
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganometallic catalysis in aqueous and biological environments: harnessing the power of metal carbenes
Sara
Gutiérrez
 ,
María
Tomás-Gamasa
* and
José Luis
Mascareñas
*
,
María
Tomás-Gamasa
* and
José Luis
Mascareñas
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15705 Santiago de Compostela, Spain. E-mail: maria.tomas@usc.es; joseluis.mascarenas@usc.es
First published on 16th May 2022
Abstract
Translating the power of transition metal catalysis to the native habitats of enzymes can significantly expand the possibilities of interrogating or manipulating natural biological systems, including living cells and organisms. This is especially relevant for organometallic reactions that have shown great potential in the field of organic synthesis, like the metal-catalyzed transfer of carbenes. While, at first sight, performing metal carbene chemistry in aqueous solvents, and especially in biologically relevant mixtures, does not seem obvious, in recent years there has been a growing number of reports demonstrating the feasibility of the task. Either using small molecule metal catalysts or artificial metalloenzymes, a number of carbene transfer reactions that tolerate aqueous and biorelevant media are being developed. This review intends to summarize the most relevant contributions, and establish the state of the art in this emerging research field.
1. Introduction
Organometallic catalysis has revolutionized the way in which chemists conceive organic synthesis. The use of transition metals as catalysts transcended classical synthetic methodologies, and has allowed chemical transformations that otherwise would not be feasible.1 Most of these organometallic reactions have been carried out in organic solvents, and usually under air and water-free conditions to avoid catalyst deactivation and the formation of side products. The fact that many organic substrates are not soluble in water has further averted the use of aqueous solvents in organic and organometallic reactions.However, there has been an increasing demonstration that many organometallic complexes and intermediates do tolerate the presence of water. Indeed, a huge number of water-compatible organometallic reactions have been recently developed.2 Work in the field has been further impelled by the growing interest in developing sustainable synthetic technologies, and by the observation that the presence of water can even accelerate some reactions.3
These advances have led scientists to wonder if organometallic reactions could be carried out in biological and even living settings, as this could open new opportunities in cell biology and biomedicine. However, the translation of organometallic chemistry to biological habitats is not straightforward, and presents important challenges. First, a cellular milieu cannot be equated to a typical aqueous solvent, owing to its gel-like nature and intrinsic crowded environment. Furthermore, the presence of numerous biological components can promote the deactivation of the metal reagents or inhibit the catalytic cycles. Moving to living environments is further hampered by potential toxicity and transport issues, and by the low concentration of reagents and reactants.
Nonetheless, with the advent of bioorthogonal chemistry, as coined by Bertozzi and coworkers,4 an increasing number of organic reactions, including metal-catalyzed processes, have been demonstrated to work in biological and living environments.5,6 A key discovery that sparked the field is the well-known copper-catalyzed azide alkyne cycloaddition (CuAAC), paradigm of click chemistry, which is usually carried out with Cu(II) reagents and external reducing agents like ascorbate. Despite the intrinsic toxicity of these reagents, an appropriate tuning of the reaction conditions has allowed its use in cellular settings.7–11 Its impressive chemoselectivity has also led to many applications for bioconjugation and post-translational modification of proteins or nucleic acids.12,13
The CuAAC entails a typical organometallic mechanism involving oxidative cyclometallation and reductive elimination steps, and it is a fundamental reference in the field of biological organometallic catalysis. Another early (2006) report that has gained increasing recognition by the chemical biology community is the discovery by Meggers et al. that Ru(II) catalysts can promote the removal of alloc protecting groups in designed substrates, even in the presence of HeLa cells.14
Although for several years there were not many new contributions in the area, in the last decade there has been an upsurge in reports dealing with transition metal-promoted reactions in biological media, and also in living organisms like bacteria.15–20 Some of these discoveries have already found very relevant biological applications.21–24 The reactions include uncaging processes, mainly those catalyzed by ruthenium or palladium complexes, such as the cleavage of N-allylcarbamates or O-allyl/propargyl ethers,14,25–33 ruthenium catalyzed bond-forming reactions (annulations between thioalkynes and azides (RuAtAC),34,35 (2 + 2 + 2) cycloadditions36) or isomerization of allylic alcohols,37 gold-catalyzed hydroarylations,38 and even palladium promoted Sonogashira39 or Suzuki–Miyaura40–43 cross couplings.
Therefore, a wide range of organometallic transformations, from deprotection to cross-coupling, cyclization or cycloaddition reactions, have been demonstrated to be compatible with biological environments, provided appropriate metal complexes and probes are employed.
What about metal carbenes? Can their reactivity be exported to biological environments? Metal carbene transfer reactions are among the most powerful and versatile transformations in catalysis and organometallic chemistry, and have been used for many synthetic applications.44,45 Translating the rich reactivity typically exhibited by metal carbenes in organic solvents to aqueous and biological media, could open important new avenues in research at the interface between chemistry and biology.
Indeed, in the last decade, there has been an increase in the number of reactions involving bioorthogonal metal-carbene transfer processes. In some cases, it has also been demonstrated that this chemistry can be performed in cells or in bacteria. In this review, we intend to cover the most significant contributions in this topic, from bioconjugations and chemoselective modifications of biopolymers to synthetic transformations of small molecules in biological settings, including bacteria or mammalian cells.
2. Catalysis involving metal carbene transfer reactions
Carbenes are highly reactive chemical entities that possess a neutral divalent carbon atom with six electrons on its valence shell. According to the spin configuration of the two nonbonding electrons in the ground state, we can consider two types of carbenes: singlet carbenes, with both electrons occupying the sp2 orbital with antiparallel spins, or triplet carbenes, with two electrons with parallel spins, occupying the sp2 and the pz orbital (Fig. 1a).46 | ||
| Fig. 1 (a) Electronic structure of carbenes. (b) Classification of metal carbenes based on the nature of the substituents adjacent to the carbene. (c) Some transformations of metal carbenes carried out in aqueous media. EDG = electron donating group; EWG = electron withdrawing group. | ||
A wide range of precursors have been employed for the generation of carbenes, such as tosylhydrazones, diaziridines, triazoles, cycloheptatrienes, alkynes or ynamides, among others.47 However, the most widely used carbene precursors are diazo compounds, which are reasonably stable but can readily decompose to carbenes under mild reaction conditions.44,48 The resulting free carbenes tend to be highly reactive, and therefore difficult to use in chemical transformations; however, upon appropriate coordination to transition metals they give metal carbenes, which present a more controllable and rich reactivity. The chemical properties of these metal complexes depend on the spin configurations of the carbene, and on the overlap with the metal orbitals.
Different criteria have been used to classify metal carbenes. Historically, they have been categorized as Fischer and Schrock carbenes, depending on the metal complex and the type of substituents at the carbene; but in the context of this review, it is useful a classification based on the nature of the substituents adjacent to the reactive carbene centre. Acceptor/acceptor (A/A) and acceptor (A) carbenes are highly reactive due to the lack of stabilization of the electrophilic carbene centre. In contrast, the presence of donor groups stabilizes the metal carbene centre and attenuates the reactivity. This is the case of donor/acceptor (D/A) carbenes, which have been widely used due to their good balance between reactivity and stability (Fig. 1b).49,50
The rich reactivity of metal carbenes is mostly related to the charge distribution along the metal–carbon bond and the electrophilicity of the carbene carbon, allowing its reaction with a variety of nucleophiles. Within the broad spectrum of reactions enabled by metal carbenes, most common transformations include cyclopropanations of alkenes,51–53 C–H functionalizations,49,54–56 insertions into X–H bonds (X = O, N, S, Si),57,58 the formation of ylides59–61 (and subsequent rearrangements) or cycloaddition processes44,61 (Fig. 1c). This reactivity is different from that exhibited by carbene precursors, usually diazocarbonyl compounds, which tend to decompose and/or engage in rearrangement or dimerization reactions.
The reactions of metal carbenes with substrates exhibiting non-polar bonds (i.e. cyclopropanation of alkenes or insertions into C–H or Si–H bonds), tend to proceed through a concerted mechanism,62 while in the case of insertions into polar X–H bonds a stepwise ylide mechanism might be operative. Beyond these general features, the characteristics of the transition metal complex play a key role in the mechanistic pathway.58
Transition metal-catalyzed carbene transformations have been well studied in organic solvents, with rhodium, copper and iron being the most explored metals. It is important to note that besides carbene transfer reactions from diazo or related precursors, metal carbenes can also be used as catalysts, e.g. in metathesis processes.
3. Metal-promoted carbene transfer reactions in aqueous media
The rich chemistry of metal carbenes has been explored essentially in organic solvents, and under water-free conditions, owing to the assumption that water might react with the carbene. However, as mentioned above, the reactivity of metal carbenes can be modulated by playing with the metal ligands and the substituents at the carbene atom, as well as with the type of substrates and reaction conditions; and thus, it can be made compatible with aqueous media. Indeed, in recent years there have been important contributions that demonstrate the viability of metal-mediated carbene transformations in aqueous media, and also in biologically relevant settings. Herein we present a summary of the most significant developments. The reactions have been organized according to the type of substrate (small molecules or biopolymers) and, where relevant, to the type of transformation (cyclopropanation, C–H insertion, X–H insertion or ylide formation). Metal carbenes have also been used as catalysts for olefin metathesis in aqueous and biorelevant environments. This topic has been covered in specific reviews,63 and thus herein we will only mention some relevant examples performed in cellulo (Section 5.2).3.1. Transformations of small molecules
The first example of a metal-catalyzed cyclopropanation performed in the presence of large amounts of water was reported by the Nishiyama group in 2001.64 The authors used EDA as carbene precursor, and a ruthenium complex exhibiting a water-soluble chiral ligand [hm-pybox: bis(hydroxymethyldihydrooxazolyl)pyridine] as catalyst, in a biphasic aqueous/organic milieu. The reaction yields the cyclopropane product with high levels of enantioselectivity (up to 94%, Fig. 2, trans/cis stereoselectivity up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Curiously, the presence of water had a marked influence in the enantioselectivity, which changed from 8% ee in pure THF to a 78% ee in THF/H2O mixtures. This result was attributed to a solvation effect of water around the hydroxy groups of the hm-pybox.
:3). Curiously, the presence of water had a marked influence in the enantioselectivity, which changed from 8% ee in pure THF to a 78% ee in THF/H2O mixtures. This result was attributed to a solvation effect of water around the hydroxy groups of the hm-pybox.
 | ||
| Fig. 2 Metal catalyzed cyclopropanations. Top: General reaction scheme. Bottom-left: Asymmetric Ru-catalyzed cyclopropanation reported by Nishiyama. Bottom-right: Rh-catalyzed cyclopropanation reported by Charette and Wurz. | ||
In 2002 Charette and Wurz reported a different approach to perform similar cyclopropanations. They found that the combination of hydrophobic rhodium catalysts such as dirhodium(II) octanoate (Rh2(Oct)4) with hydrophobic alkenes allows the formation of “small alkene/catalyst beads or micelles” in water. Then, a slow diffusion of EDA through the surface of the beads enabled a controlled formation of the desired cyclopropanated products in high yields (Fig. 2). Noticeably, water-soluble rhodium carboxylates (Rh2(OAc)4 or Rh2(O2CCF3)4) were inefficient, leading to low yields. An asymmetric version of this reaction was also studied using ruthenium(II) and cobalt(II) chiral catalysts.65
A family of catalysts that has been extensively employed for cyclopropanation reactions in organic solvents are metalloporphyrins, an interesting type of metal complexes that is also present in certain metalloenzymes with oxidative functions. In 2008, Simonneaux and coworkers prepared water-soluble ruthenium and iron porphyrins by the introduction of sulfonate groups, and used them as catalysts for the asymmetric cyclopropanation of styrene in water, with EDA as the carbene precursor (Fig. 3a). They obtained the desired products in yields up to 85%, with high diastereoselectivity (trans/cis up to 96/4) and good enantioselectivity for the trans isomer (83%).66
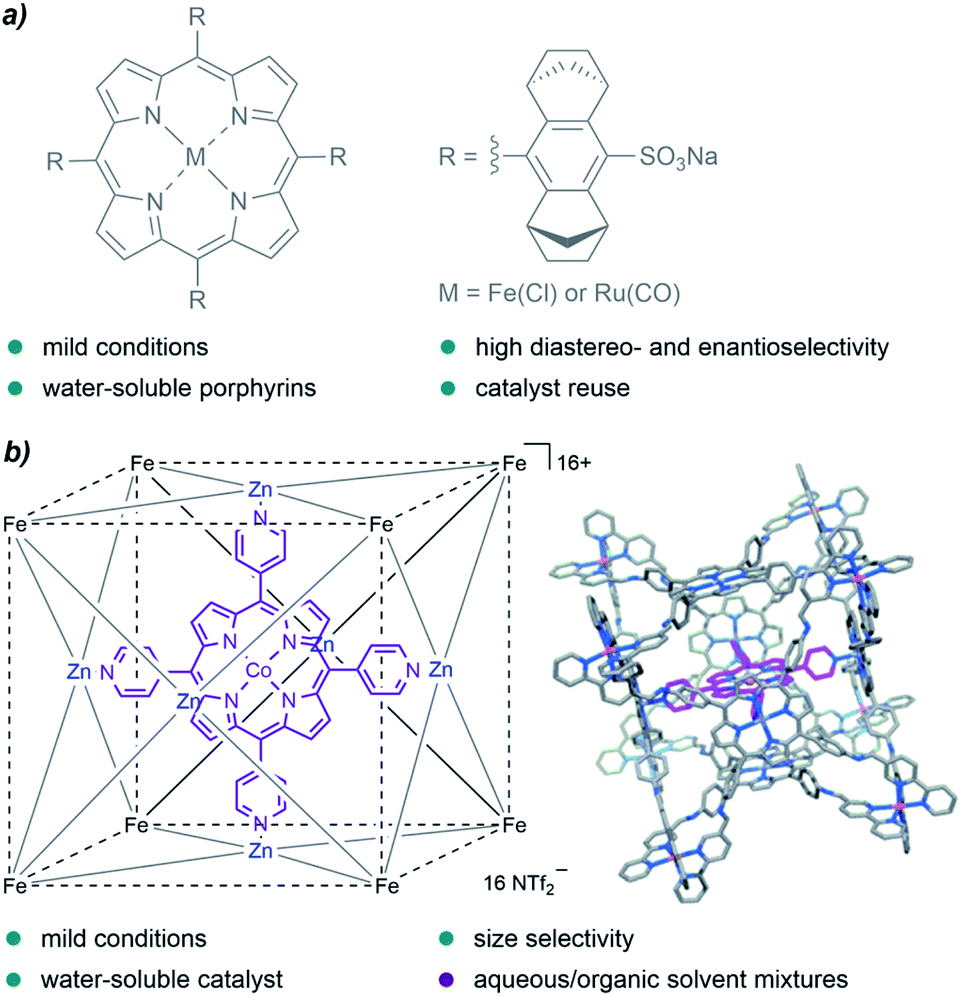 | ||
| Fig. 3 (a) Water-soluble Fe and Ru porphyrins developed by Simonneaux and coworkers for asymmetric cyclopropanations in water. (b) Cobalt porphyrin “ship in a bottle” developed by de Bruin and coworkers, reproduced from ref. 67 with permission from Wiley-VCH GmbH, copyright 2014. | ||
In 2014, de Bruin and co-authors reported the first example of size-selective cyclopropanation reactions using a soluble “molecular ship-in-a-bottle catalyst”. The bio-inspired supramolecular cage constituted by a cobalt-porphyrin catalyst encapsulated into a cubic M8L6 cage (Fig. 3b) showed excellent activity in the cyclopropanation of styrene derivatives using water/acetone mixtures (5:1).67 The reaction afforded the products in yields up to 88%, under mild reaction conditions (50 °C) and low catalyst loadings (0.25 mol%, TONs up to 351). Remarkably, the porous cage-catalyst allowed for size selectivity; bulky substrates led to lower yields likely due to the slower migration through the cage pores.
Another interesting approach based on the use of nanoreactors was reported in 2014 by van Hest et al., who immobilized chiral bis(oxazoline)–copper catalysts inside a polymersome membrane to perform asymmetric cyclopropanations of alkenes in water. Interestingly, only hydrophobic alkenes underwent the cyclopropanation, likely because of their ability to localize into the active site of the polymer.68
All these results confirm the compatibility of specific metal carbenes with water, which even plays an important role in the reactivity and selectivity of the processes by eliciting hydrophobic effects.
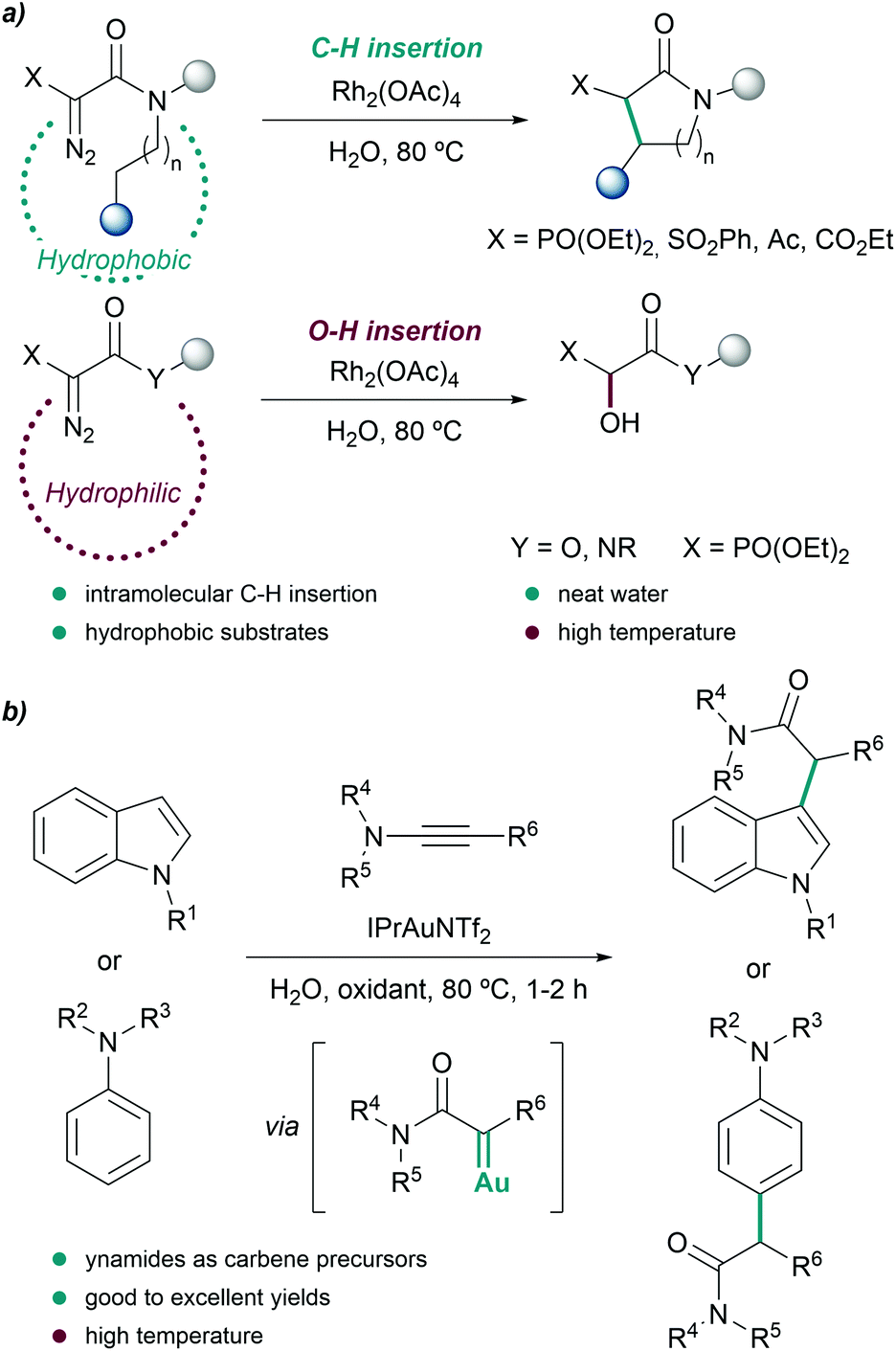 | ||
| Fig. 4 C–H insertion reactions in water. (a) Intramolecular insertion of rhodium carbenoids into aliphatic C–H bonds reported by Afonso and coworkers. (b) Functionalization of indoles and anilines by in situ generated α-oxo gold carbenes. | ||
Further studies revealed that the use of more hydrophobic catalysts such as Rh2(Oct)4 partially avoided or completely suppressed the hydroxylation reaction, even when less hydrophobic substrates were employed.70
Another interesting metal-carbene mediated C–H insertion was proposed by the group of Ye, using ynamides as carbene precursors and gold catalysts. In the presence of an oxidant, the reaction generates α-oxo gold carbenes that can be trapped by indoles or anilines (Fig. 4b). The intermolecular reactions were carried out at 80 °C using water as reaction media, providing the C-alkylation products in excellent yields, after 1–2 hours.71 Remarkably, the presence of water suppressed undesired over-oxidations of the gold carbene.
:4) mixture as reaction media, with 90% conversion after 1 h. In this case, 10% of the double insertion product was detected.72
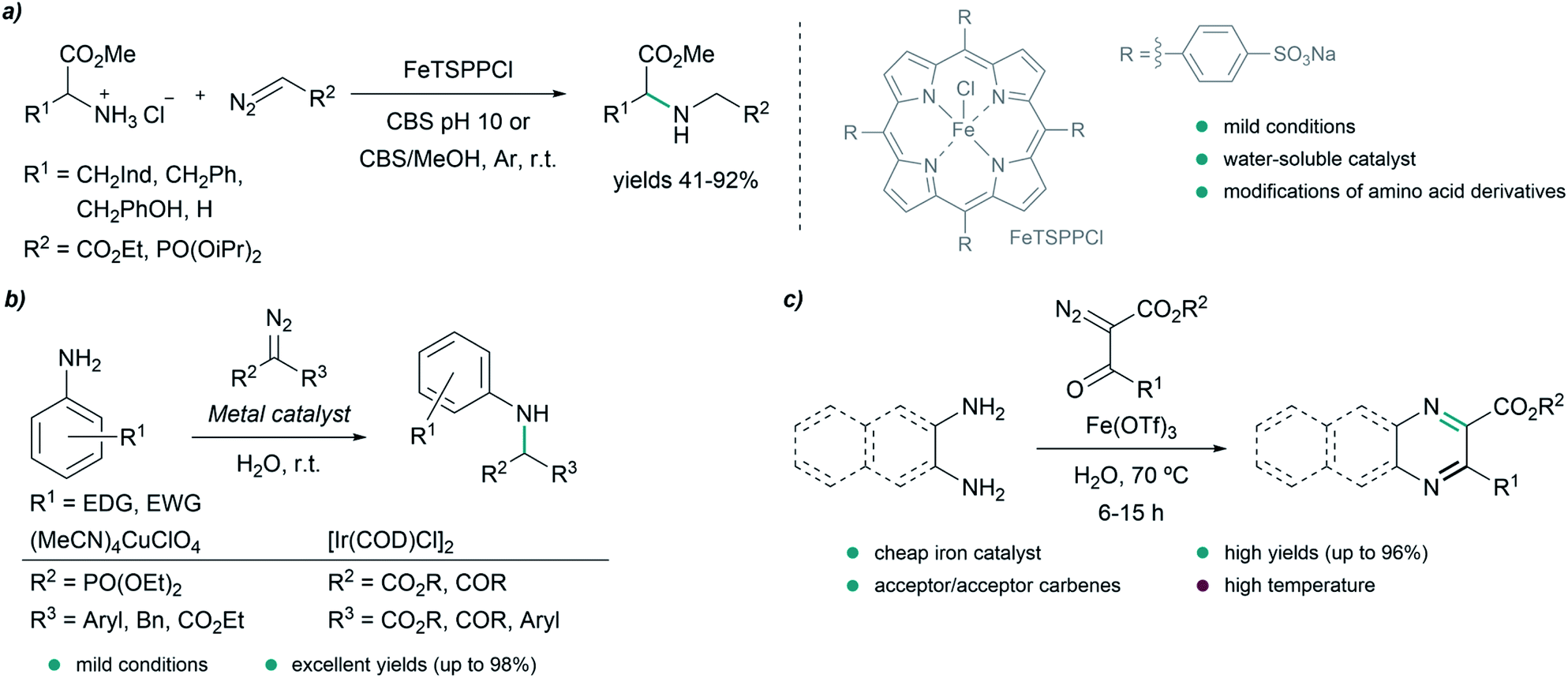 | ||
| Fig. 5 N–H insertion reactions in water. (a) N–H insertion of iron carbenoids into amino acid derivatives catalyzed by a water soluble iron porphyrin in citrate buffer (CBS) reported by Simonneaux. (b) Carbene insertions into N–H bonds of anilines catalyzed by copper or iridium complexes reported by Sivasankar. (c) Iron catalyzed annulation of 1,2-diamines and diazodicarbonyl compounds reported by Lee. | ||
In 2016, Sivasankar and coworkers reported an efficient synthesis of α-amino phosphonates in water via insertion of copper(I) carbenoids into the N–H bond of anilines under mild reaction conditions (Fig. 5b). The best results were obtained with (CH3CN)4CuClO4 as catalyst, yielding the desired N–H insertion products in short reaction times (from 15 min to 2 h in most cases).73 Later on, in 2017, they found conditions for the N–H insertion of carbenes exhibiting different electronic properties. AgOTf was the best choice for donor/donor carbene precursors, providing moderate yields after 5 minutes of reaction, while for donor/acceptor carbenes containing aryl and ester groups, Pd2(dba)3 led to the product in 90% yield after 30 minutes. In the case of acceptor/acceptor carbenes, the most efficient catalyst was found to be [Ir(COD)Cl]2, affording the N–H insertion product in 81% yield after 12 h. The authors studied in more detail the reaction for acceptor/acceptor carbenes, and used their method to synthesize a series of aniline-derivatives in good yields. However, in the case of aliphatic amines, such as benzylamine, and heteroaromatic amines like 2-aminopyridine, only traces amounts of the N–H insertion products were detected.74
In 2016, Lee and coworkers reported an elegant strategy for the synthesis of quinoxalines, pyrazines and benzoquinoxalines through the annulation of 1,2-diamines and diazodicarbonyls in water at 70 °C, using Fe(OTf)3 as catalyst (Fig. 5c). The process is initiated by the insertion of an iron carbenoid into the N–H bond of the diamine, followed by cyclization and oxidative aromatization, to afford the desired heterocycles in excellent yields.75
A relevant report by the group of Kwak in 2016 described the use of a Cu(I)-zeolite as a heterogeneous catalyst for the N–H insertion of α-diazoesters into substituted anilines. The reaction proceeds under mild conditions in a mixture of H2O/t-BuOH (1:1). The Cu(I)-zeolite catalysts were stable and could be recycled, maintaining their activity over four reaction runs.76
Gross and coworkers reported N–H insertion reactions of EDA and ethyl diazopropionate (EDP) on anilines in aqueous solutions, catalyzed by iron corrole conjugated albumins. The authors did not observe enantiomerically enriched products in any of the cases, and did not comment on the potential accelerating effect of the protein.77
In some transformations, the insertion of water into the metal carbenes is the desired process. This is the case of the work of Wang et al., who developed a water insertion/oxacyclization cascade of o-acetylenyl-substituted phenyldiazoacetates catalyzed by the gold(I) complex IPrAuCl (Fig. 6a). The process takes place through a sequence of gold carbene formation/water trapping and alcohol-alkyne 6-endo-dig cyclization. The transformation can take place in neat water providing mixtures (2:1) of 5-endo-dig and 6-endo-dig cyclization products; however, the best conditions consisted of using a mixture DMF/H2O (1:1) at 80 °C for 24 h, providing 1H-isochromenes in moderate to good yields (64–82%).78
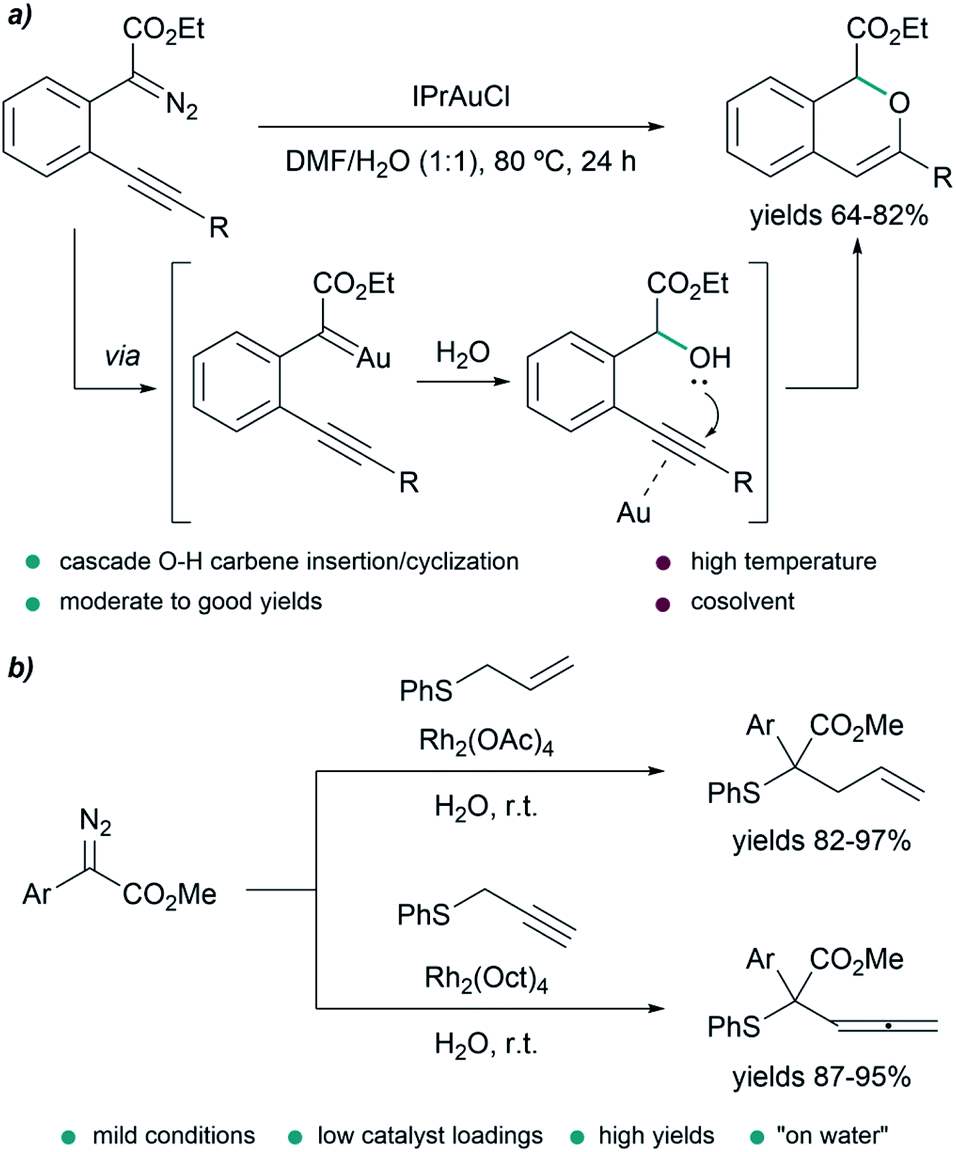 | ||
| Fig. 6 (a) Gold-catalyzed tandem O–H insertion/cyclization in DMF/H2O. (b) Doyle–Kirmse reaction of in situ generated sulfonium ylides. | ||
3.2. Chemoselective reactions of peptides and proteins
The above examples demonstrate that, in contrast to preestablished assumptions, the reactivity of metal carbenes can be harnessed in the presence of aqueous mixtures. This is especially relevant for their potential use to modify biological polymers like peptides or proteins, as most of them require aqueous solvents for an appropriate handling and solubility. As early as in 1966 it was shown that diazoketones, in the presence of copper salts, were able to inactivate pepsin, apparently due to the selective modification of a carboxylate residue in the active site of the protein.80,81However, truly designed protein modification reactions using metal carbenes were not reported until almost 40 years later. In 2004, Antos and Francis demonstrated the viability of using rhodium carbenoids for the selective modification of tryptophan side chains in myoglobin and subtilisin (Fig. 7a). The reaction was performed in water/ethyleneglycol (8:2, in the millimolar concentration range), using α-diazo esters and Rh2(OAc)4, and in the presence of hydroxylamine, providing a mixture of indole N–H and C–H insertion products. The addition of hydroxylamine dramatically enhanced the reactivity of the catalyst, presumably by binding to the distal rhodium of the bimetallic catalyst and stabilizing the reactive intermediates.82 Subsequent studies using N-(tert-butyl)hydroxylamine as additive allowed modification of peptides and proteins (lysozyme and FKBP mutants) selectively on tryptophan residues exhibiting solvent-accessible indole side chains, at mild pH.83
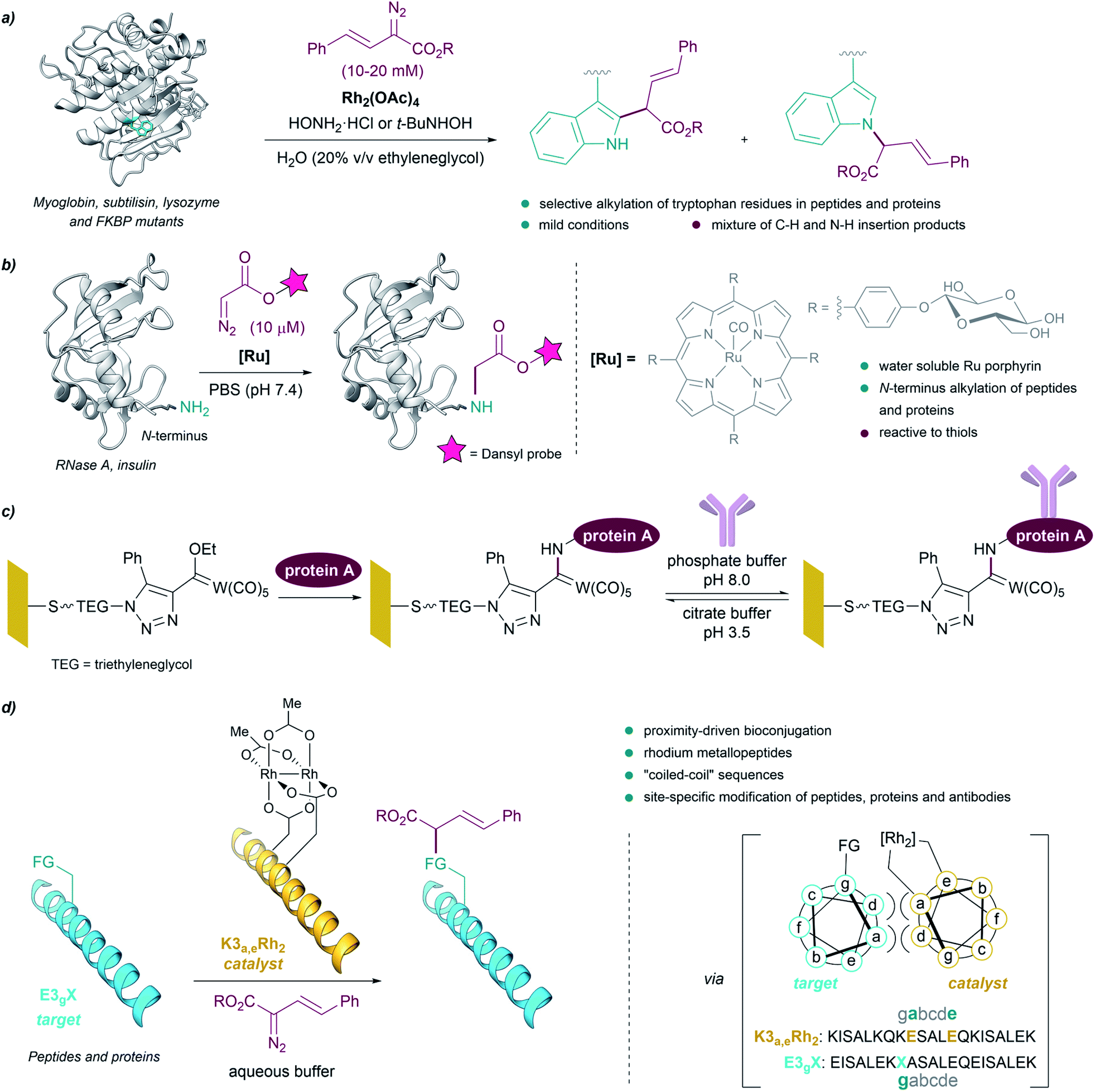 | ||
| Fig. 7 (a) Bioconjugation of peptides and proteins based on the selective alkylation of tryptophan residues using rhodium carbenoids reported by Antos and Francis. (b) Bioconjugation of proteins based on the alkylation of the N-terminus. (c) Bioconjugation using Fischer carbenes on gold or glass surfaces. (d) Proximity-driven bioconjugation using rhodium metallopeptides developed by Ball for the site-specific modification of peptides, proteins and antibodies. The figure shows the structure of subtilisin Carlsberg ((a) PDB ID: 1SBC, tryptophans in blue) and RNase A ((b) PDB ID: 3A1R, N-terminus in blue). | ||
In 2013 Ball demonstrated the feasibility of performing selective cysteine modification under mild reaction conditions, using a N-(tert-butyl)hydroxylamine-containing buffer employed by Antos and Francis, Rh2(OAc)4 and a biotin-tethered diazo substrate. The authors applied these reaction conditions to a mixture of proteins, and observed the modification of proteins with accessible cysteine thiols, while those with buried or oxidized cysteine residues remained unmodified.84
Che et al. reported the first example of a metalloporphyrin-catalyzed carbene transfer for the selective modification of proteins. A designed water-soluble ruthenium glycosylated porphyrin catalyst [RuII(4-Glc-TPP)(CO)] (1,4-Glc-TPP = meso-tetrakis(4-(β-D-glucosyl)phenyl)porphyrinato dianion) was employed for different carbene transfer reactions in aqueous media (inter- and intramolecular cyclopropanation, Doyle–Kirmse reaction, N–H insertion). Interestingly, the catalyst proved to be effective for the selective alkylation of the N-terminus of peptides and proteins using a fluorescent-tethered diazo compound (Fig. 7b). However, when the protein presented free thiols (cysteines) in its structure, the insertion of the metal carbene took place into the S–H bond rather than in the terminal N–H bond.85
Interestingly, Fischer carbenes have also been employed for bioconjugation reactions. Jaouen and coworkers performed a organotungsten labelling of bovine serum albumin using a Fischer carbene with the general formula L(CO)4W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OR1)R2.86 The authors observed a side-chain specific labelling in the amino group borne of some lysine residues, leading to stable aminocarbene adducts. On the other hand, Sarkar et al. modified self-assembled monolayers (SAMs) on gold or glass with Fischer carbenes to immobilize protein A on surface, through reaction of pendant lysine residues with the electrophilic Fischer carbene (Fig. 7c).87
C(OR1)R2.86 The authors observed a side-chain specific labelling in the amino group borne of some lysine residues, leading to stable aminocarbene adducts. On the other hand, Sarkar et al. modified self-assembled monolayers (SAMs) on gold or glass with Fischer carbenes to immobilize protein A on surface, through reaction of pendant lysine residues with the electrophilic Fischer carbene (Fig. 7c).87
Despite these successes, the reactions tend to be low yielding, and chemoselectivities are, in many cases, modest. As an alternative to these methods, Ball and coworkers developed an elegant strategy based on proximity-driven bioconjugations that combines molecular recognition with metallocarbene reactivity. This approach is based on the use of a dirhodium metallopeptide made of a rhodium(II) catalyst conjugated to a peptide that is able to recognize another specific protein (via a coiled-coil assembly, Fig. 7d).88
Using this approach, the authors performed site-specific modification of peptides and proteins at physiological pH, and in biologically relevant buffers.89–91 The use of biotin–diazo conjugates as reagents allowed for affinity tagging of the target proteins. They were even able to use designed metallopeptide catalysts to perform selective protein modifications in E. coli lysate. Moreover, they developed a strategy for the site-specific functionalization of antibodies based on the use of a hexarhodium metallopeptide catalyst with affinity for the Fc fragment of the antibody.92 Based on molecular recognition of the Fc region, this approach enabled the introduction of orthogonal alkyne handles into mono- or polyclonal antibodies, opening the door to the quick production of antibody conjugates.
3.3. Chemoselective modification of nucleic acids
Pioneering work by Gillingham et al. demonstrated the potential of metal carbenes to perform selective post-synthetic modifications of oligonucleotides. In their first report, in 2012, they used Rh2(OAc)4 to catalyze the structure-selective N–H insertion of in situ generated rhodium(II) carbenoids into exocyclic amine groups of purine nucleobases under mild conditions. The reaction was carried out in aqueous buffers using donor/acceptor substituted carbenes at concentrations of 50 mM. This approach allows strategic targeting of solvent exposed nucleobases in double-stranded nucleic acids, as well as in single strands, bulge regions or overhangs (Fig. 8a, left).93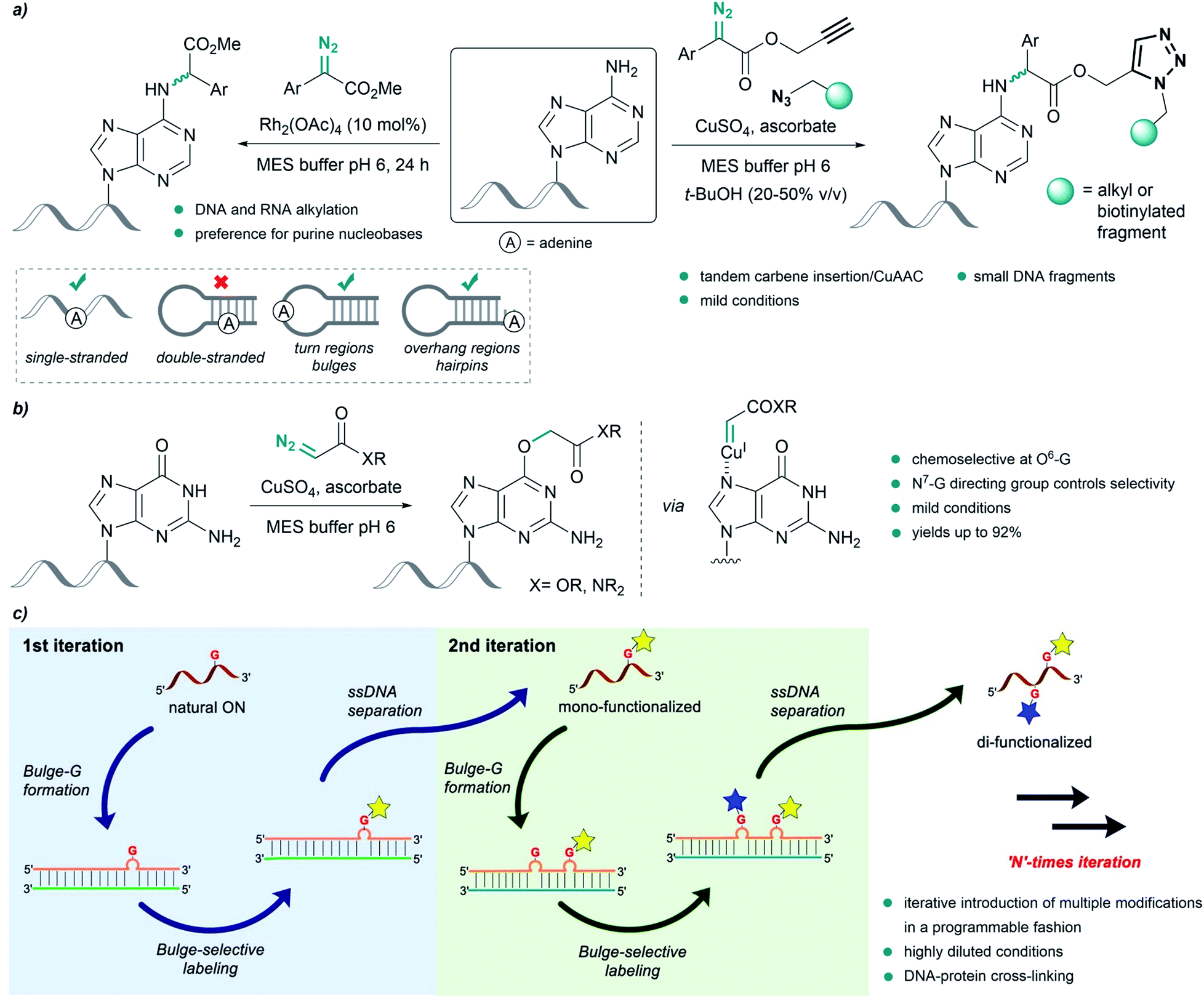 | ||
| Fig. 8 Nucleic acid alkylation using metal carbenes. (a) Structure-selective catalytic alkylation of DNA and RNA (left) and tandem carbene N–H insertion/CuAAC (right) reported by Gillingham. (b) Chemoselective alkylation at guanine O6-G using copper carbenes reported by Gillingham. (c) Site-selective functionalization of oligonucleotides using rhodium carbenes reported by Park, reproduced with permission from ref. 96, licensed under a Creative Commons Attribution (CC BY) license. It is attributed to Park, and the original version can be found here (https://www.nature.com/articles/s41467-021-21839-4). | ||
Shortly thereafter, they demonstrated the effectiveness of Cu(I) salts, generated from copper sulfate and sodium ascorbate, to catalyze a similar insertion of donor/acceptor carbenes into the N–H bond of adenine in MES buffer. The diazo compound was further modified to contain an alkyne or azide handle, thus developing a tandem carbene insertion/CuAAC reaction (Fig. 8a, right).94
Interestingly, the use of acceptor carbenes generated from α-diazoesters or diazoacetamides and copper(I) led to chemoselective alkylation of the O6 position in guanine (O6-G) in mono- and oligonucleotides, attributed to the pre-coordination of the catalyst to the N7 of guanine (Fig. 8b).95 However, with complex oligonucleotides containing multiple chelation sites, reaction rates are low, likely due to unproductive catalyst sequestration.
Very recently, Park and coworkers reported an elegant site-selective post-synthetic modification of oligonucleotides at unpaired guanosines. The use of a coordinatively saturated Rh(I) catalyst like [Rh(COD)Cl]2 circumvented the chelation issues related to the deactivation of the catalyst observed by Gillingham, while maintaining the chemoselectivity towards base-unpaired guanosines in single- and double-stranded oligonucleotides. They exploited this feature and introduced guanosine-bulge loops in duplexes, which resulted in high regioselectivity, and allowed iterative introduction of multiple modifications in a programmable fashion (Fig. 8c).96 The utility of this approach was further showcased by performing DNA-protein cross-linking in cell lysate.
4. Carbene transfer reactions catalyzed by metallobiopolymers
A major challenge in research at the interface of catalysis and chemical biology is the design of artificial metalloenzymes (ArMs) that can reproduce the catalytic efficiency and selectivity of natural enzymes, but promoting new-to-nature reactions. In recent years, and largely thanks to the advent of protein engineering, and specially directed evolution, there has been impressive progress in the field, and a variety of catalytic metalloproteins capable of performing transition-metal catalyzed reactions have been developed.97–100 These reactions have been carried out mainly in vitro, in aqueous buffers, but in recent years a number of metalloenzymes that also work in bacteria have been reported.101–103 Importantly, some of the most relevant contributions in this area deal with carbene transfer reactions, mainly because of the ability of metalloheme-containing proteins to generate metal carbene intermediates.The design and development of ArMs has been largely based on repurposing natural metalloenzymes or on anchoring a metal cofactor to a protein-binding pocket. In the first approach (Fig. 9, left), a natural metalloenzyme is modified by directed mutagenesis and/or by introducing a non-natural metal-containing cofactor. In the second approach (Fig. 9, right), the metal can be either covalently bound to protein residues, or anchored into a protein cavity through supramolecular association (e.g. biotin-modified catalysts in streptavidin, strategy primarily developed by Ward).
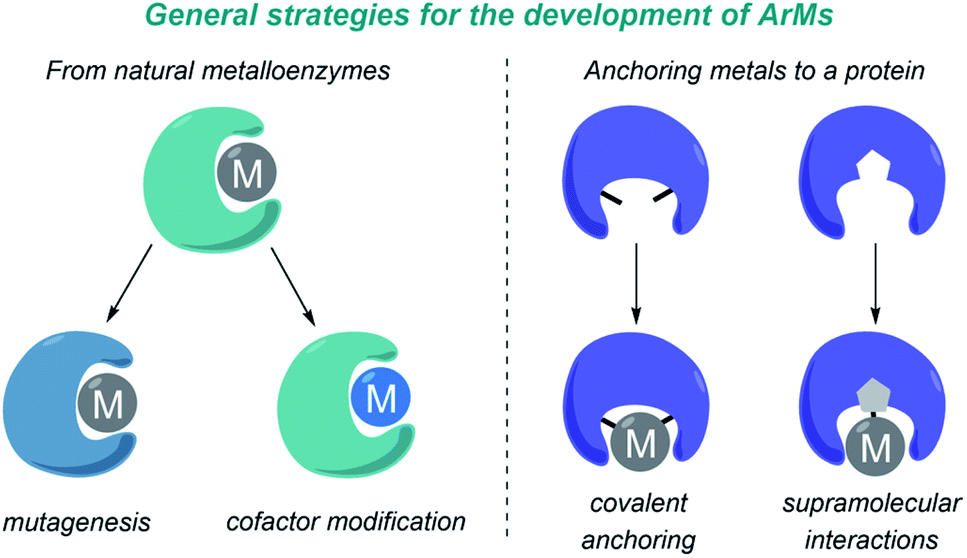 | ||
| Fig. 9 Main strategies for the development of ArMs. | ||
Although the overwhelming number of reports of the use of ArMs to perform carbene transfer reactions cannot be covered in this review, we present some selected contributions. In this first section, we will mainly focus on the strategies employed to prepare new ArMs and their reactivity profiles, while the following section spotlights the compatibility of the systems with living cells. Besides, we will also disclose some examples of the use of nucleic acids instead of protein as coatings for the catalytic metals in carbene transfer processes.
4.1. Reactions promoted by heme-containing artificial metalloenzymes
Arnold and coworkers, envisioning the structural similarity between the iron-oxo intermediate involved in cytochrome P450-catalyzed oxidations and an iron carbene, repurposed the system to work as a carbene transferase. In 2013, using as benchmark reaction the cyclopropanation of styrene with EDA, they demonstrated that variants of P450BM3 from Bacillus megaterium can promote the carbene transfer, forming cyclopropane products with high levels of diastereo- and enantioselectivity.104,105 Additional studies using directed evolution to introduce mutations at the binding pocket of different P450 cytochrome variants allowed to tune the selectivity and activity. Therefore, in the last decade, the group has successfully engineered new variants that catalyze cyclopropanation106,107 and cyclopropenation108 reactions, as well as N–H,109 Si–H,110 B–H111 and C–H112,113 insertions, with high rates and turnovers, and exquisite regio- and stereocontrol.
In 2014, Fasan et al. extended this strategy to other heme proteins, developing evolved variants of myoglobin (Mb) and horseradish peroxidase. Furthermore, they demonstrated that it is possible to replace the heme group by porphyrin cofactors with abiotic metals (i.e. Mn or Co), to give metalloenzymes with similar levels of activity and selectivity.114 Using directed evolution, they reported Mb mutants that successfully catalyze the cyclopropanation of a variety of aryl-substituted olefins with EDA, with excellent levels of diastereo- and enantioselectivity.115–120 They also developed variants that catalyze carbene transfer reactions such as insertions into N–H,121–123 S–H124 or Si–H125 bonds, and ylide formations.126
Other pivotal contributions in the field were made by the laboratory of Hartwig in 2016, by replacing iron with iridium (exhibiting a methyl group as axial ligand) in myoglobins. After directed evolution, they identified versions, such as [Ir(Me)–PIX–Mb], that display carbene transfer activity and can even promote stereoselective Csp3–H bond insertions.127 The same strategy was applied to variants of the cytochrome P450 enzyme CYP119, to give ArMs (Ir(Me)–MPIX–CYP119) with high activity and excellent selectivity (C–H bond insertions with up to 98% enantiomeric excess, 35000 turnovers, and 2550 per hour turnover frequency, Fig. 10).128 In addition to the improved rates and the enantioselectivities that can be achieved, some ArMs allow challenging regioselectivities to be controlled, such as in C–H insertions in a range of 4-substituted phthalan derivatives.129
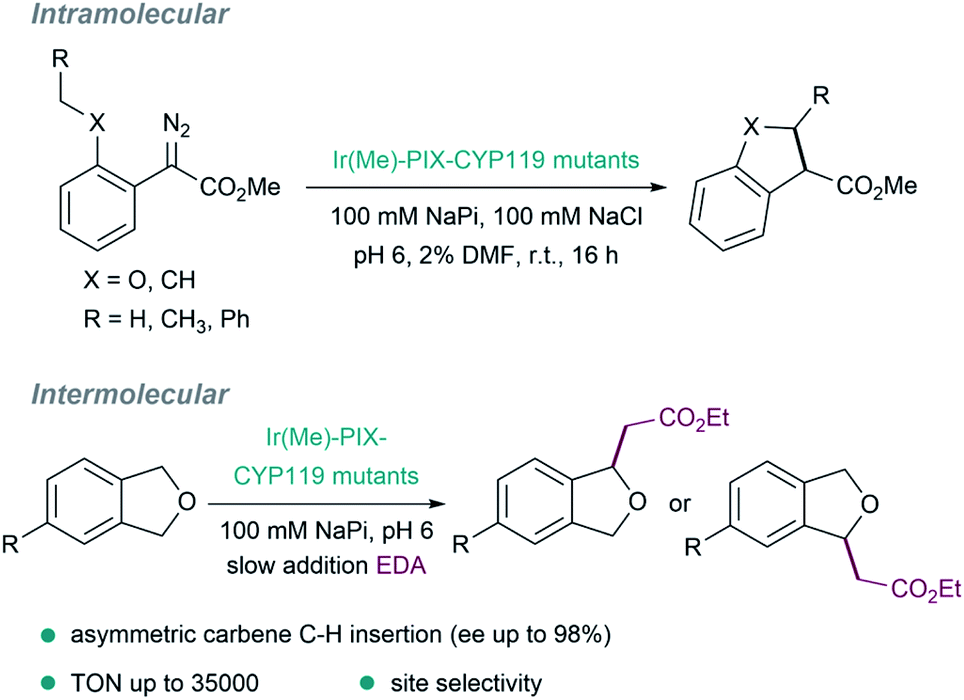 | ||
| Fig. 10 Insertion of carbenes into C(sp3)–H bonds catalyzed by several metal–PIX reconstituted myoglobins containing a single mutation at the axial residue. | ||
It is also possible to build ArMs with modified heme scaffolds, such as the one developed by Lehnert with mesoporphyrin IX (RuMpIX) incorporated into wild-type (wt) Mb and Mb mutants (Fig. 11). However, these modified proteins exhibited moderate catalytic activity towards the cyclopropanation of styrene derivatives, and in N–H modifications of aniline derivatives.130 Hayashi et al. selected an iron porphycene cofactor to reconstitute myoglobin, and demonstrated that the modified protein significantly accelerates a catalytic cyclopropanation reaction, which turned to be 615-fold faster than that obtained using native myoglobin (Fig. 11). Interestingly, the authors performed mechanistic studies and reported the first spectroscopic observation of the active metallocarbene species in this type of reconstituted proteins.131
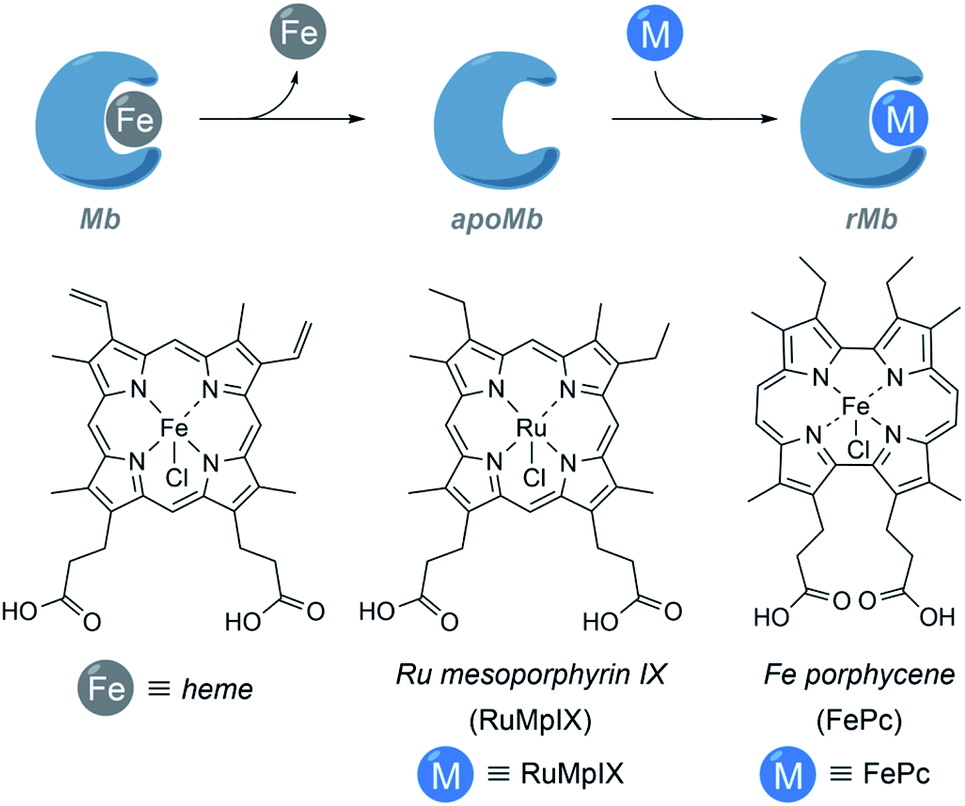 | ||
| Fig. 11 Schematic representation of the reconstitution of myoglobin with RuMpIX and FePc. rMb = reconstituted myoglobin. | ||
All these impressive contributions demonstrate the enormous power of ArMs to perform carbene transfer reactions in aqueous buffers, with rates and selectivities much better than those achieved under standard organometallic conditions with discrete catalysts.
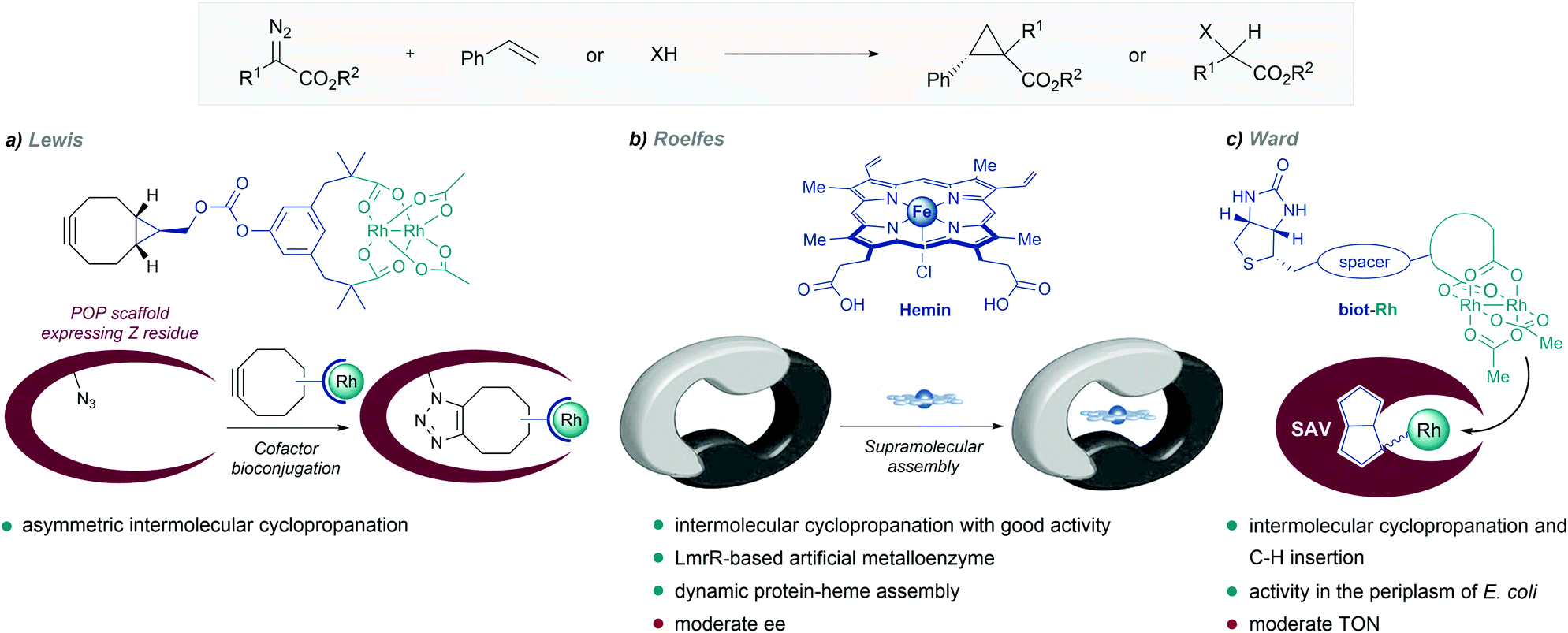 | ||
| Fig. 12 De novo designed metalloenzymes. (a) Rhodium ArM developed by Lewis. (b) Self-assembled LmrR-heme metalloenzyme reported by Roelfes, reproduced with permission from ref. 135, licensed under a Creative Commons Attribution (CC BY-NC 4.0) license, by Roelfes. Discolored from original. The original version can be found here (https://onlinelibrary.wiley.com/doi/full/10.1002/anie.201802946). (c) Streptavidin-biotin strategy followed by Ward. | ||
In 2018, Roelfes and coworkers reported the de novo design of a non-native heme protein based on the large, promiscuous, hydrophobic binding pocket of the lactococcal multidrug resistance regulator (LmrR) (Fig. 12b). By self-assembly of hemin and LmrR in a buffered solution, they created a biocatalyst for the cyclopropanation of styrene derivatives with EDA. The reaction requires an inert atmosphere and the addition of sodium dithionate as a reductant agent to generate the active Fe(II) heme complex. Among the different mutants synthesized, LmrR_M8A delivered the product with good yield (45%) and reasonable enantiomeric excess (ee = 51% [1R,2R enantiomer]), and TTNs up to 449. Interestingly, the crystal structure showed that the iron centre is apparently inaccessible for the substrates, as it is sandwiched between two tryptophan residues, and therefore, the dynamic nature of the enzyme seems to be responsible for the observed activity and enantioselectivity.135
Inspired by the work of Wilson and Whitesides,136 several groups, and especially Ward's, have designed artificial enzymes based on the use of biotin–avidin interactions to tether the metal cofactor to the protein. In 2018, the group demonstrated that covalently anchoring a dirhodium catalyst to a biotin, and embedding it within engineered streptavidin, gives a hybrid catalyst for olefin cyclopropanation and carbene C–H insertion reactions, albeit with modest performances (Fig. 12c).137 Importantly, they demonstrated that the strategy can be implemented to work in the periplasm of E. coli (vide infra).
4.2. Carbene transformations promoted by metals coated on DNA scaffolds
Although much less than proteins, nucleic acids have also been used to build metalloderivatives capable of performing carbene transfer reactions in aqueous buffers. DNA can be considered as a privileged scaffold that can provide a protective microenvironment for metal carbene intermediates, while also serving as a source of chirality.Pioneering work in this field of DNA-based organometallic catalysis was performed by Roelfes et al. (Fig. 13a). Using copper complexes with intercalating ligands (dppz derivatives), and salmon testes duplex DNA as a source of chirality, they achieved asymmetric intramolecular cyclopropanation of α-diazo-β-ketosulfones, with enantiomeric excesses up to 84%.138 Likely, the microenvironment resulting from the stacking of the ligands between the DNA bases may favour cyclopropanation over water addition to the carbene.
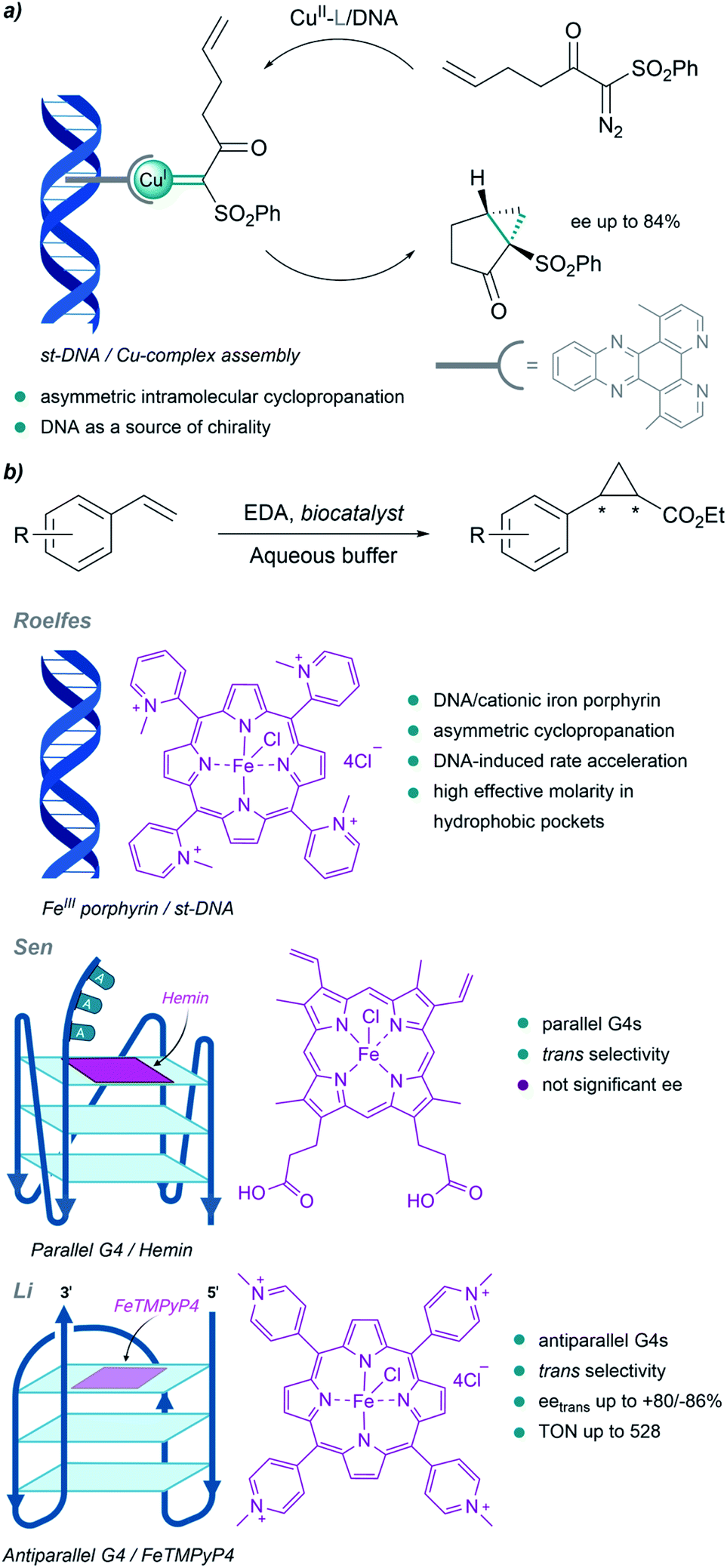 | ||
| Fig. 13 DNA-based carbene transfer reactions. (a) Intramolecular cyclopropanation catalyzed by assembled DNA/Cu complexes. (b) Cyclopropanation of styrene with EDA using DNA-based biocatalysts. | ||
They also implemented the use of porphyrin metal ligands in this type of carbene transfer reactions. In 2016 they reported a DNA-accelerated cyclopropanation of styrene derivatives with EDA, with a moderate enantiomeric excess of 53% in the presence of the salmon testes DNA/cationic iron porphyrin hybrid catalyst (Fig. 13b). An ortho-substituted N-methylpyridinium porphyrin that predominantly interacts with DNA through groove binding, and makes the catalytic site accessible for substrates, was shown to be the best catalyst.139 The authors proposed that the rate acceleration observed may be related to the formation of hydrophobic pockets in the DNA upon binding of the iron porphyrin, thus providing a high effective molarity that benefits reactivity.
DNA G-quadruplexes (G4s) have recently been incorporated as scaffolds for transition-metal promoted carbene transfer reactions. In 2019, Sen's group reported the cyclopropanation of styrene with EDA using heme·DNAzymes as catalysts (Fig. 13b). They exploited the ability of hemin to target and bind by stacking parallel G4 DNAs with three unpaired adenines or thymines at their 3′-end (G4-AAA or G4-TTT). The resulting complex was used to perform cyclopropanation reactions in yields up to 80%, and with good cis/trans ratios (8:92); but not significant enantiomeric excesses. Interestingly, the G4s provided faster kinetics and better product turnover than when using disaggregated hemes, both in the absence and in the presence of other DNA folds (e.g. double-stranded DNA).140
Li et al. built hybrid systems using intramolecular antiparallel G-quadruplex variants of thrombin-binding aptamer (TBA) G4-DNAs and cationic Fe porphyrin FeTMPyP4 (Fe-meso-tetra(N-methyl-4-pyridyl)porphyrin, Fig. 13b). After adequate mutations, two efficient stereocomplementary catalysts were identified. Specific residues in the G4 structure, spatially close to the active site, were crucial for obtaining good selectivities in cyclopropanation reactions.141
5. Exporting metal carbene reactions into biological environments and living cells
5.1. Organometallic reactions in living contexts
Nature exploits the efficiency and selectivity of native metalloenzymes to regulate a plethora of biochemical transformations in living cells and organisms. As stated above, artificial metalloenzymes have demonstrated impressive activities in abiotic processes in vitro; however, their use in living cells has been essentially limited to bacteria, and usually with metal cofactors that are very similar to those occurring in nature. This contrasts with discrete transition metal catalysts, that have demonstrated to work in complex biological environments and even within living mammalian cells, albeit with very modest activities.6,14,15,17–43As indicated in the previous sections, reactions involving metal carbenes can be performed in aqueous media, using specifically tailored conditions. A more challenging issue is related with their bioorthogonality, and the possibility of exporting them to complex biological environments and living settings. Although advances in this area have been scarce, a number of reports increasingly show the potential of metal-promoted carbene transfer reactions in the fields of bioorthogonal and biological chemistry.
5.2. Carbene transfer reactions in cellular settings and in bacteria
In addition to the considerations mentioned above, exporting metal carbene chemistry to living environments poses important challenges associated with the activity and, especially, stability of the catalyst and/or reactive intermediates, and the selectivity towards abiotic substrates in the presence of different biomolecules (thiols, amines, amino acids…).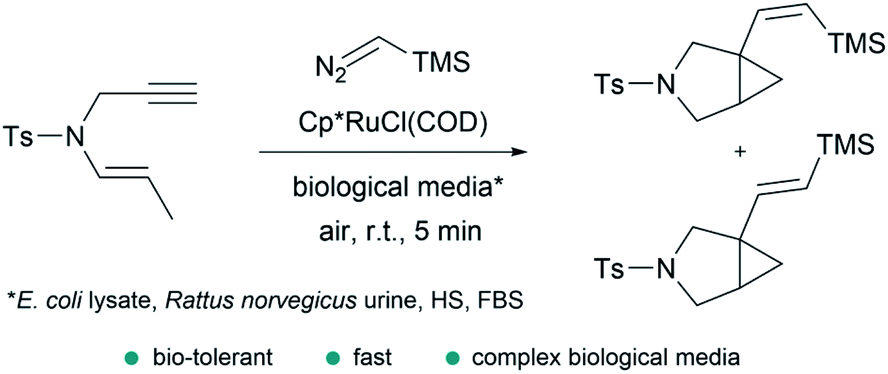 | ||
| Fig. 14 Ru-catalyzed fusion of a diazo with an enyne in complex biological media to provide bicyclic cyclopropanes reported by Teply and coworkers. | ||
A particularly remarkable contribution in the area was disclosed by Balskus et al., who demonstrated that metallocarbene chemistry can be interfaced with bacterial metabolism. They engineered E. coli to generate styrene from D-glucose, and integrated metallocarbene intermediates into the metabolic pathway using a biocompatible iron(III) phthalocyanine catalyst (FePcCl) that transformed styrene into non-natural phenyl cyclopropanes in single-vessel fermentations (Fig. 15).143
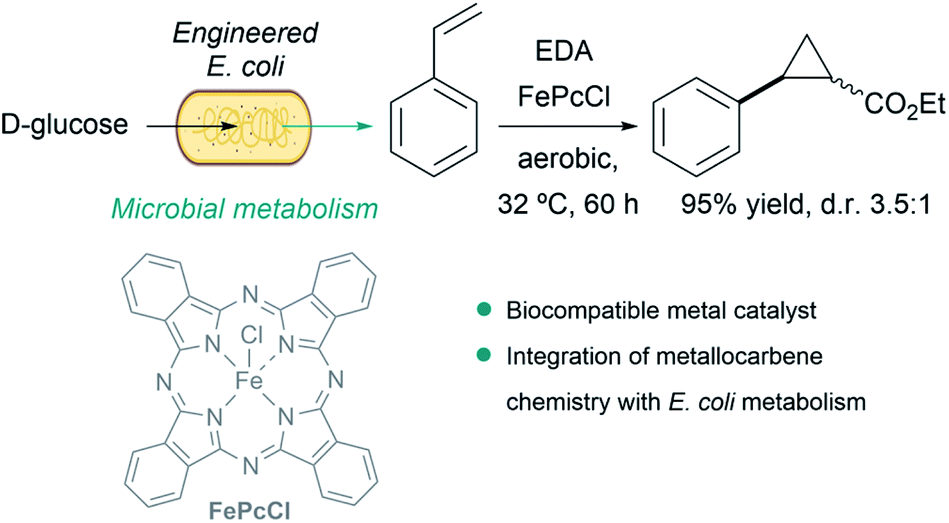 | ||
| Fig. 15 Combination of microbial metabolism and metallocarbene chemistry to generate cyclopropanes from D-glucose in E. coli reported by Balskus. | ||
Very recently, Mascareñas and coworkers reported the first example of a metal-carbene transfer reaction within live mammalian cells (Fig. 16). Using a common copper salt (Cu(OAc)2), they synthesized benzoquinoxalines in cellulo, in a one-pot process that is initiated by an N–H carbene insertion of the in situ generated copper carbenoid into ortho-amino arylamines. The intracellular generation of a fluorescent benzoquinoxaline allowed for in vivo monitoring of the reaction by fluorescence microscopy. Remarkably, the methodology could be applied to elicit biological effects through the intracellular synthesis of Tyrphostin AG1385, a tyrosine phosphorylation inhibitor that disrupts mitochondrial functions, producing mitochondrial fragmentation and depolarization. Moreover, the authors were able to elicit cell-selective responses by equipping the copper catalyst with a targeting unit (RGD motif).144
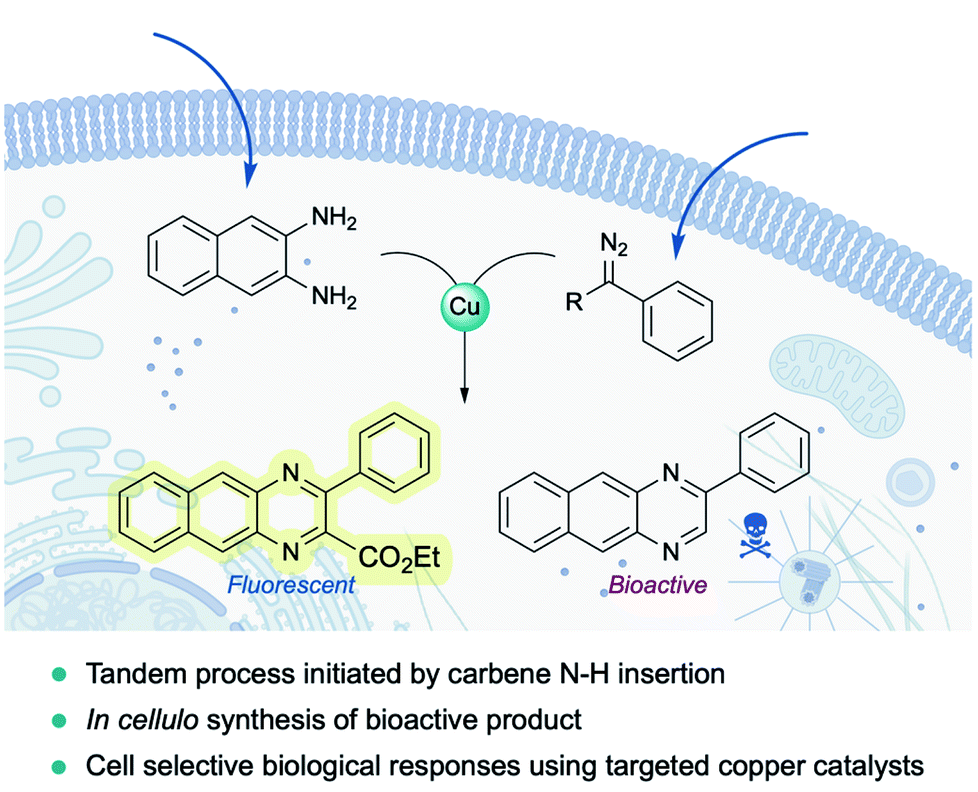 | ||
| Fig. 16 Exporting metal carbenes to live mammalian cells. Copper catalyzed synthesis of quinoxalines enabled by N–H carbene insertion. | ||
Metalloenzymes derived from natural counterparts. As previously indicated, some of the metalloenzymes capable of promoting carbene transfer reactions have been used directly in vivo, in the bacteria in which they are generated. The first example was reported by Arnold's group in 2013. Following their initial studies on directed evolution of cytochrome P450, they demonstrated that the introduction of an axial cysteine-to-serine mutation in the iron cofactor abolishes monooxygenation activity of P450 while maintaining carbene transfer activity (Fig. 17). They performed the cyclopropanation of styrene with EDA in whole-cells using E. coli expressing a serine-mutated P450 (also called P411), obtaining good results in terms of yield, diastereo- and enantioselectivity, as well as good turnover numbers.105
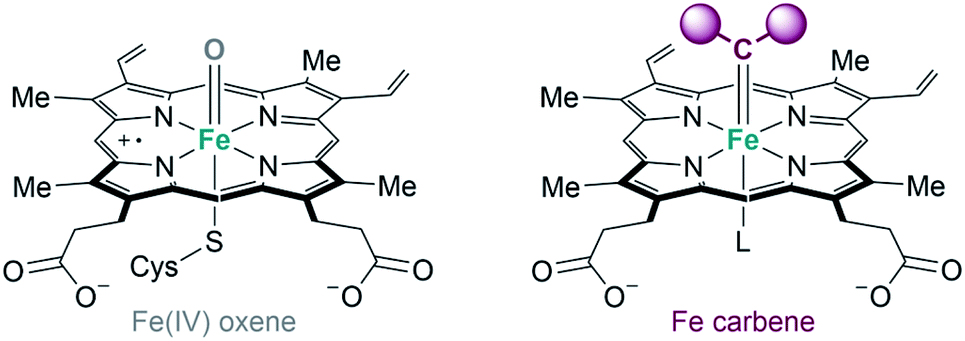 | ||
| Fig. 17 Comparison of Fe-oxene and Fe-carbene intermediates in heme-based metalloenzymes proposed by Arnold.104,105 | ||
It is worth noting the strategy reported by Arnold in 2016, which allowed the in vivo production of organosilicon compounds (Fig. 18a).110 In this case, cytochrome c from Rhodothermus marinus (Rma cyt c) was shown to be the most active hemoprotein. Importantly, high levels of selectivity were achieved for Si–H carbene insertions. Even in the presence of other functionalities such as alcohols or primary amines, styryl olefins or electron rich double bonds, susceptible of undergoing O–H, N–H insertion or cyclopropanations, the C–Si bond formation was predominant. Interestingly, by selecting the adequate ArMs, the authors were able to selectively catalyze the insertion of the carbene either into a Si–H or N–H bond of the same substrate (Fig. 18b).145
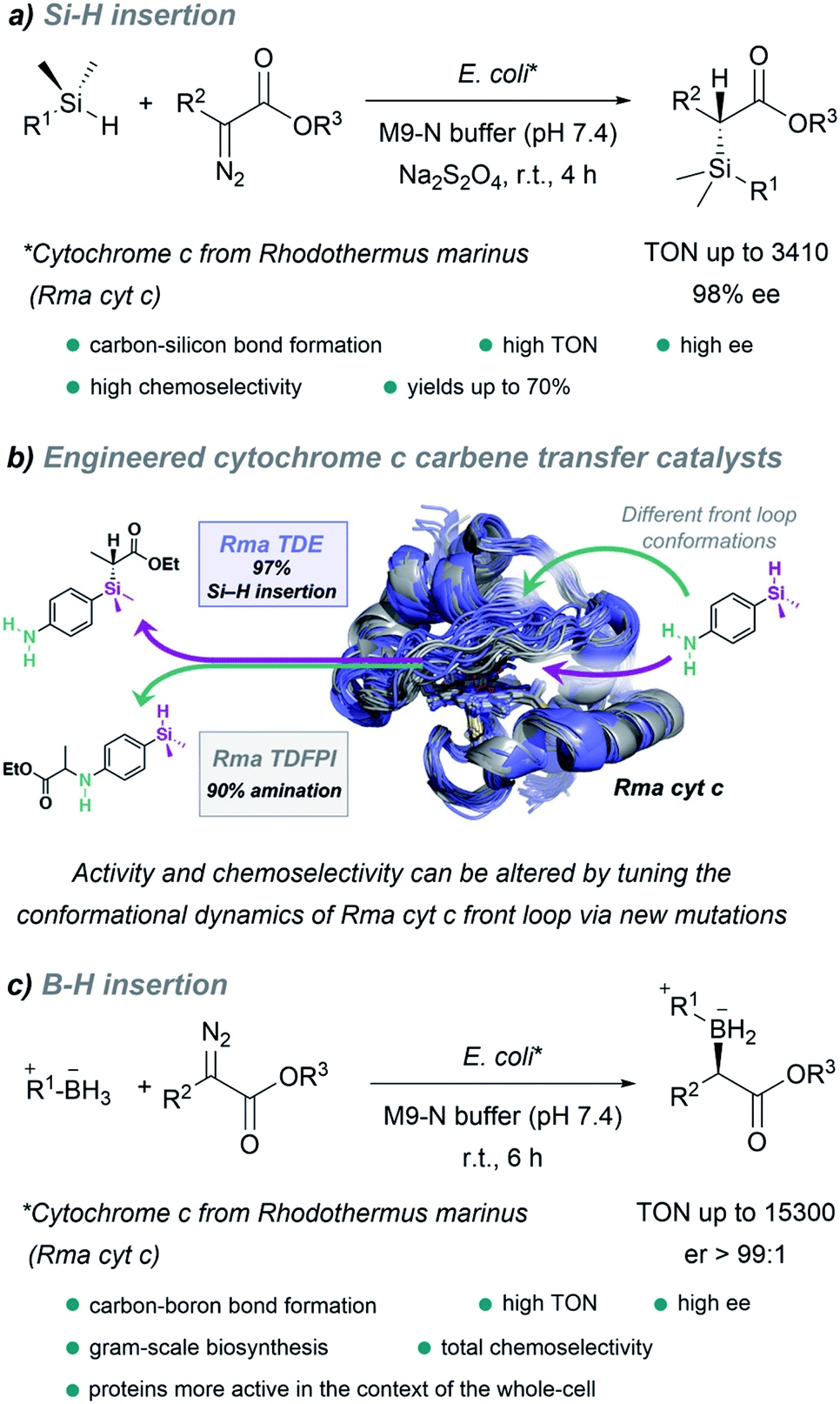 | ||
| Fig. 18 Expanding nature's carbene catalytic repertoire for in vivo (E. coli) reactions. (a) Synthesis of chiral silicon compounds. (b) Chemoselectivity of carbene transfer: Si–H versus N–H insertion, reproduced from ref. 145 with permission from American Chemical Society, copyright 2021. (c) Synthesis of chiral boron compounds. | ||
Using directed evolution, they could enhance the catalytic function of this ArM for B–H insertions, reporting, one year later, a fully genetically encoded platform for producing chiral organoboranes in bacteria (Fig. 18c).111 These enzymes were found to form carbon–boron bonds in the presence of borane–Lewis base complexes, through carbene insertion into the boron–hydrogen bond. Surprisingly, the variants demonstrated to be even more productive when the reactions were carried out using whole Escherichia coli cells expressing this enzyme.
The P411 scaffold has been further optimized by directed evolution, and demonstrated to work in E. coli for other reactions, such as the synthesis of bicyclobutanes, via successive carbene addition to unsaturated carbon–carbon bonds.146
Fasan and coworkers reported the first example of intramolecular olefin cyclopropanations with allyl α-diazoacetate derivatives (with high stereocontrol and complementary enantioselectivities), in whole cells, using engineered Mb based catalysts.147 The same strategy was applied to the synthesis of symmetrically fused cyclopropane-γ-lactams starting from allyldiazoacetamides.148
Very recently, Arnold et al. evolved a hemoprotein (P411) that exhibits a mechanism entailing an N–H insertion and a protonation at the active site of the protein. This engineered active site also places the necessary water molecules for rapid and stereoselective proton rearrangement prior to product release. The ArM works in whole cells, exhibiting high activity and enantioselectivity (up to 98% ee).149
All the above reactions are based on the modification of native metalloprotein systems. The intracellular assembly of metalloproteins equipped with non-natural metal cofactors is more difficult.
An interesting example from Brustad's group deals with the engineering of cytochrome P450 to selectively incorporate Ir(Me)–deuteroporphyrin IX (Ir(Me)–DPIX), in lieu of natural heme, directly in bacteria. The strategy required the introduction of mutations within the heme binding pocket. The resulting “in-cell” assembled iridium complex promoted olefin cyclopropanation reactions by EDA, and showed enhanced activity for aliphatic and electron-deficient olefins compared to the native heme enzyme.150
Hartwig and coworkers recently demonstrated the viability of assembling an iridium containing heme metalloenzyme in bacteria, using the HUG system to transport the Ir(Me)MPIX cofactor into engineered cells expressing CYP119. They even demonstrated that it is possible to combine natural and artificial metalloenzymes in E. coli to afford cyclopropanated products from simple sugars (Fig. 19).151 Very recently, they identified a second system for transporting iridium porphyrin into Nissle 1917 (a non-pathogenic E. coli strain), based on the presence of an outer-membrane receptor of these cells.152
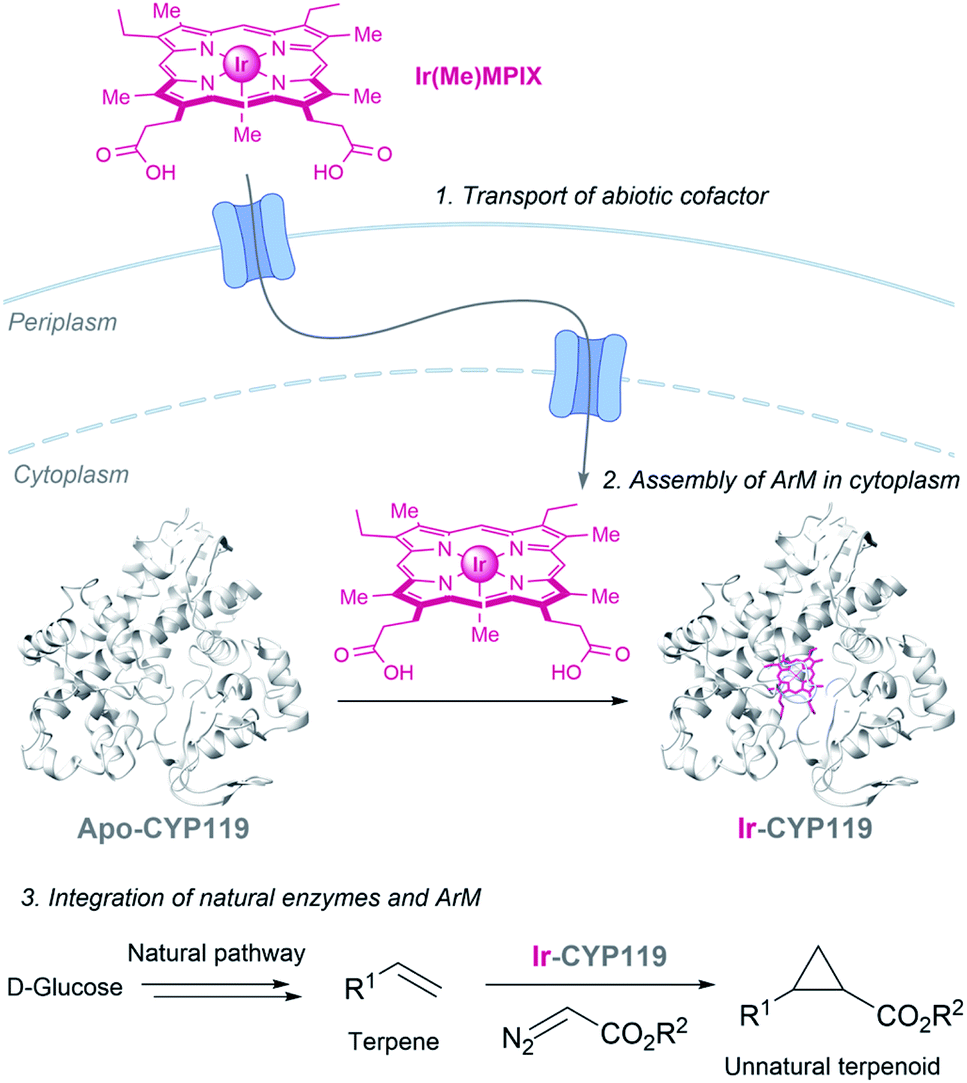 | ||
| Fig. 19 Combination of microbial metabolism and artificial metalloenzyme cyclopropanation to generate cyclopropanes from D-glucose in E. coli reported by Hartwig (CYP119 PDB ID: 14FU). | ||
Metalloenzymes based on non-native protein scaffolds. As discussed above, it is possible to assemble artificial metalloenzymes using a biotin–streptavidin recognition strategy. Ward demonstrated that a dirhodium complex tethered to a biotin moiety can interact with streptavidin proteins located in the periplasm of E. coli, and the resulting complex performed carbene transfer reactions in vivo.137 The reactions are only effective in the periplasm, owing to the absence of thiols and other components that could deactivate the catalyst.
In addition to carbene transfer reactions from diazo precursors, metal carbenes are also key reagents and intermediates in alkene metathesis processes, which can also be catalyzed by artificial metaloenzymes.101 Ward and coworkers designed a biotinylated Hoveyda–Grubbs second-generation catalyst (biot-Ru) that binds to streptavidin in the periplasm of E. coli, to give an effective artificial metalloenzyme that promotes a ring-closing-metathesis (Fig. 20, left).
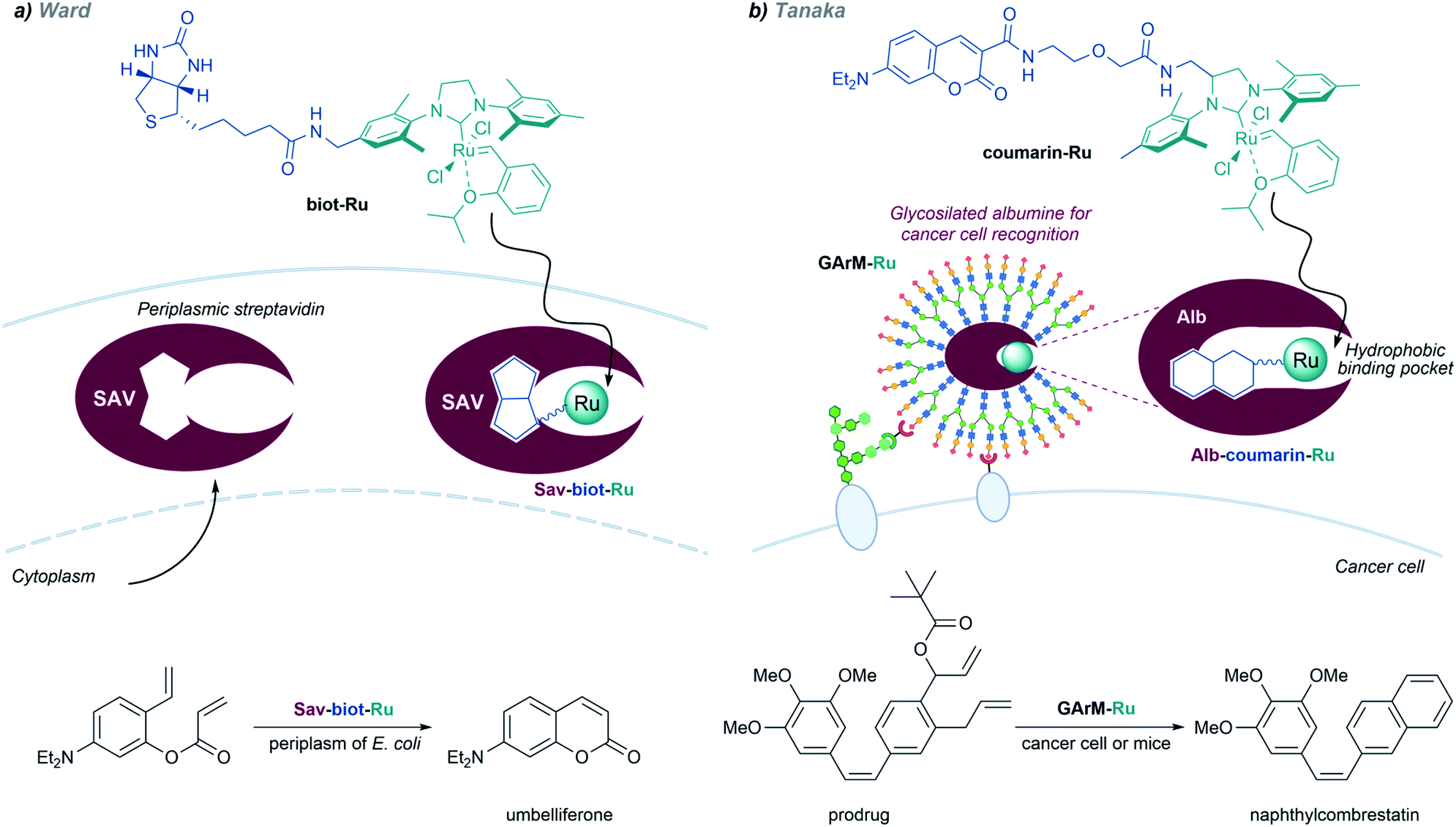 | ||
| Fig. 20 In vivo metathesis reactions developed by (a) Ward and (b) Tanaka. | ||
A similar approach, based on the affinity of coumarin for albumin, was employed by the group of Tanaka to build ArMs that perform “in cell” metathesis reactions (Fig. 20, right).153
6. Conclusions
What seemed unfeasible at first, achieving metal-catalyzed carbene transfer reactions in water mixtures, and even under the stringent conditions of a biological environment, has now become a reality. Exporting metal carbene chemistry to these reaction environments is possible, provided the substrates, catalysts and conditions are appropriately tuned.Considering the rich and versatile chemistry of metal carbenes, we foresee a bright future for further expanding the applications of these intermediates in chemical and cell biology, as well as biomedicine.
Current challenges include, among others, the creation of versatile artificial metalloenzymes that can perform chemoselective and bioorthogonal metal-carbene transfer reactions in mammalian cells, increasing rates and turnover with discrete metal catalysts, the development of metallocatalysts embedded in nanoscaffolds that facilitate the reactivity, or the development of biological applications.
Author contributions
JLM conceived the idea of the review and together with MTG supervised the project. SG prepared the draft, and MTG made the initial revision. JLM revised and edited the manuscript. All the authors participated in the discussion of the drafts and gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received financial support from Spanish grants (PID2019-108624RB-I00, RTI2018-093813-J-I00, R&C2020-029150-I and ORFEO-CINQA network CTQ2016-81797-REDC), the Consellería de Cultura, Educación e Ordenación Universitaria (ED431C-2021/25 and Centro Singular de Investigación de Galicia accreditation 2019–2022, ED431G 2019/03), the European Union (European Regional Development Fund-ERDF corresponding to the multiannual financial framework 2014–2020), and the European Research Council (Advanced Grant No. 340055). Figures have been created with https://BioRender.com.Notes and references
- J. F. Hartwig, Organotransition metal chemistry: from bonding to catalysis, University Science Books: Mill Valley, CA, 2010 Search PubMed.
- P. H. Dixneuf and V. Cadierno, Metal-catalyzed reactions in water, Wiley-VCH Verlag GmbH & Co. KGaA, 2013 Search PubMed.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- A. J. Link and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 11164–11165 CrossRef CAS PubMed.
- A. J. Link, M. K. S. Vink and D. A. Tirrell, J. Am. Chem. Soc., 2004, 126, 10598–10602 CrossRef CAS PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa, A. Olmos, P. J. Pérez and J. L. Mascareñas, Chem. Sci., 2018, 9, 1947–1952 RSC.
- C. Uttamapinant, M. I. Sanchez, D. S. Liu, J. Z. Yao, K. A. White, S. Grecian, S. Clark, K. R. Gee and A. Y. Ting, Nat. Protoc., 2013, 8, 1620–1634 CrossRef PubMed.
- L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS PubMed.
- M. Martínez and J. L. Mascareñas, Coord. Chem. Rev., 2018, 359, 57–79 CrossRef.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106–114 CrossRef CAS PubMed.
- A. H. Ngo, S. Bose and L. H. Do, Chem.–Eur. J., 2018, 24, 10584–10594 CrossRef CAS PubMed.
- Y. Liu and Y. Bai, ACS Appl. Bio Mater., 2020, 3, 4717–4746 CrossRef CAS PubMed.
- P. Destito, C. Vidal, F. López and J. L. Mascareñas, Chem.–Eur. J., 2021, 27, 4789–4816 CrossRef CAS PubMed.
- T.-C. Chang and K. Tanaka, Bioorg. Med. Chem., 2021, 46, 116353 CrossRef CAS PubMed.
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- M. O. N. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef PubMed.
- W. Wanga, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef PubMed.
- S. Fedeli, J. Im, S. Gopalakrishnan, J. L. Elia, A. Gupta, D. Kimade and V. M. Rotello, Chem. Soc. Rev., 2021, 50, 13467–13480 RSC.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Organometallics, 2012, 31, 5968–5970 CrossRef CAS.
- M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- T. Volker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef PubMed.
- R. Das, R. F. Landis, G. Y. Tonga, R. Cao-Milan, D. C. Luther and V. M. Rotello, ACS Nano, 2019, 13, 229–235 CrossRef CAS PubMed.
- S. Learte-Aymamí, C. Vidal, A. Gutiérrez-González and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 9149–9154 CrossRef PubMed.
- X. Zhang, S. Fedeli, S. Gopalakrishnan, R. Huang, A. Gupta, D. C. Luther and V. Rotello, ChemBioChem, 2020, 21, 2759–2763 CrossRef CAS PubMed.
- P. Destito, J. R. Couceiro, H. Ferreira, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2017, 56, 10766–10770 CrossRef CAS PubMed.
- A. Gutiérrez-González, P. Destito, J. R. Couceiro, C. Pérez-González, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 16059–16066 CrossRef PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 17628–17633 CrossRef PubMed.
- C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascareñas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS PubMed.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1913 CrossRef PubMed.
- N. Li, C. P. Ramil, R. K. V. Lim and Q. Lin, ACS Chem. Biol., 2015, 10, 379–384 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS PubMed.
- L. Lercher, J. F. McGouran, B. M. Kessler, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2013, 52, 10553–10558 CrossRef CAS PubMed.
- P. Destito, A. Sousa-Castillo, J. R. Couceiro, F. López, M. A. Correa-Duarte and J. L. Mascareñas, Chem. Sci., 2019, 10, 2598–2603 RSC.
- J. Wang, C.-M. Che and M. P. Doyle, Transition Metal-Catalyzed Carbene Transformations, WILEY-VCH Verlag GmbH, 2021 Search PubMed.
- F. Zaragoza Dörwald, Metal carbenes in organic synthesis, WILEY-VCH Verlag GmbH, 1999 Search PubMed.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2016, 55, 8452–8456 CrossRef CAS PubMed.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS PubMed.
- M. Brookhart and W. B. Studabaker, Chem. Rev., 1987, 87, 411–432 CrossRef CAS.
- D. Intrieri, A. Caselli and E. Gallo, Eur. J. Inorg. Chem., 2011, 5071–5081 CrossRef CAS.
- H. M. L. Davies and E. G. Antoulinakis, Org. React., 2001, 57, 1–326 CAS.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- M. Álvarez, R. Gava, M. R. Rodríguez, S. G. Rull and P. J. Pérez, ACS Catal., 2017, 7, 3707–3711 CrossRef.
- S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed.
- D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC.
- W. Kirmse and M. Kapps, Chem. Ber., 1968, 101, 994–1003 CrossRef CAS.
- M. P. Doyle, J. H. Griffin, M. S. Chinn and D. van Leusen, J. Org. Chem., 1984, 49, 1917–1925 CrossRef CAS.
- S. Jana, Y. Guo and R. M. Koenigs, Chem.–Eur. J., 2021, 27, 1270–1281 CrossRef CAS PubMed.
- M. C. Pirrung, H. Liu and A. T. Morehead, J. Am. Chem. Soc., 2002, 124, 1014–1023 CrossRef CAS PubMed.
- For specific reviews on olefin metathesis in aqueous and biological environments, see: M. S. Messina and H. D. Maynard, Mater. Chem. Front., 2020, 4, 1040–1051 RSC; J. B. Binder and R. T. Raines, Curr. Opin. Chem. Biol., 2008, 12, 767–773 CrossRef CAS PubMed.
- S. Iwasa, F. Takezawa, Y. Tuchiya and H. Nishiyama, Chem. Commun., 2001, 59–60 RSC.
- R. P. Wurz and A. B. Charette, Org. Lett., 2002, 4, 4531–4533 CrossRef CAS PubMed.
- I. Nicolas, P. Le Maux and G. Simonneaux, Tetrahedron Lett., 2008, 49, 5793–5795 CrossRef CAS.
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2014, 20, 4880–4884 CrossRef CAS PubMed.
- M. C. M. van Oers, L. K. E. A. Abdelmohsen, F. P. J. T. Rutjes and J. C. M. van Hest, Chem. Commun., 2014, 50, 4040–4043 RSC.
- N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, Chem. Commun., 2005, 391–393 RSC.
- N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, J. Org. Chem., 2006, 71, 5489–5497 CrossRef CAS PubMed.
- L. Li, C. Shu, B. Zhou, Y.-F. Yu, X.-Y. Xiao and L.-W. Ye, Chem. Sci., 2014, 5, 4057–4064 RSC.
- H. F. Srour, P. Le Maux, S. Chevance, D. Carrié, N. Le Yondre and G. Simonneaux, J. Mol. Catal. A: Chem., 2015, 407, 194–203 CrossRef CAS.
- K. Ramakrishna, J. M. Thomas and C. Sivasankar, J. Org. Chem., 2016, 81, 9826–9835 CrossRef CAS PubMed.
- K. Ramakrishna and C. Sivasankar, Org. Biomol. Chem., 2017, 15, 2392–2396 RSC.
- R. P. Pandit, S. H. Kim and Y. R. Lee, Adv. Synth. Catal., 2016, 358, 3586–3599 CrossRef CAS.
- P. Saha, H. Jeon, P. K. Mishra, H.-W. Rhee and J. H. Kwak, J. Mol. Catal. A: Chem., 2016, 417, 10–18 CrossRef CAS.
- I. Aviv and Z. Gross, Chem.–Eur. J., 2008, 14, 3995–4005 CrossRef CAS PubMed.
- L. Zhou, Y. Liu, Y. Zhang and J. Wang, Beilstein J. Org. Chem., 2011, 7, 631–637 CrossRef CAS PubMed.
- M. Liao and J. Wang, Green Chem., 2007, 9, 184–188 RSC.
- T. G. Rajagopalan, W. H. Stein and S. Moore, J. Biol. Chem., 1966, 241, 4295–4297 CrossRef CAS PubMed.
- G. R. Delpierre and J. S. Fruton, Proc. Natl. Acad. Sci. U. S. A., 1966, 1817–1822 CrossRef CAS PubMed.
- J. M. Antos and M. B. Francis, J. Am. Chem. Soc., 2004, 126, 10256–10257 CrossRef CAS PubMed.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS PubMed.
- R. Kundu and Z. T. Ball, Chem. Commun., 2013, 49, 4166–4168 RSC.
- C.-M. Ho, J.-L. Zhang, C.-Y. Zhou, O.-Y. Chan, J. J. Yan, F.-Y. Zhang, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS PubMed.
- M. Salmain, E. Licandro, C. Baldoli, S. Maiorana, H. Tran-Huy and G. Jaouen, J. Organomet. Chem., 2001, 617–618, 376–382 CrossRef CAS.
- P. Dutta, S. Sawoo, N. Ray, O. Bouloussa and A. Sarkar, Bioconjugate Chem., 2011, 22, 1202–1209 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, J. Am. Chem. Soc., 2010, 132, 6660–6662 CrossRef CAS PubMed.
- B. V. Popp and Z. T. Ball, Chem. Sci., 2011, 2, 690–695 RSC.
- Z. Chen, F. Vohidov, J. M. Coughlin, L. J. Stagg, S. T. Arold, J. E. Ladbury and Z. T. Ball, J. Am. Chem. Soc., 2012, 134, 10138–10145 CrossRef CAS PubMed.
- F. Vohidov, J. M. Coughlin and Z. T. Ball, Angew. Chem., Int. Ed., 2015, 54, 4587–4591 CrossRef CAS PubMed.
- J. Ohata and Z. T. Ball, J. Am. Chem. Soc., 2017, 139, 12617–12622 CrossRef CAS PubMed.
- K. Tishinov, K. Schmidt, D. Häussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 CrossRef CAS PubMed.
- K. Tishinov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401–4406 RSC.
- S. N. Geigle, L. A. Wyss, S. J. Sturla and D. G. Gillingham, Chem. Sci., 2017, 8, 499–506 RSC.
- Y.-H. Lee, E. Yu and C.-M. Park, Nat. Commun., 2021, 12, 1681 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- M. Diéguez, J.-E. Bäckvall and O. Pàmies, Artificial metalloenzymes and metalloDNAzymes in catalysis: from design to applications, Wiley-VCH. Weinheim, 2018 Search PubMed.
- M. Jeschek, S. Panke and T. R. Ward, Trends Biotechnol., 2018, 36, 60–72 CrossRef CAS PubMed.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120–1136 CrossRef CAS PubMed.
- M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed.
- M. Wittwer, U. Markel, J. Schiffels, J. Okuda, D. F. Sauer and U. Schwaneberg, Nat. Catal., 2021, 4, 814–827 CrossRef CAS.
- S. Chordia, S. Narasimhan, A. Lucini Paioni, M. Baldus and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 5913–5920 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307–310 CrossRef CAS PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS PubMed.
- H. Renata, Z. J. Wang, R. Z. Kitto and F. H. Arnold, Catal. Sci. Technol., 2014, 4, 3640–3643 RSC.
- K. E. Hernandez, H. Renata, R. D. Lewis, S. B. J. Kan, C. Zhang, J. Forte, D. Rozzell, J. A. McIntosh and F. H. Arnold, ACS Catal., 2016, 6, 7810–7813 CrossRef CAS PubMed.
- K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 6891–6895 CrossRef CAS PubMed.
- Z. J. Wang, N. E. Peck, H. Renata and F. H. Arnold, Chem. Sci., 2014, 5, 598–601 RSC.
- S. B. J. Kan, R. D. Lewis, K. Chen and F. H. Arnold, Science, 2016, 354, 1048–1051 CrossRef CAS PubMed.
- B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132–136 CrossRef PubMed.
- J. Zhang, X. Huang, R. K. Zhang and F. H. Arnold, J. Am. Chem. Soc., 2019, 141, 9798–9802 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed.
- M. Bordeaux, R. Singh and R. Fasan, Bioorg. Med. Chem., 2014, 22, 5697–5704 CrossRef CAS PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744–1748 CrossRef CAS PubMed.
- A. Tinoco, V. Steck, V. Tyagi and R. Fasan, J. Am. Chem. Soc., 2017, 139, 5293–5296 CrossRef CAS PubMed.
- A. L. Chandgude and R. Fasan, Angew. Chem., Int. Ed., 2018, 57, 15852–15856 CrossRef CAS PubMed.
- A. L. Chandgude, X. Ren and R. Fasan, J. Am. Chem. Soc., 2019, 141, 9145–9150 CrossRef PubMed.
- V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967–10977 CrossRef CAS PubMed.
- D. Nam, V. Steck, R. J. Potenzino and R. Fasan, J. Am. Chem. Soc., 2021, 143, 2221–2231 CrossRef CAS PubMed.
- G. Sreenilayam and R. Fasan, Chem. Commun., 2015, 51, 1532–1534 RSC.
- V. Steck, G. Sreenilayam and R. Fasan, Synlett, 2020, 31, 224–229 CrossRef CAS PubMed.
- V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967–10977 CrossRef CAS PubMed.
- V. Tyagi, R. B. Bonn and R. Fasan, Chem. Sci., 2015, 6, 2488–2494 RSC.
- R. L. Khade, A. L. Chandgude, R. Fasan and Y. Zhan, ChemCatChem, 2019, 11, 3101–3108 CrossRef CAS PubMed.
- V. Tyagi, G. Sreenilayam, P. Bajaj, A. Tinoco and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 13563–13566 Search PubMed.
- H. M. Key, P. Dydio, D. S. Clark and J. F. Hartwig, Nature, 2016, 534, 534–537 CrossRef CAS PubMed.
- P. Dydio, H. M. Key, A. Nazarenko, J. Y.-E. Rha, V. Seyedkazemi, D. S. Clark and J. F. Hartwig, Science, 2016, 354, 102–106 CrossRef CAS PubMed.
- Y. Gu, S. N. Natoli, Z. Liu, D. S. Clark and J. F. Hartwig, Angew. Chem., Int. Ed., 2019, 58, 13954–13960 CrossRef CAS PubMed.
- M. W. Wolf, D. A. Vargas and N. Lehnert, Inorg. Chem., 2017, 56, 5623–5635 CrossRef CAS PubMed.
- K. Oohora, H. Meichin, L. Zhao, M. W. Wolf, A. Nakayama, J. Hasegawa, N. Lehnert and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 17265–17268 CrossRef CAS PubMed.
- H. Yang, P. Srivastava, C. Zhang and J. C. Lewis, ChemBioChem, 2014, 15, 223–227 CrossRef CAS PubMed.
- P. Srivastava, H. Yang, K. Ellis-Guardiola and J. C. Lewis, Nat. Commun., 2015, 6, 7789 CrossRef CAS PubMed.
- D. M. Upp, R. Huang, Y. Li, M. J. Bultman, B. Roux and J. C. Lewis, Angew. Chem., Int. Ed., 2021, 60, 23672–23677 CrossRef CAS PubMed.
- L. Villarino, K. E. Splan, E. Reddem, L. Alonso-Cotchico, C. Gutiérrez de Souza, A. Lledós, J.-D. Maréchal, A.-M. W. H. Thunnissen and G. Roelfes, Angew. Chem., Int. Ed., 2018, 57, 7785–7789 CrossRef PubMed.
- M. E. Wilson and G. M. Whitesides, J. Am. Chem. Soc., 1978, 100, 306–307 CrossRef CAS.
- J. Zhao, D. G. Bachmann, M. Lenz, D. G. Gillingham and T. R. Ward, Catal. Sci. Technol., 2018, 8, 2294–2298 RSC.
- J. Oelerich and G. Roelfes, Chem. Sci., 2013, 4, 2013–2017 RSC.
- A. Rioz-Martínez, J. Oelerich, N. Ségaud and G. Roelfes, Angew. Chem., Int. Ed., 2016, 55, 14136–14140 CrossRef PubMed.
- H. Ibrahim, P. Mulyk and D. Sen, ACS Omega, 2019, 4, 15280–15288 CrossRef CAS PubMed.
- J. Hao, W. Miao, Y. Cheng, S. Lu, G. Jia and C. Li, ACS Catal., 2020, 10, 6561–6567 CrossRef CAS.
- L. Adriaenssens, L. Severa, J. Vávra, T. Šálová, J. Hývl, M. Čížková, R. Pohl, D. Šaman and F. Teplý, Collect. Czech. Chem. Commun., 2009, 74, 1023–1034 CrossRef CAS.
- S. Wallace and E. P. Balskus, Angew. Chem., Int. Ed., 2015, 54, 7106–7109 CrossRef CAS PubMed.
- S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 22017–22025 CrossRef PubMed.
- M. Garcia-Borràs, S. B. J. Kan, R. D. Lewis, A. Tang, G. Jimenez-Osés, F. H. Arnold and K. N. Houk, J. Am. Chem. Soc., 2021, 143, 7114–7123 CrossRef PubMed.
- K. Chen, X. Huang, S. B. J. Kan, R. K. Zhang and F. H. Arnold, Science, 2018, 360, 71–75 CrossRef CAS PubMed.
- A. L. Chandgude, X. Ren and R. Fasan, J. Am. Chem. Soc., 2019, 141, 9145–9150 CrossRef PubMed.
- X. Ren, A. L. Chandgude and R. Fasan, ACS Catal., 2020, 10, 2308–2313 CrossRef CAS PubMed.
- Z. Liu, C. Calvó-Tusell, A. Z. Zhou, K. Chen, M. García-Borràs and F. H. Arnold, Nat. Chem., 2021, 13, 1166–1172 CrossRef CAS PubMed.
- E. W. Reynolds, T. D. Schwochert, M. W. McHenry, J. W. Watters and E. M. Brustad, ChemBioChem, 2017, 18, 2380–2384 CrossRef CAS PubMed.
- J. Huang, Z. Liu, B. J. Bloomer, D. S. Clark, A. Mukhopadhyay, J. D. Keasling and J. F. Hartwig, Nat. Chem., 2021, 13, 1186–1191 CrossRef CAS PubMed.
- Z. Liu, J. Huang, Y. Gu, D. S. Clark, A. Mukhopadhyay, J. D. Keasling and J. F. Hartwig, J. Am. Chem. Soc., 2022, 144, 883–890 CrossRef CAS PubMed.
- I. Nasibullin, I. Smirnov, P. Ahmadi, K. Vong, A. Kurbangalieva and K. Tanaka, Nat. Commun., 2022, 13, 39 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |