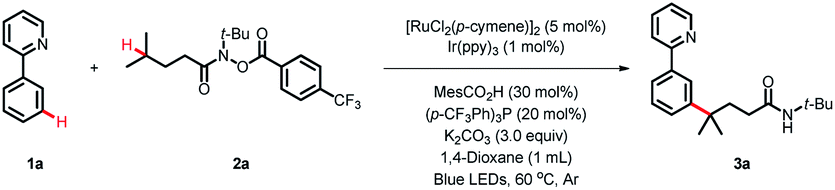DOI:
10.1039/D2SC00764A
(Edge Article)
Chem. Sci., 2022,
13, 5382-5389
Site-selective coupling of remote C(sp3)–H/meta-C(sp2)–H bonds enabled by Ru/photoredox dual catalysis and mechanistic studies†
Received
7th February 2022
, Accepted 11th April 2022
First published on 12th April 2022
Abstract
Construction of C(sp2)–C(sp3) bonds via regioselective coupling of C(sp2)–H/C(sp3)–H bonds is challenging due to the low reactivity and regioselectivity of C–H bonds. Here, a novel photoinduced Ru/photocatalyst-cocatalyzed regioselective cross-dehydrogenative coupling of dual remote C–H bonds, including inert γ-C(sp3)–H bonds in amides and meta-C(sp2)–H bonds in arenes, to construct meta-alkylated arenes has been accomplished. This metallaphotoredox-enabled site-selective coupling between remote inert C(sp3)–H bonds and meta-C(sp2)–H bonds is characterized by its unique site-selectivity, redox-neutral conditions, broad substrate scope and wide use of late-stage functionalization of bioactive molecules. Moreover, this reaction represents a novel case of regioselective cross-dehydrogenative coupling of unactivated alkanes and arenes via a new catalytic process and provides a new strategy for meta-functionalized arenes under mild reaction conditions. Density functional theory (DFT) calculations and control experiments explained the site-selectivity and the detailed mechanism of this reaction.
Introduction
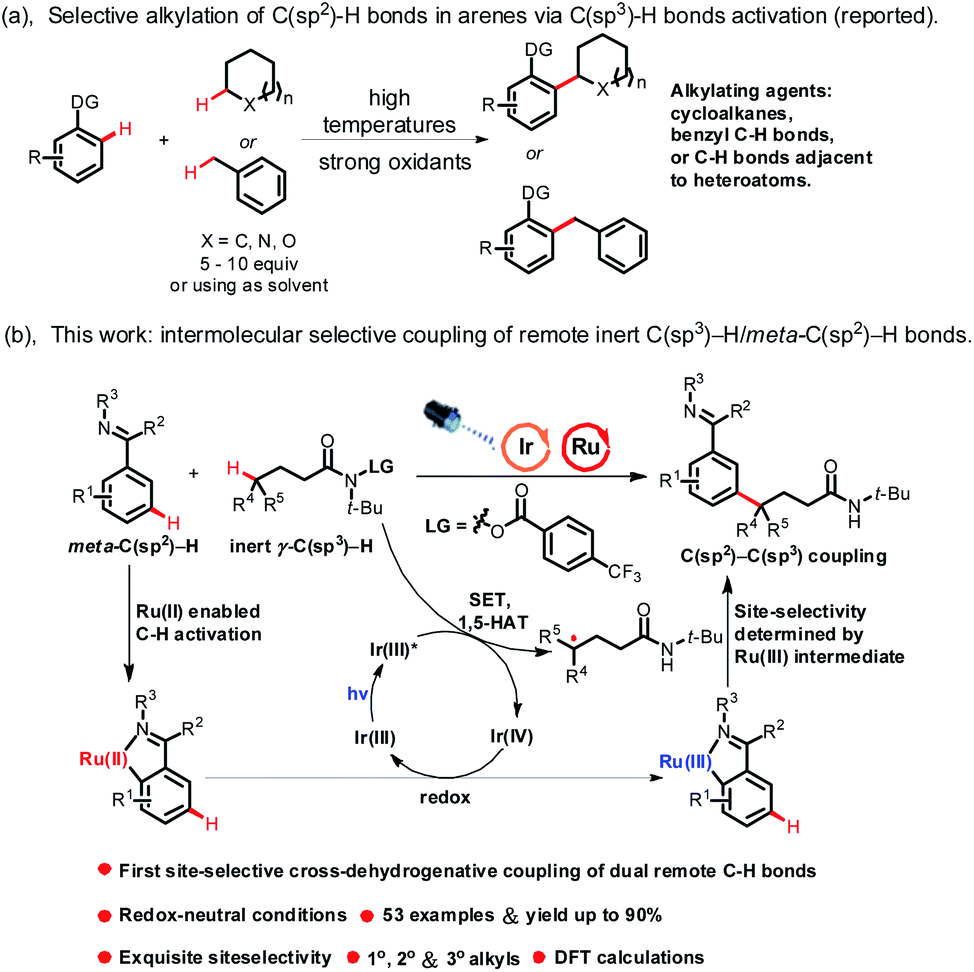
C–C bond construction is a long-standing target in organic chemistry. In recent years, construction of C–C bonds via cross-dehydrogenative coupling (CDC) has made great progress due to its atom- and step-economy.1 In a pioneering study, Fagnou and co-workers reported the catalytic coupling of indole derivatives and aromatic derivatives enabled by a palladium catalyst.2 Subsequently, various cross-dehydrogenative coupling reactions forming C(sp2)–C(sp2) bonds were developed.3 However, apart from Minisci-type reactions, C(sp2)–C(sp3) bond construction via coupling of C(sp2)–H bonds in arenes (excluding heteroaromatic rings, the same below) and C(sp3)–H bonds in alkanes is rare. In fact, Li developed the Ru(II)-catalyzed oxidative coupling of (sp3)–H bonds in cycloalkanes with ortho-(sp2)–H bonds in 2-arylpyridines.4 Though excellent work has been done in this field, coupling of C(sp2)–H bonds in arenes and C(sp3)–H bonds mainly focused on the use of simple cycloalkanes or hydrocarbons with extraordinarily reactive C(sp3)–H bonds as alkylating agents, such as benzyl C(sp3)–H bonds and C(sp3)–H bonds adjacent to heteroatoms (Scheme 1a),5 and their transformations were centralized on using dangerously strong oxidants (such as peroxides) or high temperatures to enable oxidative cleavage of the C(sp3)–H bonds to generate C(sp3)-centered radicals, meaning that this kind of reaction suffers from limited substrate scope.6 What's more, CDC of C(sp2)–H bonds in arenes and unactivated C(sp3)–H bonds (except C(sp3)–H bonds in simple cycloalkanes, the same below) remained challenging and was difficult to achieve due to the low reactivity and regioselectivity of unactivated C(sp3)–H bonds. In addition, alkylated arenes have found numerous applications in inter alia natural product synthesis, medicinal chemistry and materials science. Thus, a new methodology that could effectively construct C(sp2)–C(sp3) bonds via coupling of C(sp2)–H bonds in arenes and unactivated C(sp3)–H bonds with high regioselectivity under mild reaction conditions is in high demand.
 |
| | Scheme 1 (a) General coupling of C–H bond reaction modes; (b) our work. | |
The strategy of hydrogen atom transfer (HAT) has emerged as an effective tool to activate C(sp3)–H bonds. However, activation of C(sp3)–H bonds via this strategy generally lacked regioselectivity. The Hofmann–Löffler–Freytag (HLF) reaction,7 radical translocation processes triggered by nitrogen-centered radicals, such as 1,5-hydrogen atom transfer (1,5-HAT), was well documented and has been proven to be an effective strategy for the controllable and selective functionalization of remote inert C(sp3)–H bonds in complex molecules. Excellent work has been done in the past decades.8 Until now, the HLF reaction has mainly focused on halogenation,9 Michael addition,10 Minisci reaction11 and coupling with nucleophiles.12 However, C-centered radical coupling with intermolecular C(sp2)–H bonds of arenes has not been developed yet. Therefore, a method that could effectively construct C(sp2)–C(sp3) bonds by site-selective coupling of remote inert C(sp3)–H bonds and C(sp2)–H bonds in arenes would be of great importance, and by this method, remote C(sp3)–H bond arylated products could be obtained in an atom- and step-economical approach.
Despite considerable advances in ortho-arene C–H bond functionalization, meta-arene C–H bond transformation remains elusive. The strategy of ruthenium-catalyzed σ-activation devised by Ackermann, Frost, and others is a powerful technique for meta-arene C–H bond transformation.13 So far, excellent work has been done in this area, such as bromination,14 sulfonation,15 nitration16 and alkylation.17 However, meta-alkylation of arenes mainly focused on the use of alkyl halides as alkylating agents, and to the best of our knowledge, Ru-catalyzed meta-alkylation of arenes through regioselective activation of unactivated C(sp3)–H bonds has not yet been reported, which was challenging to realize due to both the reactivity and regioselectivity needing to be simultaneously controlled in the reaction. In addition, meta-alkylation reactions mainly employed substrates that could produce radicals via single electron transfer (SET) with Ru-catalysts as alkylating agents, which severely restricted the development of meta-alkylation reactions. Therefore, a new methodology that could produce radicals via a new catalytic process rather than through SET with Ru-catalysts would expand the scope of meta-arene C–H bond functionalization reactions, and would provide a new synthetic platform for meta-functionalized arenes. Continuing our research interest in the meta-C(sp2)–H bond functionalization reaction18 and remote C(sp3)–H bond activation reaction,19 here we disclosed a novel example of dual Ru/photoredox-catalyzed meta-alkylation of arenes via site-selective coupling of dual remote C–H bonds, including unactivated γ-C(sp3)–H bonds in amides and meta-C(sp2)–H bonds in arenes. This reaction represents a novel example of employing a photocatalyst/Ru as a co-catalyst for regioselective CDC of unactivated alkanes and arenes via a new catalytic process, and provides a new strategy to synthesize meta-functionalized arenes under mild reaction conditions (Scheme 1b).
Results and discussion
We commenced our study using 2-phenylpyridine 1a and N-(tert-butyl)-4-methyl-N-((4-(trifluoromethyl)benzoyl)oxy)pent-anamide 2a as model substrates (Table 1). With [RuCl2(p-cymene)]2/Ir(ppy)3 as the cocatalyst, the desired product 3a was obtained in 84% yield under blue LED irradiation at 60 °C after 36 h (entry 1). Further investigation disclosed that the phosphine ligand and carboxylic additive were the key to accomplishing this reaction (entries 2 and 3). It is noteworthy that Ir(ppy)3 is of great importance in this reaction, as other photocatalysts, such as 4CzIPN, Ir[dF(CF3)ppy]2(dtbbpy)PF6 and Eosin, failed to trigger the coupling reaction (entries 4–6). Variation in other solvents fell short of 1,4-dioxane (entries 7–10). The Ru-catalyst was also investigated, and the results indicated that [RuCl2(p-cymene)]2 was more efficient than the other catalysts tested (entries 1, 11 and 12). Control experiments verified the essential roles of the photocatalyst, Ru-catalyst and light (entries 13–15).
Table 1 Optimization of the reaction conditionsa
|
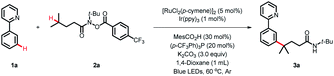
|
| Entry |
Deviation from standard condition |
Yieldb (%) |
|
Reaction conditions: 1a (0.2 mmol), 2a (0.5 mmol, 2.5 equiv), [RuCl2(p-cymene)]2 (5 mol%), Ir(ppy)3 (1 mol%), MesCO2H (30 mol%), (p-CF3Ph)3P (20 mol%), K2CO3 (3.0 equiv), 1,4-dioxane (1.0 mL), blue LEDs, 60 °C, Ar, 36 h.
Isolated yield. [Ir] = Ir[dF(CF3)ppy]2(dtbbpy)PF6, [Ru] = [RuCl2(p-cymene)]2.
|
| 1 |
None |
84 |
| 2 |
No (p-CF3Ph)3P |
<10 |
| 3 |
No MesCO2H |
65 |
| 4 |
4CzIPN instead of Ir(ppy)3 |
0 |
| 5 |
Eosin instead of Ir(ppy)3 |
0 |
| 6 |
[Ir] instead of Ir(ppy)3 |
0 |
| 7 |
DME as solvent |
40 |
| 8 |
DMSO as solvent |
Trace |
| 9 |
DMF as solvent |
Trace |
| 10 |
PhMe as solvent |
19 |
| 11 |
Ru3(CO)12 instead of [Ru] |
<10 |
| 12 |
RuCl3 instead of [Ru] |
Trace |
| 13 |
No [Ru] |
0 |
| 14 |
No Ir(ppy)3 |
0 |
| 15 |
No light (100 °C) |
0 |
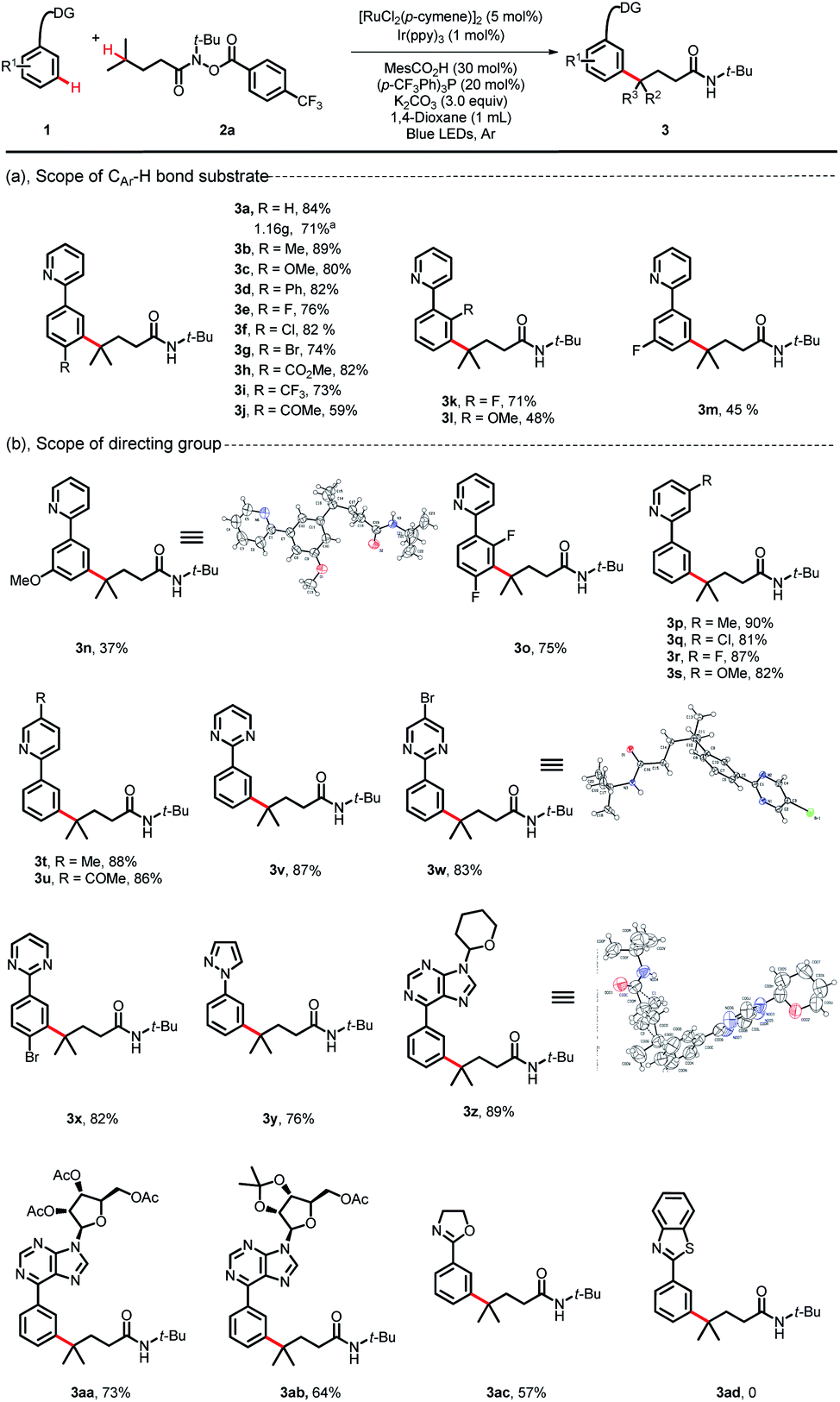
With the optimized reaction conditions established, we explored the versatility of this reaction. The scope of 2-phenylpyridines was first investigated using substrate 2a as a characteristic counterpart (Scheme 2). A wide range of synthetically useful functional groups on the arenes were well tolerated and delivered the C(sp3)–H/C(sp2)–H bond coupling products in moderate to high yields. Substrates bearing electron-neutral/electron-donating substituent groups at the para-position of aromatic rings proceeded well with 2a (3a–3d, 80–89%). Indeed, electrophilic functional groups, such as a valuable ester substituent, a carbonyl substituent and halides (F, Cl and Br), were well tolerated under these reaction conditions (3e–3j, 59–82%). Although the process by which ruthenium participated in the C–H activation approach may be shut down by the steric encumbrance of ortho-substituted phenylpyridines, this coupling reaction could also proceed in moderate yields when substituents such as F and OMe were at the ortho-position on phenylpyridines (3k, 71%; 3l, 48%; 3o, 75%). The scope of meta-substituted phenylpyridines was more limited, but with small groups such as F and OMe, the coupling reaction could also furnish the corresponding products in moderate yields (3m, 45%; 3n, 37%). With various substituents on the pyridyl, the desired products were obtained in high yields (3p–3u, 81–90%). We observed that with other types of directing groups, such as pyrimidine-, pyrazole-, and oxazoline-, the reaction delivered the desired products in moderate to good yields (3v–3y, 76–87%; 3ac, 57%). It is worth noting that when the bioactive purine derivative was used as the directing group, the corresponding product 3z was obtained in 89% yield. More appealingly, we observed that highly reactive bioactive nucleosides, even those substrates bearing sensitive protecting groups, were all well tolerated and generated the regioselective C(sp2)–H/C(sp3)–H bond coupling products 3aa and 3ab in 73% and 64% yields, respectively. However, the reaction could not proceed when benzothiazole was used as the directing group (3ad, 0). A gram-scale reaction was also conducted. When this transformation was performed at the 5.0 mmol scale using 0.5 mol% Ir(ppy)3, the product 3a could be obtained in 71% yield upon isolation (1.16 g). The structures of 3n, 3w and 3z were identified unambiguously by X-ray diffraction.
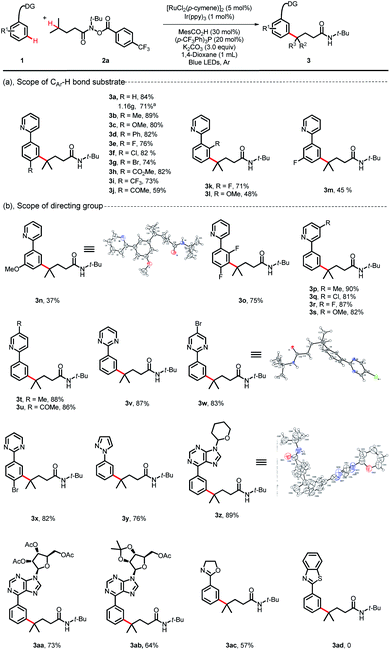 |
| | Scheme 2 Substrate scope of the arenes. Reaction conditions: 1 (0.2 mmol), 2a (0.5 mmol, 2.5 equiv), [RuCl2(p-cymene)]2 (5 mol%), Ir(ppy)3 (1 mol%), MesCO2H (30 mol%), (p-CF3Ph)3P (20 mol%), K2CO3 (3.0 equiv), 1,4-dioxane (1 mL), blue LEDs, 60 °C, Ar, 36 h, isolated yield. aGram-scale reaction, Ir(ppy)3 (0.5 mol%) for 48 h. | |
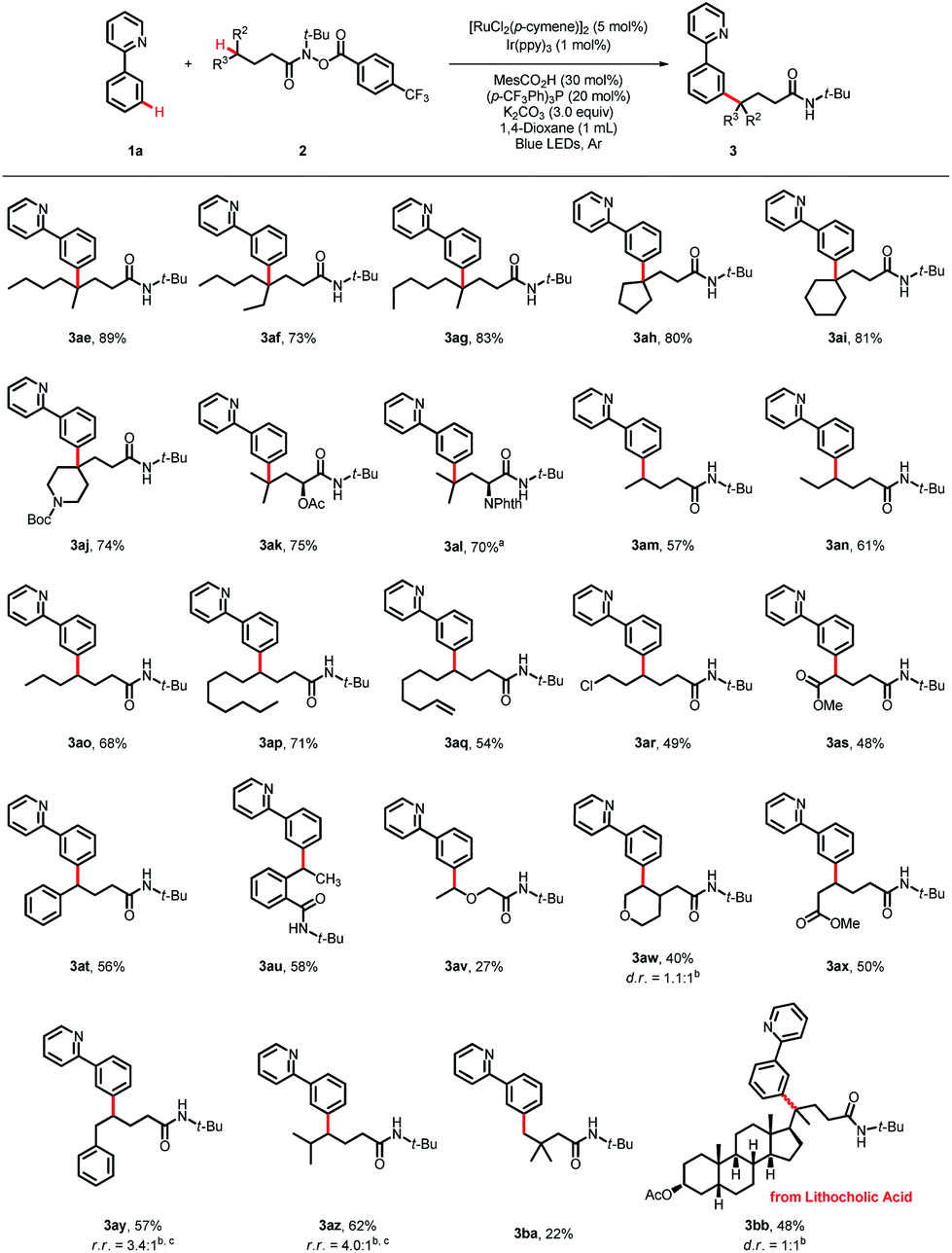
The scope of the hydroxamide compounds was then investigated using 2-phenylpyridine (1a) as the coupling partner (Scheme 3). Hydroxamide compounds bearing γ-tertiary C(sp3)–H bonds were well tolerated and furnished the desired products in good yields (3ae–3al, 70–89%). The coupling reaction could also proceed using hydroxamide compounds with secondary C(sp3)–H bonds at the γ-position, and the γ-arylated products were obtained in moderate to good yields. Substrates bearing diverse chain lengths showed considerable compatibility, and only γ-arylated products were obtained, revealing that the 1,5-HAT process was dominant (3am–3ap, 57–71%). Hydroxamide derivatives bearing various functional groups, such as a double bond, halides and esters, could also react with 1a and deliver the coupling products in moderate yields (3aq–3as, 48–54%). The γ-C(sp3)–H bond next to the oxygen atom could serve as an appropriate coupling partner, giving the product 3av in 27% yield. For reactions with δ-benzylic or tertiary C(sp3)–H bonds, the 1,6-HAT products were detected, giving products 3ay and 3az in 57% and 62% yields, and the ratios of 1,5-HAT products and 1,6-HAT products were 3.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4.0:1, respectively. The protocol was also applicable to hydroxamide compounds with the γ-primary C(sp3)–H bond, providing the desired product 3ba in 22% yield. Late-stage functionalization of natural product derivatives was also conducted, as verified by lithocholic acid, and the corresponding product 3bb was obtained in 48% yield.
:1 and 4.0:1, respectively. The protocol was also applicable to hydroxamide compounds with the γ-primary C(sp3)–H bond, providing the desired product 3ba in 22% yield. Late-stage functionalization of natural product derivatives was also conducted, as verified by lithocholic acid, and the corresponding product 3bb was obtained in 48% yield.
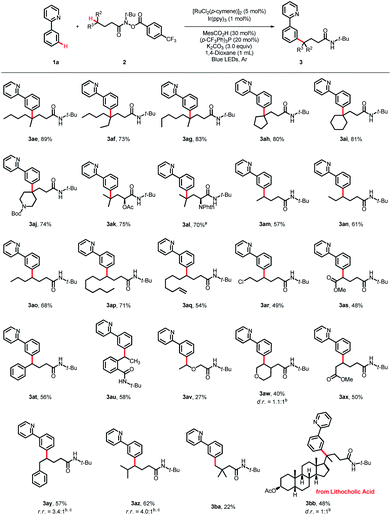 |
| | Scheme 3 Substrate scope of the hydroxamide compounds. Reaction conditions: 1a (0.2 mmol), 2 (0.5 mmol, 2.5 equiv), [RuCl2(p-cymene)]2 (5 mol%), Ir(ppy)3 (1 mol%), MesCO2H (30 mol%), (p-CF3Ph)3P (20 mol%), K2CO3 (3.0 equiv), 1,4-dioxane (1 mL), blue LEDs, 60 °C, Ar, 36 h, isolated yield. aIr(ppy)3 (2 mol%) for 5 days. bDetermined by 1H NMR. cThe ratio of 1.5-HAT and 1,6-HAT products. | |
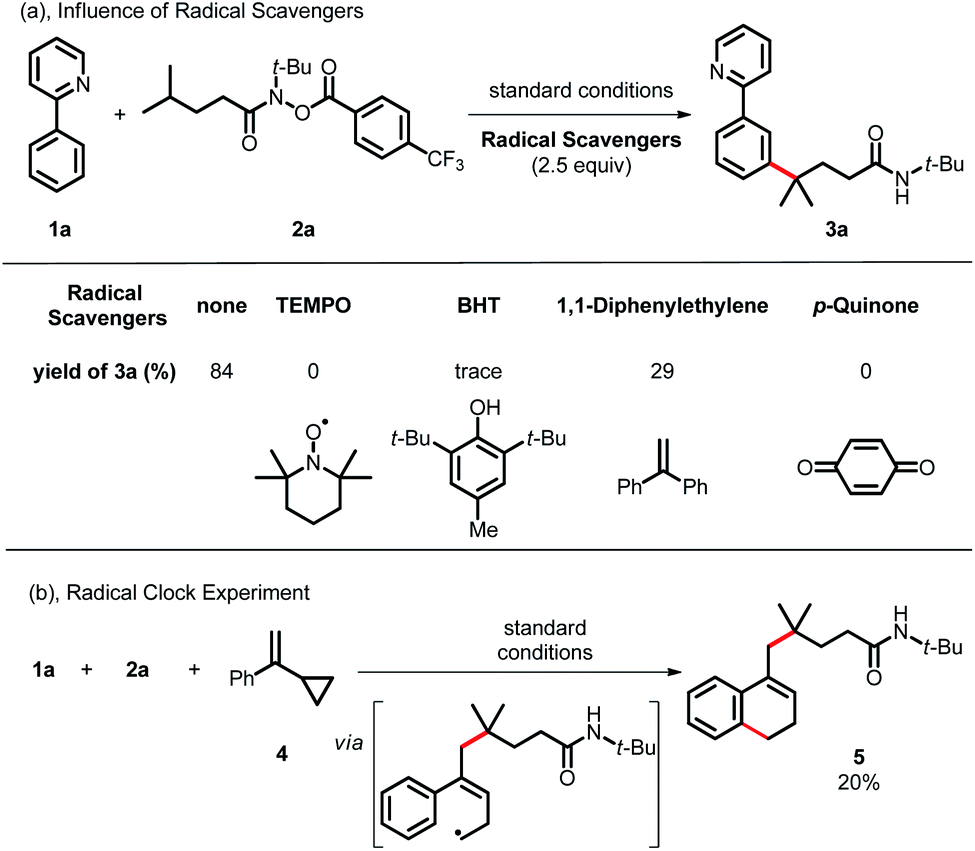
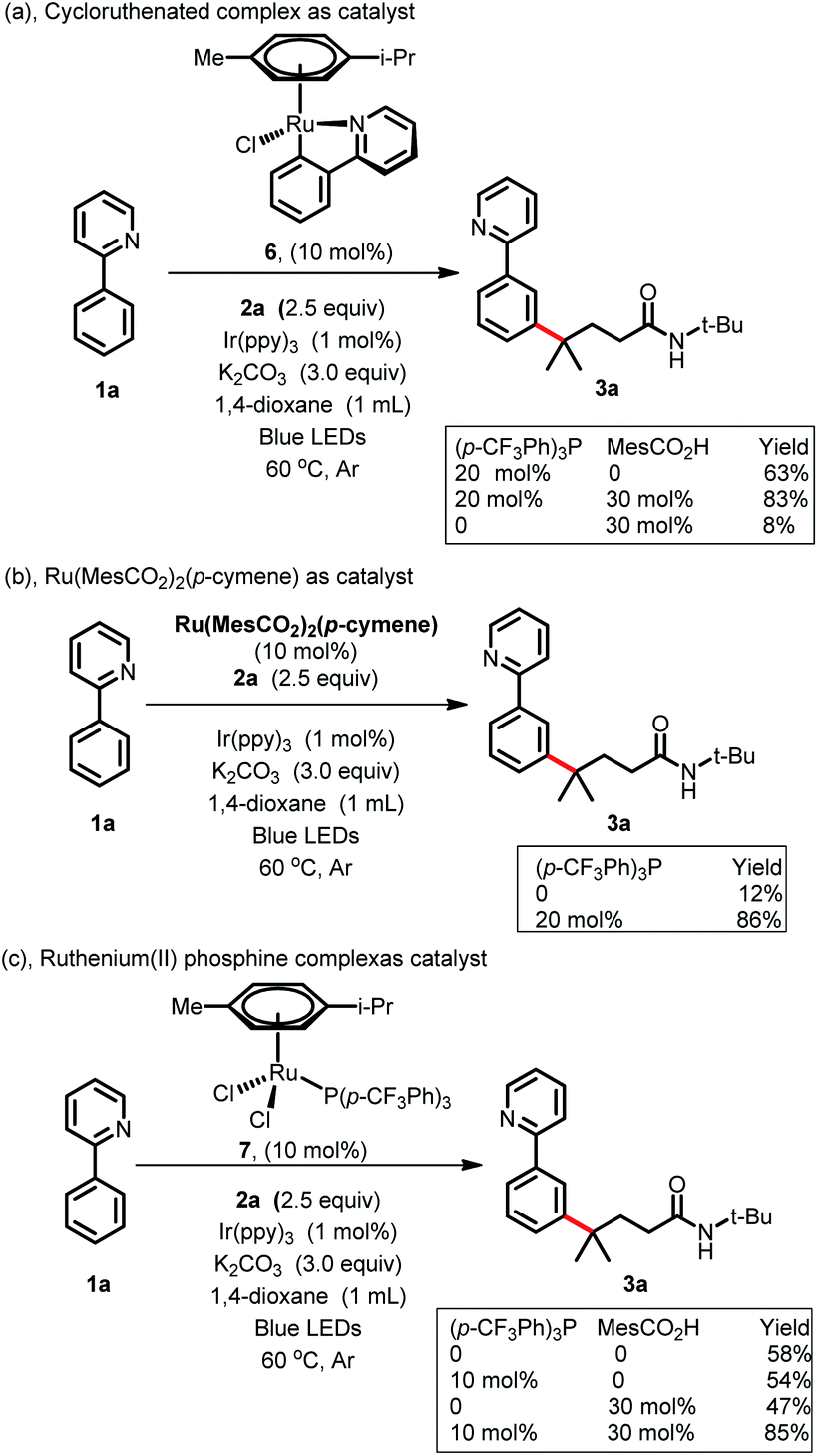
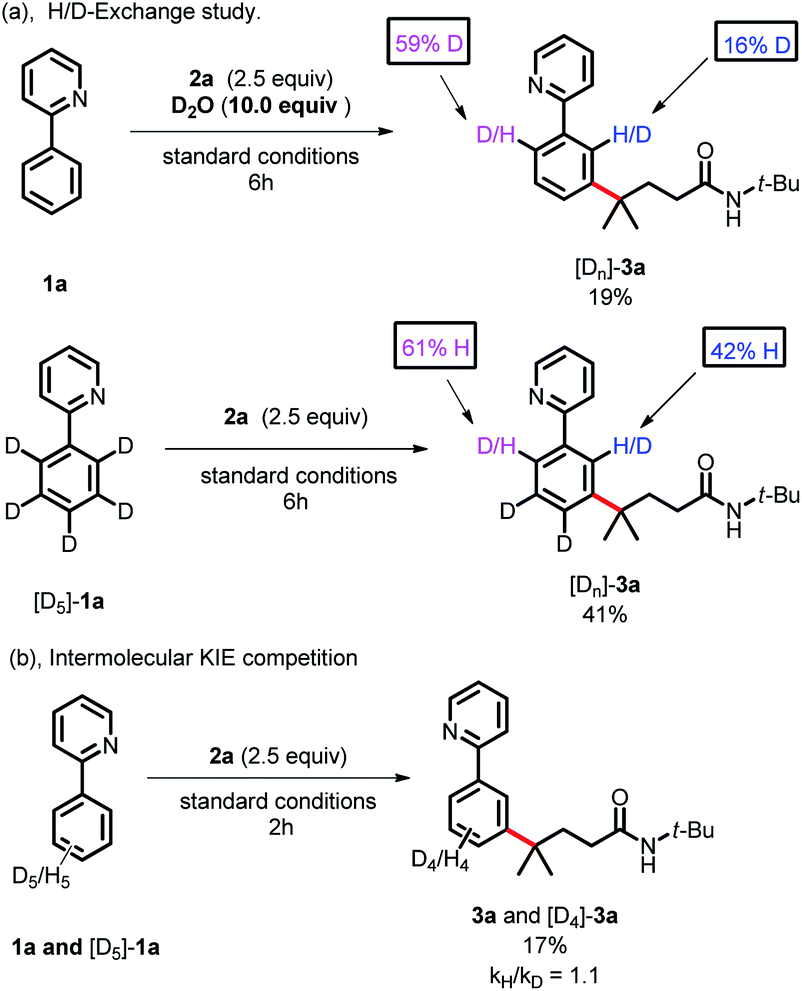
To gain more insight into the reaction mechanism, various control experiments were conducted. First, the reaction was completely inhibited when radical scavengers, such as 1,4-benzoquinone, butylated hydroxytoluene (BHT) or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), were added, indicating that this coupling reaction presumably proceeded via a radical pathway (Scheme 4a). Second, radical clock experiments were also carried out to probe the generation of the C-centered radical. The olefin 4 with a cyclopropyl moiety next to a double bond, which would rearrange in a radical reaction, was found to open under the reaction conditions, producing the corresponding ring closure product 5 in 20% yield (Scheme 4b). These radical-trap experiments verified that a radical pathway was involved in this coupling reaction. Furthermore, when the pre-synthesized cycloruthenated complex 6 was used as the catalyst instead of [RuCl2(p-cymene)]2, it was found to be an efficient catalyst (Scheme 5a). Likewise, when other types of ruthenium complexes, for example, Ru(MesCO2)2(p-cymene) and carboxylate-free ruthenium(II) phosphine complex 7, were used as catalysts, the reaction proceeded well (Scheme 5b and c). All of these reactions proceeded poorly in the absence of (p-CF3Ph)3P; in contrast, the reaction could still occur without MesCO2H, demonstrating that the phosphine ligand played a decisive role in this reaction and that the carboxylate assistance acted as a coaccelerator (Scheme 5). Notably, isotopic labeling experiments were also conducted to explore the possible ortho-C–H bond cyclometalation of the arenes. When D2O was added to the reaction, significant H/D scrambling was observed in the ortho-position of arenes (Scheme 6a). In addition, when the isotopically labeled derivative [D5]-1a was employed under the standard conditions, it was found that the desired product exhibited obvious H/D scrambling (Scheme 6a). Clearly, different degrees of H/D scrambling in each ortho-position of the arenes provided evidence for the remote C(sp2)–H bond functionalization of arenes occurring at the position para to the C–Ru bonds. Intermolecular kinetic isotope effects using substrates 1a and [D5]-1a indicated that the C(sp2)–H bond cleavage process at the ortho-position of arenes was not kinetically relevant (Scheme 6b).
 |
| | Scheme 4 Radical process experiments. | |
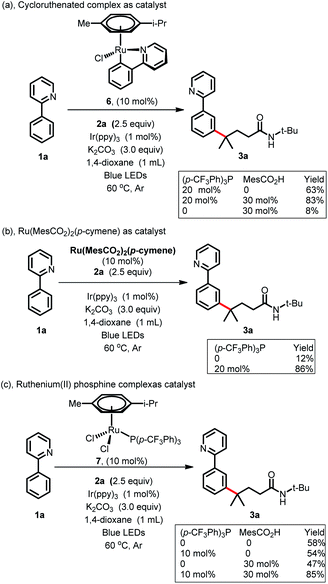 |
| | Scheme 5 Control experiments of the Ru-catalyst. | |
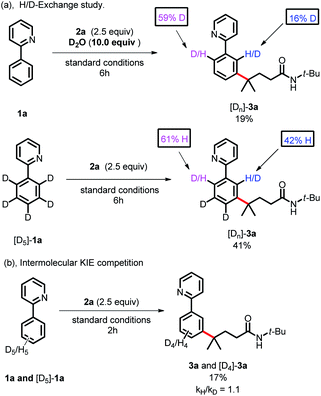 |
| | Scheme 6 Mechanistic studies by isotopic labeling experiments. | |
Furthermore, a luminescence quenching experiment revealed that the hydroxamide 2a could efficiently quench the excited-state photocatalyst Ir(III)* (Fig. S1†). In addition, cyclic voltammetry studies showed that the reduction potential of 2a was −1.56 V (vs. SCE) in MeCN (Fig. S2†), thus indicating that 2a can be easily reduced by the excited-state photocatalyst Ir(III)* (E1/2IV/*III = −1.73 V vs. SCE).
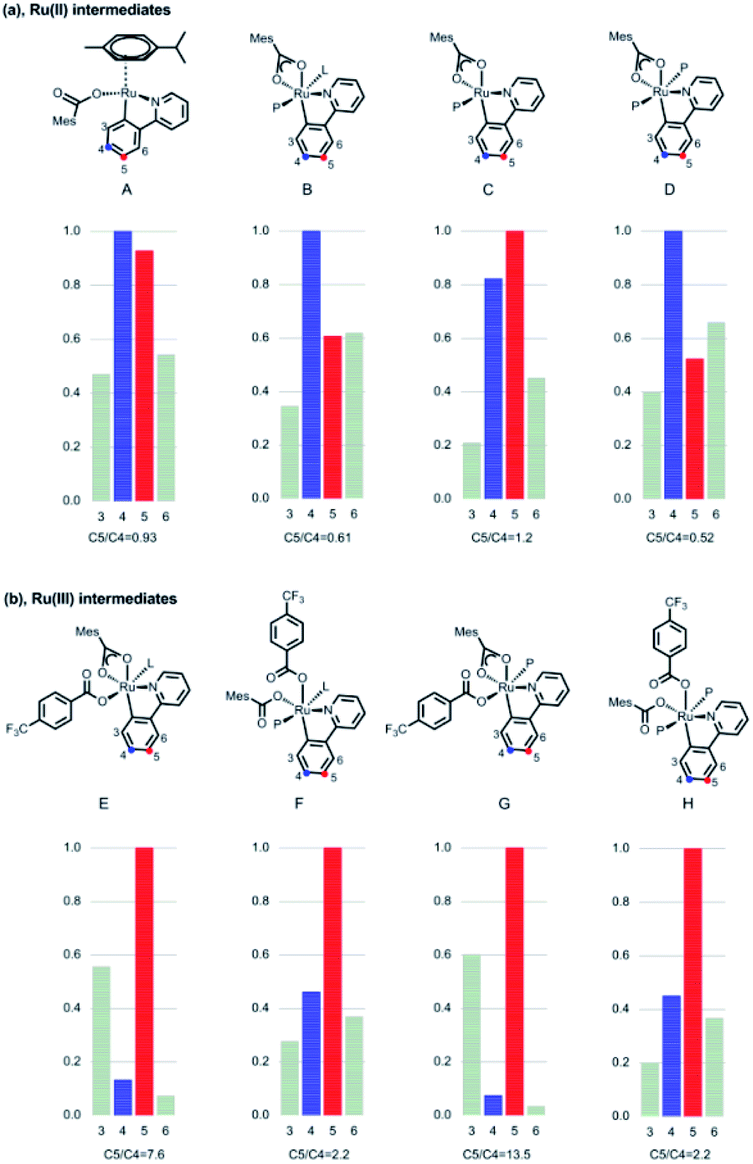
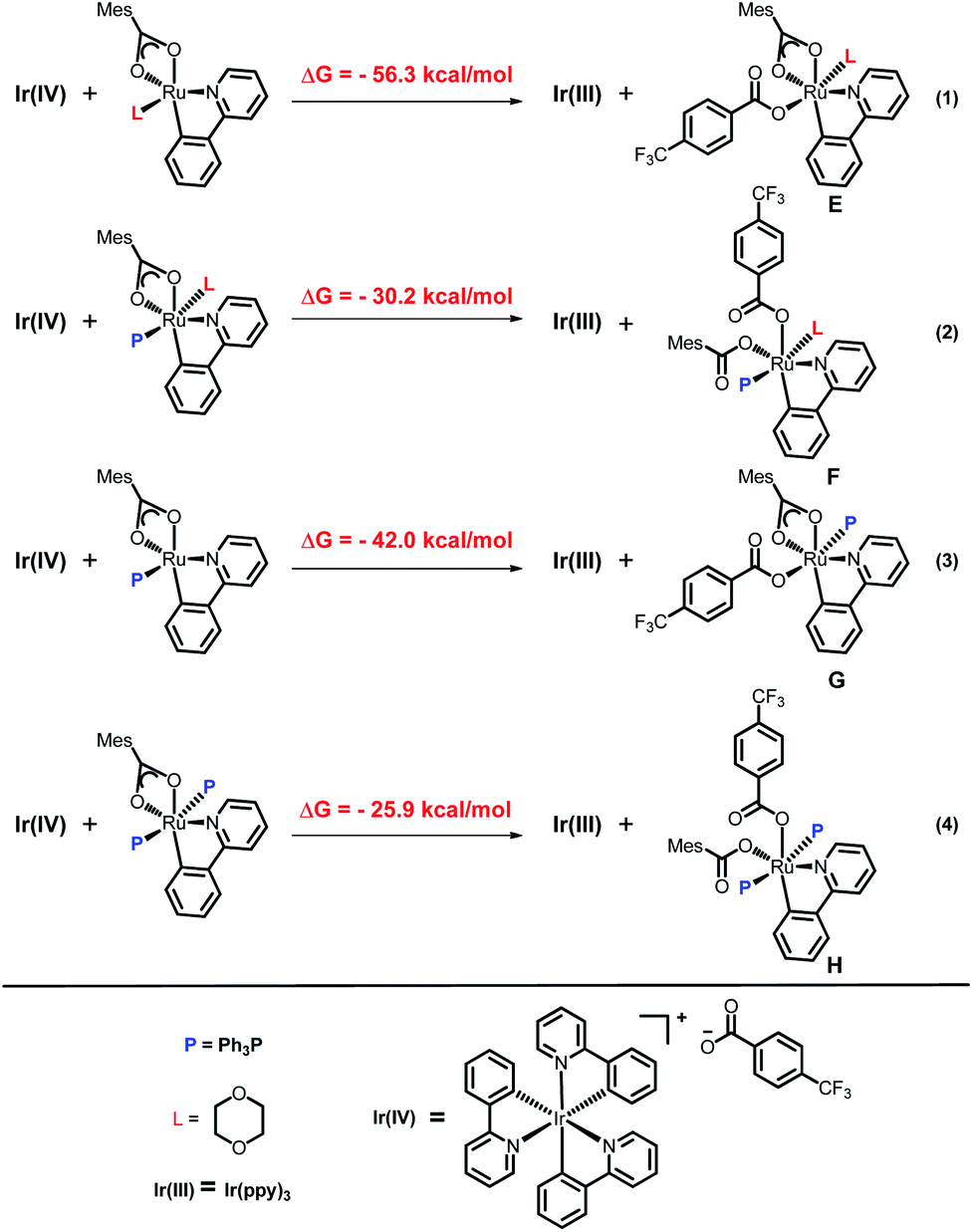
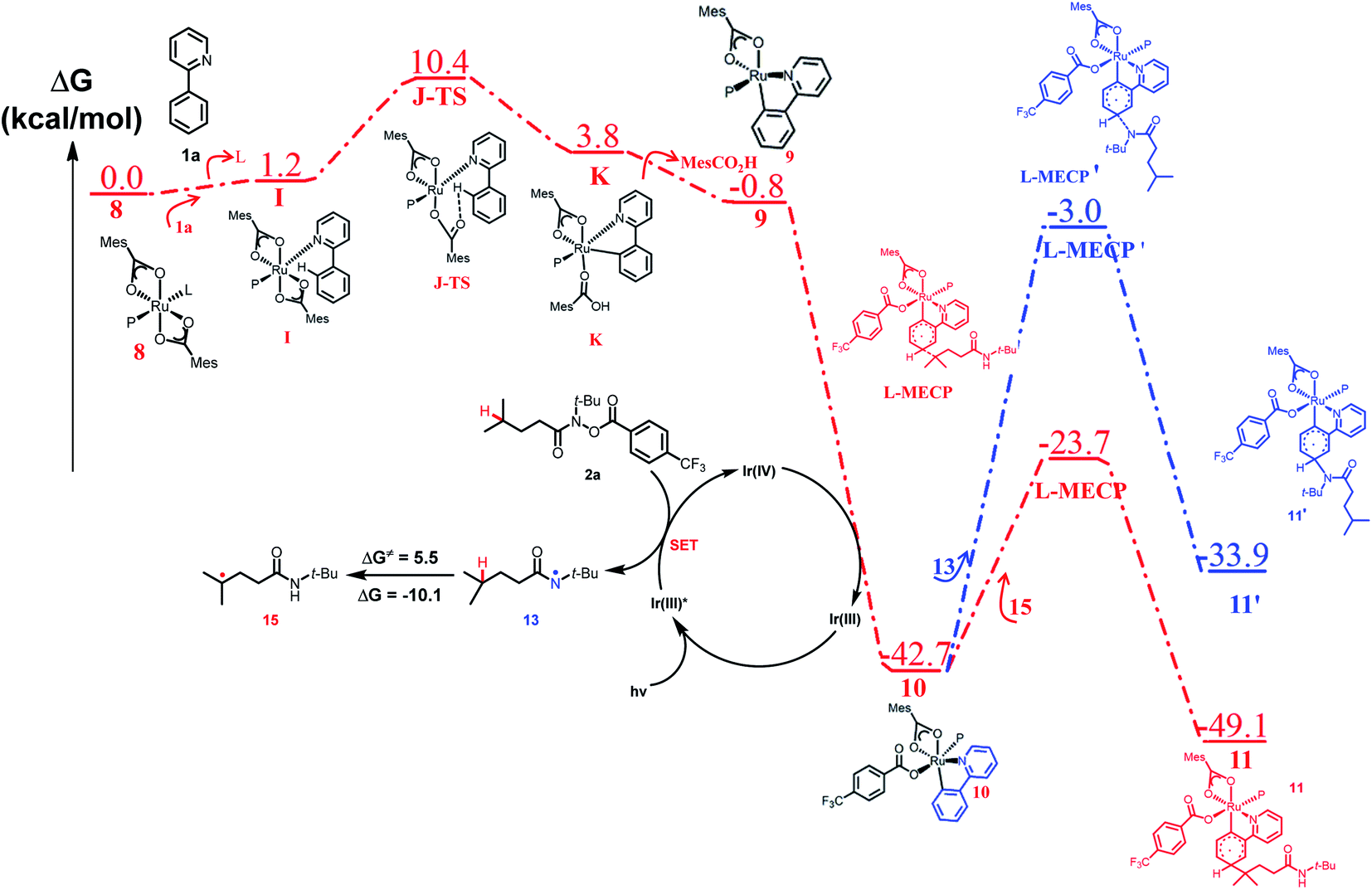
To explain the site-selectivity of the C(sp2)–C(sp3) bond formation, Fukui indices were calculated by density functional theory (DFT). The relative Fukui indices for possible Ru(II) intermediates A–D and Ru(III) intermediates E–H are shown in Scheme 7. On the whole, the relative Fukui indices of the Ru(III) complexes were generally better than those of Ru(II) complexes. This result agreed with previous theoretical and experimental studies that Ru(III) intermediates may be the key intermediates in meta-C(sp2)–H bond functionalization reactions. Then, the products of Ru(III) intermediates in this reaction were investigated. Considering that the Ru(II) complex was used as the catalyst and the Ir(IV) complex could form in our experiments, whether Ru(III) complexes could generate during the redox reaction was assessed by reaction Gibbs free energies (Scheme 8). The calculated results of the Gibbs free energy profile for the redox reaction between Ru(II)/Ir(IV) and Ru(III)/Ir(III) were negative (ΔG < 0), indicating that the possibly formed Ru(II) intermediates in this reaction could be easily oxidized to Ru(III) complexes by the Ir(IV) complex. All of these results demonstrated that Ru(III) complexes were generated in this reaction and played the key role in site-selectivity of this reaction. On the basis of the above results, Ru(III) complex G was chosen as the reaction intermediate in the following calculated reaction mechanism (Scheme 9). The catalytically active Ru(II) complex 8 transformed into I by the ligand exchange, and the Gibbs free energy change of this step was only 1.2 kcal mol−1. Next, the intermediate K was formed by the MesCOO− assisted C–H bond activation process from IviaJ-TS with an energy barrier of 9.2 kcal mol−1, and was 3.8 kcal mol−1 compared with 8. Then, MesCO2H left forming intermediate 9 and the energy decreased to −0.8 kcal mol−1 by the entropy effect. Subsequently, the Ru(II) intermediate 9 was oxidized to Ru(III) intermediate 10 by the Ir(IV) complex resulting from single electron transfer (SET) from Ir(ppy)* to 2a, releasing about 42 kcal mol−1. Finally, the alkyl radical 15 underwent addition to the benzene ring of 10 to form 11 through L-MECP with an energy barrier of 19.0 kcal mol−1, releasing 6.4 kcal mol−1 energy. In contrast, the nitrogen-centered radical 13 underwent addition to the same site through L-MECP′ with an energy barrier of 39.7 kcal mol−1 to form 11′. In addition, N-centered radical 13 could easily transform to C-centered radical 15 through intramolecular 1,5-HAT with an energy barrier of only 5.5 kcal mol−1. Hence, the intermediate 11 was dominant, and the calculated results were consistent with the experimental results that no meta aminated arenes were observed. After that, 11 underwent the LMCT step, rearomatization and demetalation to form the meta-alkylated arenes, which has been reported by previous reports. The computational results implied that the reaction mechanism was plausible under our reaction conditions.
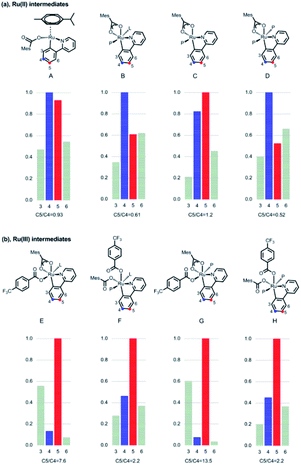 |
| | Scheme 7 Calculated Fukui indices of Ru(II)/Ru(III) intermediates. aL = 1,4-dioxane, P = PPh3. | |
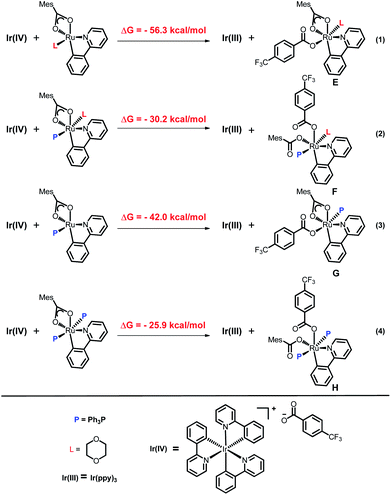 |
| | Scheme 8 Calculated Gibbs free energy profile for the redox reaction between Ru(II)/Ir(IV) and Ru(III)/Ir(III). | |
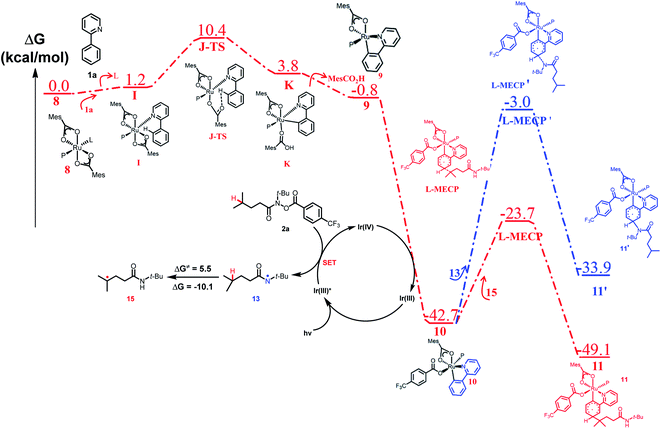 |
| | Scheme 9 Computed free energy profile of site-selective coupling of unactivated C(sp3)–H/meta-C(sp2)–H bonds. | |
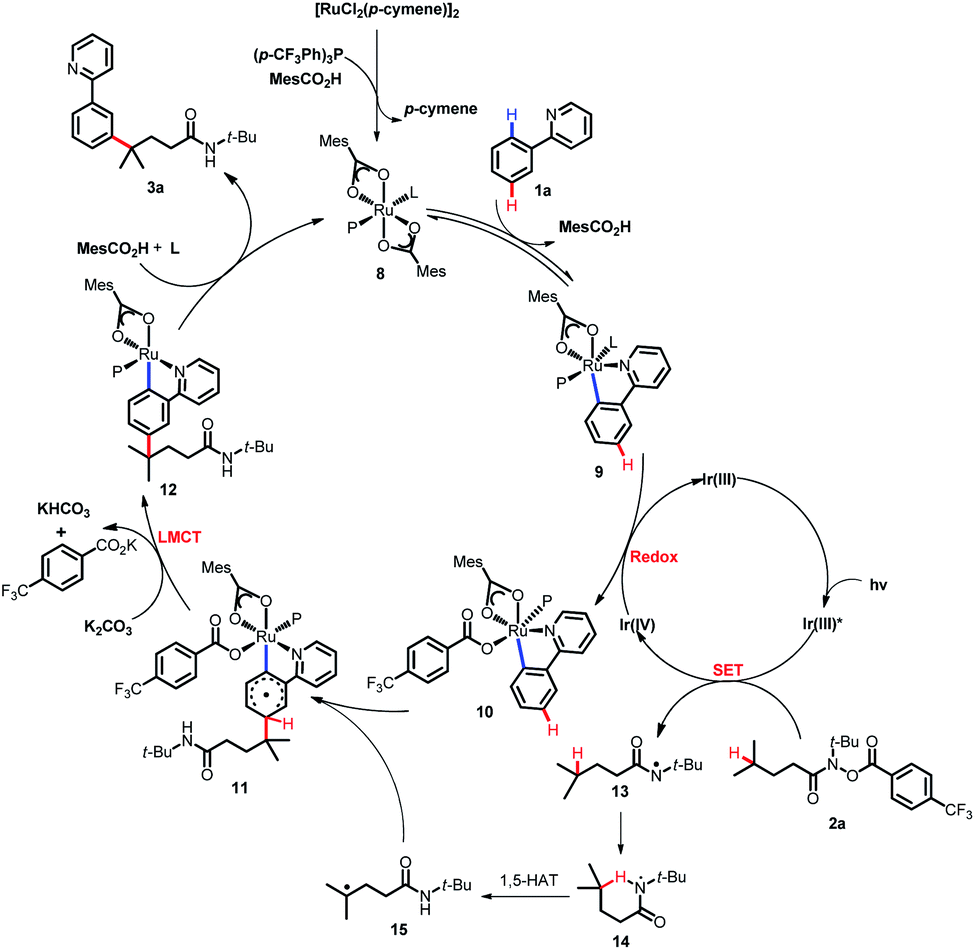
Based on the previous reports,11a,17b-d DFT calculations and control experiments, a plausible mechanism was proposed for this coupling reaction as depicted in Scheme 10. SET from the photoexcited Ir(ppy)3 catalyst (Ir(III)*) to the hydroxamide substrate 2a and subsequent N–O bond cleavage generated the nitrogen-centered radical 13 and Ir(IV). Then, intramolecular 1,5-HAT of 13 occurred, forming the carbon-centered radical 15. The cyclometalated Ru(II) complex 9 formed from the reversible carboxylate-assisted C–H bond ruthenation of the arene was then oxidized by the formed Ir(IV) complex to produce a Ru(III) complex 10, and regenerated the Ir(ppy)3 catalyst simultaneously. The formed carbon-centered radical 15 underwent CAr–H bond addition at the position para to the C–Ru(III) bond of Ru(III) complex 10, generating the species 11, which underwent subsequent rearomatization via the LMCT step to afford the Ru(II) complex 12. Eventually, the anticipated coupling product 3a was delivered by demetalation, and the catalytically active ruthenium(II) complex 8 was regenerated.
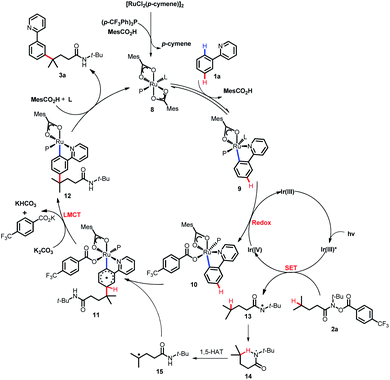 |
| | Scheme 10 Proposed mechanism. | |
Conclusions
In conclusion, we developed a novel example of metallaphotoredox-enabled regioselective CDC of dual remote C–H bonds, including unactivated γ-C(sp3)–H bonds in amides and meta-C(sp2)–H bonds in arenes, to construct challenging meta-alkylated arenes. This reaction represented a novel case of employing Ru/photoredox dual catalysis for regioselective coupling of unactivated C(sp3)–H bonds in amides and C(sp2)–H bonds arenes via a new catalytic process, and provided a new strategy to synthesize meta-functionalized arenes under mild reaction conditions. The reaction worked well with primary, secondary and tertiary C(sp3)–H bonds in amides and a broad range of arenes were well tolerated. Moreover, excellent regioselectivity was observed in terms of inert C(sp3)–H bonds in hydroxamides and meta-C(sp2)–H bonds in arenes. Detailed mechanistic studies, including DFT calculations and control experiments, explained the high regioselectivity of C(sp3)–H bonds and C(sp2)–H bonds, and provided a theoretical basis for this site-selective coupling reaction.
Data availability
Detailed synthetic procedures, and complete characterization data for all new compounds can be found in the ESI.†
Author contributions
Methodology, H.-C. L. and X. K.; investigation, H.-C. L. and X. K.; writing original draft, H.-C. L. and X. K; writing review & editing, X.-P. G., Y. L., Z.-J. N., X.-Y. G., X.-S. L., Y.-Z. W., W.-Y. S., Y.-C. H., X.-Y. L., and Y.-M. L.; Y. L. is responsible for the calculation studies; funding acquisition, Y.-M. L.; supervision, Y.-M. L. All authors co-wrote the paper, discussed the results, analyzed the data and commented on the paper.
Conflicts of interest
The authors declare no competing interests.
Acknowledgements
We thank the National Natural Science Foundation of China (NSF 22171114 and 21772075) and Henan Key Scientific Research Projects (22A150001) for support.
Notes and references
-
(a) W. Xie, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 20588–20593 CrossRef CAS PubMed;
(b) X. Wang, D. Leow and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 13864–13867 CrossRef CAS PubMed;
(c) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed;
(d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed;
(e) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- D. R. Stuart and K. Fagnou, Science, 2007, 316, 1172 CrossRef CAS PubMed.
-
(a) B.-J. Li, S.-L. Tian, Z. Fang and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1115–1118 CrossRef CAS PubMed;
(b) T. W. Lyons, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 4455–4464 CrossRef CAS PubMed;
(c) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837–5844 CrossRef CAS PubMed;
(d) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- G. Deng, L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 6278–6282 CrossRef CAS PubMed.
-
(a) G. Li, D. Li, J. Zhang, D.-Q. Shi and Y. Zhao, ACS Catal., 2017, 7, 4138–4143 CrossRef CAS;
(b) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817–3820 CrossRef CAS PubMed;
(c) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
-
(a) S. J. Blanksb and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef PubMed;
(b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
-
(a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef;
(b) R. S. Neale, Synthesis, 1971, 1971, 1–15 CrossRef;
(c) M. E. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef CAS;
(d) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC.
-
(a) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed;
(b) B. J. Groendyke, D. I. AbuSalim and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 12771–12774 CrossRef CAS PubMed;
(c) W. Guo, Q. Wang and J. Zhu, Chem. Soc. Rev., 2021, 50, 7359–7377 RSC;
(d) E. Nobile, T. Castanheiro and T. Besset, Angew. Chem., Int. Ed., 2021, 60, 12170–12191 CrossRef CAS PubMed;
(e) A. F. Prusinowski, R. K. Twumasi, E. A. Wappes and D. A. Nagib, J. Am. Chem. Soc., 2020, 142, 5429–5438 CrossRef CAS PubMed;
(f) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561–3565 CrossRef CAS PubMed;
(g) S. M. Rafferty, J. E. Rutherford, L. Zhang, L. Wang and D. A. Nagib, J. Am. Chem. Soc., 2021, 143, 5622–5628 CrossRef CAS PubMed;
(h) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC;
(i) L. Wang, Y. Xia, V. Derdau and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 18645–18650 CrossRef CAS PubMed;
(j) Y. Xia, K. Jana and A. Studer, Chem. - Eur. J., 2021, 27, 16621–16625 CrossRef CAS PubMed;
(k) L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed.
-
(a) A. N. Herron, D. Liu, G. Xia and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 2766–2770 CrossRef CAS PubMed;
(b) S. P. Morcillo, E. M. Dauncey, J. H. Kim, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 12945–12949 CrossRef CAS PubMed;
(c) S. M. Thullen, S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2019, 141, 14062–14067 CrossRef CAS PubMed;
(d) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef CAS PubMed.
-
(a) D.-F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897–14900 CrossRef CAS PubMed;
(b) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed;
(c) W.-J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS PubMed.
-
(a) H. Chen, W. Fan, X.-A. Yuan and S. Yu, Nat. Commun., 2019, 10, 4743 CrossRef PubMed;
(b) N. Kim, C. Lee, T. Kim and S. Hong, Org. Lett., 2019, 21, 9719–9723 CrossRef CAS PubMed;
(c) G.-X. Li, X. Hu, G. He and G. Chen, Chem. Sci., 2019, 10, 688–693 RSC.
-
(a) X. Zeng, W. Yan, M. Paeth, S. B. Zacate, P.-H. Hong, Y. Wang, D. Yang, K. Yang, T. Yan, C. Song, Z. Cao, M.-J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 19941–19949 CrossRef CAS PubMed;
(b) A. Modak, E. N. Pinter and S. P. Cook, J. Am. Chem. Soc., 2019, 141, 18405–18410 CrossRef CAS PubMed;
(c) Z. Li, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2018, 57, 13288–13292 CrossRef CAS PubMed.
-
(a) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed;
(b) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC;
(c)
J. Li, S. De Sarkar and L. Ackermann, Meta- and para-Selective C–H Functionalization by C–H Activation, in C–H Bond Activation and Catalytic Functionalization, ed. I. Dixneuf, H. Pierre and H. Doucet, Springer International Publishing, Cham, 2016, pp. 217–257 Search PubMed.
- C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677–11680 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef CAS PubMed.
-
(a) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS PubMed;
(b) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed;
(c) K. Korvorapun, R. Kuniyil and L. Ackermann, ACS Catal., 2020, 10, 435–440 CrossRef CAS;
(d) J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed;
(e) Z. Ruan, S.-K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 2045–2049 CrossRef CAS PubMed;
(f) A. Sagadevan and M. F. Greaney, Angew. Chem., Int. Ed., 2019, 58, 9826–9830 CrossRef CAS PubMed.
-
(a) X.-G. Wang, Y. Li, H.-C. Liu, B.-S. Zhang, X.-Y. Gou, Q. Wang, J.-W. Ma and Y.-M. Liang, J. Am. Chem. Soc., 2019, 141, 13914–13922 CrossRef CAS PubMed;
(b) X.-Y. Gou, Y. Li, Y.-Y. Luan, W.-Y. Shi, C.-T. Wang, Y. An, B.-S. Zhang and Y.-M. Liang, ACS Catal., 2021, 11, 4263–4270 CrossRef CAS.
- H.-C. Liu, Y. Li, X.-P. Gong, Z.-J. Niu, Y.-Z. Wang, M. Li, W.-Y. Shi, Z. Zhang and Y.-M. Liang, Org. Lett., 2021, 23, 2693–2698 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Zhi-Jie
Niu
a,
Xue-Ya
Gou
a,
Xue-Song
Li
a,
Yu-Zhao
Wang
a,
Wei-Yu
Shi
a,
Yan-Chong
Huang
a,
Xue-Yuan
Liu
c,
Zhi-Jie
Niu
a,
Xue-Ya
Gou
a,
Xue-Song
Li
a,
Yu-Zhao
Wang
a,
Wei-Yu
Shi
a,
Yan-Chong
Huang
a,
Xue-Yuan
Liu