
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular basis for turnover inefficiencies (misses) during water oxidation in photosystem II†
Guangye
Han‡
 a,
Petko
Chernev
a,
Stenbjörn
Styring
a,
Johannes
Messinger
ab and
Fikret
Mamedov
*a
a,
Petko
Chernev
a,
Stenbjörn
Styring
a,
Johannes
Messinger
ab and
Fikret
Mamedov
*a
aMolecular Biomimetics, Department of Chemistry, Ångström Laboratory, Uppsala University, Box 523, 751 20 Uppsala, Sweden. E-mail: fikret.mamedov@kemi.uu.se
bDepartment of Chemistry, Umeå University, 901 87 Umeå, Sweden
First published on 5th July 2022
Abstract
Photosynthesis stores solar light as chemical energy and efficiency of this process is highly important. The electrons required for CO2 reduction are extracted from water in a reaction driven by light-induced charge separations in the Photosystem II reaction center and catalyzed by the CaMn4O5-cluster. This cyclic process involves five redox intermediates known as the S0–S4 states. In this study, we quantify the flash-induced turnover efficiency of each S state by electron paramagnetic resonance spectroscopy. Measurements were performed in photosystem II membrane preparations from spinach in the presence of an exogenous electron acceptor at selected temperatures between −10 °C and +20 °C and at flash frequencies of 1.25, 5 and 10 Hz. The results show that at optimal conditions the turnover efficiencies are limited by reactions occurring in the water oxidizing complex, allowing the extraction of their S state dependence and correlating low efficiencies to structural changes and chemical events during the reaction cycle. At temperatures 10 °C and below, the highest efficiency (i.e. lowest miss parameter) was found for the S1 → S2 transition, while the S2 → S3 transition was least efficient (highest miss parameter) over the whole temperature range. These electron paramagnetic resonance results were confirmed by measurements of flash-induced oxygen release patterns in thylakoid membranes and are explained on the basis of S state dependent structural changes at the CaMn4O5-cluster that were determined recently by femtosecond X-ray crystallography. Thereby, possible “molecular errors” connected to the e− transfer, H+ transfer, H2O binding and O2 release are identified.
Introduction
Oxygen is an essential part of our atmosphere and a major component of cellular respiration. It appeared about 2.7 billion years ago as a by-product of oxygenic photosynthesis evolved in cyanobacteria.1 Now oxygenic photosynthesis is present in all three kingdoms which possess photosynthetic organisms such as cyanobacteria, algae and plants. Together, these organisms provide the chemical energy for essentially all life in the biosphere. Oxygenic photosynthesis is driven by two photosystems, PSII and PSI, that are both localized in the thylakoid membrane and work in sequence to utilize sun light and to provide reducing equivalents for the CO2 fixation.Photosystem II (PSII) is a multisubunit pigment–protein complex that forms the starting point of the photosynthetic electron flow by catalysing the light-induced oxidation of water and the reduction of plastoquinone (PQ)2–5 (Fig. 1). The first step of the photosynthetic water splitting is light excitation of the primary electron donor chlorophylls (Chl), P680. After the initial charge separation between P680 and pheophytin, e− is transferred via the two quinone acceptors, QA and QB, to the PQ pool to be utilized in subsequent reactions in the thylakoid membrane. QB serves as an exchangeable carrier and is reduced by two electrons (e−) and two protons (H+) before exchange with another plastoquinone from the PQ pool. These electron transfer reactions are usually referred to as the acceptor side reactions in PSII (Fig. 1).2–4,6–8
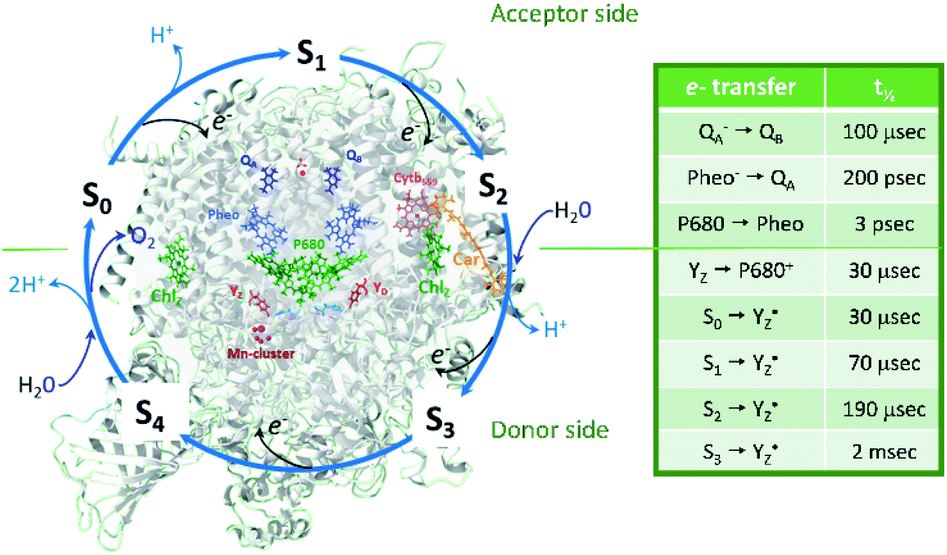 | ||
| Fig. 1 Protein and cofactor organization of PSII based on 6DHE pdb file, S1-enriched state,52 S cycle and sequence of events involving e−, H+ and H2O leading to oxygen evolution in the WOC. The half-times of the individual electron transfer steps are indicated in the table and are reviewed in ref. 2–4, 6, 7 and 9. | ||
P680+ is a highly oxidizing species (Em = +1.35 V) and extracts an electron from the nearby redox-active tyrosine residue, YZ. YZ together with the CaMn4O5-cluster and its water and protein environment compose the water oxidizing complex (WOC), the catalytic site where oxidation of water occurs. 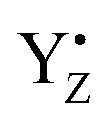 subsequently oxidizes the CaMn4O5-cluster with bound water molecules. The reactions in the WOC are referred to as the donor side reactions (Fig. 1).4,9–11
subsequently oxidizes the CaMn4O5-cluster with bound water molecules. The reactions in the WOC are referred to as the donor side reactions (Fig. 1).4,9–11
The quest to solve the photosynthetic water oxidation mechanism started with the famous Joliot's experiment where O2 release, for the first time, was studied under a train of short (10 μs) light flashes.12 When applied to the dark-adapted algal suspension, the first O2 release peak appeared on the 3rd flash with the next maxima appearing after every 4 flashes until these oscillations were damped and oxygen yields became equal on each flash. These flash-induced oxygen oscillation patterns (FIOPs) were explained by Kok and coworkers in a model, which postulates that during water oxidation, the WOC cycles through five intermediate redox states, collectively called the S states, labelled S0–S4 (Fig. 1).13,14 S0 is the most reduced state while S1, S2 and S3 represent sequentially higher oxidation states in the WOC. S1 is the dominating state in the dark while the S2 and S3 states are metastable and decay back to the S1 state in a few minutes at room temperature.15–18 In addition, the S0 state can be slowly (in tens of minutes) oxidized to the S1 state by 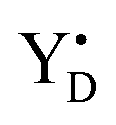 , the second redox active tyrosine in PSII.19,20 O2 is released during the S3 → [S4] → S0 transition, where S4 is a transient state (Fig. 1).4,9 Thus, four consecutive charge separations and accompanying electron transfer events are necessary in PSII to oxidize two water molecules to molecular oxygen and four protons (Fig. 1), which accounts for the period of four oscillations in FIOPs.9,13,14
, the second redox active tyrosine in PSII.19,20 O2 is released during the S3 → [S4] → S0 transition, where S4 is a transient state (Fig. 1).4,9 Thus, four consecutive charge separations and accompanying electron transfer events are necessary in PSII to oxidize two water molecules to molecular oxygen and four protons (Fig. 1), which accounts for the period of four oscillations in FIOPs.9,13,14
In Kok's S state cycle model, the dampening of the FIOP oscillations with the increasing flash number is explained by “double hits” and “misses”. The double hit parameter accounts for centers that perform two consecutive charge separations and S state transitions as a result of a single flash excitation.13,14 Double hits are produced by the long flash tails from Xe flash-lamps and broad LEDs light pulses (tens to hundreds of μs long) and can be eliminated by using short nanosecond laser flashes which produce only a single charge separation in PSII.21 The miss parameter is the most important factor in understanding the S state cycle advancement and dampening of the FIOPs. It represents a probability of the WOC of not advancing to the next S state when the PSII center is exposed to a single flash, and is connected to the turnover efficiency by the formula: miss = 1 − turnover efficiency. Misses are routinely used in the analysis of the S state cycle, mostly in the analysis of FIOPs, where they were first introduced.13,14,17,18,22–29 However, this concept has also been used in the analysis of S state cycle intermediates studied by almost any technique, such as variable and delayed fluorescence,25,30–36 transient optical,37 EPR,38–42 FTIR43–45 and EXAFS spectroscopies.46–50 These analyses were done on many types of photosynthetic species with different degree of purification ranging from intact leaves and cells to PSII core complexes.
The first crystal structure of dark adapted PSII from thermophilic cyanobacteria has been solved almost two decades ago and the highest resolution available now is at 1.9 Å.51 Recently, Kok's model was used in analysis of the crystal structures obtained from the advanced S states of the cycle by serial femtosecond X-ray crystallography.52 In this study, changes in the WOC structure were superimposed with the S states of the full S cycle for the first time.
Under saturating light excitation, the origin of misses in PSII could be acceptor or donor side related. A limitation on the acceptor side electron transfer arises from blocked or slow oxidation of QA− by QB (or QB−). The presence of QA− at the time of light excitation results in the failure of charge separation. A small contribution to misses may also arise under such conditions from a charge recombination between QA− and P680+ that occurs in the 100 microseconds time range before P680+ could be reduced by YZ.53,54 More interesting and mechanistically more important are the donor side induced misses, that originate from the molecular chemistry of the S state transitions. These “actual misses” are in the focus of our present study.
In most studies, the miss parameter was obtained by measurements of only one probe, such as oxygen release during the S3 → [S4] → S0 transition17,18,22–28 or the S2 multiline EPR signal during the S1 → S2 transition.19,39,41,42 This type of analysis, however, has the internal limitation that it allows only determination of the average miss parameter for all S state transitions per cycle. Determination of the misses specific to the individual S state transition in this case is not trivial and requires additional measurements of the S state decay times, estimation of contribution from the secondary electron donors in PSII such as YD and Cytochrome b559 and extensive modelling.29,55–58
In our previous studies, we developed an alternative approach where the individual misses were determined from the exact distribution of S states in the PSII sample after each laser flash advancement.21,40 For this purpose we used well-known, S state-specific electron paramagnetic resonance (EPR) probes such as split S1, S3 and S0 signals and the S2 state multiline signal. We found that the misses during the water oxidation process in PSII are S state-dependent.40 Here we extend the previous study and determined the individual misses over a wide temperature range and at the different frequencies of the advancing (turnover) laser flashes. We also compare our EPR data to FIOPs data obtained under similar conditions and conclude the analysis by relating the origin of the S state dependent misses to events at the CaMn4O5-cluster elaborated by femtosecond X-ray crystallography.52 This allowed us, for the first time, to pinpoint possible “molecular errors” that cause the S state dependent misses during the water oxidizing Kok cycle.
Experimental procedures
Preparation of thylakoid and PSII membranes
Spinach thylakoid membranes were isolated as described in ref. 20 and PSII-enriched membrane fragments (BBY-type) as in ref. 86 and stored at −80 °C until used.FIOP measurements
The FIOP experiments were performed with an unmodulated in-house build Joliot-type electrode embedded in the thermostated, buffer flowing stainless steel cell as described in ref. 20. 10 μL of thylakoid membranes at concentration of 2.4 mg Chl per mL in a measuring buffer, containing 25 mM 2-(N-morpholino)ethanesulfonic acid (Mes) – NaOH (pH 6.3), 400 mM sucrose, 5 mM MgCl2 and 10 mM NaCl, were given 2 saturating pre-flashes at frequency of 1 Hz and dark adapted for 15 min at 20 °C. No artificial electron acceptors were present. Thereafter, 16 saturating flashes were given at indicated temperature (−10 °C, 1 °C, 10 °C and 20 °C) and flash frequency (1.25 and 5 Hz) and FIOPs were recorded. Flashes were provided by a Nd:YAG G100 laser from Spectra Physics (6 ns, 100 mJ, 532 nm). FIOPs analysis was done by using in-house developed software routine.EPR sample preparation
PSII membranes were diluted to 2 mg Chl per ml in the measuring buffer and filled into the calibrated EPR tubes of 4 mm outer diameter. The EPR samples were exposed to room light at 20 °C for 5 minutes to fully oxidize YD, and were then dark adapted for 15 min. Thereafter, PSII in the samples was synchronized to the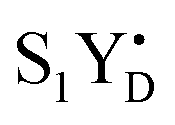 state by the application of two saturating pre-flashes given at 1 Hz frequency followed by a dark adaptation for 30 min at 20 °C.21,40 Then PpBQ in dimethylsulfoxide (DMSO) was added to a final concentration of 0.5 mM. 30 s after the addition of PpBQ, samples were transferred to an ethanol bath at the indicated temperature (−10 °C, 1 °C, 10 °C and 20 °C). After sample temperature equilibration for 3 min, the samples were immediately exposed to saturating turnover flashes (from 0 to 6) at indicated flash frequency (1.25, 5 or 10 Hz) and frozen within 1–2 s in an ethanol–dry ice bath at 198 K and then transferred to liquid N2. Flashes were provided by a Nd:YAG G200 laser from Spectra Physics (6 ns, 840 mJ, 532 nm).
state by the application of two saturating pre-flashes given at 1 Hz frequency followed by a dark adaptation for 30 min at 20 °C.21,40 Then PpBQ in dimethylsulfoxide (DMSO) was added to a final concentration of 0.5 mM. 30 s after the addition of PpBQ, samples were transferred to an ethanol bath at the indicated temperature (−10 °C, 1 °C, 10 °C and 20 °C). After sample temperature equilibration for 3 min, the samples were immediately exposed to saturating turnover flashes (from 0 to 6) at indicated flash frequency (1.25, 5 or 10 Hz) and frozen within 1–2 s in an ethanol–dry ice bath at 198 K and then transferred to liquid N2. Flashes were provided by a Nd:YAG G200 laser from Spectra Physics (6 ns, 840 mJ, 532 nm).
EPR spectroscopy
EPR measurements were performed with a Bruker BioSpin GmbH (Germany) ELEXYS E500 spectrometer with a SuperX ER049X microwave bridge and high Q ER4122SHQE-LC cavity. The spectrometer was equipped with an ESR 900 cryostat and ITC 503 temperature controller from Oxford Instruments, UK. The radical signal and the S2 state multiline signal were measured directly after the flashing. Then illumination into the EPR cavity at 5 K with visible light (160 W m−2, white light lamp projector, 4 min) to induce the split S1, S3 and S0 EPR signals, and with 830 nm light (280 W m−2, LQC830-135E laser diode, Newport, USA, 10 min) to induce only the split S3 EPR signal, were carried out as described in.21,40 In addition, after the initial measurements, each sample was illuminated at 198 K in dry ice/ethanol bath (white light, 4 min) and the S2 state multiline was recorded again. Each experiment of the flash series at a defined temperature and flash frequency was carried out at least twice with deviation of the determined S state distributions by less than 5%.
radical signal and the S2 state multiline signal were measured directly after the flashing. Then illumination into the EPR cavity at 5 K with visible light (160 W m−2, white light lamp projector, 4 min) to induce the split S1, S3 and S0 EPR signals, and with 830 nm light (280 W m−2, LQC830-135E laser diode, Newport, USA, 10 min) to induce only the split S3 EPR signal, were carried out as described in.21,40 In addition, after the initial measurements, each sample was illuminated at 198 K in dry ice/ethanol bath (white light, 4 min) and the S2 state multiline was recorded again. Each experiment of the flash series at a defined temperature and flash frequency was carried out at least twice with deviation of the determined S state distributions by less than 5%.
Quantification of the S state distribution after turnover flashes
Quantification of the S state distribution after the application of turnover flashes was done as in ref. 21 and 40. All EPR data were normalized to the amplitude of the fully induced radical signal of each sample. The fraction of centers in the S1, S2, S3 and S0 states in the samples exposed to 0–6 flashes, were determined from the split S1, S2 multiline, split S3, and split S0 EPR signals (Fig. 2) correspondingly. In the dark sample, after application of our pre-flash protocol, all PSII centers stay in the S1 state. Only the split S1 signal was observed and thus, its intensity represents 100% of the PSII centers. Our early results have shown that all centers in the S1 state are turned over to the S2 state by one saturating laser flash provided at 1 °C. Thus, the S2 multiline signal intensity induced by a single flash at 1 °C also represents 100% of the PSII centers. The application of two flashes at 1 °C resulted in the appearance of S3 state but with some PSII centers remaining in the S2 state. The population of the S3 state was determined by measuring the split S3 EPR signal induced by near-infrared (NIR) or visible light. Similarly, the amplitude of the split S0 EPR signal induced by visible light was defined by the PSII centers in S0 state in three-flash sample. Illumination at 198 K allowed estimation of PSII centers remaining in the S1 state and to ensure the maximal size of the S2 state multiline EPR signal in each sample. It should be noted that in samples that give rise to an EPR spectrum with mixed split EPR signals, a weighted deconvolution of these as described in ref. 21 and 40 was used to obtain a pure split EPR signals from the different S states to enable the analysis. Finally, the fraction of PSII centers in each S state was defined by the corresponding size of respective EPR signal, making possible the quantification of S state distribution in all samples. The procedure described here is used for analysis of experiments with turnover flashes given at 1 °C but was also applied for experiments at other temperatures. All spectral analysis was done by Xepr 2.6b Bruker BioSpin software.
radical signal of each sample. The fraction of centers in the S1, S2, S3 and S0 states in the samples exposed to 0–6 flashes, were determined from the split S1, S2 multiline, split S3, and split S0 EPR signals (Fig. 2) correspondingly. In the dark sample, after application of our pre-flash protocol, all PSII centers stay in the S1 state. Only the split S1 signal was observed and thus, its intensity represents 100% of the PSII centers. Our early results have shown that all centers in the S1 state are turned over to the S2 state by one saturating laser flash provided at 1 °C. Thus, the S2 multiline signal intensity induced by a single flash at 1 °C also represents 100% of the PSII centers. The application of two flashes at 1 °C resulted in the appearance of S3 state but with some PSII centers remaining in the S2 state. The population of the S3 state was determined by measuring the split S3 EPR signal induced by near-infrared (NIR) or visible light. Similarly, the amplitude of the split S0 EPR signal induced by visible light was defined by the PSII centers in S0 state in three-flash sample. Illumination at 198 K allowed estimation of PSII centers remaining in the S1 state and to ensure the maximal size of the S2 state multiline EPR signal in each sample. It should be noted that in samples that give rise to an EPR spectrum with mixed split EPR signals, a weighted deconvolution of these as described in ref. 21 and 40 was used to obtain a pure split EPR signals from the different S states to enable the analysis. Finally, the fraction of PSII centers in each S state was defined by the corresponding size of respective EPR signal, making possible the quantification of S state distribution in all samples. The procedure described here is used for analysis of experiments with turnover flashes given at 1 °C but was also applied for experiments at other temperatures. All spectral analysis was done by Xepr 2.6b Bruker BioSpin software.
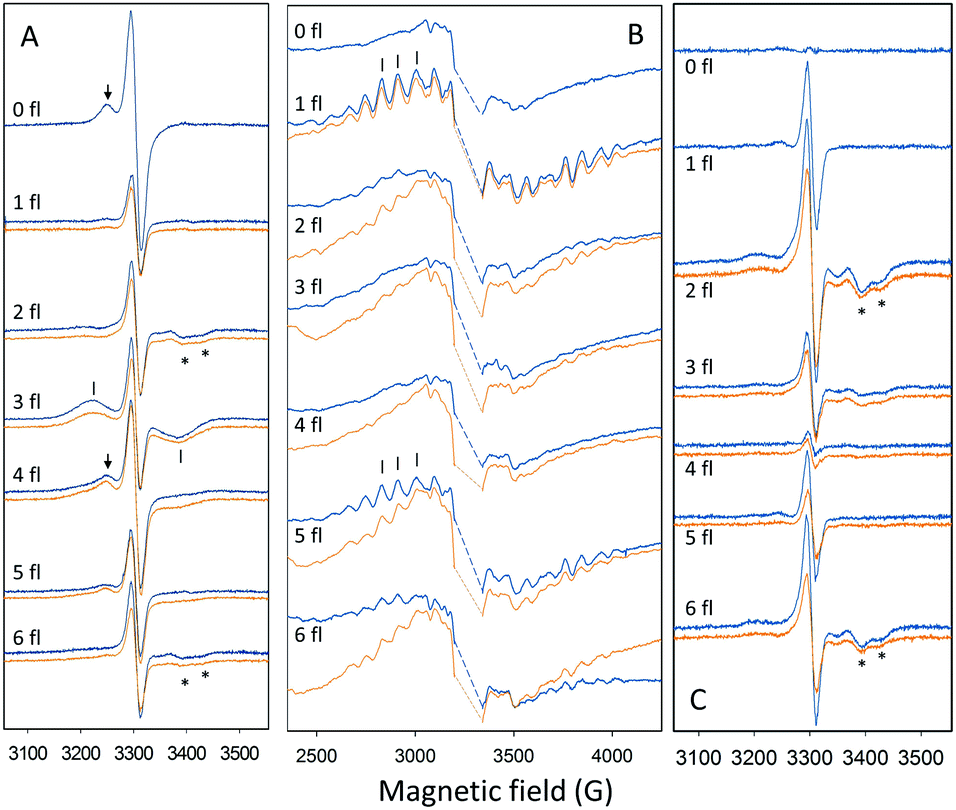 | ||
Fig. 2 EPR spectra used to quantify the fraction of PSII centers in the different S states after 0–6 saturating flashes with 1.25 Hz frequency at −10 °C (orange) and 10 °C (blue). (A) Flash-dependent oscillation of the split S1, split S3 and split S0 EPR signals. EPR signals were induced by illumination by visible light for 4 min at 5 K. The spectra are light minus dark difference spectra. (B) Flash-dependent oscillation of the S2 multiline EPR signal. The large intensity from 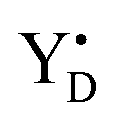 in the center has been removed for clarity (dashed line). (C) Flash-dependent oscillation of the split S3 EPR signal induced by illumination by NIR light for 10 min at 5 K. The spectra presented are light minus dark difference spectra. Peaks used to quantify the EPR signals are indicated by arrows (S1, A), bars (S0, A and S2, B) and stars (S3, A and C). EPR conditions: for (A and C): microwave power 25 mW, microwave frequency 9.27 GHz, modulation amplitude 10 G, temperature (T) 5 K; for (B): microwave power 10 mW, microwave frequency 9.27 GHz, modulation amplitude 20 G, T 10 K. in the center has been removed for clarity (dashed line). (C) Flash-dependent oscillation of the split S3 EPR signal induced by illumination by NIR light for 10 min at 5 K. The spectra presented are light minus dark difference spectra. Peaks used to quantify the EPR signals are indicated by arrows (S1, A), bars (S0, A and S2, B) and stars (S3, A and C). EPR conditions: for (A and C): microwave power 25 mW, microwave frequency 9.27 GHz, modulation amplitude 10 G, temperature (T) 5 K; for (B): microwave power 10 mW, microwave frequency 9.27 GHz, modulation amplitude 20 G, T 10 K. | ||
Analysis of the changes in the CaMn4O5-cluster during the S cycle
Changes in the CaMn4O5-cluster and its immediate surrounding during the S state cycle were visualized by using 6DHE, 6DHF, 6DHO and 6DHP pdb files from the Protein Data Bank.52 Molecular visualization was done with YASARA view program (YASARA Biosciences GmbH).Results
S state distribution after the application of turnover flashes at different temperatures
It is well-known that the S state transitions during the water oxidation cycle are strongly temperature dependent.15,18,28,40,47,59 Previously, we used EPR spectroscopy to determine the S state dependence of the misses at two temperatures, 1 °C and 20 °C.40 In this study, we have now extended these data to two other temperatures: −10 °C and 10 °C. The resulting EPR spectra and corresponding S state distribution after each turnover flash are shown in Fig. 2 and Table 1. From these data the miss parameter after each flash was determined directly from the fraction of PSII centers that did not advance to the next S state as described in ref. 40 and presented in Table 2. Table 2 also shows our earlier data obtained at 1 °C and 20 °C for comparison.| Temp. | Fl. no. | S1 | S2 | S3 | S0 | S1(2nd) | S2(2nd) | S3(2nd) | Total |
|---|---|---|---|---|---|---|---|---|---|
| a The fraction of the Si state was determined from the EPR spectra as described in the text and in ref. 40. | |||||||||
| −10 °C | 0 | 100 | 100 | ||||||
| 1 | 100 | 100 | |||||||
| 2 | 38 ± 2 | 62 ± 1 | 100 | ||||||
| 3 | 18 ± 1 | 30 ± 1 | 52 ± 1 | 100 | |||||
| 4 | 13 ± 1 | 12 ± 1 | 32 ± 1 | 43 ± 1 | 100 | ||||
| 5 | 5 ± 1 | 15 ± 1 | 25 ± 3 | 54 ± 2 | 99 | ||||
| 6 | 12 ± 1 | 5 ± 1 | 42 ± 2 | 39 ± 1 | 98 | ||||
| 10 °C | 0 | 100 | 100 | ||||||
| 1 | 3 ± 1 | 97 ± 1 | 100 | ||||||
| 2 | 21 ± 1 | 79 ± 1 | 100 | ||||||
| 3 | 4 ± 1 | 21 ± 1 | 75 ± 1 | 100 | |||||
| 4 | 3 ± 1 | 6 ± 1 | 20 ± 2 | 70 ± 1 | 99 | ||||
| 5 | 3 ± 1 | 9 ± 2 | 19 ± 1 | 70 ± 1 | 101 | ||||
| 6 | 4 ± 1 | 8 ± 1 | 33 ± 1 | 56 ± 1 | 101 | ||||
| Temp. | S1 → S2 | S2 → S3 | S3 → S0 | S0 → S1 | S1 → S2(2nd)b | S2 → S3(2nd)b | Totalc | Averaged |
|---|---|---|---|---|---|---|---|---|
| a The miss factor is given in % of total PSII in the corresponding Si state that didn't proceed to the next Si+1 state after the flash. b Miss parameters of the transitions in the second turnover of S cycle. c Sum of misses for all S transition of the first turnover of the S cycle (total miss). d Average miss for single transition of the first turnover of the S cycle. e Data from ref. 40. nd - not determined. Accuracy is <5% (standard error). | ||||||||
| −10 °C | 0 | 38 | 16 | 17 | nd | nd | 71 | 17.75 |
| 1 °Ce | 0 | 23 | 7 | 10 | 0 | 20 | 40 | 10 |
| 10 °C | 3 | 19 | 5 | 7 | 0 | 20 | 34 | 8.5 |
| 20 °Ce | 10 | 16 | 3 | 11 | 8 | 15 | 40 | 10 |
We observed no misses in the S1 to S2 transition at −10 °C. This is similar to the situation at 1 °C. The miss factor for the S2 to S3 state transition at −10 °C was found to be very high, 38%. For the S3 to S0 and S0 to S1 transitions the miss parameter was also found to be higher than at higher temperatures – 16% and 17% respectively (Tables 1 and 2).
At 10 °C, the miss in the S1 to S2 transition was no longer zero, instead a very small miss (3%) was found. For the rest of the S state transitions the miss factor was smaller than at lower temperatures. The miss parameter in the S2 to S3 state transition, 19%, was again highest if compared to the other transitions at the same temperature. At 10 °C, the miss parameters for the S3 to S0 and S0 to S1 transitions were 5% and 7%, respectively (Tables 1 and 2).
The temperature dependence of the miss parameter for each individual transition in the S cycle between −10 °C and 20 °C from the present and previous study40 are compiled in Table 2 and Fig. 3. Two different trends can be observed. First, the S1 to S2 transition showed an increase in the miss parameter with increase of the temperature. No miss was observed in the S1 to S2 transition at −10 °C and 1 °C. Above this temperature this transition was less effective, the miss parameter reaching 10% at 20 °C (Fig. 3).40
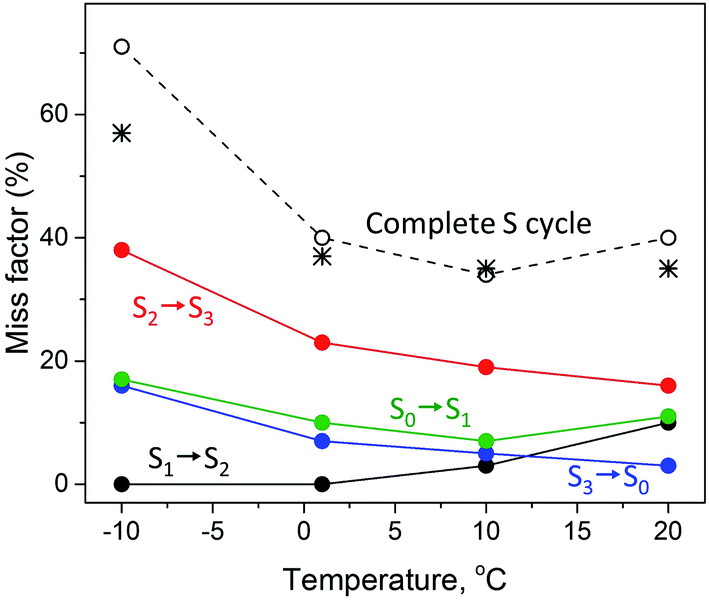 | ||
| Fig. 3 Temperature dependence of the miss factor occurring in the S1 → S2 (black), S2 → S3 (red), S3 → S0 (blue) and S0 → S1 (green circles) state transitions as determined for a flash frequency of 1.25 Hz by EPR. The miss factor is the fraction of PSII centers that did not advance to the next S state after a turnover flash and was calculated from the data on the S state composition provided in Table 2 and ref. 40. The black dotted line (open black circles) represents the accumulated total miss of the complete S state cycle, i.e. is the sum of the misses during the individual S state transitions at the corresponding temperature. The black stars represent the total miss obtained with FIOP measurements (Fig. 4, Table 3). | ||
The S2 to S3 and S3 to S0 state transitions in the S cycle were different. The highest miss factor in the entire temperature range studied was found for the S2 to S3 transition at −10 °C – almost 40% (Table 2). It gradually decreased with increasing temperature reaching 16% at 20 °C (Fig. 3). The miss parameter in the S3 to S0 transition was lower than in the S2 to S3 transition but its temperature dependence was very similar. At −10 °C it was found to be 16% and decreased to only 3% at 20 °C (Table 2, Fig. 3). In the S0 to S1 transition, the miss is first decreased from 17% to 7% when temperature was changed from −10 °C to 10 °C and then slightly increased to 11% at 20 °C (Table 2, Fig. 3).
It is also worth to mention that under our experimental conditions the overall S state turnover was found to be most effective at 10 °C (Table 2, Fig. 3). The sum of the miss factors for the entire S cycle (the total miss) was the lowest at this temperature (34%) and the average miss factor was 8.5%. At 1 °C and 20 °C temperature, the total and average misses were slightly higher than those at 10 °C. However, at −10 °C, the total miss was more than 70%, mostly due to the contribution of the high miss during the S2 to S3 state transition (Table 2, Fig. 3).
FIOP measurements at different temperatures
In order to verify our EPR data we performed classical Joliot-type experiments by measuring FIOPs at different temperatures. These measurements were performed on the thylakoid membranes which retain an active PQ-pool. There are two differences in the experimental conditions in these measurements if compared to EPR: (i) more intact PSII containing membranes were used (ii) in the absence of exogenous electron acceptor. These differences were necessary in order to perform measurements on the bare electrode and to obtain lasting O2 release oscillations. The rest of the conditions were similar to our EPR measurements and the data are shown in Fig. 4 and Table 3. It is known that FIOPs show significant temperature dependence.18,28 It is clear that at 1.25 Hz flash frequency oscillations were running deep in the temperature range between 1 °C and 20 °C, reflecting good WOC turnover (Fig. 4A–C). At −10 °C oscillations were damped after the second cycle (first 8 flashes), indicating high misses during S sate turnover at this temperature (Fig. 4D). This tendency was even more pronounced in measurements at flash frequency of 5 Hz (Fig. 4E–G). With this higher flash frequency at −10 °C, oscillations were damped already after the first cycle (first 4 flashes, Fig. 4G).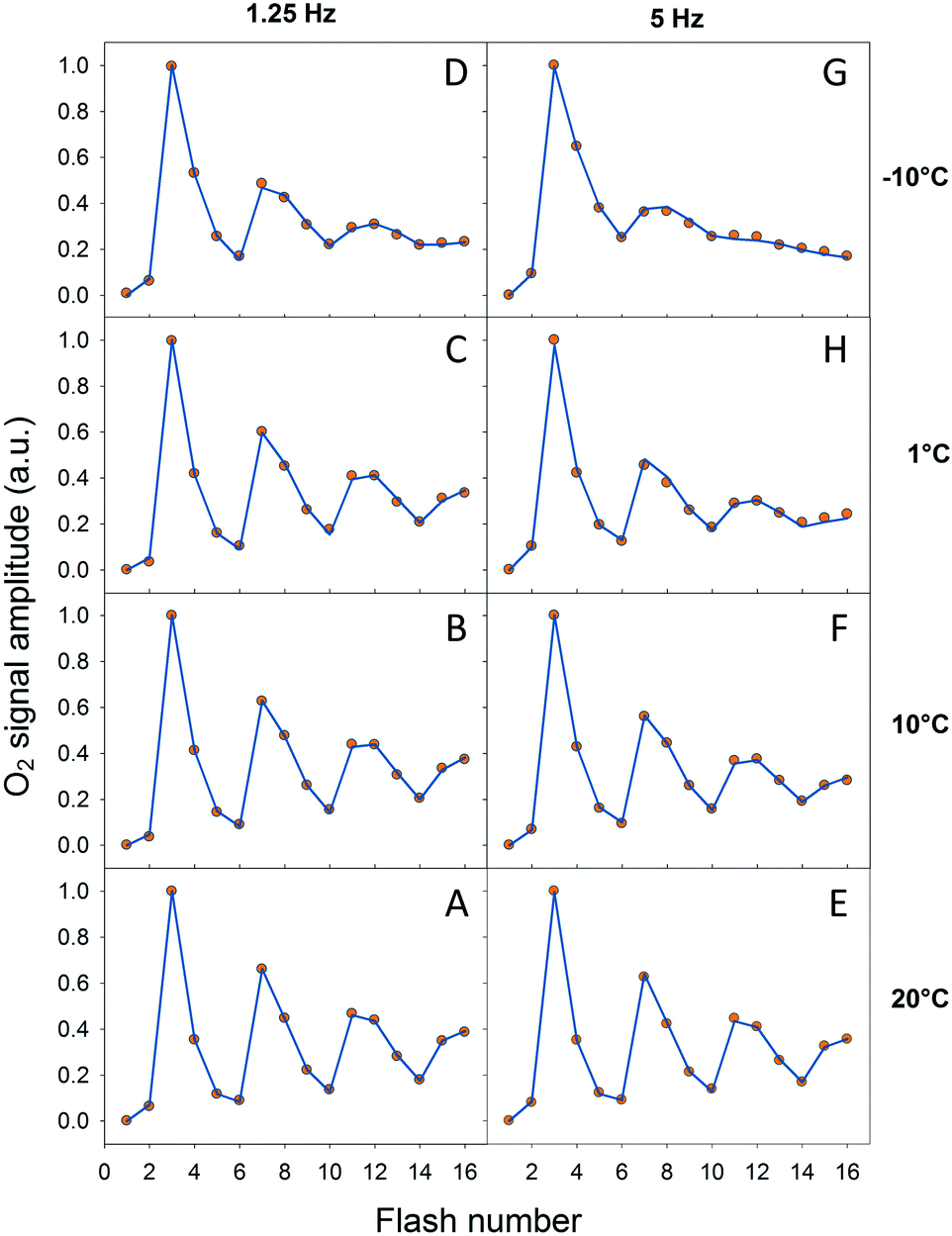 | ||
| Fig. 4 Normalized FIOPs obtained from the thylakoid membranes at different temperatures (−10 °C, 1 °C, 10 °C and 20 °C) and flash frequencies (1.25 Hz and 5 Hz). Measured oxygen yield is shown by orange circles and simulation by blue lines. | ||
| Temp. | Fl. freq. | Totalb | Averagec | Total EPRd | Average EPRe |
|---|---|---|---|---|---|
| a Flash frequencies are given in Hz. b Sum of misses for all S transition of the S cycle (total miss). c Average miss for single transition of the S cycle. d Total miss obtained from EPR measurements (Tables 2 and 6). nd – not determined. e Average miss obtained from EPR measurements (Tables 2 and 6). nd – not determined. | |||||
| −10 °C | 1.25 | 57 ± 4 | 14.4 | 71 | 17.75 |
| 5 | 69 ± 5 | 17.4 | nd | nd | |
| 1 °C | 1.25 | 37 ± 2 | 9.3 | 40 | 10 |
| 5 | 44 ± 3 | 11.0 | 61 | 15.25 | |
| 10 °C | 1.25 | 35 ± 2 | 8.8 | 34 | 8.5 |
| 5 | 37 ± 3 | 9.2 | nd | nd | |
| 20 °C | 1.25 | 35 ± 1 | 8.8 | 40 | 10 |
| 5 | 35 ± 1 | 8.8 | 50 | 12.5 | |
It was not possible to determine the miss factor for each individual S state transition with our FIOP measurements, however our analysis allowed us to determine the average and total miss for each measurement (Table 3). The average miss at 1.25 Hz was found to be about 9% at 1 °C, 10 °C and 20 °C, while at −10 °C it was about 14%. Correspondingly, the total miss of the complete S state turnover was almost 60% at −10 °C, while at the higher temperatures it was only 35–37% (Table 3). In addition, the steady-state O2 yield during flash sequence at both frequencies declined by almost 20% when temperature was decreased from 20 °C to −10 °C (Fig. 4).
The temperature dependence of average miss parameter and total miss determined from the FIOP measurements was found to be very similar to the ones determined by EPR measurements (Fig. 3, Table 3). The only discrepancy we observed at −10 °C (almost 15% difference). This lower FIOP miss most probably reflects a combination of few factors, the most important of which is the more intact acceptor side in PSII in thylakoids. Otherwise, the similarity is much better at higher temperatures and the overall trend in both EPR and FIOP measurement is the same (Fig. 3 and Table 3). This confirms our EPR measurements and quantitative analysis of the miss parameter in each individual S transition.
S state distribution after the application of turnover flashes at different frequencies
EPR experiments described at first were performed at flash frequency of 1.25 Hz. We performed another set of the experiments using the flash frequencies of 5 and 10 Hz. The experiments were performed at two temperatures, 1 °C and 20 °C, and the results are shown in Tables 4–6, Fig. 5 and ESI Fig. 1 and 2.†| Fl. freq. | Fl. no. | S1 | S2 | S3 | S0 | S1(2nd) | S2(2nd) | S3(2nd) | Total |
|---|---|---|---|---|---|---|---|---|---|
| a The fraction of the Si – state was determined from the EPR spectra as described in the text and in ref. 40. | |||||||||
| 5 Hz | 0 | 100 | 100 | ||||||
| 1 | 100 | 100 | |||||||
| 2 | 35 ± 1 | 65 ± 1 | 100 | ||||||
| 3 | 16 ± 2 | 29 ± 2 | 57 ± 1 | 102 | |||||
| 4 | 11 ± 2 | 13 ± 1 | 26 ± 1 | 49 ± 1 | 99 | ||||
| 5 | 6 ± 2 | 15 ± 1 | 28 ± 1 | 53 ± 3 | 102 | ||||
| 6 | 6 ± 1 | 10 ± 1 | 44 ± 2 | 40 ± 1 | 100 | ||||
| 10 Hz | 0 | 100 | 100 | ||||||
| 1 | 100 | 100 | |||||||
| 2 | 43 ± 2 | 57 ± 1 | 100 | ||||||
| 3 | 24 ± 2 | 28 ± 1 | 48 ± 1 | 100 | |||||
| 4 | 17 ± 1 | 13 ± 2 | 33 ± 3 | 37 ± 3 | 100 | ||||
| 5 | 6 ± 1 | 22 ± 1 | 23 ± 3 | 46 ± 1 | 97 | ||||
| 6 | 14 ± 1 | 14 ± 1 | 41 ± 1 | 29 ± 3 | 98 | ||||
| Fl. freq. | Fl. no. | S1 | S2 | S3 | S0 | S1(2nd) | S2(2nd) | S3(2nd) | Total |
|---|---|---|---|---|---|---|---|---|---|
| a The fraction of the Si – state was determined from the EPR spectra as described in the text and in ref. 40. | |||||||||
| 5 Hz | 0 | 100 | 100 | ||||||
| 1 | 10 | 90 | 100 | ||||||
| 2 | 29 ± 1 | 71 ± 2 | 100 | ||||||
| 3 | 11 ± 3 | 23 ± 1 | 66 ± 1 | 100 | |||||
| 4 | 10 ± 3 | 10 ± 1 | 22 ± 2 | 58 ± 1 | 100 | ||||
| 5 | 4 ± 1 | 9 ± 1 | 26 ± 1 | 62 ± 1 | 101 | ||||
| 6 | 6 ± 1 | 7 ± 1 | 38 ± 1 | 46 ± 3 | 97 | ||||
| 10 Hz | 0 | 100 | 100 | ||||||
| 1 | 10 | 90 | 100 | ||||||
| 2 | 33 ± 2 | 64 ± 1 | 97 | ||||||
| 3 | 17 ± 2 | 26 ± 1 | 57 ± 1 | 100 | |||||
| 4 | 15 ± 1 | 7 ± 1 | 28 ± 1 | 49 ± 2 | 99 | ||||
| 5 | 3 ± 1 | 9 ± 1 | 30 ± 2 | 58 ± 2 | 100 | ||||
| 6 | 3 ± 1 | 14 ± 1 | 39 ± 2 | 42 ± 1 | 99 | ||||
| Temp. | Fl. freq. | S1 → S2 | S2 → S3 | S3 → S0 | S0 → S1 | Totalb | Averagec | Total FIOPe | Average FIOPf |
|---|---|---|---|---|---|---|---|---|---|
| a The miss factor is given in % of total PSII in the corresponding Si state that didn't proceed to the next Si+1 state after the flash. b Sum of misses for all S transition of the first turnover of the S cycle (total miss). c Average miss for single transition of the first turnover of the S cycle. d Data from ref. 40. e Total miss obtained from FIOP measurements (Table 3) is shown for comparison. f Average miss obtained from FIOP measurements (Table 3) is shown for comparison. nd – not determined. Accuracy is <5% (standard error). | |||||||||
| 1 °C | 1.25d | 0 | 23 | 7 | 10 | 40 | 10 | 37 | 9.3 |
| 5 | 0 | 35 | 12 | 14 | 61 | 15.25 | 44 | 11 | |
| 10 | 0 | 43 | 16 | 23 | 82 | 20.5 | nd | nd | |
| 20 °C | 1.25d | 10 | 16 | 3 | 11 | 40 | 10 | 35 | 8.8 |
| 5 | 10 | 21 | 7 | 12 | 50 | 12.5 | 35 | 8.8 | |
| 10 | 10 | 29 | 11 | 14 | 64 | 16 | nd | nd | |
 | ||
| Fig. 5 The miss occurring in the S1 → S2, S2 → S3, S3 → S0 and S0 → S1 transitions at 1 °C (A) and 20 °C (B) after application of the turnover flashes at different frequencies: 1.25 Hz (black bars), 5 Hz (blue bars) and 10 Hz (orange bars). The miss was calculated from the fraction of PSII centers that did not advance to the next S state after a turnover flash. | ||
Application of flashes with higher frequency had a profound effect on the miss parameter at 1 °C (Table 4, Fig. 5). For the S2 to S3 transition, the miss parameter rose from 23% at 1.25 Hz to 35% at 5 Hz and to more than 40% at 10 Hz. The effect on the S3 to S0 and S0 to S1 transition was similar – more than two times increase in the miss parameter with increasing frequency of the applied flashes (Table 6 and Fig. 5).
At 20 °C (Table 5, Fig. 5), there was a similar increase of the miss factor with higher flash frequency (1.25 Hz vs. 10 Hz) for the S2 to S3 and S3 to S0 transitions, in this case from 16% to 29% and from 3% to 11%. In contrast to the results at 1 °C, the increase of the miss parameter during the S0 to S1 transition was almost negligible at 20 °C (Table 6, Fig. 4).
Thus, the miss parameter in each S state transitions was not only temperature but also frequency dependent. There was a profound increase in the miss parameter with the increasing frequency of the applied turnover flashes at 1 °C as determined by both EPR and FIOP measurements (Table 6). The effect was also present at 20 °C. Largest increase was again observed for the S2 to S3 transition. It should be noted that determination of the miss parameter based on the FIOP measurements at 10 Hz flash frequency was not possible due to the overlap of the O2 release peaks (not shown).
Discussion
Miss parameter originates from the WOC
The discovery of the period-four oscillation of the oxygen evolution by PSII and the introduction of the S cycle concept laid important foundation for understanding of photosynthetic water splitting.12–14 The dampening of the oscillation with increasing flash number is routinely explained with misses and double hits. Double hits, which normally originate from double turnover in the S cycle during a long flash, currently are easily eliminated by using shorter nanosecond laser flashes thus, limiting the charge separations in PSII to one per flash.21 Our measurements confirm this since we did not observe, for example, any EPR signals from the S3 state after one flash, or from the S0 state after two flashes, and further on (Tables 1, 4 and 5 and Fig. 2, ESI Fig. 1 and 2†).At saturating flash excitation, the miss parameter may have many components.9 As we will outline below, two of the trivial ones we have excluded for the optimal conditions. Firstly, not all QA- could be oxidized between the flashes. In this situation, no stable charge separation can be obtained and thus a miss would be the consequence. Secondly, at very long times between flashes, especially if reduced YD is present, charge recombination between already formed S2 and S3 states can occur and thereby increase the miss parameter. The remaining “actual misses” are a consequence of 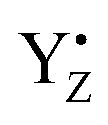 reduction by components other than the CaMn4O5-cluster. This most commonly happens by electrons from the acceptor side, but also other PSII cofactors may occasionally be oxidized, or reactive oxygen species may be formed. The following discussion outlines why we can exclude the trivial reasons for misses, and then aims to clarify the reasons for why the recombination reaction can win over the forward reaction more frequently in the S2–S3 transition than in all other transitions.
reduction by components other than the CaMn4O5-cluster. This most commonly happens by electrons from the acceptor side, but also other PSII cofactors may occasionally be oxidized, or reactive oxygen species may be formed. The following discussion outlines why we can exclude the trivial reasons for misses, and then aims to clarify the reasons for why the recombination reaction can win over the forward reaction more frequently in the S2–S3 transition than in all other transitions.
In our EPR experiments, both trivial reasons for misses were, in most cases, completely eliminated by the use of excess concentration of the exogenous electron acceptor PpBQ. Addition of 0.5 mM PpBQ efficiently keeps the QA–Fe–QB non-heme iron quinone complex in PSII completely oxidized, effectively eliminating recombination reactions with the S2 and S3 states. In the same way, it eliminates also the presence of QA− at the time of the flash and overcomes the acceptor side limitations of non-QB reducing PSII centers.53,54,60 This is confirmed by the maximal induction of the S2 state multiline signal and similar misses observed during the second S cycle (Tables 1 and 2), as well as the deep oscillation of the S2 multiline signal with almost no S2 state population after flashes 3 and 4 (Table 1, 10 °C). The only exception were measurements at low temperature where electron transfer to PpBQ was less efficient and some blocked centers may occur (less than 5%).16 This can be seen as a partial contribution to the higher miss at −10 °C (Table 2, Fig. 3).
Thus, it is clear that at 1.25 Hz flash frequency the acceptor side of PSII kept efficiently oxidized (Tables 1 and 2, also see ref. 40). However, our experiments at 5 and 10 Hz frequency of the turnover flashes, especially at lower temperatures, indicate that some limitations appear on the acceptor side of PSII. At these conditions there is not enough time to complete the electron transfer from QA− to PpBQ before the next flash arrives, and increase in misses during all S transitions is observed (Tables 3 and 6). It is worth to mention that the complete S cycle turnover takes place within 1–2 ms (Fig. 1 and see ref. 4 and 9 for the review). The highest frequency we used, 10 Hz or one flash per 100 ms is far too low to exert any limitations on these transitions. Therefore, it is very likely that the effect we observe in this experiment originates from the lingering electron on the acceptor side of PSII (most likely QA−) at least in a fraction of the PSII centers (so-called closed or inactive PSII centers) making charge separation and therefore, the next state transition impossible. This is in agreement with the earlier results that demonstrated that the reactions at the PSII acceptor side are rate limiting at high flash frequencies.61 There are indications, however, that the turnover efficiency of PSII is not affected by flash frequency up to 30 Hz when PpBQ was used as exogenous electron acceptor at 10 °C.62
As mentioned above, another potential factor which can contribute to the increased miss parameter is the redox state of secondary tyrosine, YD. If present, reduced YD, can be oxidized by the S2 and S3 states in the dark in the pH dependent manner.17,56,58 Due to these reactions, if a flash is given in the S1 or S2 state in the presence of reduced YD, the transition to the S2 and S3 state is reversed in a fraction of PSII centers within 1 s between the flashes.56 However, our pre-flash treatment kept YD fully oxidized and thus its interference with produced S states in the time frame of our experiments (i.e. sample freezing, 1–2 s) is negligible. The latter argument also holds for QB−, Cyt b559 or other electron donor/acceptor cofactors in PSII, for all of which the redox reactions with the S states are even slower (Fig. 1).57
Thus, taking into account implementation of the pre-flash protocol in the presence of PpBQ in EPR measurements, our starting sample was 100% in the S1 state with 100% oxidized YD (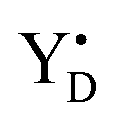 ). During S state advancement there was thus no contribution from recombination reactions with the S2 or S3 states or of blocked centers to the miss parameter. Therefore, we can safely conclude that at our measuring conditions, at least at low flash frequencies and temperatures above 0 °C, the misses we observed in PSII membrane samples in the EPR and FIOPs measurements are actual misses originated from the molecular events in the WOC during S state transitions. While simple kinetic competition between forward and charge recombination reactions certainly also contribute to misses,35,36 we propose that the actual misses are mainly a consequence of “molecular miss” events at the water oxidation (donor) side of PSII that do not allow an S state dependent fraction of centers to advance to the next S state, thereby leading to reduction of
). During S state advancement there was thus no contribution from recombination reactions with the S2 or S3 states or of blocked centers to the miss parameter. Therefore, we can safely conclude that at our measuring conditions, at least at low flash frequencies and temperatures above 0 °C, the misses we observed in PSII membrane samples in the EPR and FIOPs measurements are actual misses originated from the molecular events in the WOC during S state transitions. While simple kinetic competition between forward and charge recombination reactions certainly also contribute to misses,35,36 we propose that the actual misses are mainly a consequence of “molecular miss” events at the water oxidation (donor) side of PSII that do not allow an S state dependent fraction of centers to advance to the next S state, thereby leading to reduction of 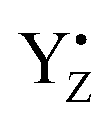 by QA− or QB−.
by QA− or QB−.
The highest miss was found in the S2 → S3 transition
Understanding nature of miss factor during S state transitions is important not only for elucidating the reaction mechanism of water oxidation, but also for analysis of spectroscopic, electronic and geometric structure of the WOC. Our current and previous EPR study40 and more recent investigation,55 where global fit analysis of the FIOPs was used and where life-times of the all metastable S states and redox states of QA, QB and YD were considered, showed that the misses during water oxidation are S state-dependent and the highest miss was found in the S2 → S3 transition, whereas the S3 → S0 transition, where O2 formation and release occur, showed a relatively low miss factor (Table 2, Fig. 3).However, these results are in contradiction with studies by de Wijn et al.25 and by Suzuki et al.43 On the basis of the fluorescence studies on the thylakoid membranes from spinach, de Wijn and van Gorkom reported a lack of misses in the S0 to S1 transition, whereas the largest miss was found for the S3 → S0 transition.25 Similarly, the largest miss in the S3 → S0 transition was also reported by Suzuki et al. after using FTIR difference spectroscopy in the PSII core complexes from Thermosynechococcus elongatus and PSII membranes from spinach.43 Both these studies relied on the acceptor side components for dissemination of the individual miss factors. In the first study the flash dependent Chl fluorescence transient yield, which depends on the redox state of QA was used as a probe to deconvolute the miss parameters. In the second study, the electron flow in PSII detected by monitoring the CN stretching bands of ferri/ferrocyanide (used as an exogenous electron acceptor), was used with the same purpose.
In contrast, our measurements only rely on the signals originated from the WOC. We directly measure the S state distribution after the flash by EPR spectroscopy. Each EPR signal we measured, originates from the CaMn4O5-cluster and is easily distinguishable from others (Fig. 2). Moreover, neither inactive PSII centers or acceptor side reactions or other components in PSII interfered with in our experiments. In addition, measurements at low temperature, where misses are amplified significantly, support our finding that the highest miss during the S cycle takes place during the S2 → S3 transition. These facts are now well-established and clearly observable (Table 2, Fig. 3).
WOC miss parameters vary with temperature
It is known that temperature has a profound effect on the S state transitions by tuning of electron and proton transfer.15,18,28,35,40,47,59 Our measurements show different temperature behavior of misses during these transitions (Table 2, Fig. 3). Two trends were observed. The S1 → S2 transition proceeds with a 100% efficiency at low temperatures but occurred with 10% misses at 20 °C, i.e. misses were increasing with increasing temperature (Table 2, Fig. 3). The miss parameter in the S0 → S1, S2 → S3 and S3 → S0 transitions shown the opposite effect, i.e. they decrease with increasing temperature. The only exception is S0 → S1 transitions which increased by 4% from 10 °C to 20 °C (Table 2, Fig. 3). Typical difference in the miss factor was about 10% in the investigated temperature range, except for the S2 → S3 which was more than twice higher.Our understanding is that these two trends reflect the different nature of molecular events during the S state cycle. The S1 → S2 transition is unique among the reactions in the S state cycle in many aspects. It is the only transition which involves only an electron transfer step, i.e. it is not coupled to a proton release from the WOC63,64 and is thus largely pH independent.37,39 It is also operational at much lower temperatures than all other transition.15 At our measured low temperatures (−10 °C to 10 °C), the S1 → S2 transition proceeds without any miss with transition efficiency of almost 100%. Only at higher temperatures misses started to occur. This reflects the fact that the WOC is unaltered and the surrounding protein requires only a small readjustment during this transition. We thus propose that the misses at 20 °C at least partially originate from the higher disorder of the protein matrix between the CaMn4O5-cluster and 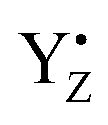 .
.
The opposite temperature dependence of misses in the other S state transitions reflects their more complicated chemical nature. They all include de-protonation events, show strong pH dependence in the physiological region and exhibit a much higher temperature limit.15 In addition, the S2 → S3 and S3 → [S4] → S0 transitions involve water binding events and the later one also the O2 formation and release event (Fig. 1). Decrease of miss factors with increased temperature during these transitions indicate that these events are promoted at high temperatures. We are addressing the molecular nature of these events in the section below.
Molecular origin of the miss factor at the WOC during the S cycle
Much spectroscopic, biophysical and biochemical information that has been accumulated over the years allow to certain extent understand the chemical nature of S state transitions and to pinpoint some possible missteps from which misses could be originated. We have attempted this exercise in our first publication.40 Much more changed since then. First of all, the first high resolution structure of PSII and the CaMn4O5-cluster became available.51 More importantly, with the development of the femtosecond XFEL methods it became possible to obtain structures of all four stable and metastable S states of the WOC.52 This allows us to discuss the origin of the misses based on the molecular structures and most feasible mechanism of water oxidation presently available.The electron transfer only event in the S1 → S2 transition implies no significant structural change in the CaMn4O5-cluster. This is corroborated by a number of EXAFS spectroscopy studies.47,65–69 More recent structural studies also show no fundamental change upon this transition.52 The CaMn4O5-cluster remained in the open, non-cubane geometry corresponding to the low spin configuration (Fig. 6).70,71 Change of Mn4(III) to Mn4(IV) oxidation state might induce small structural readjustments in the first coordination sphere, which in our opinion could contribute to the appearance of the miss at high temperature (20 °C). Otherwise, there is no chemical reason for miss in this transition.
 | ||
| Fig. 6 Possible changes in the structure of the CaMn4O5-cluster, e− transfer, H2O binding, H+ and O2 release during the S cycle. Sites of the miss origin during S1 → S2, S2 → S3, S3 → S0 and S0 → S1 transitions are correspondingly highlighted by blue, orange, yellow and green colored areas. | ||
The situation is dramatically changed during the S2 → S3 transition where we found the lowest transition efficiency at all temperatures. This transition is pH dependent and shows the highest deuterium isotope effect on the kinetics for electron transfer from  .31,72,73 Both effects reflect large proton movements around YZ and from the CaMn4O5-cluster to the bulk water. In addition, EXAFS studies have revealed major structural rearrangements in the CaMn4O5-cluster involving a shift in coordination number for one of the Mn atoms.47,65–69,74 This was recently confirmed by XFEL experiments that show that a new oxygen (Ox), likely from the Ca-bound water (W3), is inserted and forms an additional hydroxo-bridge between Ca and Mn1, which is oxidized from Mn1(III) to Mn1(IV) (Fig. 6). It is suggested that the insertion is accompanied by the removal of the first proton from W3 and binding of a new water to Ca to fill the empty W3 binding site.52,75 This well-orchestrated sequence of events requires a structurally well-defined hydrogen and/or water network around the CaMn4O5-cluster, especially surrounding the Ca, YZ and Mn1 site (the O1 channel52,75,76) and immediate protein ligands. This correlates with changes in the Mn1 coordination and distances to Mn3 and Mn4. Even a small hindrance of any of these many steps during the S2 → S3 transition will results in either complete failure or at least slowness of the transition and as a result in an increased miss parameter (charge recombination of
.31,72,73 Both effects reflect large proton movements around YZ and from the CaMn4O5-cluster to the bulk water. In addition, EXAFS studies have revealed major structural rearrangements in the CaMn4O5-cluster involving a shift in coordination number for one of the Mn atoms.47,65–69,74 This was recently confirmed by XFEL experiments that show that a new oxygen (Ox), likely from the Ca-bound water (W3), is inserted and forms an additional hydroxo-bridge between Ca and Mn1, which is oxidized from Mn1(III) to Mn1(IV) (Fig. 6). It is suggested that the insertion is accompanied by the removal of the first proton from W3 and binding of a new water to Ca to fill the empty W3 binding site.52,75 This well-orchestrated sequence of events requires a structurally well-defined hydrogen and/or water network around the CaMn4O5-cluster, especially surrounding the Ca, YZ and Mn1 site (the O1 channel52,75,76) and immediate protein ligands. This correlates with changes in the Mn1 coordination and distances to Mn3 and Mn4. Even a small hindrance of any of these many steps during the S2 → S3 transition will results in either complete failure or at least slowness of the transition and as a result in an increased miss parameter (charge recombination of 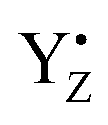 with acceptor side). Our results clearly suggest that this complicated chemistry is reflected in a lowered transition efficiency.
with acceptor side). Our results clearly suggest that this complicated chemistry is reflected in a lowered transition efficiency.
It must be mentioned that alternative conformations of the S2 state and possibly S3 state were proposed to exist and are manifested, for example, by the high spin EPR signal in the S2 state. This structure has been proposed to represent a closed cubane in contrast to the low spin open cubane structure shown in Fig. 6. In an alternative scenario for water insertion during the S2 → S3 transition, the so-called pivot or carousel pathways, the dominant, low-spin conformation of the S2 state, often referred to S2A, needs to convert after YZ oxidation into the closed cube, high-spin S2B conformation, in which Mn4 attains the Mn(III) oxidation state instead of Mn1, before water can be inserted at Mn4(III) and the S3 state is reached by 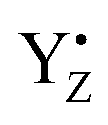 reduction.71,77–79 This scenario involves even more steps and sensitive equilibria, fully consistent with our finding that the S2 → S3 transition, which has a low-driving force, has the highest miss. Both conformations were proposed to be interconvertible and energetically similar (discussed in ref. 78).
reduction.71,77–79 This scenario involves even more steps and sensitive equilibria, fully consistent with our finding that the S2 → S3 transition, which has a low-driving force, has the highest miss. Both conformations were proposed to be interconvertible and energetically similar (discussed in ref. 78).
Very large chemical changes and protein structural rearrangements take place during the S3 → [S4] → S0 transition, involving the final step in the water oxidation cycle – the formation and release of O2 molecule. This final step in water oxidation is pH dependent,39,43 involves two deprotonation steps63,64 and binding of the second water molecule in the cycle (Fig. 6). EXAFS studies have also revealed that the coordination chemistry around the Mn1 atom that was altered in the S2 → S3 transition is returned to its original state.47,65–69
These data correlate with the latest structural study.52 According to one possibility,87 O5 and Ox form dioxygen and concomitantly with O2 release the new water molecule binds under deprotonation so that it binds as hydroxo at the O5 binding site. Thereby, the CaMn4O5-cluster returns to the open cubane configuration and attains its lowest valence state (Fig. 6). We observed a quite low transition efficiency at −10 °C, but not at higher temperatures in this transition (Fig. 3). Our understanding is that this final step in water oxidation to molecular oxygen (O2) has a strong driving force after a critical, highly reactive intermediate (S4) is formed from  .80,81 Thus, O2 release and the binding of the second water likely do not contribute to the miss under normal conditions. However, the formation of the S4 state involves a deprotonation of the catalytic site, possibly of Ox, and the oxidation of the Mn4CaO5-cluster, to form either a Mn-oxyl radical or a Mn(V)-oxo.82–84 Furthermore, additional structural changes cannot be excluded. Thus, all molecular misses connected to the S3 → S4 → S0 transition are expected to come from these initial transformations leading to the formation of the S4 state (Fig. 6).
.80,81 Thus, O2 release and the binding of the second water likely do not contribute to the miss under normal conditions. However, the formation of the S4 state involves a deprotonation of the catalytic site, possibly of Ox, and the oxidation of the Mn4CaO5-cluster, to form either a Mn-oxyl radical or a Mn(V)-oxo.82–84 Furthermore, additional structural changes cannot be excluded. Thus, all molecular misses connected to the S3 → S4 → S0 transition are expected to come from these initial transformations leading to the formation of the S4 state (Fig. 6).
We also observed relatively low miss parameter in the S0 → S1 transition, especially at −10 °C (Fig. 3). The S0 → S1 transition is pH dependent39,85 and involves the release of one proton.37,64 The structural changes at the CaMn4O5-cluster, however, are not large and are considered to involve deprotonation of a μ-oxo-bridge between two of the Mn atoms64,66 and it is reasonable to suggest that it takes place at the O5 position (Fig. 6). This deprotonation event is the one that governs misses during this transition and is different from the change in Mn-coordination and water binding occurring in the previous transitions thus, resulting in the altered temperature dependence.
Conclusions
In this work we correlate the actual misses or transition efficiencies in the S state cycle with different degrees of failure of the specific molecular reactions during the WOC advancement. The absence of misses (100% transition efficiency) during the S1 → S2 transition reflects the only electron transfer event at the Mn4. The transition efficiency during the S2 → S3 transition and its strong temperature dependence reflect deprotonation steps, water binding and insertion of new hydroxo resulting in significant structural rearrangements at the CaMn4O5-cluster that need to be performed with a low driving force. The intermediate transition efficiency in the S0 → S1 and S3 → S0 transitions and their slight temperature dependence instead reflects that these are governed by electron and proton transfer steps that involve only minor structural changes at the WOC. Details of these reactions are depicted in Fig. 6.Data availability
The data supporting this article are available in the ESI.†Author contributions
F. M. and S. S. conceived and designed experiments. G. H., P. C., and M. F. performed experiments. G. H., P. C., S. S., J. M. and F. M. analyzed data. F. M. and J. M. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Johannes Sjöholm for help in some measurements. Financial support for this study was provided by the Swedish Research Council (2020-03809) and NordForsk (82845).References
- D. E. Canfield, Annu. Rev. Earth Planet. Sci., 2005, 33, 1–36 CrossRef CAS.
- B. A. Diner and F. Rappaport, Annu. Rev. Plant Biol., 2002, 53, 551–580 CrossRef CAS PubMed.
- G. Renger, in Primary processes of photosynthesis: Principles and apparatus, ed. G. Renger, RSC Publihing, 2008, vol. 2, pp. 237–290 Search PubMed.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- D. Shevela, J. F. Kern, G. Govindjee, J. Whitmarsh and J. Messinger, Encyclopedia of Life Sciences, 2021, vol. 2, pp. 1–20 Search PubMed.
- T. Cardona, A. Sedoud, N. Cox and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 26–43 CrossRef CAS PubMed.
- F. Müh, C. Glockner, J. Hellmich and A. Zouni, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 44–65 CrossRef PubMed.
- G. Renger and T. Renger, Photosynth. Res., 2008, 98, 53–80 CrossRef CAS PubMed.
- J. Messinger and G. Renger, in Primary processes of photosynthesis: Principles and apparatus, ed. G. Renger, RSC Publihing, 2008, vol. 2, ch. 17. Photosynthetic water splitting, pp. 291–349 Search PubMed.
- G. Renger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1164–1176 CrossRef CAS PubMed.
- N. Cox, D. A. Pantazis and W. Lubitz, in Annual Review of Biochemistry, ed. R. D. Kornberg, 2020, vol. 89, pp. 795–820 Search PubMed.
- P. Joliot, G. Barbieri and R. Chabaud, Photochem. Photobiol., 1969, 10, 309–331X CrossRef CAS.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed.
- B. Forbush, B. Kok and M. P. McGloin, Photochem. Photobiol., 1971, 14, 307–331X CrossRef CAS.
- S. Styring and A. W. Rutherford, Biochim. Biophys. Acta, Bioenerg., 1988, 933, 378–387 CrossRef CAS.
- G. Y. Chen, G. Y. Han, E. Goransson, F. Mamedov and S. Styring, Biochemistry, 2012, 51, 138–148 CrossRef CAS PubMed.
- J. Messinger and G. Renger, Biochemistry, 1994, 33, 10896–10905 CrossRef CAS PubMed.
- S. Isgandarova, G. Renger and J. Messinger, Biochemistry, 2003, 42, 8929–8938 CrossRef CAS PubMed.
- S. Styring and A. W. Rutherford, Biochemistry, 1987, 26, 2401–2405 CrossRef CAS.
- J. Messinger and G. Renger, Biochemistry, 1993, 32, 9379–9386 CrossRef CAS PubMed.
- G. Y. Han, F. M. Ho, K. G. V. Havelius, S. F. Morvaridi, F. Mamedov and S. Styring, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 496–503 CrossRef CAS PubMed.
- J. Lavorel, J. Theor. Biol., 1976, 57, 171–185 CrossRef CAS PubMed.
- P. C. Meunier, Photosynth. Res., 1993, 36, 111–118 CrossRef CAS PubMed.
- V. P. Shinkarev, Biophys. J., 2005, 88, 412–421 CrossRef CAS PubMed.
- R. de Wijn and H. J. van Gorkom, Photosynth. Res., 2002, 72, 217–222 CrossRef CAS PubMed.
- N. K. Packham, M. Hodges, A. L. Etienne and J. M. Briantais, Photosynth. Res., 1988, 15, 221–232 CrossRef CAS PubMed.
- P. C. Meunier, R. L. Burnap and L. A. Sherman, Photosynth. Res., 1996, 47, 61–76 CrossRef CAS PubMed.
- J. Messinger, W. P. Schröder and G. Renger, Biochemistry, 1993, 32, 7658–7668 CrossRef CAS PubMed.
- L. V. Pham and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 848–859 CrossRef CAS PubMed.
- G. Christen, F. Reifarth and G. Renger, FEBS Lett., 1998, 429, 49–52 CrossRef CAS PubMed.
- G. Christen, A. Seeliger and G. Renger, Biochemistry, 1999, 38, 6082–6092 CrossRef CAS PubMed.
- J. Clausen, W. Junge, H. Dau and M. Haumann, Biochemistry, 2005, 44, 12775–12779 CrossRef CAS PubMed.
- P. J. Nixon, J. T. Trost and B. A. Diner, Biochemistry, 1992, 31, 10859–10871 CrossRef CAS PubMed.
- G. Ananyev, T. Nguyen, C. Putnam-Evans and G. C. Dismukes, Photochem. Photobiol. Sci., 2005, 4, 991–998 CrossRef CAS PubMed.
- M. Grabolle and H. Dau, Physiol. Plant., 2007, 131, 50–63 CrossRef CAS PubMed.
- I. Zaharieva, J. M. Wichmann and H. Dau, J. Biol. Chem., 2011, 286, 18222–18228 CrossRef CAS PubMed.
- O. Saygin and H. T. Witt, Biochim. Biophys. Acta, Bioenerg., 1987, 893, 452–469 CrossRef CAS.
- K. A. Ahrling, S. Peterson and S. Styring, Biochemistry, 1997, 36, 13148–13152 CrossRef CAS PubMed.
- G. Bernat, F. Morvaridi, Y. Feyziyev and S. Styring, Biochemistry, 2002, 41, 5830–5843 CrossRef CAS PubMed.
- G. Y. Han, F. Mamedov and S. Styring, J. Biol. Chem., 2012, 287, 13422–13429 CrossRef CAS PubMed.
- R. G. Evelo, S. Styring, A. W. Rutherford and A. J. Hoff, Biochim. Biophys. Acta, Bioenerg., 1989, 973, 428–442 CrossRef CAS.
- J. Messinger, J. H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R. M. Cinco, K. L. McFarlane, E. Bellacchio, S. A. Pizarro, S. P. Cramer, K. Sauer, M. P. Klein and V. K. Yachandra, J. Am. Chem. Soc., 2001, 123, 7804–7820 CrossRef CAS PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2012, 51, 6776–6785 CrossRef CAS PubMed.
- W. Hillier and G. T. Babcock, Biochemistry, 2001, 40, 1503–1509 CrossRef CAS PubMed.
- Y. Kato, S. Haniu, Y. Nakajima, F. Akita, J. R. Shen and T. Noguchi, J. Phys. Chem. B, 2020, 124, 121–127 CrossRef CAS PubMed.
- T. A. Roelofs, W. C. Liang, M. J. Latimer, R. M. Cinco, A. Rompel, J. C. Andrews, K. Sauer, V. K. Yachandra and M. P. Klein, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 3335–3340 CrossRef CAS PubMed.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef CAS PubMed.
- L. Iuzzolino, J. Dittmer, W. Dorner, W. Meyer-Klaucke and H. Dau, Biochemistry, 1998, 37, 17112–17119 CrossRef CAS PubMed.
- Y. L. Pushkar, J. Yano, K. Sauer, A. Boussac and V. K. Yachandra, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1879–1884 CrossRef CAS PubMed.
- T. Ono, T. Noguchi, Y. Inoue, M. Kusunoki, T. Matsushita and H. Oyanagi, Science, 1992, 258, 1335–1337 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–65 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. N. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Nature, 2018, 563, 421–443X CrossRef CAS PubMed.
- R. de Wijn and H. J. van Gorkom, Biochim. Biophys. Acta, Bioenerg., 2002, 1553, 302–308 CrossRef CAS.
- H. Conjeaud and P. Mathis, Biochim. Biophys. Acta, Bioenerg., 1980, 590, 353–359 CrossRef CAS.
- L. V. Pham, J. D. J. Olmos, P. Chernev, J. Kargul and J. Messinger, Photosynth. Res., 2019, 139, 93–106 CrossRef CAS PubMed.
- N. Ahmadova, F. M. Ho, S. Styring and F. Mamedov, Biochim. Biophys. Acta, Bioenerg., 2017, 1858, 407–417 CrossRef CAS PubMed.
- C. A. Buser, L. K. Thompson, B. A. Diner and G. W. Brudvig, Biochemistry, 1990, 29, 8977–8985 CrossRef CAS PubMed.
- I. Vass and S. Styring, Biochemistry, 1991, 30, 830–839 CrossRef CAS PubMed.
- P. Kuhn, H. Eckert, H. J. Eichler and G. Renger, Phys. Chem. Chem. Phys., 2004, 6, 4838–4843 RSC.
- F. Mamedov, R. T. Sayre and S. Styring, Biochemistry, 1998, 37, 14245–14256 CrossRef CAS PubMed.
- G. Ananyev and G. C. Dismukes, Photosynth. Res., 2005, 84, 355–365 CrossRef CAS PubMed.
- D. Shevela and J. Messinger, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 1208–1212 CrossRef CAS PubMed.
- E. Schlodder and H. T. Witt, J. Biol. Chem., 1999, 274, 30387–30392 CrossRef CAS PubMed.
- H. Dau and M. Haumann, Coord. Chem. Rev., 2008, 252, 273–295 CrossRef CAS.
- V. J. Derose, I. Mukerji, M. J. Latimer, V. K. Yachandra, K. Sauer and M. P. Klein, J. Am. Chem. Soc., 1994, 116, 5239–5249 CrossRef CAS.
- J. H. Robblee, R. M. Cinco and V. K. Yachandra, Biochim. Biophys. Acta, Bioenerg., 2001, 1503, 7–23 CrossRef CAS.
- M. Haumann, M. Grabolle, T. Neisius and H. Dau, FEBS Lett., 2002, 512, 116–120 CrossRef CAS PubMed.
- J. Yano, Y. Pushkar, P. Glatzel, A. Lewis, K. Sauer, J. Messinger, U. Bergmann and V. Yachandra, J. Am. Chem. Soc., 2005, 127, 14974–14975 CrossRef CAS PubMed.
- H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562–583 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Inorg. Chem., 2016, 55, 488–501 CrossRef CAS PubMed.
- D. Bovi, D. Narzi and L. Guidoni, Angew. Chem., Int. Ed., 2013, 52, 11744–11749 CrossRef CAS PubMed.
- M. J. Schilstra, F. Rappaport, J. H. A. Nugent, C. J. Barnett and D. R. Klug, Biochemistry, 1998, 37, 3974–3981 CrossRef CAS PubMed.
- G. Christen and G. Renger, Biochemistry, 1999, 38, 2068–2077 CrossRef CAS PubMed.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, Chem. Sci., 2016, 7, 72–84 RSC.
- M. Suga, F. Akita, K. Yamashita, Y. Nakajima, G. Ueno, H. J. Li, T. Yamane, K. Hirata, Y. Umena, S. Yonekura, L. J. Yu, H. Murakami, T. Nomura, T. Kimura, M. Kubo, S. Baba, T. Kumasaka, K. Tono, M. Yabashi, H. Isobe, K. Yamaguchi, M. Yamamoto, H. Ago and J. R. Shen, Science, 2019, 366, 334–338 CrossRef CAS PubMed.
- M. Ibrahim, T. Fransson, R. Chatterjee, M. H. Cheah, R. Hussein, L. Lassalle, K. D. Sutherlin, I. D. Young, F. D. Fuller, S. Gul, I. S. Kim, P. S. Simon, C. de Lichtenberg, P. Chernev, I. Bogacz, C. C. Pham, A. M. Orville, N. Saichek, T. Northen, A. Batyuk, S. Carbajo, R. Alonso-Mori, K. Tono, S. Owada, A. Bhowmick, R. Bolotovsky, D. Mendez, N. W. Moriarty, J. M. Holton, H. Dobbek, A. S. Brewster, P. D. Adams, N. K. Sauter, U. Bergmann, A. Zouni, J. Messinger, J. Kern, V. K. Yachandra and J. Yano, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12624–12635 CrossRef CAS PubMed.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef CAS PubMed.
- C. de Lichtenberg and J. Messinger, Phys. Chem. Chem. Phys., 2020, 22, 12894–12908 RSC.
- M. Chrysina, E. Heyno, Y. Kutin, M. Reus, H. Nilsson, M. M. Nowaczyk, S. DeBeer, F. Neese, J. Messinger, W. Lubitz and N. Cox, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 16841–16846 CrossRef CAS PubMed.
- M. Haumann, A. Grundmeier, I. Zaharieva and H. Dau, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17384–17389 CrossRef CAS PubMed.
- D. Shevela, K. Beckmann, J. Clausen, W. Junge and J. Messinger, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3602–3607 CrossRef CAS PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
- P. L. Dilbeck, H. J. Hwang, I. Zaharieva, L. Gerencser, H. Dau and R. L. Burnap, Biochemistry, 2012, 51, 1079–1091 CrossRef CAS PubMed.
- D. J. Vinyard and G. W. Brudvig, in Annual Review of Physical Chemistry, ed. M. A. Johnson and T. J. Martinez, 2017, vol. 68, pp. 101–116 Search PubMed.
- H. Suzuki, M. Sugiura and T. Noguchi, Biochemistry, 2005, 44, 1708–1718 CrossRef CAS PubMed.
- M. Völker, T. Ono, Y. Inoue and G. Renger, Biochim. Biophys. Acta, Bioenerg., 1985, 806, 25–34 CrossRef.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00854h |
| ‡ Current address: Photosynthesis Research Center, Key Laboratory of Photobiology, Institute of Botany, Chinese Academy of Sciences, No. 20, Nanxincun, Xiangshan, Beijing, 100093, China. |
| This journal is © The Royal Society of Chemistry 2022 |