic and anticoagulating peptide compounds.61,62 With this report, we add a new, third level of application: The malpinins can be used as carrier peptides triggering the uptake of compounds into human macrophages. Macrophages are common drug targets for several disease treatments, because they are involved in cancer and inflammation processes.51 Mesoporous silica nanoparticles are currently being investigated as drug carrier, but their metabolism is still a problem that hinders successful clinical translation.63 In this respect, malpinins represent a natural, small molecular weight alternative.
Experimental section
Organisms and culture maintenance
The fungal strains Mortierella alpina ATCC 32222 and Mortierella (syn. Linnemannia) amoeboidea CBS 889.72 were purchased from the American Type Culture Collection (ATCC) and the Westerdijk Fungal Biodiversity Institute (CBS), respectively (Table S13†). Cultures were maintained on malt extract peptone (MEP) agar plates (30 g L−1 malt extract, 3 g L−1 soy peptone, 18 g L−1 agar) for 7 days at 25 °C.Liquid cultures (100 mL) for metabolite quantification and expression analyses were inoculated with five agar blocks (2 × 2 mm) and incubated at 25 °C and 140 rpm for 3, 7 or 14 days (depending on the experiment). Media were MEP, potato dextrose broth (PDB, Sigma Aldrich), yeast extract peptone dextrose medium (YPD; 20 g L−1 peptone, 10 g L−1 yeast extract, 20 g L−1 glucose), lysogeny broth (LB; 10 g L−1 tryptone, 5 g L−1 yeast extract, 10 g L−1 sodium chloride), or hay medium (HM, 25 g L−1 hay, extracted with hot water, 100 mM phosphate buffer, pH 5.6). For the precursor-directed biosynthesis, 100 mL Aspergillus minimal medium64 supplemented with 100 mM D-glucose and 20 mM ammonium nitrate was amended with 5 to 10 mM of the respective amino acid.
Escherichia coli strains (Table S13†) used for plasmid propagation or heterologous protein production were maintained in LB medium amended with carbenicillin (50 μg L−1) or kanamycin (100 μg L−1), if required.
Molecular biological techniques and microscopy
Details on isolation of nucleic acids, genome sequencing, genome annotation, cloning of DNA in expression vectors, gene expression analysis and fluorescence microscopy are provided in the ESI.† Constructed plasmids and oligonucleotides are listed in Tables S14 and S15.†Protein purification
Heterologous protein production in Escherichia coli and purification by affinity chromatography were carried out as previously described.5 For details on the purification procedure, protein yields and SDS polyacrylamide gels, see ESI (Fig. S3 and Table S3†).Adenylation enzyme activity assays
Chemical analysis of metabolites
Metabolite extraction and isolation
:12:1, 400 mL). The culture broth was extracted three times using the same amount of ethyl acetate. Extracts were pooled, dried under vacuum and dissolved in 50 mL methanol. For initial separation, crude extracts were submitted to preparative HPLC using method E (Table S16†). Fractions containing 6 and 7 were then transferred to a semipreparative HPLC and a separation from 1 as main contaminant was achieved by method F (Table S16†). Method G (Table S16†) was applied to purify 6 and 7 from 2 and 3 using a methanol gradient. Final separation of the isomers 6 (9 mg) and 7 (16 mg) was accomplished on a C18 reverse phase column using an acetonitrile gradient (method H, Table S16†).
Chemical synthesis
:water = 7:3). 2 eq. NaN3, 0.1 eq. CuI (catalyst), 0.1 eq. N,N′-dimethylethylenediamine (DMEA, ligand) and 0.2 eq. sodium ascorbate were added to a final reaction volume of 5 mL as described elsewhere.48 After incubation at 95 °C for 1 h under argon atmosphere, the reaction was evaporated in vacuo, dissolved in methanol and 26 was purified using method E (Table S16†), yielding 38 mg (85%). The structure of 26 was verified by HR-MS/MS (Tables S6, S7 and Fig. S52†).
:water = 7:3). 50 μL of 0.5 M CuSO4 and 150 μL of 0.5 M sodium ascorbate (both Jena Bioscience) were added immediately and after a reaction time of 24 h.49 After 42 h at RT under gentle agitation (100 rpm) the reaction mixture was dried by lyophilisation and dissolved in methanol. Separation was performed by HPLC using method E (Table S16†). 24 mg of 27 were recovered and submitted to NMR and HR-MS/MS structure verification (Tables S6, S7, S12 and Fig. S53–S59†).
Physicochemical and antimicrobial properties of 6 and 7
Determination of surface tension and critical micelle concentration (CMC) was conducted by the ring tear off method using a De Noüy ring tensiometer (Krüss Processor Tensiometer K12, Krüss, Hamburg, Germany) in a concentration range from 1000 to 1.95 μg mL−1 as described previously.4 Antimicrobial activity testing was carried out by agar diffusion tests.4 Ciprofloxacin dissolved in water (5 μg mL−1) and amphotericin B in DMSO/methanol (1:1) (10 μg mL −1) served as controls.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 7 software. Pearson correlation was calculated assuming Gaussian distribution with a confidence interval of 95% and a significance level of 5%.Data availability
The draft genomes of M. alpina ATCC32222 and M. amoeboidea CBS 889.72 are accessible under Genbank accession numbers JALIRG010000000 and JALIGY010000000, respectively. The sequences of the malA genes from M. alpina and M. amoeboidea are deposited under Genbank accession numbers MW984675 and MW984676, respectively.Author contributions
The manuscript was written by OW, HK and MG. JMW and AS performed the experimental work, with guidance and supervision from HK and MG. SZ performed cloning and the full-length gene sequencing of malA. PJ and KG performed the microscopical imaging. MG designed and managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Heike Heinecke, Andrea Perner and Veit Hänsch (all from the Hans Knöll Institute [HKI], Jena, Germany) for their technical assistance in recording NMR and HR-MS/MS spectra, respectively. We thank Kerstin Voigt (Jena Microbial Resource Collection, Jena, Germany) and Sandra Jungmann (FSU, Jena) for antimicrobial testings and biphasic measurements, respectively. We also thank Christian Kretzer for initial fluorescence-imaging experiments. Hajo Kries gratefully acknowledges a fellowship from the Daimler und Benz Foundation and financial support from the DFG (grant number 441781663) and the Fonds der Chemischen Industrie (FCI).Notes and references
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C. P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- H. Kikukawa, E. Sakuradani, A. Ando, S. Shimizu and J. Ogawa, J. Adv. Res., 2018, 11, 15–22 CrossRef CAS PubMed.
- F. Baldeweg, P. Warncke, D. Fischer and M. Gressler, Org. Lett., 2019, 21, 1444–1448 CrossRef CAS PubMed.
- J. M. Wurlitzer, A. Stanišic, I. Wasmuth, S. Jungmann, D. Fischer, H. Kries and M. Gressler, Appl. Environ. Microbiol., 2020, 87, e02051–02020 Search PubMed.
- S. D. Copley, Curr. Opin. Struct. Biol., 2017, 47, 167–175 CrossRef CAS PubMed.
- H. Kaljunen, S. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., 2015, 54, 8833–8836 CrossRef CAS PubMed.
- A. Stanišić and H. Kries, ChemBioChem, 2019, 20, 1347–1356 CrossRef PubMed.
- S. Meyer, J. C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Süssmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef CAS PubMed.
- L. G. Otten, M. L. Schaffer, B. R. Villiers, T. Stachelhaus and F. Hollfelder, Biotechnol. J., 2007, 2, 232–240 CrossRef CAS PubMed.
- D. J. Wilson and C. C. Aldrich, Anal. Biochem., 2010, 404, 56–63 CrossRef CAS PubMed.
- A. Stanišić, A. Hüsken and H. Kries, Chem. Sci., 2019, 10, 10395–10399 RSC.
- L. Wang, W. Chen, Y. Feng, Y. Ren, Z. Gu, H. Chen, H. Wang, M. J. Thomas, B. Zhang, I. M. Berquin, Y. Li, J. Wu, H. Zhang, Y. Song, X. Liu, J. S. Norris, S. Wang, P. Du, J. Shen, N. Wang, Y. Yang, W. Wang, L. Feng, C. Ratledge, H. Zhang and Y. Q. Chen, PLoS One, 2011, 6, e28319 CrossRef CAS PubMed.
- L. Wagner, B. Stielow, K. Hoffmann, T. Petkovits, T. Papp, C. Vagvolgyi, G. S. de Hoog, G. Verkley and K. Voigt, Persoonia, 2013, 30, 77–93 CrossRef CAS PubMed.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed.
- J. F. Tabima, I. A. Trautman, Y. Chang, Y. Wang, S. Mondo, A. Kuo, A. Salamov, I. V. Grigoriev, J. E. Stajich and J. W. Spatafora, G3: Genes, Genomes, Genet., 2020, 10, 3417–3433 CrossRef CAS PubMed.
- H. Kikukawa, E. Sakuradani, A. Ando, T. Okuda, M. Ochiai, S. Shimizu and J. Ogawa, J. Biotechnol., 2015, 208, 63–69 CrossRef CAS PubMed.
- C. Li, K. E. Roege and W. L. Kelly, ChemBioChem, 2009, 10, 1064–1072 CrossRef CAS PubMed.
- B. Diez, S. Gutierrez, J. L. Barredo, P. van Solingen, L. H. van der Voort and J. F. Martin, J. Biol. Chem., 1990, 265, 16358–16365 CrossRef CAS PubMed.
- W. Kallow, J. Kennedy, B. Arezi, G. Turner and H. von Döhren, J. Mol. Biol., 2000, 297, 395–408 CrossRef CAS PubMed.
- J. F. Martin, J. Antibiot., 2000, 53, 1008–1021 CrossRef CAS PubMed.
- M. Wittmann, U. Linne, V. Pohlmann and M. A. Marahiel, FEBS J., 2008, 275, 5343–5354 CrossRef CAS PubMed.
- H. J. Imker, D. Krahn, J. Clerc, M. Kaiser and C. T. Walsh, Chem. Biol., 2010, 17, 1077–1083 CrossRef CAS PubMed.
- L. Zhong, X. Diao, N. Zhang, F. Li, H. Zhou, H. Chen, X. Bai, X. Ren, Y. Zhang, D. Wu and X. Bian, Nat. Commun., 2021, 12, 296 CrossRef CAS PubMed.
- C. Greco, B. T. Pfannenstiel, J. C. Liu and N. P. Keller, ACS Chem. Biol., 2019, 14, 1121–1128 CrossRef CAS PubMed.
- X. Yang, P. Feng, Y. Yin, K. Bushley, J. W. Spatafora and C. Wang, mBio, 2018, 9, e01211–e01218 CAS.
- R. Iacovelli, L. Mozsik, R. A. L. Bovenberg and A. J. M. Driessen, Microbiologyopen, 2021, 10, e1145 CrossRef CAS PubMed.
- C. Liu, A. Minami, T. Ozaki, J. Wu, H. Kawagishi, J. I. Maruyama and H. Oikawa, J. Am. Chem. Soc., 2019, 141, 15519–15523 CrossRef CAS PubMed.
- C. Mehner, D. Müller, A. Krick, S. Kehraus, R. Löser, M. Gütschow, A. Maier, H. F. Fiebig, R. Brun and G. M. König, Eur. J. Org. Chem., 2008, 10, 1732–1739 CrossRef.
- I. Pergament and S. Carmeli, Tetrahedron Lett., 1994, 35, 8473–8476 CrossRef CAS.
- P. N. Leao, A. R. Pereira, W. T. Liu, J. Ng, P. A. Pevzner, P. C. Dorrestein, G. M. König, V. M. Vasconcelos and W. H. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11183–11188 CrossRef CAS PubMed.
- E. N. Zainuddin, R. Jansen, M. Nimtz, V. Wray, M. Preisitsch, M. Lalk and S. Mundt, J. Nat. Prod., 2009, 72, 1373–1378 CrossRef CAS PubMed.
- A. Mavaro, A. Abts, P. J. Bakkes, G. N. Moll, A. J. Driessen, S. H. Smits and L. Schmitt, J. Biol. Chem., 2011, 286, 30552–30560 CrossRef CAS PubMed.
- M. R. Levengood, C. C. Kerwood, C. Chatterjee and W. A. van der Donk, ChemBioChem, 2009, 10, 911–919 CrossRef CAS PubMed.
- W. H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef CAS PubMed.
- S. Wang, Q. Fang, Z. Lu, Y. Gao, L. Trembleau, R. Ebel, J. H. Andersen, C. Philips, S. Law and H. Deng, Angew. Chem., 2021, 60, 3229–3237 CrossRef CAS PubMed.
- S. Doekel and M. A. Marahiel, Chem. Biol., 2000, 7, 373–384 CrossRef CAS PubMed.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- S. Caboche, V. Leclère, M. Pupin, G. Kucherov and P. Jacques, J. Bacteriol., 2010, 192, 5143–5150 CrossRef CAS PubMed.
- T. Sano, T. Usui, K. Ueda, H. Osada and K. Kaya, J. Nat. Prod., 2001, 64, 1052–1055 CrossRef CAS PubMed.
- M. A. Rashid, K. R. Gustafson, J. L. Boswell and M. R. Boyd, J. Nat. Prod., 2000, 63, 956–959 CrossRef CAS PubMed.
- J. Kobayashi, T. Nakamura and M. Tsuda, Tetrahedron, 1996, 52, 6355–6360 CrossRef CAS.
- G. R. Pettit, B. E. Toki, J. P. Xu and D. C. Brune, J. Nat. Prod., 2000, 63, 22–28 CrossRef CAS PubMed.
- G. Christiansen, B. Philmus, T. Hemscheidt and R. Kurmayer, J. Bacteriol., 2011, 193, 3822–3831 CrossRef CAS PubMed.
- T. K. Shishido, J. Jokela, A. Humisto, S. Suurnakki, M. Wahlsten, D. O. Alvarenga, K. Sivonen and D. P. Fewer, Mar. Drugs, 2019, 17(5), 271 CrossRef CAS PubMed.
- D. L. Niquille, I. B. Folger, S. Basler and D. Hilvert, J. Am. Chem. Soc., 2021, 143, 2736–2740 CrossRef CAS PubMed.
- H. Kries, R. Wachtel, A. Pabst, B. Wanner, D. Niquille and D. Hilvert, Angew. Chem., 2014, 53, 10105–10108 CrossRef CAS PubMed.
- J. Andersen, U. Madsen, F. Björkling and X. F. Liang, Synlett, 2005, 1, 2209–2213 Search PubMed.
- M. Dörfer, D. Heine, S. König, S. Gore, O. Werz, C. Hertweck, M. Gressler and D. Hoffmeister, Org. Biomol. Chem., 2019, 17, 4906–4916 RSC.
- I. Licona-Limon, C. A. Garay-Canales, O. Munoz-Paleta and E. Ortega, J. Leukocyte Biol., 2015, 98, 85–98 CrossRef CAS PubMed.
- Z. Duan and Y. Luo, Signal Transduction Targeted Ther., 2021, 6, 127 CrossRef CAS PubMed.
- Y. Qi, X. Yan, T. Xia and S. J. Liu, Mater. Des., 2021, 198, 109388 CrossRef CAS.
- R. Sonnabend, L. Seiler and M. Gressler, J. Fungi, 2022, 8, e196 CrossRef PubMed.
- R. D. Firn and C. G. Jones, Nat. Prod. Rep., 2003, 20, 382–391 RSC.
- M. Kaniusaite, R. J. A. Goode, J. Tailhades, R. B. Schittenhelm and M. J. Cryle, Chem. Sci., 2020, 11, 9443–9458 RSC.
- K. A. J. Bozhüyük, J. Watzel, N. Abbood and H. B. Bode, Angew. Chem., Int. Ed., 2021, 60, 17531–17538 CrossRef PubMed.
- A. S. Brown, M. J. Calcott, J. G. Owen and D. F. Ackerley, Nat. Prod. Rep., 2018, 35, 1210–1228 RSC.
- C. Kegler and H. B. Bode, Angew. Chem., 2020, 59, 13463–13467 CrossRef CAS PubMed.
- H. M. Huang, P. Stephan and H. Kries, Cell Chem. Biol., 2021, 28, 221–227 CrossRef CAS PubMed.
- S. Galanie, D. Entwistle and J. Lalonde, Nat. Prod. Rep., 2020, 37, 1122–1143 RSC.
- N. Koyama, S. Kojima, T. Fukuda, T. Nagamitsu, T. Yasuhara, S. Omura and H. Tomoda, Org. Lett., 2010, 12, 432–435 CrossRef CAS PubMed.
- N. Koyama, S. Kojima, K. Nonaka, R. Masuma, M. Matsumoto, S. Omura and H. Tomoda, J. Antibiot., 2010, 63, 183–186 CrossRef CAS PubMed.
- D. Kwon, B. G. Cha, Y. Cho, J. Min, E. B. Park, S. J. Kang and J. Kim, Nano Lett., 2017, 17, 2747–2756 CrossRef CAS PubMed.
- R. W. Barratt, G. B. Johnson and W. N. Ogata, Genetics, 1965, 52, 233–246 CrossRef CAS PubMed.
- F. I. Kraas, V. Helmetag, M. Wittmann, M. Strieker and M. A. Marahiel, Chem. Biol., 2010, 17, 872–880 CrossRef CAS PubMed.
- E. H. Hashimoto, H. Kato, Y. Kawasaki, Y. Nozawa, K. Tsuji, E. Y. Hirooka and K. Harada, Chem. Res. Toxicol., 2009, 22, 391–398 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures, Figu. S1–S62 and Tables S1–S16. See https://doi.org/10.1039/d2sc00860b |
| This journal is © The Royal Society of Chemistry 2022 |