
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective and enantioselective photoredox pinacol coupling promoted by titanium complexes with a red-absorbing organic dye†
Francesco
Calogero‡
a,
Giandomenico
Magagnano‡
a,
Simone
Potenti‡
 ab,
Francesco
Pasca
a,
Andrea
Fermi
*ac,
Andrea
Gualandi
*ac,
Paola
Ceroni
ac,
Giacomo
Bergamini
a and
Pier Giorgio
Cozzi
*ac
ab,
Francesco
Pasca
a,
Andrea
Fermi
*ac,
Andrea
Gualandi
*ac,
Paola
Ceroni
ac,
Giacomo
Bergamini
a and
Pier Giorgio
Cozzi
*ac
aDipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: andrea.fermi2@unibo.it; andrea.gualandi10@unibo.it; piergiorgio.cozzi@unibo.it
bLaboratorio SMART, Scuola Normale Superiore, Piazza dei Cavalieri 7, 56126, Pisa, Italy
cCenter for Chemical Catalysis – C3, Alma Mater Studiorum – Università di Bologna, Via Selmi 2, 40126 Bologna, Italy
First published on 21st April 2022
Abstract
The pinacol coupling reaction, a reductive coupling of carbonyl compounds that proceeds through the formation of ketyl radicals in the presence of an electron donor, affords the corresponding 1,2-diols in one single step. The photoredox version of this transformation has been accomplished using different organic dyes or photoactive metal complexes in the presence of sacrificial donors such as tertiary amines or Hantzsch's ester. Normally, the homo-coupling of such reactive ketyl radicals is neither diastereo- nor enantio-selective. Herein, we report a highly diastereoselective pinacol coupling reaction of aromatic aldehydes promoted by 5 mol% of the non-toxic, inexpensive and available Cp2TiCl2 complex. The key feature that allows the complete control of diastereoselectivity is the employment of a red-absorbing organic dye in the presence of a redox-active titanium complex. Taking advantage of the well-tailored photoredox potential of this organic dye, the selective reduction of Ti(IV) to Ti(III) is achieved. These conditions enable the formation of the D,L (syn) diastereoisomer as the favored product of the pinacol coupling (d.r. > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in most of the cases). Moreover, employing a simply prepared chiral SalenTi complex, the new photoredox reaction gave a complete diastereoselection for the D,L diastereoisomer, and high enantiocontrol (up to 92% of enantiomeric excess).
:1 in most of the cases). Moreover, employing a simply prepared chiral SalenTi complex, the new photoredox reaction gave a complete diastereoselection for the D,L diastereoisomer, and high enantiocontrol (up to 92% of enantiomeric excess).
Introduction
Photoredox catalysis1 has become a powerful methodology to access radical intermediates.2 The ability to convert visible light into chemical energy, mainly by SET (single electron transfer) processes, enables the exploration of new challenging chemical transformations.3 Thus, avoiding the use of classical radical initiators or stoichiometric metal reagents, visible light-mediated photoredox catalysis can serve as a simple method to generate ketyl radicals, allowing interesting transformations and providing alternative methods to the traditional approaches for the generation of these reactive intermediates.4 The first example of generation of a ketyl radical by visible light was described in 1979 by Kellogg and co-workers, exploiting the ability of [Ru(bpy)3]Cl2 to act as a photocatalyst for the SET reduction and fragmentation of phenacyl sulfonium salts.5 After this report, different photoredox methodologies have been applied for the formation of new C–C bonds via the generation of ketyl radicals from aldehydes and ketones.6 A photocatalytic pinacol coupling was described by Rueping and co-workers.7 This simple and catalytic methodology provides access to symmetrical pinacol and diamine products with good functional group tolerance. This study attracted considerable interest in the chemistry community, and many different alternative procedures were reported. In particular, the use of [Ir(III)] and [Ru(II)] complexes,8 organic dyes,9 and semiconductors10 has been described in photoredox-mediated pinacol coupling. However, the redox potential of the photocatalyst at the excited state is often not sufficient for the direct formation of aryl ketyl radicals.11 Key studies reported by Rueping and other authors have clarified that tributylamine [(n-Bu)3N], the sacrificial stoichiometric reductant, plays another important role. The amine radical cation – formed upon reductive quenching of the photocatalyst by amine – activates the carbonyl group lowering its reduction potential, thus leading to the formation of the ketyl radical. The ketyl radicals dimerize and, after protonation, the 1,2-diol products are obtained. In almost all the described catalytic processes, the promptness of the radical–radical dimerization step is responsible for the formation of a diastereoisomeric mixture of diols, generally in 1:1 ratio (D,L:meso), and the observed diastereoselection is generally low (d.r. up to 1.4:1).
We have hypothesized that the generation of [Ti(III)] complexes under photoredox conditions could be a straightforward method to perform the pinacol coupling with high diastereocontrol, as was reported by Gansäuer more than 20 years ago, employing manganese as the stoichiometric reductant, and Me3SiCl as the scavenger.12 Recently, Gansäuer,13 our group,14 and Shi15 have reported the generation of [Ti(III)] under photoredox conditions for radical chemistry (allylation reactions,14a,15b opening of epoxides,13,15a propargylations14b).16 Unfortunately, any attempt to use organic dyes,17 as well as [Ru(II)] or [Ir(III)] photocatalysts, with [Ti(IV)] complexes, in the presence of Hantzsch's ester or amine-based reductants, gave almost a 1:1 ratio of the diastereomeric diols in a model reaction with p-chlorobenzaldehyde. This was probably due to a fast and effective reaction performed by the photoredox catalysts, with no participation of the titanium complex. In addition, Hantzsch's ester, used in stoichiometric amount in our attempts, displays a non-negligible absorption feature at 400 nm and it is a strong reductant in its excited state.18 In order to avoid the direct formation of ketyl radicals due to the photocatalyst or Hantzsch's ester, a milder photochemical system is needed. This photocatalyst needs to show the capability of reducing titanium, either in its excited state (via a direct oxidative quenching with titanium), or in its reduced form (upon reductive quenching in the presence of a sacrificial reductant). In addition, titanium complexes used in photoredox reactions can absorb visible light themselves, acting as the active photocatalysts as illustrated by Gansäuer.13b
Based on all these limitations, we decided to use a red-absorbing dye, so that the photocatalyst can be selectively excited without concomitant absorption of light by titanium complexes and Hantzsch's ester. Excitation in the low-energy portion of the visible spectrum (orange and red) brings the advantages of decreased harmfulness and high penetration depth through various media, as evidenced by Rovis, who employed red-absorbing osmium complexes to perform metallaphotoredox catalysis.19 However, due to the potential toxicity of osmium and the difficulties related to the preparation of the complexes, we investigated the use of an organic dye for this metallaphotoredox chemistry.20 Among all organic dyes with the capability of absorbing red light, dimethoxyquinacridinium (DMQA+), a helical carbenium ion, presents interesting photophysical21 and electrochemical22 properties. Lacour et al. used N,N′-dipropyl-1,13-dimethoxyquinacridinium tetrafluoroborate ([nPr-DMQA]+[BF4]−) in aerobic photooxidation of benzylamine.23 More recently, the use of [nPr-DMQA]+[BF4]− as a new organic photoredox catalyst for photoreduction and photooxidation upon excitation with red light (λmax = 640 nm) with broader applications has been reported.24 The use of this dye in metallaphotoredox catalysis, combining a photoredox catalytic cycle with metal catalysts such as gold and palladium complexes, has also been reported (Fig. 1A).24a The organic dye was prepared in a few steps, and it was robust, and could be isolated by simple recrystallization. In addition, the redox potential measured for the dye is potentially suitable for the reduction of [Ti(IV)] complexes (Fig. 1).25 On these bases, we decided to use ([nPr-DMQA]+[BF4]−) in photocatalytic titanium mediated processes, and herein, we illustrate the implementation of our idea in a highly diastereoselective (>20:1 D,L:meso) pinacol coupling promoted by 5 mol% of the available Cp2TiCl2. Moreover, the application of a simply prepared and available chiral titanium Salen complex gave, in the presence of several aromatic aldehydes, satisfactory yields of the desired pinacol product with a complete diastereoselection in favour of the D,L (syn) diastereoisomer, and high enantiomeric excess (up to 92%).
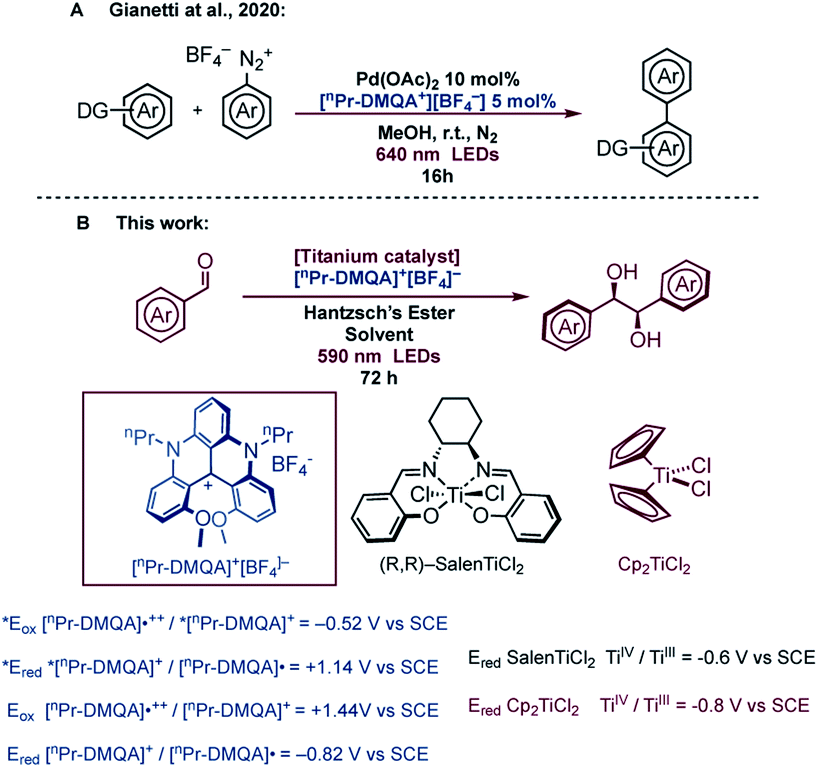 | ||
| Fig. 1 (A) Use of organic red-absorbing dye [nPr-DMQA]+[BF4]− in palldium mediated metallaphotoredox catalysis; (B) this work: photoredox pinacol coupling promoted by titanium complexes with a red-absorbing organic dye. | ||
Results and discussion
In order to study all the parameters for the reaction, we chose p-chlorobenzaldehyde (1a) as the model substrate. The reaction was optimized by varying conditions and in Table 1 we report the salient features about the optimization process.|
|
|||
|---|---|---|---|
| Entrya | Deviation from standard conditions | Yieldsb | d.r. (D,L:meso)c |
| a Reactions performed on a 0.1 mmol scale. b Determined by 1H NMR analysis using the internal standard method. c Determined by 1H NMR analysis of reaction crude. d Reaction performed on a 0.2 mmol scale; the value in parentheses is the isolated yield after chromatographic purification; 4+ was recovered (>90%) after chromatographic purification. e Irradiation at 456 nm. f Reaction time 72 h. | |||
| 1 | None | >99(89) |
>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 2 | No photocatalyst | No reaction | — |
| 3 | No 3 | Traces | 1:1 |
| 4 | No light | No reaction | — |
| 5 | 7 instead of 4+ | >99 | 1:1 |
| 6 | 8 instead of 4+ | >99 | 1:1 |
| 7 | 9 instead of 4+ | >99 | 1:1 |
| 8 | 10 instead of 4+ | >99 | 1:1 |
| 9 | 11 instead of 4+ | No reaction | — |
| 10 | 12 instead of 4+ | No reaction | — |
| 11 | 7 Instead of 4+ and No 3 | >99 | 1:1 |
| 12 , | No photocatalyst, No 3 | 50 | 1:1 |
| 13 | Toluene instead of PhCF3 | >99 | 7:1 |
| 14 | Benzene instead of PhCF3 | No reaction | — |
| 15 | DMF instead of PhCF3 | 53 | 1:2 |
| 16 | MeCN instead of PhCF3 | Traces | — |
| 17 | PhCN instead of PhCF3 | Traces | — |
| 18 | PhF instead of PhCF3 | 23 | 3:1 |
| 19 | THF instead of PhCF3 | Traces | — |
| 20 | 5 Instead of 3 | No reaction | — |
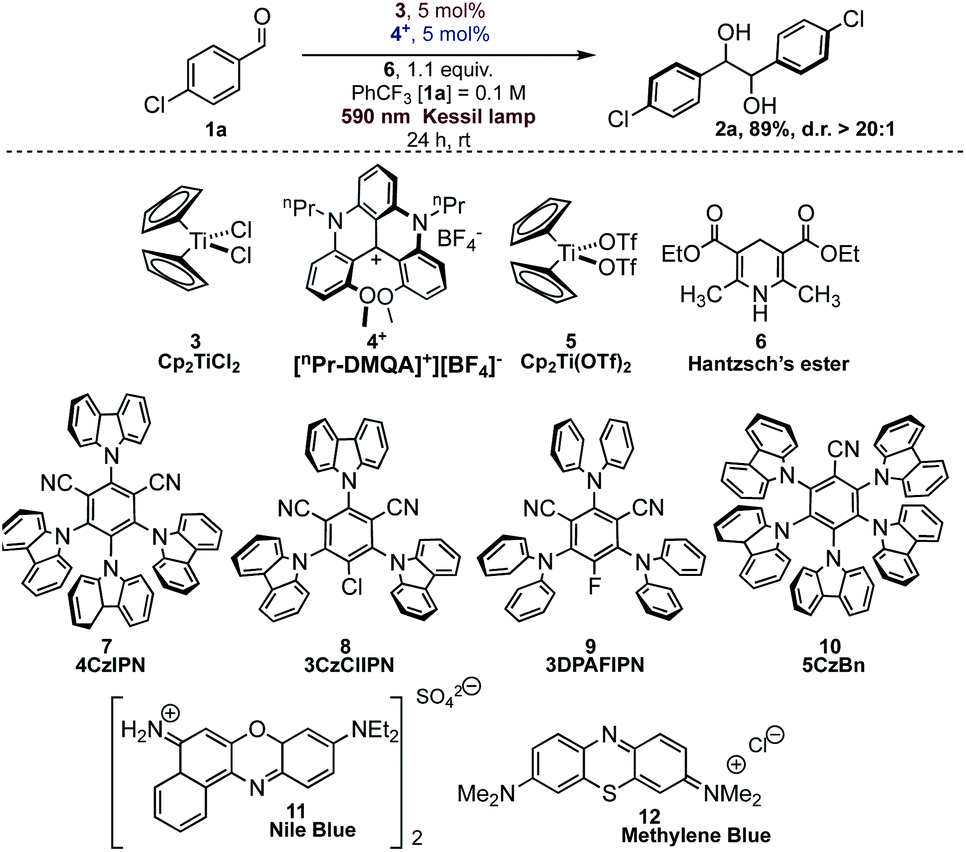
Hantzsch's ester 6 (HE), photocatalyst [nPr-DMQA]+[BF4]−4+ and Cp2TiCl23 gave the best results in trifluorotoluene as solvent under orange light irradiation (Table 1, entry 1). The concurrent presence of the photocatalyst, the titanium complex, and light is mandatory for the successful outcome of the reaction (Table 1, entries 2–4). As we have already mentioned, the reaction promoted by different organic dyes such as 7–10 gave complete conversion but with complete lack of diastereocontrol, leading to a 1:1 diastereoisomeric mixture of products (Table 1, entries 5–8). Other commercially available organic dyes (11 and 12, Table 1, entries 9 and 10), absorbing in the red region of the visible spectrum, were tested under the reaction conditions and no conversion of aldehyde 1a was observed. The reaction with dye 7 under blue light irradiation proceeds smoothly both in the presence and in the absence of the titanium complex (Table 1, entries 5 and 11), likely due to the direct formation, via SET from the photocatalyst, of the ketyl radical on the activated carbonyl substrate (see a detailed discussion below).26 Furthermore, Hantzsch's ester itself is capable of promoting the pinacol coupling reaction under blue light irradiation (Table 1, entry 12). The success of the reaction with dye 4+ and orange light irradiation is also highly dependent on the solvent (Table 1, entries 13–19).
While THF or MeCN gave only traces of the desired product, the reaction proceeded with good diastereoselection in aromatic solvents (benzene excluded). Among all the aromatic solvents, yield and diastereoselectivity were optimal in trifluorotoluene due to its documented inertness towards free-radical intermediates.27 When these reactions were conducted in more conventional solvents, such as MeCN, benzene, or THF, yields are diminished, or no reaction is observed.
No formation of the pinacol product was observed for the titanium complex 5, suggesting that the presence of stronger O-based ligands in place of weakly bound chloride is detrimental for the titanium complex reactivity (Table 1, entry 20).
The reaction conditions were found to be suitable with a variety of aromatic aldehydes (Scheme 1) but a prolonged reaction time of 72 h was necessary for full conversion of the substrates. Some limitations of the methodology were observed with heterocyclic aldehydes, due to reaction conditions (formation of acidic oxidized Hantzsch's ester) or inherent reduced reactivity, while ketones or aliphatic aldehydes were found to be unreactive (see Scheme 1 and Fig. S6† for details).
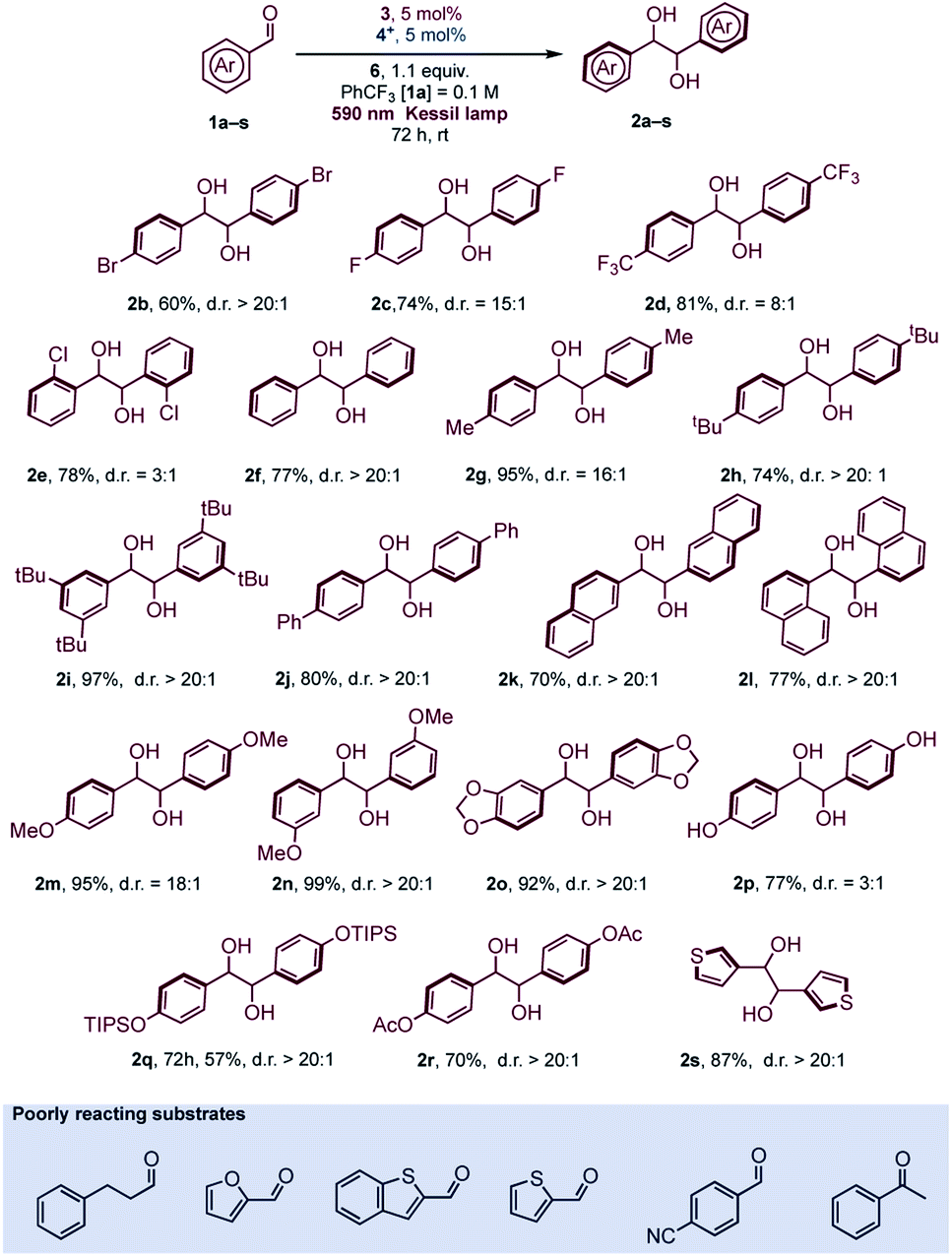 | ||
| Scheme 1 Scope of the diastereoselective pinacol coupling promoted by Cp2TiCl2. | ||
In general, aromatic aldehydes bearing alkoxy groups (R = alkyl, Scheme 1) at the para position were found to be more reactive and the products were isolated in high yields. This behavior was also observed with moderate activating groups such as a methyl substituent. For aromatic aldehydes, the protection of the oxygen at the para position is not required since aldehyde 1p was found to be reactive under the reaction conditions. Introduction of hindered substituent (1q) or electron withdrawing substituent (1r) onto the para OH group was found to reduce the reactivity. In any case, the diastereoselective outcome with these substrates was always excellent. Naphthyl aldehydes (α 1l and β 1k) were both reactive substrates, and, as reported in various studies with titanium mediated pinacol coupling,28 no hindrance was observed with these two substrates. Some aldehydes bearing electron withdrawing groups gave satisfactory yields but a slight drop of diastereoselectivity was observed. This was particularly evident with ortho-chlorobenzaldehyde (1e), probably for a concomitant sterical hindrance. Limitations due to the presence of bulky groups close to carbonyl were also observed in Ti-mediated pinacol coupling with overstoichiometric Zn as sacrificial reductant.28a
Enantioselective variant
Different examples of enantioselective catalytic pinacol coupling of aldehydes were reported in the literature, based on the pioneering work developed by Gansäuer.12 Although chiral titanium cyclopentadienyl complexes were employed in enantioselective pinacol couplings with moderate results,29 titanium-Schiff base complexes30 were found to be quite suitable promoters for catalytic titanium-mediated pinacol coupling reactions,31 and good scope and high enantioselectivity were obtained using different types of these complexes.32Riant32a and Joshi28a have reported interesting pinacol coupling protocols that gave high enantioselectivities using a titanium-Schiff base complex. Joshi published a particularly noteworthy study, featuring the use of an unsubstituted chiral SalenTiCl2 complex (13, Table 2), simply prepared from Ti(OiPr)4 and the bis-imine ligand by treatment with TMSCl. The titanium complex was converted in situ into the active Ti(III) species, through reduction with a stoichiometric amount of Zn. As well evidenced by Gansäuer, the presence of TMSCl in the reaction mixture was also crucial in order to close the catalytic cycle, upon pinacol silylation and concomitant liberation of Ti(IV) for a successive reduction. Recently, Ti(Salen) was found to be active in other radical mediated processes.33
|
|
||||
|---|---|---|---|---|
| Entrya | Deviation from standard conditions | Yieldsb | d.r. (D,L:meso)c |
e.r.d (R,R/S,S) |
| a Reactions performed on a 0.1 mmol scale. b Determined by 1H NMR analysis using the internal standard method. c Determined by 1H NMR analysis of reaction crude. d Determined by HPLC analysis on a column with a chiral stationary phase. e Reaction performed on a 0.2 mmol scale. The value in parentheses is the isolated yield after chromatographic purification. f 5 mol% of titanium catalyst was used in the reaction. g Reaction time 16 h. | ||||
| 1 | None | 74(67)e |
>20:1
|
96:4
|
| 2 | 14 instead 13, [1a] 0.1 M | Traces | — | — |
| 3 | 15 instead 13, [1a] 0.1 M | n.r. | 1:1 |
— |
| 4 | No photocatalyst | n.r. | — | — |
| 5 | No titanium | Traces | 1:1 |
50:50 |
| 6 | No light | n.r. | 1:1 |
— |
| 7 | 10 mol% of (S,S) 11 | 49 | 1:1 |
3:97 |
| 8 | Toluene instead of PhCF3 | Traces | 1:1 |
— |
| 9 | MeCN instead of PhCF3 | n.r. | 1:1 |
— |
| 10 | Benzene instead of PhCF3 | n.r. | 1:1 |
— |
| 11 | THF instead of PhCF3 | Traces | 7:1 |
— |
| 12 | DME instead of PhCF3 | Traces | — | — |
| 13 | 25 °C instead 6–12 °C | 92 | 9:1 |
64.5:35.5 |
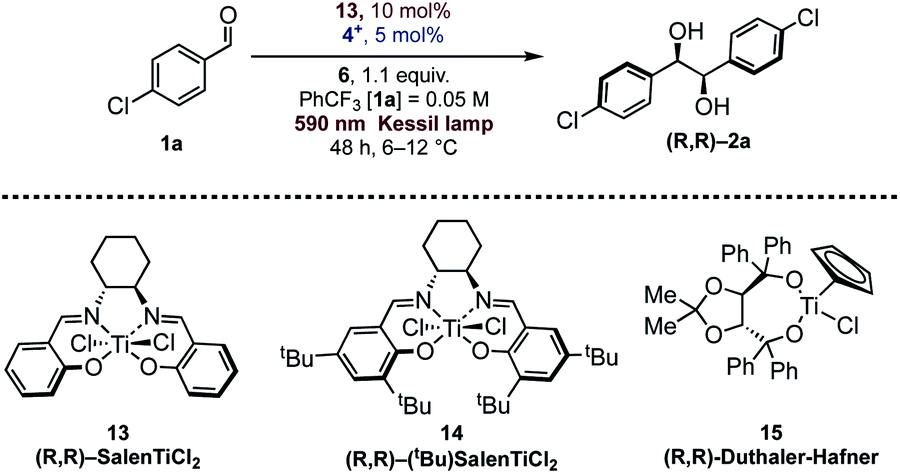
Since Schiff bases are modular and simply prepared from commercially available diamines, and both complexation and isolation of titanium complexes are quite straightforward, we selected two chiral Salen complexes 13 and 14 for a detailed investigation of our photoredox pinacol coupling. We were pleased to find promising results with 13, while 14 and 15 were found to be unreactive (Table 2, entries 1–3). The conditions were optimized by varying the reaction parameters, as illustrated in Table 2. The presence of light, the titanium complex, and the photocatalyst is crucial for the reaction (Table 2, entries 4–6). It is worth mentioning that the reaction proceeds smoothly in trifluorotoluene, while other solvents are ineffective (Table 2, entries 8–12). The stereoselectivity depends on the temperature, whose optimal value was found to be close to 10 °C (Table 2, entry 13). With these indications, a series of aromatic aldehydes were subjected to our reaction, and we were pleased to observe good results, reported in Scheme 2. In general, the reaction was found to be effective with unhindered aldehydes. Electronic effects seem not to play a relevant role in the reactions, contrary to Ti(Salen)/Zn mediated reactions.28a Sterical effects, in contrast, are detrimental for the enantioselectivity, as ortho substituted aromatic aldehydes were found to be less reactive. Hindrance in the para position lowers the stereoselectivity of the process. Compared to Cp2TiCl2, the SalenTiCl2 mediated protocol was found to feature a slightly reduced reactivity, probably due to the scarce solubility of the complex in the reaction solvent.
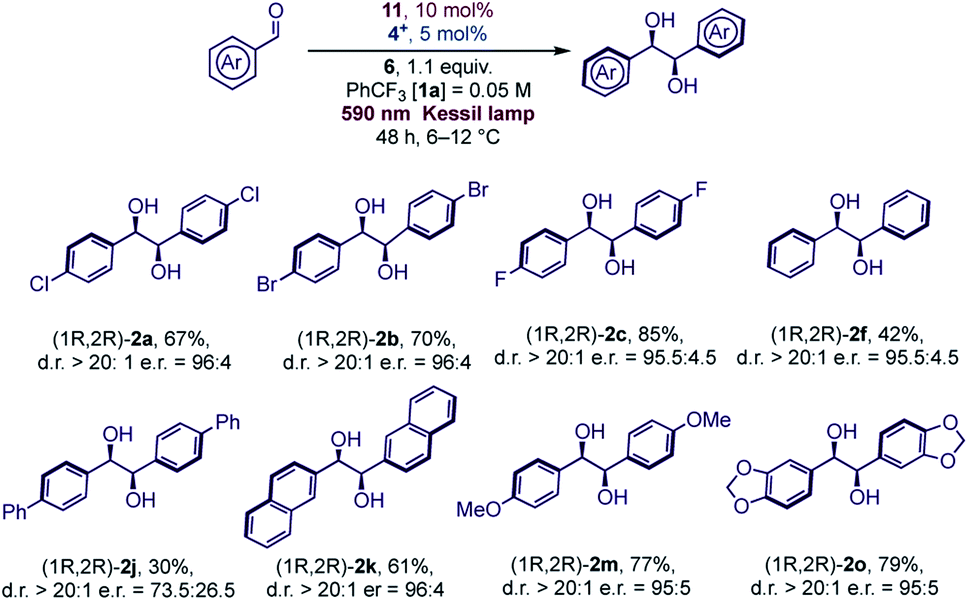 | ||
| Scheme 2 Scope of the enantioselective photoredox pinacol coupling with (R,R)-SalenTiCl2. | ||
Photophysical and mechanistic analyses
To shed light on the reaction mechanism, as for the synthetic protocol, PhCF3 was chosen as the main solvent for the photophysical characterization of photocatalyst 4+. In agreement with published results,24a compound 4+ shows a low energy-lying absorption band with the maximum at 614 nm (ε614 nm = 10300 M−1 cm−1 in CH2Cl2, λonset ∼ 680 nm; see Fig. 2A and S8†) allowing its excitation with orange light (LED source at λmax = 590 nm, see Fig. S1†). The emission band of 4+ is peaked at 648 nm – with a lifetime of 12.8 ns and a quantum yield of 0.26 in air-equilibrated PhCF3 solution – corresponding to a spectroscopic energy E0–0 of 1.96 eV. The emission decay is not dramatically affected by the presence of molecular oxygen, as expected for fluorescent dyes: a lifetime of 13.8 ns is determined in oxygen-free solutions (Fig. 2B). No phosphorescence has been detected in the visible and NIR spectral regions from deoxygenated PhCF3 solutions at r.t. or in a rigid matrix at 77 K in diethylether:ethanol:PhCF3 mixtures (2:1:1 v/v/v). The compound shows excellent photostability in solution with no degradation upon prolonged irradiation at 570 nm and 590 nm (see ESI†), making it a good candidate for an active initiator in photoredox processes. Since 4+ is isolated and used as a racemic mixture, no investigations on electronic circular dichroism nor circular polarized luminescence have been carried out.
 | ||
| Fig. 2 (A) Absorption (blue line) and fluorescence spectra (red line) of a PhCF3 solution of 4+ (31 μM) at r. t. (λex = 575 nm). (B) Comparison between fluorescence decays of air-equilibrated (blue dots) and N2-saturated (red dots) PhCF3 solutions of 4+ (ca. 30 μM) at r.t. The corresponding monoexponential fitting functions are shown as solid lines. The instrument response function (IRF) is also reported (grey dots). | ||
With this in hand, the analysis of the luminescence quenching in the presence of the reaction components has been undertaken to draw a mechanistic picture of the chemical reaction occurring upon visible-light excitation of 4+. The reduction potentials of 4+ have been reported as: E0(4+/4˙) = −0.82 V vs. SCE, and E0(42+/4+) = +1.44 V vs. SCE in CH2Cl2,24a whose polarity is comparable to that of PhCF3.34 It follows that the excited state of 4+ is able, on a thermodynamic basis, to oxidize substrates whose potential does not exceed +1.14 V vs. SCE and to reduce others whose potential is as low as −0.52 V vs. SCE. No relevant quenching mechanism has been observed upon addition of p-chlorobenzaldehyde (1a, up to ca. 0.06 M, see Fig. S10†), as expected by its low reduction potential (Epc = −1.82 V vs. SCE, irreversible electron transfer process).11 On the other hand, quenching processes have been observed for the two [Ti(IV)] complexes (3 and 13) and Hantzsch's ester 6 in air-equilibrated solutions, with compound 6 displaying the highest quenching constant (kq = 1.5 × 109 M−1 s−1; see Fig. S11–S13†). By taking into account the concentrations used for the photocatalytic reaction, comparison between contributions of all substrates to the quenching efficiencies indicates that 6 is the most efficient quencher with a quenching efficiency of ca. 67% compared to ca. 3% in the case of the Ti(IV) complexes (see Fig. S14†). 6 can act as an efficient reductant of the dye's excited state (E0(6˙+/6) = 1.0 V vs. SCE),18 leading to the formation of radical 4˙, which is able, in a second stage, to reduce the Ti(IV) complexes and ultimately triggering the redox activity of the titanium centre towards aldehydes (Fig. 3A). Indeed, Ti(IV) species are characterized by the following reduction potentials: ca. −0.8 V vs. SCE for 3 in CH2Cl2;25ca. −0.6 V vs. SCE for 11 in CH2Cl2,28a (see Fig. S16†), compatible with the reduction potential E0(4+/4˙) = −0.82 V vs. SCE.
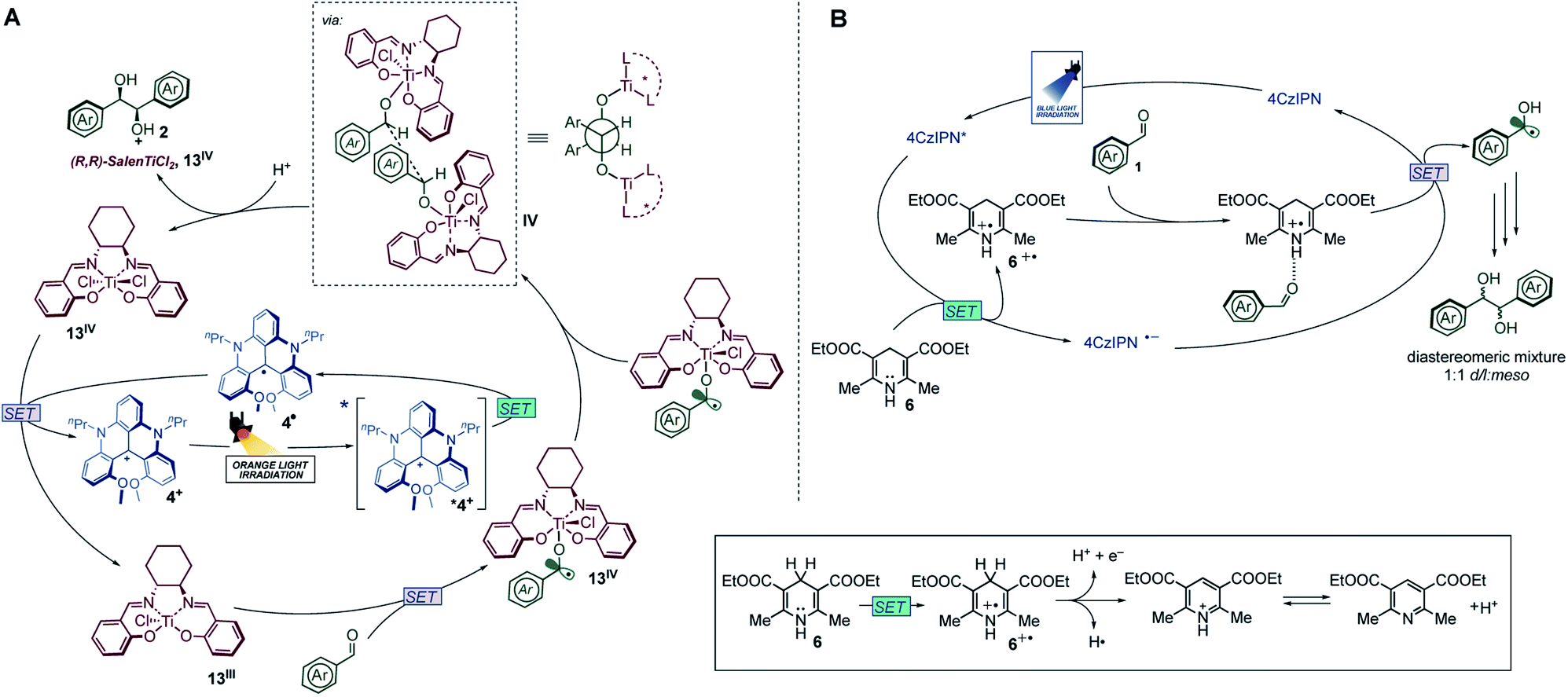 | ||
| Fig. 3 Mechanistic proposal for the photoredox pinacol coupling catalyzed by 4+ in the presence of titanium complexes (A) and by TADF dye 7 (B). | ||
We can now compare the above-mentioned photochemical mechanism upon orange light excitation (Fig. 3A) with the one responsible for the photoreaction in the presence of photocatalysts 7–12 upon blue light excitation (Fig. 3B and Table 1, entries 5–9). In the latter case, the excited state of the photocatalyst is reductively quenched by Hantzsch's ester 6 and its reduced form can reduce the aldehyde activated by the oxidized species 6˙+, as previously reported in the literature.26 Therefore, the titanium complex is not involved in the catalytic mechanism (Table 1, entry 9) and the reaction does not show any stereoselectivity.
In the case of the carbenium photocatalyst 4+, the first photochemical event is again the reductive quenching by 6, but the reduced photocatalyst 4˙ can reduce only the titanium complex and not the aldehyde, giving a high stereocontrol (Fig. 3A). The stereoselective process involves the coupling of two ketyl radicals mediated by two molecules of Ti(Salen). In Fig. 3A we have depicted the diastereoselective/enantioselective step suggesting the encounter of two ketyl radicals. Based on sterical reasons, the approach in which the oxygen and phenyl substituent are in anti-positions is the most favourable, but it was not considered as it would lead to the meso product. Formation of D/L products could be rationalized by a transition state in which the two aryl groups are in a gauche relationship, and the titanium complexes are in the anti-position.35
As pointed out in a recent calculation carried out by Streuff36 on the reaction of carbonyl compounds with nitrile, the radical–radical coupling is the favoured pathway and, as far as the relative position of the titanium complex (Cp2TiCl) coordinated fragment is concerned, the anti-coordination of the titanium is favoured from the energetic point of view. We assume that this coordination is favourable also in the case of pinacol coupling due to the positive non-linear effect (two molecules of titanium are involved in the stereoselective process). This led to the depicted transition states with anti-titanium and gauche configurations for the phenyl.
Since many stereoselective protocols involving two M(Salen) complexes (where M is either Co, Cr or Ti) usually display non-linear effects,37 due to cooperative bimetallic mechanisms,38 we have conducted a non-linear effect study by preparing the enantiomerically pure (R,R)- and (S,S)-SalenTiCl2 complexes, and using different ratios of the two complexes to promote the reaction. The result of the study is depicted in Fig. 4. Clearly, a non-linear effect is registered in our reaction. Since Ti(III) complexes are involved in associative or dissociative equilibria (in the case of Cp2TiCl2, the loss of the chloride ligand with the formation of the corresponding cationic complex),26,39 the effective concentration of active titanium species can be hardly estimated.40 Although it is difficult to attribute the suitable model to fit the data reported in Fig. 4, the best fitting model to describe the observed curve is (ML)2, indicating a cooperative role played by two Ti(Salen) complexes in the enantiodetermining step.41
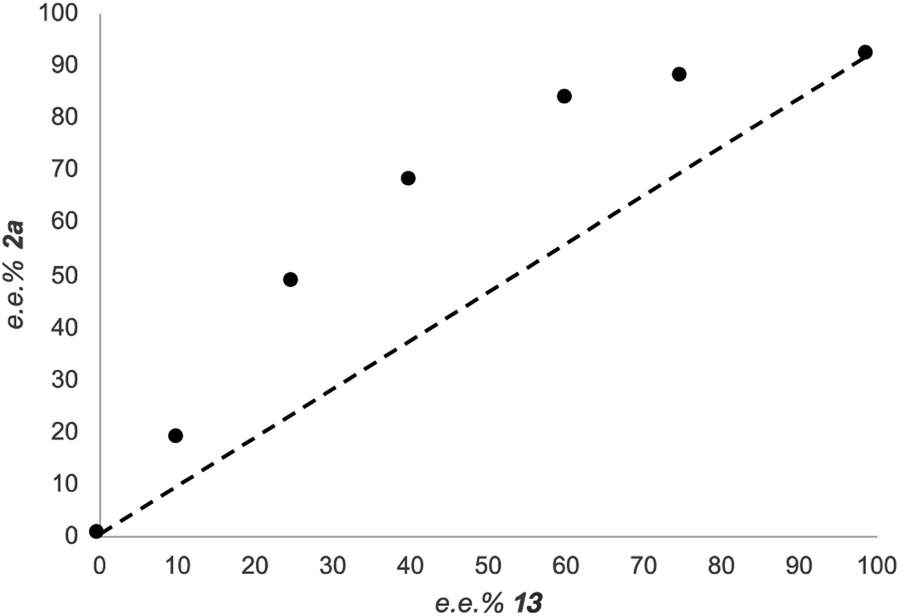 | ||
| Fig. 4 Enantiomeric excess of the product 2avs. enantiomeric excess of 13 (see ESI† for the reaction setup and conditions). | ||
Conclusions
In conclusion, we have described an effective photoredox diastereo- and enantioselective pinacol coupling mediated by titanium complexes. The application of a red-absorbing organic dye, readily available and stable under the reaction conditions, is crucial to obtain excellent yields and avoid the use of stoichiometric amounts of metals as the reductants. The use of orange light in combination with this dimethoxyquinacridinium organic dye 4+ allows the stereoselective coupling of aldehydes mediated by titanium, avoiding undesired processes lacking stereocontrol. The enantioselective process is mediated by a simply prepared Ti(Salen) complex, and good enantiomeric excesses are obtained. These results open new directions and possibilities to many stereoselective photoredox reactions promoted by titanium or other metals with low reduction potentials. In addition, titanium complexed ketyl radicals generated under these conditions could be used in different types of couplings. These directions will be subjected to further investigations in our laboratory.Author contributions
P. G. C. and A. G. conceived the study. Photoredox reactions and preparation of chiral SalenTiCl2 were carried out by F. P., F. C., G. M., and S. P. The latter three authors contributed equally to the experimental studies. Photophysical analysis and interpretation of photophysical data were carried out by A. F., P. C. and G. B. The manuscript was written with the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. G. C. acknowledges the National Project (PRIN 2017 ID: 20174SYJAF) SURSUMCAT “Raising up Catalysis for Innovative Developments” for financial support of this research. Chiesi Farmaceutici S.p.A. is fully acknowledged for financial support of photoredox research in our group and for the supply of Kessil lamps used for these investigations. The University of Bologna is gratefully acknowledged for financial support.Notes and references
- For key contribution: D. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed . For selected reviews on photoredox catalysis, see: (a) T. P. Yoon, M. A. Du and J. Ischay, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (d) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (e) X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC; (f) S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320–1330 CrossRef CAS PubMed; (g) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (h) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 RSC; (i) K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093–13112 RSC; (j) Y. – Q. Zou, F. M. Hoermann and T. Bach, Chem. Soc. Rev., 2018, 47, 278–290 RSC; (k) C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef CAS.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- G. C. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- Á. Péter, S. Agasti, O. Knowles, E. Pyea and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC.
- T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 26, 4953–4962 CrossRef.
- Q. Xia, J. Dong, H. Song and Q. Wang, Chem.–Eur. J., 2019, 25, 2949–2961 CAS.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- For the use of ruthenium complexes in pinacol coupling, see: R. Naumann and M. Goez and, Green Chem., 2019, 21, 4470–4474 RSC.
- (a) A. Gualandi, A. Nenov, M. Marchini, G. Rodeghiero, I. Conti, E. Paltanin, M. Balletti, P. Ceroni, M. Garavelli and P. G. Cozzi, ChemCatChem, 2021, 13, 981–989 CrossRef CAS; (b) A. Gualandi, G. Rodeghiero, E. Della Rocca, F. Bertoni, M. Marchini, R. Perciaccante, T. P. Jansen, P. Ceroni and P. G. Cozzi, Chem. Commun., 2018, 54, 10044–10047 RSC; (c) W. D. Rouch, M. Zhang and R. D. McCulla, Tetrahedron Lett., 2012, 53, 4942–4945 CrossRef CAS; (d) T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef CAS; (e) S. Okamoto, R. Ariki, H. Tsujioka and A. A. Sudo, J. Org. Chem., 2017, 82, 9731–9736 CrossRef CAS PubMed.
- (a) G. Han, X. Liu, Z. Cao and Y. Sun, ACS Catal., 2020, 10, 9346–9355 CrossRef CAS; (b) M. Liu, L. Tan, R. T. Rashid, Y. Cen, S. Cheng, G. Botton, Z. Mi and C.-J. Li, Chem. Sci., 2020, 11, 7864–7870 RSC.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- (a) A. Gansäuer, Chem. Commun., 1997, 457–458 RSC; (b) A. Gansäuer, Synlett, 1997, 363–364 CrossRef; (c) A. Gansäue and D. Bauer, J. Org. Chem., 1998, 63, 2070–2071 CrossRef.
- (a) Z. Zhang, R. B. Richrath and A. Gansäuer, ACS Catal., 2019, 9, 3208–3212 CrossRef CAS; (b) Z. Zhang, T. Hilche, D. Slak, N. Rietdijk, U. N. Oloyede, R. A. Flowers II and A. Gansäuer, Angew. Chem., Int. Ed., 2020, 59, 9355–9359 CrossRef CAS PubMed.
- (a) A. Gualandi, F. Calogero, M. Mazzarini, S. Guazzi, A. Fermi, G. Bergamini and P. G. Cozzi, ACS Catal., 2020, 10, 3857–3863 CrossRef CAS; (b) F. Calogero, A. Gualandi, S. Potenti, M. Di Matteo, A. Fermi, G. Bergamini and P. G. Cozzi, J. Org. Chem., 2021, 86, 7002–7009 CrossRef CAS PubMed.
- (a) S. Lin, Y. Chen, F. Li, C. Shia and L. Shi, Chem. Sci., 2020, 11, 839–844 RSC; (b) F. Li, S. Lin, Y. Chen, C. Shi, H. Yan, C. Li, C. Wu, L. Lin, C. Duan and S. Shi, Angew. Chem., Int. Ed., 2021, 60, 1561–1566 CrossRef CAS PubMed.
- For a review, see: A. Fermi, A. Gualandi, G. Bergamini and P. G. Cozzi, Eur. J. Org. Chem., 2020, 6955–6965 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS PubMed.
- For a review about application of organic dyes in metallaphotoredox catalysis, see: A. Gualandi, M. Anselmi, F. Calogero, S. P. E. Bassan, P. Ceroni and P. G. Cozzi, Org. Biomol. Chem., 2021, 19, 3527–3550 RSC.
- (a) B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS; (b) I. Hernández Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- T. J. Sørensen, M. F. Nielsen and B. W. Laursen, ChemPlusChem, 2014, 79, 1030–1035 CrossRef.
- C. Nicolas, C. Herse and J. Lacour, Tetrahedron Lett., 2005, 46, 4605–4608 CrossRef CAS.
- (a) L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 CrossRef CAS PubMed; (b) S. M. Stull, L. Mei and T. L. Gianetti, Synlett, 2021, 32, 337–343 CrossRef; (c) L. Mei, J. Moutet, S. M. Stull and T. L. Gianetti, J. Org. Chem., 2021, 86, 10640–10653 CrossRef CAS PubMed.
- T. Liedtke, T. Hilche, S. Klare and A. Gansäuer, ChemSusChem, 2019, 12, 3166–3171 CrossRef CAS PubMed.
- (a) G. S. Yedase, M. John and V. R. Yatham, Asian J. Org. Chem., 2021, 10, 2916–2920 CrossRef CAS; (b) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed.
- J. J. Maul, P. J. Ostrowski, G. A. Ublacker, B. Linclau and D. P. Curran, in Modern Solvents in Organic Synthesis, ed. P. Knochel, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, vol. 206, pp. 79–105 Search PubMed.
- (a) A. Chatterjee, T. H. Bennur and N. N. Joshi, J. Org. Chem., 2003, 68, 5668–5671 CrossRef CAS PubMed; (b) Y.-G. Li, Q.-S. Tian, J. Zhao, Y. Feng, M.-J. Li and T.-P. You, Tetrahedron: Asymmetry, 2004, 15, 1707–1710 CrossRef CAS.
- Nicholas reported a single example of the use of Brintzinger's [(R,R)-ethylenebis(tetrahydroindeny1)titanium dichloride] in the reaction with benzaldehyde (7:1 D,L:meso, 60% e.e.); M. S. Dunlap and K. M. Nicholas, Synth. Commun., 1999, 29, 1097–1106 CrossRef CAS.
- For reviews on Schiff base metal complexes in asymmetric catalysis, see: (a) S. Shaw and J. D. White, Chem. Rev., 2019, 119, 9381–9426 CrossRef CAS PubMed; (b) K. Matsumoto, B. Saitom and T. Katsuki, Chem. Commun., 2007, 3619–3627 RSC; (c) P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC; (d) J. F. Larrow and E. N. Jacobsen, Top. Organomet. Chem., 2004, 6, 123–152 CrossRef CAS; (e) L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–93 RSC.
- M. Bandini, P. G. Cozzi, S. Morganti and A. Umani-Ronchi, Tetrahedron Lett., 1999, 40, 1997–2000 CrossRef CAS.
- For a catalytic non-photoredox enantioselective pinacol coupling mediated by titanium Salen metal complexes, see ref. 28a and b; see also: (a) A. Bensari, J. L. Renaud and O. Riant, Org. Lett., 2001, 3, 3863–3965 CrossRef CAS PubMed; (b) Y.-G. Li, C. Jiang, J. Zhao, Q.-S. Tian and T.-P. You, Chin. J. Chem., 2004, 22, 950–952 CrossRef CAS; (c) J. Wen, J. Zhao, X. Wang, J. Dong and T. You, J. Mol. Catal. A: Chem., 2006, 245, 242–247 CrossRef CAS . For other titanium(III) complexes:; (d) J. Wen, J. Zhao and T. You, J. Mol. Catal. A: Chem., 2006, 245, 278–280 CrossRef CAS; for catalytic pinacol coupling with other metals: (e) J. Sun, Z. Dai, C. Li, X. Pan and C. Zhu, J. Organomet. Chem., 2009, 694, 3219–3221 CrossRef CAS; (f) H. Yamamoto and G. Xia, Chem. Lett., 2007, 36, 1082–1087 CrossRef CAS; (g) H. Yang, H. Wang and C. Zhu, J. Org. Chem., 2007, 72, 10029–10034 CrossRef CAS PubMed.
- For radical reactions mediated by Ti(III) metal complexes, see: (a) J. Streuff, M. Feurer, P. Bichovski, G. Frey and U. Gellrich, Angew. Chem., Int. Ed., 2012, 51, 8661–8664 CrossRef CAS PubMed; (b) S. G. Robinson, X. Wu, B. Jiang, M. S. Sigman and S. Lin, J. Am. Chem. Soc., 2020, 142, 18471–18482 CrossRef CAS PubMed; (c) T. McCallum, X. Wu and S. Lin, J. Org. Chem., 2019, 84, 14369–14380 CrossRef CAS PubMed; (d) X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836–14843 CrossRef CAS PubMed; (e) W. Hao, J. H. Harenberg, X. Wu, S. N. Macmillan and S. Lin, J. Am. Chem. Soc., 2018, 140, 3514–3517 CrossRef CAS PubMed.
- B. K. Freed, J. Biesecker and W. J. Middleton, J. Fluorine Chem., 1990, 48, 63–75 CrossRef CAS.
- (a) R. J. Enemærke, J. Larsen, G. H. Hjøllund, T. Skrydstrup and K. Daasbjerg, Organometallics, 2005, 24, 1252–1262 CrossRef. For further studies, see: (b) K. Daasbjerg, H. Svith, S. Grimme, M. Gerenkamp, C. Mück-Lichtenfeld, A. Gansäuer, A. Barchuk and F. Keller, Angew. Chem., Int. Ed., 2006, 45, 2041–2044 CrossRef CAS PubMed.
- J. Streuff, D. Himmel and S. R. Jounas, J. Chem. Soc., Dalton Trans., 2018, 47, 5072–5082 RSC.
- T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456–494 CrossRef CAS PubMed.
- (a) L. E. Martinez, J. L. Leighton, D. H. Carsten and E. N. Jacobsen, J. Am. Chem. Soc., 1995, 117, 5897–5898 CrossRef CAS; (b) J. F. Larrow, S. E. Schaus and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 7420–7421 CrossRef CAS; (c) K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118(44), 10924–10925 CrossRef CAS; (d) R. G. Konsler, J. Karl and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 10780–10781 CrossRef CAS.
- T. Hilche, P. H. Reinsberg, S. Klare, T. Liedtke, L. Schäfer and A. Gansäuer, Chem.–Eur. J., 2021, 27, 4903–4912 CrossRef CAS PubMed.
- Active titanium catalyst is also in equilibria with dimeric species. See ref. 34 and: (a) J. R. Enemærke, G. H. Hjøllund, K. Daasbjerg and T. Skrydstrup, C. R. Acad. Sci., Ser. IIc: Chim., 2001, 4, 435–438 CrossRef; (b) J. R. Enemærke, J. Larsen, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2004, 126, 7853–7864 CrossRef PubMed.
- Lin recently performed a non-linear effect study to verify whether a Ti(III)(Salen) promoted catalytic reaction operates through a monomeric or multiple centre; see ESI of ref. 31b.†.
Footnotes |
| † Electronic supplementary information (ESI) available: Screening tests, and further substrates studied. Photophysical measurements, and electrochemical and quenching studies. Detailed procedures, description and copies of NMR spectra for all compounds. See https://doi.org/10.1039/d2sc00800a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |