
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional amyloids from bacterial biofilms – structural properties and interaction partners
Ümit
Akbey
*a and
Maria
Andreasen
 *b
*b
aDepartment of Structural Biology, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15261, USA. E-mail: umitakbey@pitt.edu
bDepartment of Biomedicine, Aarhus University, Wilhelm Meyers Allé 3, 8000 Aarhus, Denmark. E-mail: mariaj@biomed.au.dk
First published on 6th May 2022
Abstract
Protein aggregation and amyloid formation have historically been linked with various diseases such as Alzheimer's and Parkinson's disease, but recently functional amyloids have gained a great deal of interest in not causing a disease and having a distinct function in vivo. Functional bacterial amyloids form the structural scaffold in bacterial biofilms and provide a survival strategy for the bacteria along with antibiotic resistance. The formation of functional amyloids happens extracellularly which differs from most disease related amyloids. Studies of functional amyloids have revealed several distinctions compared to disease related amyloids including primary structures designed to optimize amyloid formation while still retaining a controlled assembly of the individual subunits into classical cross-β-sheet structures, along with a unique cross-α-sheet amyloid fold. Studies have revealed that functional amyloids interact with components found in the extracellular matrix space such as lipids from membranes and polymers from the biofilm. Intriguingly, a level of complexity is added as functional amyloids also interact with several disease related amyloids and a causative link has even been established between functional amyloids and neurodegenerative diseases. It is hence becoming increasingly clear that functional amyloids are not inert protein structures found in bacterial biofilms but interact with many different components including human proteins related to pathology. Gaining a clear understanding of the factors governing the interactions will lead to improved strategies to combat biofilm associated infections and the correlated antibiotic resistance. In the current review we summarize the current state of the art knowledge on this exciting and fast growing research field of biofilm forming bacterial functional amyloids, their structural features and interaction partners.
Introduction
Historically the formation of protein aggregates or amyloids has been associated with various diseases such as neurodegenerative diseases.1 More recently it has been found that amyloids can have biological functions in the organism producing them, hence differing from the pathological amyloids and coining the term functional amyloids.2 Amyloid fibrils and functional amyloids are a topic of great interest and research, and the exponential growth of the field, research output and citations are shown in Fig. 1. Functional amyloids are found in various organisms ranging from bacteria to humans and currently a whole range is known, Fig. 2.2 They provide different types of functionality ranging from virulence during infection3 to structural scaffolds in biofilms,4 to storage of peptide hormones,5 to formation of melanin granula in melanocytes,6 to memory storage7 and during cell death,8 and many others.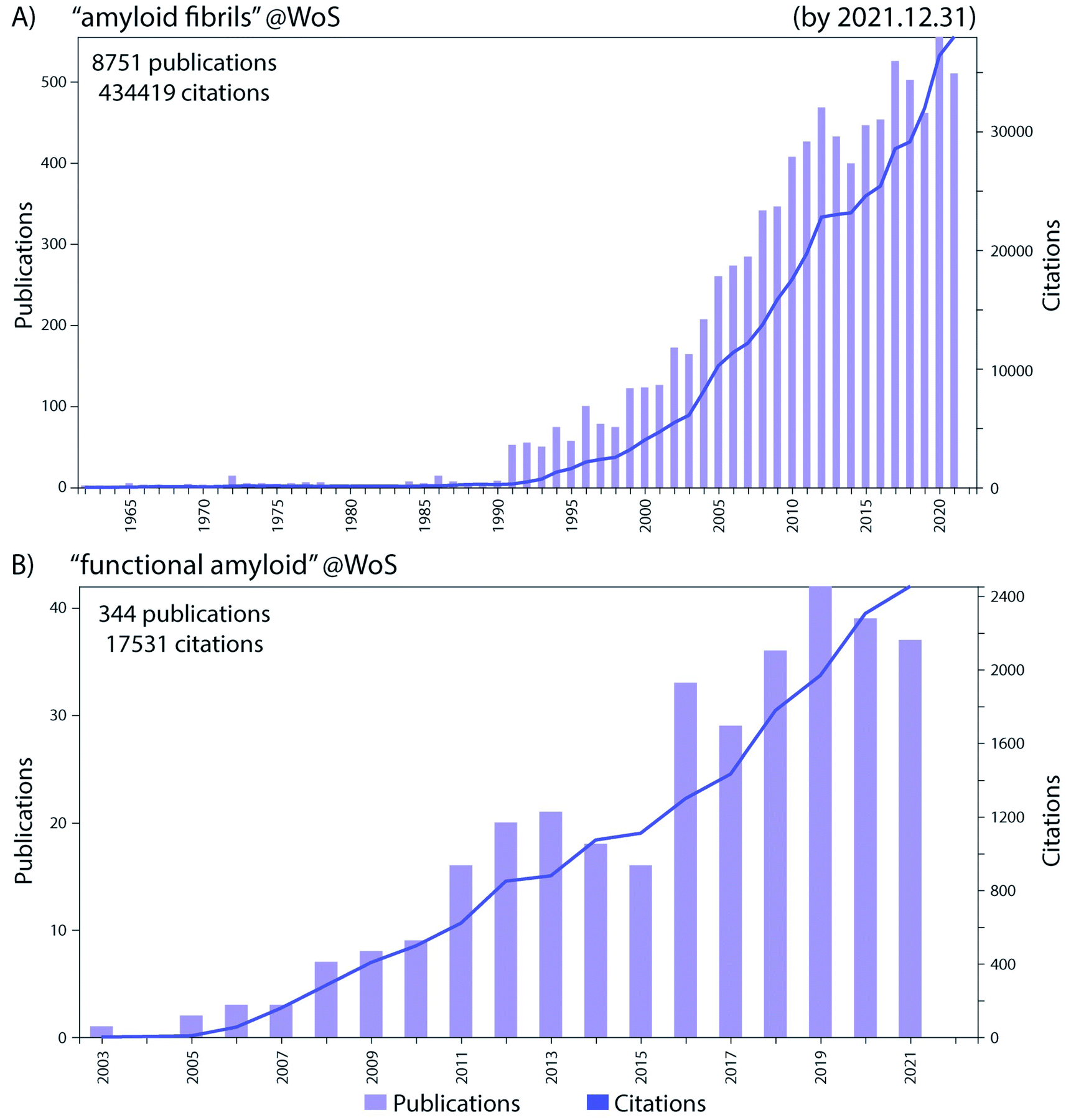 | ||
| Fig. 1 The number of publications per year (left y-axis) and total citations (right y-axis) in the Web of Science (WoS) in the research fields of (A) “amyloid fibrils” and (B) “functional amyloid”, by the end of 2021. Remarkable progress in these fields has been made in the recent years as evident from the increase in the research output in those fields. | ||
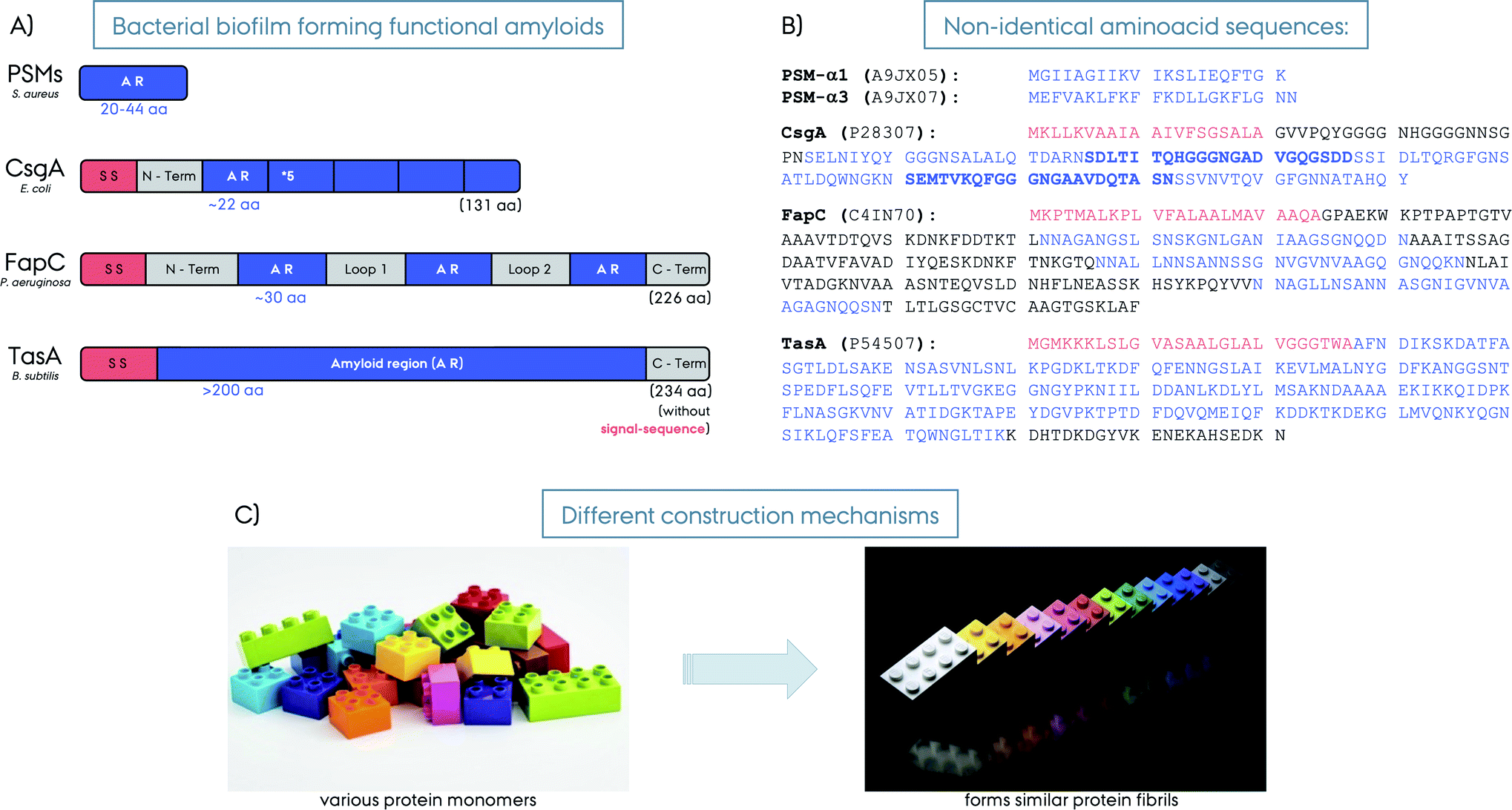 | ||
| Fig. 2 (A) Schematic representation of different bacterial biofilm forming functional amyloids: PSMs from S. aureus, CsgA from E. coli, FapC from P. aeruginosa and TasA from B. subtilis. The signal sequences are shown in red, the amyloid regions (AR) in blue and loop/linker and C-terminus regions in grey. The number of amino acids forming the amyloid region is given for each protein in blue. The lengths of the full-length proteins are given in parenthesis in black. The PSMs are short proteins and AR themselves. The CsgA is composed of 5 AR repeats, FapC from 5 AR repeats, whereas, TasA is considered to be composed of a large AR without repeats (note that other hypotheses exist on the nature of TasA amyloid region). (B) The amino acid composition of the five functional amyloids. The Uniprot identification numbers for each protein are given in parenthesis. The consecutive AR repeats are indicated as bold versus normal font for CsgA. (C) Illustration of amyloid fibril construction mechanisms by using different building blocks to the similar final amyloid fibril fold. | ||
Many bacteria form biofilms to survive under stress-inducing conditions to achieve communal living. Biofilms are microbial communities where the microorganisms live in a matrix of hydrated extracellular polymeric substances (EPS) that forms the scaffold of the biofilm.9 The EPS is produced and secreted by the bacterial cells living in the biofilm, and it is composed of biopolymers such as polysaccharides, proteins and nucleic acids in the form of extracellular DNA (eDNA), Fig. 3. The formation of biofilm by bacteria enables adhesion to surfaces and other bacteria cells, retention of nutrients and water, and additionally acts as a protective barrier. Biofilm-associated pathogenic microbes are protected from antimicrobial agents and host immune system attacks, as a result they are more infectious and difficult to treat. Eighty percent of all chronic infections are related to bacterial biofilms.10,11
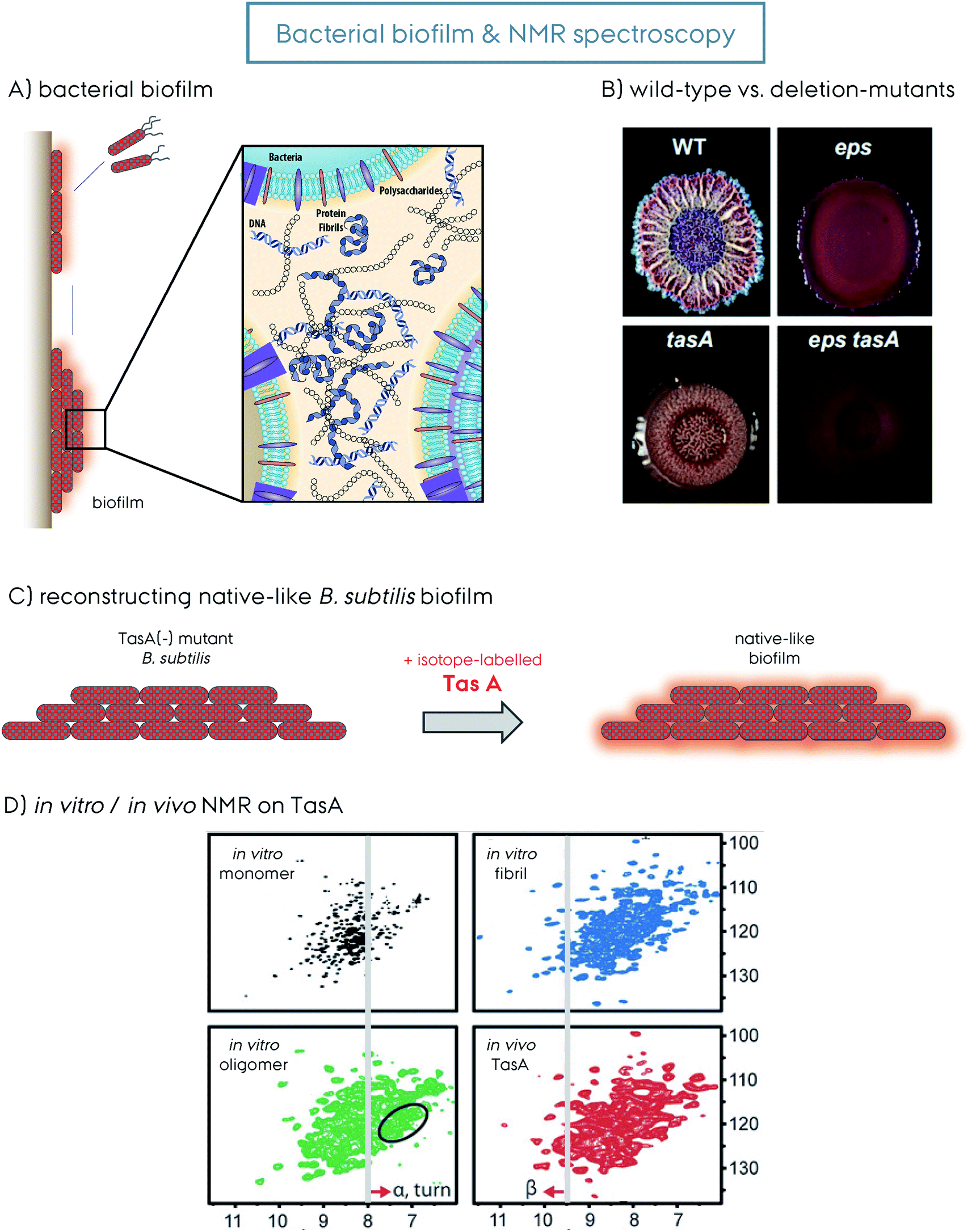 | ||
| Fig. 3 (A) Representation of bacteria mediated biofilm formation, initiated by the motile species gathering together into the mature solid biofilm matrix. The molecular details of the biofilm composed of bacteria themselves, polysaccharides, extracellular DNA and protein amyloid fibrils are additionally shown. This figure is reproduced with permission.9 (B) The wild-type (WT) versus the deletion-mutants (Δ) of B. subtilis bacteria and the biofilm formed by them. The WT, Δeps, ΔtasA and Δeps–tasA. The deletion mutants form bacterial communities that are healthy but with a weakened/disrupted biofilm. This figure is reproduced with permission.63 (C) Reconstruction of a native-like B. subtilis biofilm from a ΔtasA deletion mutant that cannot form biofilm by itself. The supplementation of recombinantly produced TasA protein to the growth medium of the ΔtasA mutant B. subtilis strain reconstitutes the native-like biofilm. This allows incorporation of isotope-labeled TasA into the biofilm for in vivo NMR spectroscopy without any background signal. (D) 2D 1H–15N NMR spectra of different TasA samples prepared in vitro, soluble monomer, oligomer and fibril, as well as in vivo TasA in biofilm. Spectral differences indicate structural differences and shed light on the biofilm TasA form. For more details on the figure and explanation of the additional indications, the reader is referred to the original article. This figure is reproduced with permission.81 | ||
Aggregated proteins in the form of amyloid fibrils play a key role in maintaining the structural integrity of biofilms and when these proteins are mutated, the biofilms are disrupted and bacteria become accessible to, e.g., antibiotic treatments. Functional amyloids strengthen biofilms and are a major threat to human health, since the (chronic) infections they cause are difficult to treat due to the biofilm structural integrity and insufficient penetration of drugs, thus promoting antibiotic resistance (antimicrobial resistance, AMR).12
Targeting biofilms and their amyloid components could be a novel approach to fight AMR, which results in ∼million casualties per year, and is estimated to cause more death than cancer by 2050.13 However, very little structural information exists about biofilms and their fibrillar components and how these components interact with other proteins. Determining these unknown structures and unraveling the structure–activity relationship of such fibrils will help to develop better therapies.
In this current review we focus on functional amyloids related to bacterial biofilm formation or functional bacterial amyloids (FuBAs), more specifically curli from E. coli, FapC from Pseudomonas, TasA from B. subtilis and phenol-soluble modulins (PSMs) from S. aureus, Fig. 2. For an extensive list of various functional amyloids along with coverage of the broader theme of amyloids, functional amyloids, prions and functional prions we refer to other recent excellent reviews.14–22
Other functional amyloids related to bacteria but not directly involved in the biofilm formation have also been observed. These include but are not limited to S. coelicolor chaplins which facilitates the rising of aerial hyphae,23,24M. tuberculosis amyloid pili (MTP) which mediates host interactions during pathogenesis,25 and X. axonopodis harpins which act as virulence factors.3,26 Recently other functional amyloids from bacterial biofilms such as the Esp from E. faecalis27 have also been discovered but due to the recent discovery these are still poorly understood and will not be described in this paper. Moreover, due to the homology between the Salmonella Tafi system and E. coli curli,28 the Tafi system will also not be addressed here.
Curli from E. coli
Curli was the first discovered biofilm forming functional amyloid fibril and hence curli fibrils from E. coli are the most extensively characterized FuBAs.4 Curli fibrils have been found to be essential for biofilm formation and attachment to surfaces including plant cells, stainless steel, glass and plastics.29–33 They also play key roles in interaction with host proteins and invasion of host cells.34–36 The curli biogenesis involves the expression of two divergently evolved operons, csgBA and csgDEF.35 CsgA is the major biofilm forming amyloid protein component of curli and CsgB is the minor amyloid protein. CsgA and CsgB are secreted as unstructured monomeric proteins to the cell surface to self-assemble into the final amyloid fibrils. CsgB facilitates CsgA fibrillation as a nucleator and is required for the complete assembly of curli fibrils.35,37 CsgC is a chaperon-like accessory protein that prevents fibrillation in the periplasm to ensure that CsgA and CsgB are secreted as monomeric units to the extracellular compartment.38 CsgE is another chaperone-like accessory protein that reversibly interacts with CsgA (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) while the pore-forming protein CsgG promotes translocation, thereby achieving specificity for secretion.39 CsgF and also CsgB help localize CsgA to the cell surface40 while CsgD regulates the expression of the csgABC operon.19 This amazingly regulated protein system controls and directs curli amyloid formation and maturation, and prevents the formation of toxic oligomers or fibrils inside the cells that may otherwise harm the bacteria itself. CsgA and CsgB are homologs and share ∼50% sequence identity. They both consist of five imperfect repeats of ∼20 amino acids and the imperfect repeats of CsgA contain highly conserved glutamine and asparagine residues, which are important for the amyloid formation,37,41Fig. 2. Repeating the amyloidogenic region several times likely comprises an effective way of fibrillation compared to non-repeat containing proteins.42 This may explain the fast fibril formation without a lag-phase compared to other amyloids. The repeats R1, R3 and R5 of CsgA are amyloidogenic on their own and can form fibrils similar to wild type (WT) CsgA, whereas R2 and R4 do not form fibrils on their own and are dispensable for WT fibril formation.40 Interestingly, the amyloidogenic R1 and R5 contain sequences that contribute significantly to the ability of CsgA to bind human proteins such as fibronectin, plasminogen, tissue plasminogen activator, and β2-microglobulin.43In vitro in the absence of the other curli components CsgA aggregates in a nucleation dependent manner.44
:1 ratio) while the pore-forming protein CsgG promotes translocation, thereby achieving specificity for secretion.39 CsgF and also CsgB help localize CsgA to the cell surface40 while CsgD regulates the expression of the csgABC operon.19 This amazingly regulated protein system controls and directs curli amyloid formation and maturation, and prevents the formation of toxic oligomers or fibrils inside the cells that may otherwise harm the bacteria itself. CsgA and CsgB are homologs and share ∼50% sequence identity. They both consist of five imperfect repeats of ∼20 amino acids and the imperfect repeats of CsgA contain highly conserved glutamine and asparagine residues, which are important for the amyloid formation,37,41Fig. 2. Repeating the amyloidogenic region several times likely comprises an effective way of fibrillation compared to non-repeat containing proteins.42 This may explain the fast fibril formation without a lag-phase compared to other amyloids. The repeats R1, R3 and R5 of CsgA are amyloidogenic on their own and can form fibrils similar to wild type (WT) CsgA, whereas R2 and R4 do not form fibrils on their own and are dispensable for WT fibril formation.40 Interestingly, the amyloidogenic R1 and R5 contain sequences that contribute significantly to the ability of CsgA to bind human proteins such as fibronectin, plasminogen, tissue plasminogen activator, and β2-microglobulin.43In vitro in the absence of the other curli components CsgA aggregates in a nucleation dependent manner.44
FapC from Pseudomonas
Functional amyloids in Pseudomonas (Fap) have more recently been discovered and are encoded by a single operon with six genes, fapA–F.45 In addition to the original strain of Pseudomonas and P. fluorescence many other pseudomonas strains also express Fap amyloids, including the pathologically relevant P. aeruginosa. This strain is involved in cystic fibrosis pathology, where it is considered the major pathogen in the development of chronic airway infections and linked to declining lung function.45–47 In Pseudomonas the Fap system is found by phylogenetic analysis in the beta-, delta-, and gammaproteobacteria where a high fraction of the identified strains are pathogens (39%) or rhizobacteria (36%). This has led to the suggestion that, although the Fap system is not required for pathology as not all pathogens in the identified genera had the Fap genes, the Fap system could be a virulence enhancing factor in the pathogen strains and could also aid in the association with plant roots. Analogous to curli, FapC is the major biofilm forming amyloid protein, whereas FapB is the minor component and a nucleator for FapC.45,48,49 They are secreted as unfolded monomers similar to CsgA–B, and then fibril formation happens at the cell surface through self-assembly.46 FapA is a transcription regulator for fapAB and controls the amyloid formation, and interestingly the deletion of it leads to fibrils composed solely of FapB.48 FapD is a protease making essential modifications to the related Fap proteins. FapE is a minor component playing a role in the amyloid formation, it is found to be part of the mature Fap-fibrils.48 Finally, FapF is a trimeric β-barrel membrane transporter having an extended domain in the periplasm and gated by a helical plug and it secretes FapC through the outer membrane.50,51 FapC contains three repeat motifs of ∼37 amino acids that stack into the β-sheet structure of the amyloids, Fig. 2. The three repeat motifs are separated by highly flexible linkers and furthermore also contain highly conserved glutamine and asparagine residues.45 The three imperfect repeats in FapC are crucial for the stability of functional amyloids,52 and the presence of imperfect repeats in FapC has been shown to promote aggregation efficiency by decreasing the lag-time of aggregation. Also the repeats reduce the tendency of the formed aggregates to fragment. Interestingly, even a construct missing all three imperfect repeats could still aggregate although the aggregation kinetics observed were irregular and the structures of the aggregates formed were altered and also destabilized.53 Removal of the repeats through deletion has been shown to increase the degree of compaction of the protein during the activation step of fibril growth through elongation.54PSMs from S. aureus
The functional amyloids comprising the structural scaffold of S. aureus biofilms are composed of various phenol-soluble modulins (PSMs).55 PSMs are amphipathic peptides that have multiple functions in pathogenesis. They are small peptides of 20–44 amino acids in size, Fig. 2. The genes encoding the core family of PSM peptides are highly conserved and located in psmα operon (αPSM1–αPSM4) and psmβ operon (βPSM1 and βPSM2), and the δ-toxin is encoded within the coding sequence of RNAIII.56 PSMs have been recognized as a crucial factor for S. aureus biofilm formation, as PSMs strengthen the S. aureus biofilms. Experiments performed on strains that cannot produce α/β-PSMs resulted in biofilm disruption.55 In their soluble monomeric form they hinder host immune response by recruiting, activating and lysing human neutrophils while also promoting biofilm dissociation.55 However, PSMs also self-associate to form the amyloid fibrils that fortify the biofilm matrix to better resist disassembly by mechanical stress and matrix degrading enzymes.57 Surprisingly, when a mature S. aureus biofilm was exposed to “soluble” PSMα1, the biofilm amount was significantly reduced, however, the polymerized fibrillar PSMα1 had no such effect.55 This indicates a biofilm maturity regulation by the very same peptides producing the biofilm. High expression of αPSMs increases the virulence potential of methicillin-resistant S. aureus.58 Moreover, PSMα3, the most cytotoxic and lytic PSM, enhances its toxicity to human cells upon fibrillation.59 Despite lower concentrations, the larger βPSMs, ∼44 residues in length, seem to have the most pronounced impact on biofilm structuring.60 Finally, PSMs can undergo truncations in vivo to form even smaller peptides (∼6 amino acids) and have an altered function as antimicrobial agents and different structural properties compared to the full-length PSMs.61TasA from B. subtilis
The extrapolymeric matrix of B. subtilis biofilms is mainly composed of exopolysaccharides and the protein TasA,62Fig. 3. The gene encoding TasA is located in the tapA–sipW–tasA operon and the polymerization of TasA in vivo requires the expression of all three members of the operon,62Fig. 2. TasA is the major biofilm forming functional amyloid fibril and TapA is the minor component. They are both secreted as unfolded monomers, in a mechanism not fully understood yet. SipW is the general secretory pathway, responsible for the secretion of these proteins. When TasA or TapA is mutated (as well as the exopolysaccharide), the native biofilm structure is disrupted, and the bacteria colony loses the wrinkled topology,62–66Fig. 3B. Processing through a signal peptidase activity and secretion of TasA and TapA are regulated by SipW, which also regulates expression of genes involved in exopolysaccharide production.67–69 Recombinantly purified TasA forms amyloid fibers in vitro and can also restore wild-type morphology to the biofilm of a TasA deletion mutant and hence the fibers formed by TasA provide structural integrity to B. subtilis biofilm.63 The fibers formed by TasA were found to connect individual cells forming a network while also possibly organizing other components in the biofilm matrix. In addition to TasA, the protein BslA (formerly named YuaB) is also found in B. subtilis biofilms where it forms a hydrophobic coat of the surface of the biofilm by self-assembly.70–72Functional amyloid structural features
Currently, to the best of our knowledge, there is no high-resolution structure of any full-length cross-β functional amyloid protein in its fibrillar conformation, except PSMα3 that forms a unique cross-α fibrillar structure.59 Despite this, there are structures available from small peptide fragments from functional amyloids, monomeric protein forms as well as some structural models. This lack of structural information limits our understanding of functional amyloids and the formation of biofilms to a large extent, and hampers the strategic fight against, e.g., biofilm related infections, pathogenesis and antimicrobial resistance (AMR).73,74General structural features of (functional) amyloid fibrils
The function of a protein in living organisms is directed by its three-dimensional (3D) structure, entailing a structure–function relationship. Every protein has to adopt its correct fold to function, which is uniquely determined by the amino acid sequence. However, one protein can adopt multiple structures if the conditions (buffer, pH, salt, concentration etc.) are manipulated towards different local/globular minima in the protein folding energy landscape. The amyloid fold derived by intermolecular contacts, rather than intramolecular contacts, is one of these possible structures and with its high stability and lower total energy, once formed they are usually trapped in this conformation.75Amyloid fibrils can be obtained for many protein sequences, e.g. via misfolding along the folding pathway towards a native conformation. Protein misfolding and/or fibrillation is usually associated with biological anomalies and diseases.1,76 Neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD) are known examples that are caused by pathological amyloid proteins. The loss of normal protein function and subsequent gain of toxicity are key phenotypes for this type of pathology. Functional amyloids, in contrast, have a distinct biological function in vivo.1,77,78 By applying harsh conditions such as extreme pH or denaturing conditions, many globular or unfolded proteins can be directed to amyloid fibrils.79 Starting from a natively unfolded protein state (intrinsically disordered protein, IDP), rather than a well-defined native fold, highly stable amyloid structures can be formed as seen in AD and PD or from functional amyloids like CsgA and FapC.20,42 Amyloid formation has been observed from proteins with all types of native structures, e.g. in an α-helical structure from serum amyloid, β-sheet rich structure from transthyretin, or α- and β-rich structure from lysozyme,80 as well as in a more controlled manner from a globular protein fold.63,81 When amyloid fibril formation occurs from an initial folded protein extensive conformational rearrangements are required, which may require unfolding to an intermediate that then forms the final amyloid fibril structure.82,83 Despite many initial forms of fibril forming proteins, the final structure consists of cross-β-sheets as explained below.84
Amyloid fibrils are insoluble protein aggregates and are rich in β-sheet content if not composed predominantly by them. For a globular protein the β-strands form intramolecular β-sheets, however, for amyloid fibrils the β-strands form intermolecular β-sheets which are arranged orthogonal to the fiber axis. The longer β-strands can be bent into a β-hairpin motif connecting two β-strands. This cross-β-sheet structure is stabilized by hydrogen bonds running parallel to the fiber axis, Fig. 4A. Steric-zippers formed from tightly packed protein sidechains between the β-sheets increases the amyloid fibril stability particularly for the small-peptide amyloid examples. One or more cross-β-sheets then form one or more protofilaments. Due to this protein architecture a typical X-ray diffraction pattern is observed, with two typical reflections at ∼4.7 Å (vertical axis) corresponding to the inter-strand spacing and at ∼10 Å (horizontal axis) corresponding to the inter-sheet spacing due to the sidechains,85–87Fig. 4. The kinetics of amyloid formation is typically studied using the amyloid binding dye Thioflavin T (ThT). The fluorescence intensity of the dye increases due to binding of ThT in grooves at the surface of the amyloid fibrils perpendicular to the fibril axis. This results in rotational immobilization of the ThT molecules changing the quantum yield of the dye.88–90 Typical aggregation curves yield a sigmoidal shape comprising an initial lag-phase followed by exponential growth ending with a stationary phase which characterizes a nucleation dependent pathway,91Fig. 5. Later studies shed light on the long-range organization of the amyloid fibrils forming the cross-β-sheet structure and their helical symmetry.92–94 Usually the amyloid fibrils are not straight but have a helical symmetry with a small twist angle to minimize the overall free energy of the structure,95 which is particularly true for structures bearing several protofilaments.96
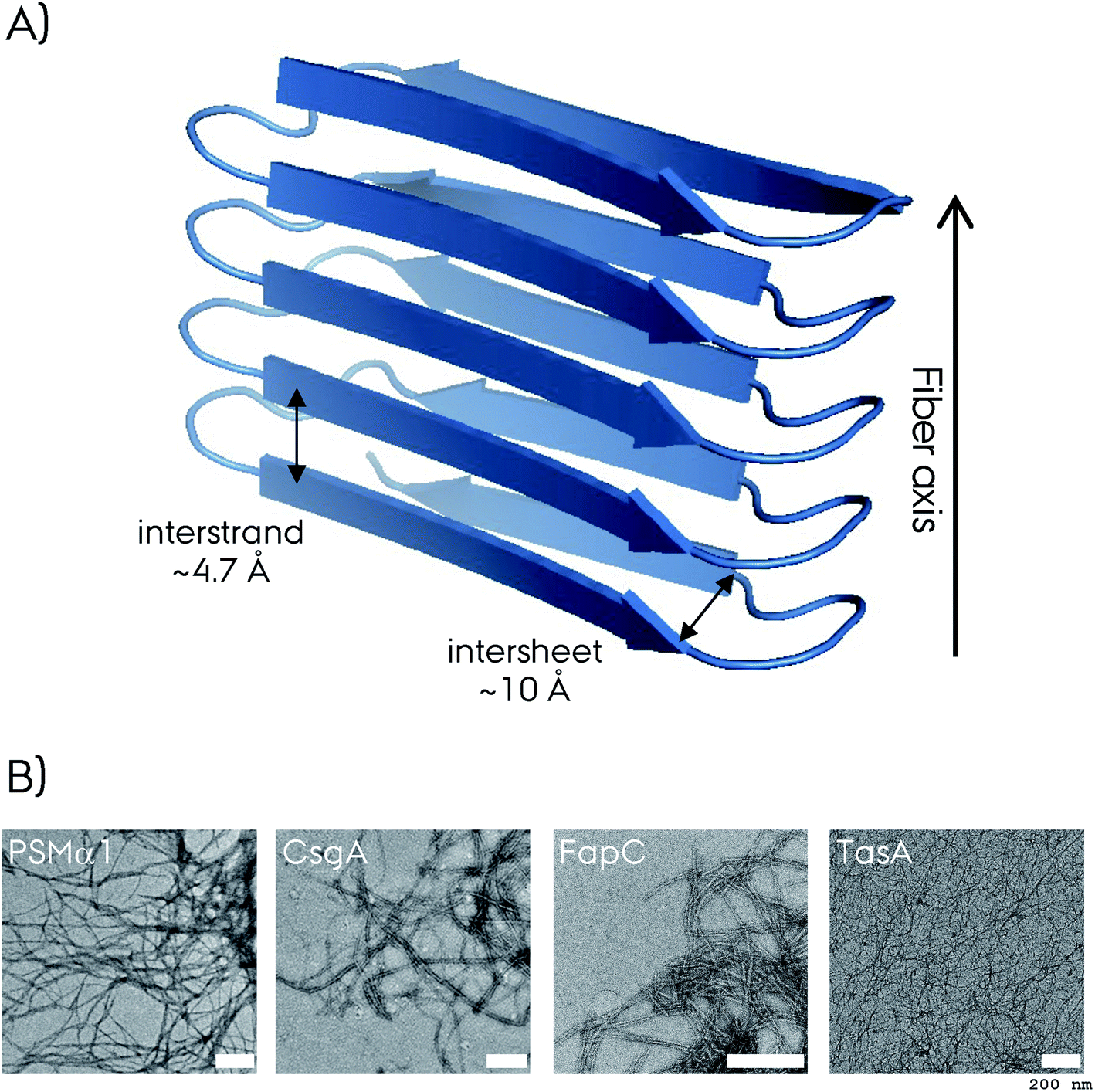 | ||
| Fig. 4 (A) Representation of the cross-β-sheet amyloid fibril fold. The interstrand and intersheet distances of ∼4.7 and ∼10 Å are depicted. Here the β-strands form intermolecular β-sheets and are orthogonal to the fiber axis. (B) The negative-stain electron microscopy (EM) micrograph of four different biofilm forming functional amyloids. The white scale bar represents 200 nm at each micrograph. The CsgA EM is reproduced with permission.148 | ||
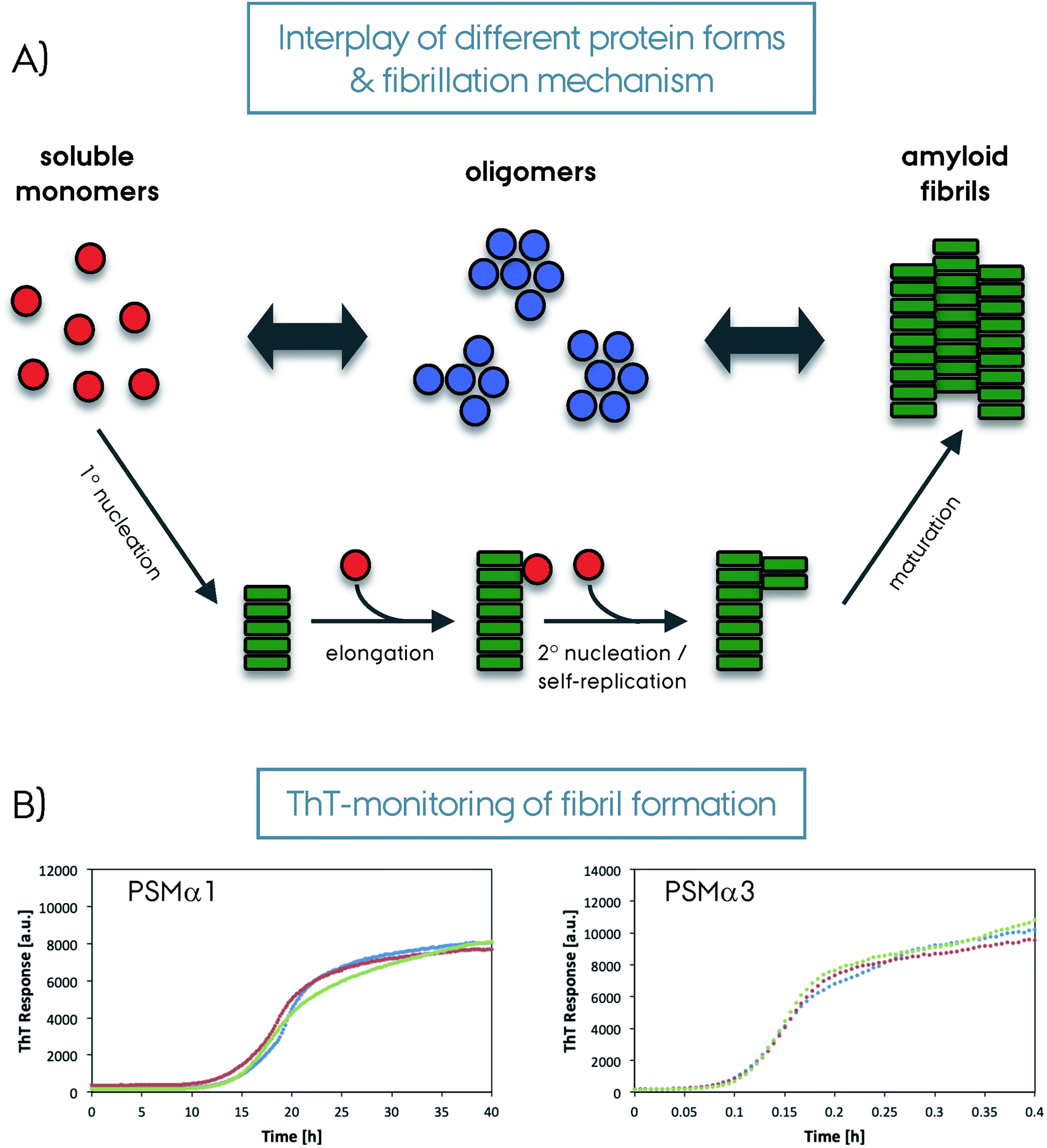 | ||
| Fig. 5 (A) Schematic representation of amyloid fibril formation. The soluble protein fold (in red) can form oligomers (in blue) or amyloid fibrils (in green) depending on the system and condition. The oligomers then form amyloid fibrils. The amyloid fibril maturation can happen with different steps/mechanisms as depicted in the figure; 1° nucleation, elongation, 2° nucleation/self-replication and a final maturation. (B) Amyloid binding Thioflavin-T dye results monitoring the formation of amyloid fibrils from soluble monomers for PSMα1 and PSMα3 proteins from S. aureus (repeated three times for each protein, please note the different time scales). | ||
Despite the unique amino acid sequences seen in functional and pathological amyloid proteins structural similarities are seen from the known atomic resolution structures.16,97,98 For example, a comparison of the functional amyloid HET-s with α-synuclein (αSyn) or amyloid-β (Aβ)42,99–101 indicates that these structures are stabilized by hydrophobic contacts, as well as intermolecular hydrogen-bonds along the cross-β-sheets. Hydrophobicity or polarity of the surfaces as a whole or by patches is a difference between these two classes. HET-s displays a polar surface with no hydrophobic areas, whereas the pathological amyloids typically expose hydrophobic areas. This difference may have an effect on the regulation/interactions of these proteins. Similarly the display of hydrophobic patches or lack thereof on the surface of structurally similar protein oligomers along with the degree of flexibility has been linked to the cytotoxicity where increased exposure of hydrophobic patches was linked to cell membrane disruption and toxicity.102 Moreover, the functional amyloids do not contain frustrated structural segments, in contrast to pathological ones that are not evolutionarily optimized towards fibril formation.16,103 Structural polymorphism is also another factor, in which functional amyloid HET-s adopts a single conformation,99,104 whereas polymorphism is commonly observed for pathological fibrils such as αSyn and Aβ.105–107
A quantitative comparison of the amino acid composition of functional versus pathological amyloids reveals more details on the evolutionary optimization of amino acids and their selection accordingly.17 A comparison of four pathological and six functional amyloid sequences indicated that despite high-frequency occurrence of nonpolar residues in both, the polar residues were more dominant for functional amyloids. The polar residues can form ladders of hydrogen bonds along the fibril, with higher frequency in functional amyloids. Positively charged residues such as lysine, arginine and histidine are observed more frequently in pathological amyloids, whereas, negatively charged residues are observed for both types to a similar degree (except glutamate showing higher frequency for pathological amyloids). This phenomenon has alternatively been described as gatekeeper residues composed of arginine, aspartate, glycine, lysine or proline.42 Overall, more frequent occurrence of glycine, serine and glutamine in functional amyloids correlates with them being more reversible with more hydrophilic and flexible interfaces, in contrast to the hydrophobic, dry and tight interfaces observed for pathological amyloids.16 The aliphatic residues constituting highly stable steric-zipper conformations are observed often in small-peptide amyloid structures and in pathological amyloids. There is strong amino acid specificity for the formation of steric-zippers. Homo-zippers by same protein sequence fragments are observed mostly in small-peptide amyloid structures. This homo-zipper motif is observed at the fibril interfaces of different protofilaments for the full-length amyloid fibril proteins. Otherwise, for full-length protein fibrils hetero-zippers are formed from two different amino acid sequences.20 The functional amyloids were also found to contain low-complexity aromatic-rich, kinked segments (LARKS) which are more reversible due to a weaker packing and a kinked backbone which reduces the stability of the complex.108,109 A recent three-fold symmetric structure of the functional neuronal amyloid Orb2 revealed a hydrophilic steric-zipper interface which highlights its functional role in Drosophila in contrast to the hydrophobic core of pathological amyloids.110
The control of fibrillation by other protein machinery is a remarkable difference between functional and pathological amyloids. For example, FapC fibrillation strongly depends on a nucleator protein FapB and others in the same operon (FapA–F) for tight regulation of the localization, secretion, and fibrillation.45 The optimized sequence properties and accessory protein machinery of functional amyloids makes the aggregation process both fast and controlled compared to the pathological ones.111 Along these lines, the aggregation kinetics and mechanism of the pathological and functional amyloids is noticeably different, Fig. 5. Aggregations of CsgA and FapC mostly progress via primary nucleation and elongation without self-replication, making the process more controlled and directed.111 On the other hand, the fibrillation of pathological amyloids can be mediated by secondary processes such as secondary nucleation and fragmentation which are more stochastic.112
High-resolution structure determination methods
Historically, negative staining EM allowed the visualization of amyloid fibrils at low resolution, Fig. 4B. In favorable situations, it's possible to differentiate the straight filaments from twisted ones or the protofilaments, singles from multiple ones. This approach combined with mass-per-length (MPL) measurements allowed the determination of the number of protofilaments per cross-section. The first atomic insights into the amyloids came from the X-ray structures of the small spine regions of amyloids that could be crystallized unlike the full-length proteins.84,113 High-resolution reconstruction of density maps at the atomic level came into play with the cryo-EM in the last decade or so, with new technology available such as direct-electron detectors and software packages for efficient and fast helical reconstruction.114 In the last decade fast frozen specimens with class averaging of many images resulted in high resolution that allowed atomic modeling of the full-length protein structures, which is referred to as the “resolution revolution”.115–117Solution NMR spectroscopy has been applied to study functional amyloids in their monomeric soluble form.81,118–120 Since most of the known functional amyloids are unfolded proteins, the use of this technique is limited to understanding the structural changes during amyloid fibril formation. Nevertheless, it provides valuable information, e.g. by hydrogen/deuterium exchange experiments to understand slowly exchanging sites.121,122 Higher molecular weight oligomers or fibrils cannot be observed directly by solution NMR, due to their slower tumbling compared to monomers which decrease both the intensity of NMR signals and the resolution. For such insoluble oligomeric and fibrillar forms of functional amyloids, ssNMR is the method of choice to determine structural information and atomic resolution structures.123,124 Several beautiful examples have been presented in the recent years, including non-biofilm forming functional and pathological fibrils. These are described in detail in recent reviews and we refer the reader to these for a full overview.114,123,124
ssNMR has been used for understanding the molecular details and structures of amyloid fibrils since the 2000s.96 The breakthrough in the application of this method was initially slowed down by general difficulties in sample preparation of fibrillar samples due to polymorphism. Extensive studies and progress in this field, including seeding approaches, resulted in high-resolution ssNMR spectra of amyloids composed of a single polymorph for various systems, such as αSyn.125,126 Amyloid fibrils have local order with irregularities in long-range order due to bends or curves.123,124 This is not a problem for ssNMR studies since the local order is sufficient to result in high-resolution NMR spectra.
ssNMR spectroscopy historically relied on 2D/3D carbon-detection experiments for chemical shift dispersion and sequential assignment,99,127–129 requiring ∼10 mg of fully/selective/sparse isotope labeled protein. More recently, proton-detection experiments relying on protein deuteration and/or ultra-fast sample spinning (MAS) have expanded the scope of ssNMR by improving the sensitivity significantly and reducing the requirement for the protein amount needed to <1 mg.130–134 Nowadays, these proton-detecting ssNMR experiments at high magnetic fields above 1 GHz combined with MAS frequencies above 100 kHz allow a fast and sensitive structure determination of amyloids or other protein forms.135–138 It is important to note that combining long-range structural information from cryo-EM and atomic resolution of a single unit from ssNMR can be a very powerful experimental approach. In addition, dynamic nuclear polarization based (DNP) ssNMR increases the sensitivity by several orders of magnitude, and opens up the possibility to study systems otherwise very difficult or impossible, e.g. proteins in complex environments at low concentrations.139–143
Structural studies on FuBAs
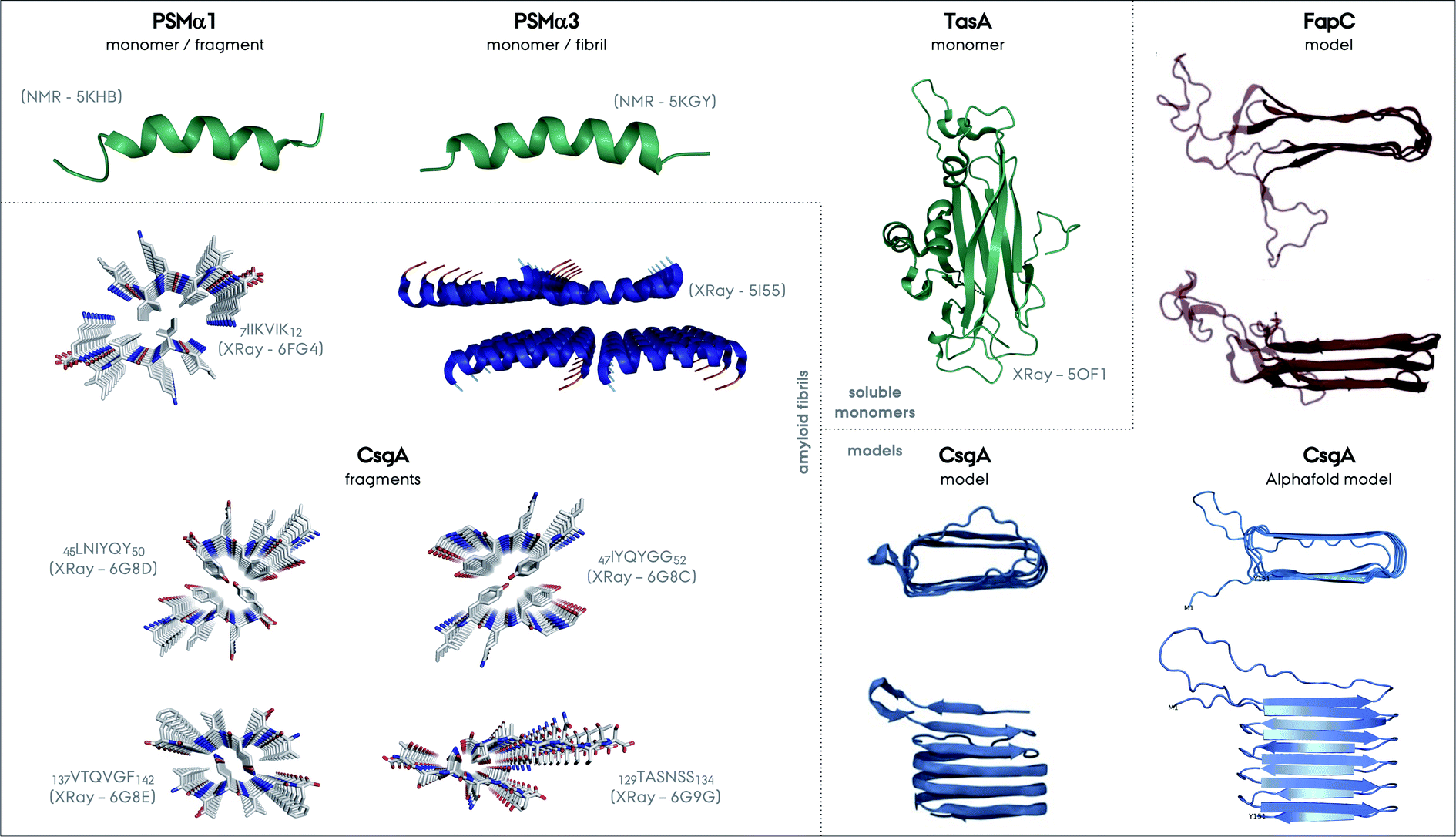 | ||
| Fig. 6 Structures of different functional amyloids discussed in this review as their full length or truncated fragments. The structure determination method and corresponding PDB code are given in parenthesis for each system. The structures determined experimentally as soluble monomers are shown in green, for PSMα1, PSMα3 and TasA as one group. Fibrillar structures of the fragment of PSMα1 and full length PSMα3 are additionally shown. There is not yet a CsgA structure from full-length protein determined experimentally, nevertheless the structures of the four different small fragments exist and are shown. Please note the parallel versus antiparallel arrangements of individual β-strands in the amyloid fragments of CsgA. The color codes on the structures indicate red for oxygen, blue for nitrogen and grey for carbon. Moreover, the two structural models exist for CsgA and are shown in the Models section in the figure. One of the models relies on amino acid contacts from a multiple sequence alignment of homologous proteins and the other one is from Alphafold (PDB-like file was kindly provided by Kresten Lindorff-Larsen and presented elsewhere earlier).152 The structural model of FapC is also based on a multiple sequence alignment of homologous proteins, reproduced from the work by Dueholm and coworkers.46 | ||
Truncation mutants of PSMα3 were studied for structure determination.61 In contrast to the cross-α structure of the full length PSMα3, the 7LFKFFK12 fragment forms two different atypical cross-β amyloid fibrils composed of anti-parallel strands, Fig. 6. One polymorph is composed of hexameric architecture with cylindrical cavities along the fiber axis, and the other one is composed of out-of-register β-sheets that are −50° off from the fibril axis in contrast to the 90° found for in-register sheets. These structures were found to be less stable compared to the classic cross-β structures containing steric-zipper elements. In contrast to this picture, the structures of truncation spine elements of PSMα1 7IIKVIK12 and PSMα4 7IIKIIK12 form cross-β fibrils with steric-zippers and have classical X-ray reflections at 4.6 and 10 Å. Full length PSMα1 and α4 undergo a structural transition from helical to β-sheet in solution during aggregation within several days, in contrast to PSMα3.146,147 These transitions or non-transitions could be related to the functions of these proteins, which are strikingly colorful. More structural studies on full length PSMs are needed to unravel the full picture of these systems.
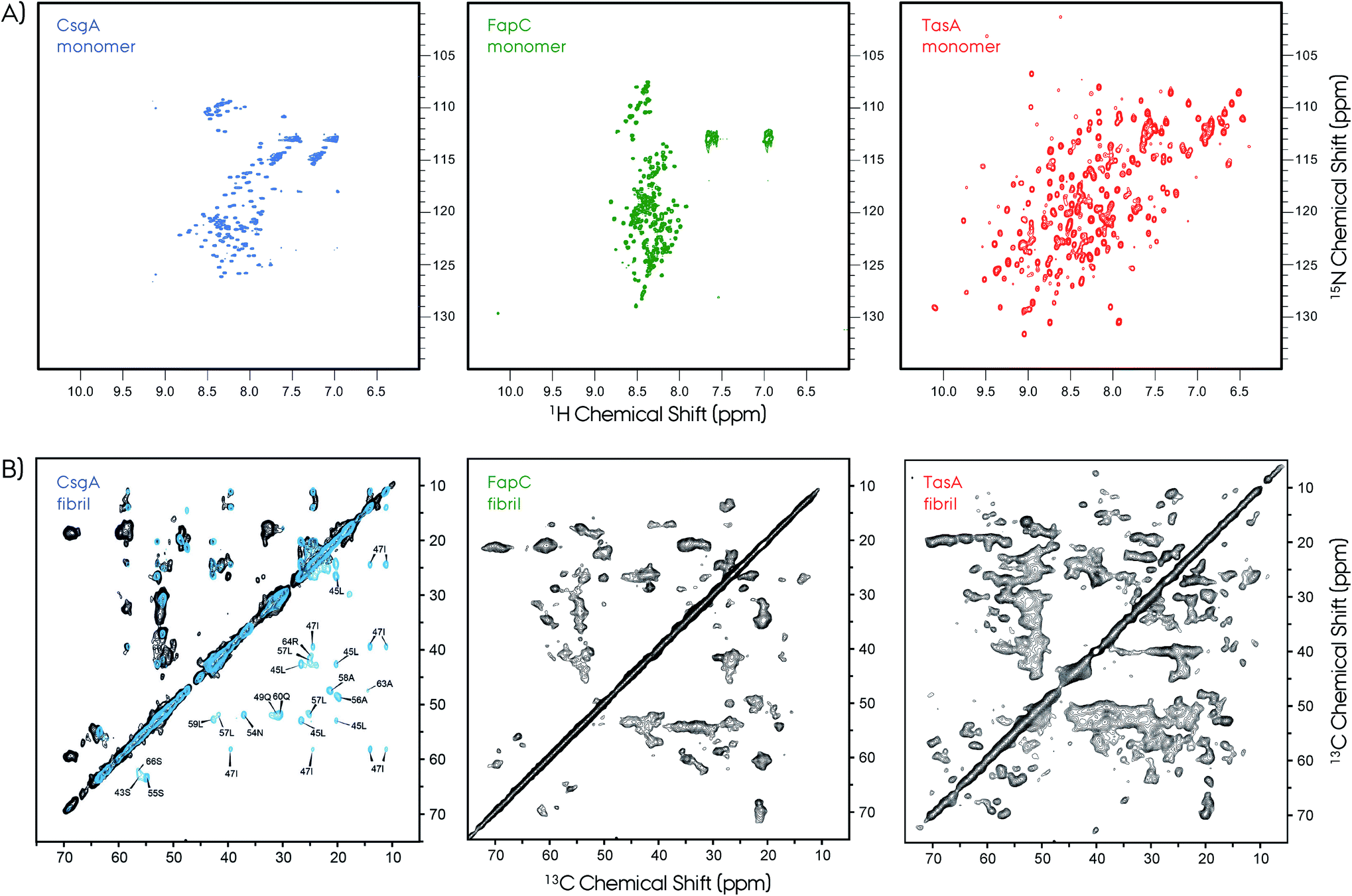 | ||
| Fig. 7 NMR is a powerful technique to study protein structures of soluble and insoluble forms. (A) 2D 1H–15N HSQC NMR spectra of soluble monomeric CsgA (in DMSO), FapC (in DMSO) and TasA (in H2O) functional amyloids. The unfolded nature of the CsgA and FapC can be seen directly from the narrow chemical shift dispersion in the proton dimension in the HSQC spectra. In contrast, the TasA has a well-dispersed HSQC spectrum, which is an indication of its folded globular protein fold. (B) The 2D 13C–13C MAS NMR spectra of CsgA, FapC (R3C construct) and TasA are shown. Solid-state NMR spectroscopy is used to study structures of functional amyloids in their insoluble fibrillar form. The NMR spectra show high quality that is suitable for structure determination. Sparse/segmental labeling combined with proton-detection experiments will additionally increase the full structure determination possibilities for these kinds of difficult proteins. Such segmental labeling of only a single repeat for CsgA results in superior spectral resolution, isotope labeling of full-length is shown in black and single-repeat is shown in blue with several signal assignments. Frans Mulder kindly provided the FapC HSQC spectrum. The 2D 13C–13C ssNMR spectrum of CsgA is reproduced with permission.149 | ||
Typical X-ray diffraction patterns were observed for both CsgA and CsgB indicating cross-β-sheet spacings of ∼4.7 and ∼9 Å. Secondary structure contributions were determined by circular dichroism (CD) to be ∼16% α-helix, ∼40% β-sheet, ∼13% β-turn and ∼31% others. These in vitro prepared fibrils of CsgA were more resistant to proteinase-K as compared to the soluble form. A comparison of the observed chemical shifts with the random-coil values150,151 indicates the presence of major and minor contributions for many of the selectively labeled sites. Quite surprisingly, these chemical shifts indicate both β-sheet and non-β-sheet contributions, which is a major difference between CsgA and other pathological amyloids that are composed of in-register β-sheet fibrils. This situation was similar to that of the HET-s structure which was found to adopt a unique β-solenoid structure.99 The distance measurements based on dipolar-dephasing experiments were not definitive but indicated a non in-register β-sheet structure. This was supported by the MPL measurements and EM images showing the fibrils having a narrow (∼3–4 nm) width. Proposed flexible loops that may be connecting the rigid repeat units were not observed by NMR.
A CsgA structural model based on amino acid contacts from a multiple sequence alignment of homologous proteins suggests that the hairpins stack on top of each other to form a β-helical structure, Fig. 6,48,152 similar to HET-s and the recently determined structure of a HET-s homolog HELLF functional amyloid fibrils.99,153 The model is consistent with the repeats identified for CsgA, Fig. 2. The Alphafold structural model is overall similar to this model, and both are consistent with the NMR data described above.154
A more recent ssNMR study revealed more insights on the structural fold of CsgA and supports the proposed models, Fig. 7.149 This work presents evidence via ssNMR that the biofilm-extracted and recombinant produced proteins adopt a similar homogeneous structure. The spectral resolution (∼0.35 ppm) of these preparations is superior compared to that of the lyophilized samples,148 and similar to that of HET-s or αSyn.99,129,155 Sample preparation of amyloid fibrils remains a challenge and needs to be optimized carefully for each system. The segmental labeling strategy of a single repeat unit R1 (21–69 amino acids) greatly simplifies the NMR spectrum and allows sequential assignment and secondary structure propensity determination.156 These results support the strand–loop–strand structural motif with two β-strands from S43–Y50 and S55–T61 separated by a short rigid glycine loop. Moreover, the R1 repeat unit and the full CsgA adopts a similar β-solenoid structure, and most probably all the repeats are structurally equivalent. Observation of long-range distance restrains defines a tight turn between the individual β-strands of the repeats. Finally, ssNMR spectra monitoring only mobile segments contain signals from the N-terminus and the loop that connects repeats.
A recent study by Pedersen and coworkers expands the macromolecular structural details of FapC amyloid fibrils.159 In this work, an MPL value of ∼33 kDa nm−1 was determined by EM and small angle X-ray scattering (SAXS), indicating that the mature fibrils consisted of two-protofilaments per fibril. The protofilaments were formed without any intermediate species besides monomers and oligomers, which then mature into more compact fibrils.
2D ssNMR spectra revealed further insights about FapC.157 First of all, the spectral quality is decent compared to the CsgA, however, it was significantly lower compared to HET-s. Nevertheless, the R3C construct gives an opportunity to extract qualitative information due to its reduced complexity. Experiments based on dipolar-coupling probe only the rigid protein parts, and the Thr, Ser and Ala regions indicate chemical shifts characteristic of β-sheet, α-helix and coil segments. This situation is similar to that of CsgA and TasA.81,148,149 The NMR cross-peaks from the reduced-size R3C construct containing only one repeat and C-terminal resulted in the observation of more number of peaks than that present in the sequence. In R3C there are 1*I, 2*S and 6*A in the rigid repeat region, and 4*T, 3*S and 3*A in the flexible C-terminal. Ile showed more than one cross-peak in the 2D spectrum, although there is a single Ile in the repeat, indicating multiple conformations. Moreover, there are threonine cross-peaks in the spectrum, which only are in the C-terminus part. Overall, these results indicate a complex interplay of the R3C fibril forming region and its polymorphism expressing as cross-peak multiplication. To further elaborate on this picture, INEPT-based NMR was performed in order to detect only very flexible amino acids. The 2D 1H–13C spectrum of this type results in an almost full amino acid coverage indicating that most of the protein can be perceived as flexible, including the Ile from the repeat region. This is in contrast to CsgA observations, where INEPT signals indicate residues only from the highly flexible N-terminal signals.149 Overall, these NMR findings indicate extreme polymorphism in the FapC system, resulting in reduced resolution and transient flexibility/rigidity. Lack of spectral resolution may indicate that the homogeneous fibrillation may need accessory proteins, such as FapB and/or others.
As a final note, the FapC structural model obtained is similar to the CsgA model, highlighting the stacking of β-hairpins.48,158 One major difference is that the linker/loop regions forming disordered/flexible parts are extending from the fibril core, since they are much longer compared to the CsgA hairpin turns, Fig. 6.
Freshly prepared TasA28–261 and TasA28–239 samples at pH 7 are mostly monomeric with a small oligomeric fraction, whereas TasA261 favors oligomer formation to a greater extent. The C-terminus of TasA (∼240–261 aa) is unstructured as determined by NMR spectroscopy. The crystal structure was obtained at 1.56 Å for the monomeric TasA239 at pH 4.6. The jellyroll-fold consists of antiparallel β-sheets separated by short helices and longer loops, with surprisingly differential rigidity observed among the proteins.
To understand the high-MW components from a fresh and mostly monomeric sample at t = 0 point, two different preparations were done. First, the fresh sample was kept for a long time to obtain a gel-like sample. Second, the pH was shifted to 3 resulting in the formation of fibrils, giving typical cross-β-sheet reflections at ∼4.8 and ∼10.4 Å. From pH 7, a slow pH shift towards the acidic condition retains the folded structure, whereas a rapid pH shift results in unfolding of the monomer. ssNMR of the fibrillar TasA261 sample resulted in a well-resolved proton-detected HSQC-type spectrum, indicating high β-sheet content and a substantial structural change compared to the solution NMR spectrum. On the other hand, the ssNMR spectrum of the gel-like sample produced from TasA239 at pH 7 is remarkably different from the fibrillar ssNMR spectrum. The gel-like sample resembles the soluble monomeric HSQC spectrum with noticeable alterations as more helical/loop fractions.
A later study on Bacillus biofilms shows additional aspects of the TasA fibrillar structure.160 In this work fibrillar B. subtilis TasA samples were compared to their B. cereus homolog. The B. cereus TasA indicates an unfolded monomer, in contrast to the TasA from B. subtilis. The ssNMR analyses revealed the co-existence of β-sheets, α-helices and loops as fibrillar structural elements, as previously shown by Diehl et al.81 The proteinase-K digestion assay indicated a shorter ∼110 amino acid long resistance part of the TasA assigned to the amyloid core between residues 35 and 144. Surprisingly, when TasA fibrils are prepared under different conditions (pH, salt and protein concentrations)161 both fibrillar and ‘oligomeric’ samples with different morphologies are observed as seen in the EM and CD spectra. All these indicate the complex biophysics of TasA and how tightly it has to be controlled for production of different protein forms. It remains to be proven which of these in vitro forms will be in vivo relevant.
As a final note here, despite the observation of both TasA oligomers,81,162 no oligomers were captured for CsgA and FapC unless they are forced by using fibrillation inhibitors.163,164 This is consistent with the fast-fibrillation hypothesis of functional amyloids made out of repeats for efficient self-templating and simple primary-nucleation and elongation mechanism. This is another interesting research avenue that needs more experimental demonstrations.
In contrast to the three different TasA forms obtained for the recombinant protein in vitro, a pure fibrillar form is obtained in vivo.149 The model B. subtilis system that cannot produce TasA (ΔtasA) was used for understanding insights into the native biofilm. This strain can grow healthy, but cannot produce biofilm. Externally supplied recombinant TasA protein reconstitutes the native-like biofilm, Fig. 3C.63,81 A DCN-labeled TasA261 was used in this model system and a fingerprint HSQC-type ssNMR spectrum was obtained, Fig. 3D. This experimental setup is the first demonstration of in vivo (in the biofilm) ssNMR spectroscopy, to the best of our knowledge. The resulting NMR spectrum with decent resolution contains all the features from the spectrum of the fibrillar sample. In vivo biofilm and in vitro fibrillar spectra overlap to a large extent, however they still have small differences which indicate that additional factors within the biofilm can modulate the protein structure. This remarkable observation indicates that the biofilm contains homogeneous TasA fibrils that are resistant to proteases.
In summary, biofilm forming functional amyloids and functional amyloids in general have many properties similar to pathological amyloids. These include, e.g., a common cross-β structure, X-ray diffraction pattern and limited proteolysis results. The details of how these two classes of amyloids form structures are still surprisingly unique. As unique features, functional amyloids have a control over the mechanism that regulates their in vivo properties and fibrillation, whereas pathological ones has not been evolutionary optimized in the same manner due to their lack of function. Structurally we have limited information and except from a special crystalline case of PSMα3, no high-resolution structure is available. Nevertheless, studies unraveling structural features exist as we have summarized above and comprise a solid basis for the future full structure determination studies. With the advancements in structural biology techniques, we expect a bright future with many high-resolution structures enlightening the molecular mechanisms of functional amyloids and biofilm formation in vitro and in vivo.
Interactions between functional amyloids and components present in the biofilm matrix
As mentioned earlier the functional amyloids in bacterial biofilms are formed extracellularly. The assembly process therefore happens in close proximity to both cell membranes and the biopolymers constituting the EPS of the biofilm matrix. The interactions between functional amyloids and other biofilm components during the formation of functional amyloids and maturation of biofilms are therefore crucial to understand the biogenesis of biofilms and have thus been a topic of interest.Interactions between functional amyloids and lipids
During a study on how the cell membrane of the bacterial cell affects the formation of curli amyloids in E. coli it was found that in the presence of lipopolysaccharides (LPS), which are found in the outer membrane of Gram-negative bacteria, the ability of CsgB to nucleate and seed the formation of CsgA is enhanced.165 This was found to be due to electrostatic interactions between the lipid bilayer and the CsgB protein. Hence the bacterial cell membrane adds a layer of cooperativity to the formation of curli amyloids ensuring that the formation of the functional amyloids occurs at the cell membrane to tether the amyloid structures to the cells and hence keeping the amyloid structures in place in the biofilm.LPS have also been found to accelerate the aggregation of FapC by bypassing the nucleation lag-phase.164 This is achieved through reduction of the population of intermediates during aggregation in addition to rearrangement of early aggregates. A similar effect on aggregation is observed for the biosurfactant rhamnolipid which is produced in Pseudomonas species alongside the FapC protein. At high concentrations of the rhamnolipid micelles, they were found to be decorated with FapC fibrils indicating a more long-term interaction between FapC and rhamnolipids.
TasA also interacts differently with bacterial cell membranes compared to eukaryotic cell membranes.166 In the presence of bacterial cell membranes the fibers formed by TasA are disordered and have a different β-sheet signature than fibers formed in the absence of membranes. In the presence of eukaryotic cell membranes TasA fibers display the same morphology and β-sheet signature as in the absence of membranes indicating that the presence of eukaryotic cell membranes does not affect the TasA fiber formation. Similarly the bacterial membranes were found to deform significantly upon interaction with TasA. Again this effect was not observed for eukaryotic cell membranes suggesting that TasA penetrates the bacterial cell membranes but not eukaryotic cell membranes.166
Other functional amyloids have also been found to interact with lipid cell membranes. S. aureus PSM peptides are able to cause cell lysis and hence interact with the cell membrane in a variety of eukaryotic cells, including monocytes, leukocytes, erythrocytes, epithelial cells, endothelial cells, and osteoblasts.58,167,168 The lytic activity of the PSM peptides is closely linked to the degree of α-helicity of the peptides in the monomeric form169 and can furthermore be counteracted by serum lipoproteins.170 Besides the α-helicity of the peptides the presence of cholesterol also increases the susceptibility to lysis in vesicles containing between 10 and 30 mol% cholesterol.169 However, when a shift from liquid-disordered to liquid-ordered state occurs in the lipid bilayer upon addition of higher amounts of cholesterol, a decrease in lytic activity from the PSM peptides occurs. Based on these results it was speculated that differences in cholesterol concentration in phagosomes from different cell types and different cell species could impact PSM-mediated phagosome escape and subsequent survival of S. aureus during an infection.
The ability of lipid bilayers to alter the aggregation of proteins is not limited to functional amyloids as lipids have also been found to play a key role in the aggregation αSyn. Recently, Galvagnion and coworkers showed that lipid bilayers enhance the rate of heterogeneous primary nucleation of αSyn by three orders of magnitude.171
Interactions between functional amyloids and extracellular polymeric substances
Since the formation of functional amyloids occurs extracellularly, other biomolecules of the biofilm matrix come in close proximity with functional amyloids. Interactions with the other matrix components could therefore play a role during the formation of the functional amyloids. The EPS of the biofilm shows high biodiversity with distinct biomolecules being expressed in different bacterial species however eDNA is emerging as a nearly universal component of the biofilm across species even including fungi.172 eDNA is found to play a role in facilitating adhesion173 and promoting intercellular aggregation174 along with providing structural integrity to the biofilm architecture.175–177During in vivo biofilm formation curli fibers have been found to bind eDNA forming curli–eDNA complexes that are highly immunogenic. The complexes caused cytokine production through activation of immune cells. When administered systemically the complexes caused immune activation and formation of autoantibodies thus triggering autoimmunity. Furthermore, the interaction with eDNA and DNA from other sources (genomic and from salmon sperm) accelerated the aggregation of CsgA in vitro.178 Additionally, it has been shown that the presence of eDNA is required for functional amyloid formation in S. aureus biofilm formation in vivo.179 Analogous to CsgA, eDNA promotes the aggregation of PSMα1 in vitro.
Interactions between functional amyloids and other extracellular components
The highly sulphonated glycosaminoglycan, heparin, has frequently been used as a soluble mimic of the human extracellular matrix component heparan sulphate. Additionally, heparin is used as an anticoagulant in catheters and hence it is a molecule potentially encountered by functional amyloids in bacterial biofilms. Heparin has been found to promote S. aureus biofilm formation180,181 and it has been suggested that heparin can mimic and even act as a substitute for eDNA in S. aureus biofilm formation.182 In an in vitro study using heparin, a complex mechanism of interaction is seen with PSM peptides. Heparin was found to accelerate the aggregation of all αPSM peptides except PSMα2 but simultaneously decelerates the aggregation of βPSM peptides. In the case of PSMβ2 heparin is even capable of altering the dominating molecular mechanism of aggregation from primary nucleation and elongation dominated to being dominated by secondary nucleation.183In summary, the formation of functional amyloids in the extracellular environment of the biofilm appears to be optimized ensuring that the biomolecules in the biofilm are generally beneficial for the aggregation process.
Functional amyloids and cross-seeding
Cross-seeding between different functional amyloids
The interactions between functional amyloids and other amyloids have been gaining attention in recent years due to the recent more detailed understanding. For the E. coli curli fibrillogenesis it has been investigated how the major curli subunit CsgA interacts with the minor curli subunit CsgB. In vitro, both CsgA and CsgB are capable of forming amyloid fibers which morphologically resemble the fibers found in vivo,4,37 however in vivo CsgA alone cannot form amyloid fibers. CsgA is secreted away from the cell as a soluble protein in the absence of CsgB,184 and CsgA is not assembled into insoluble fibers in the absence of CsgB in vivo.185 CsgB was found in vivo to aggregate on its own as overexpression of CsgB in E. coli resulted in self-assembly of CsgB into extracellular fibers located at the bacterial surface.186 The nucleating ability of CsgB towards CsgA was confirmed through several studies. In inter-bacterial complementation surface localized CsgB from one mutant strain can convert soluble secreted CsgA from another mutant strain into amyloid fibers and also nucleate endogenously added recombinant purified CsgA.4,40,185 Substoichiometric concentrations of CsgB have been found to alter the mechanism of aggregation of CsgA. In the absence of CsgB, CsgA was found to aggregate through the formation of amorphous aggregates but in the presence of CsgB ordered oligomers with a β-sheet structure were seen, thus resembling the mechanism seen for CsgB in the absence of CsgA.187Through investigation of the individual domains in the proteins the C-terminal parts of both CsgA and CsgB are found to play a crucial role in the nucleation mechanism. Besides being amyloidogenic, the C-terminal repeat 5 (R5) in CsgA has been found to be critical for in vitro nucleation of CsgA.40 The R4 and R5 of CsgB are required to tether the CsgA aggregates to the lipid membrane, and mutants lacking these domains are no longer localized to the membrane but are secreted into the extracellular space. The C-terminal repeat domain, R5, is required to anchor the protein to the cell membrane although this domain in CsgB is not amyloidogenic on its own.188 In addition, CsgA nucleation by a CsgB truncation mutant lacking the C-terminal 19 amino acids is less efficient than full-length CsgB in vivo.37
While TasA constitutes the major components of the fibers present in B. subtilis biofilm, TapA has been found to co-purify as a minor component of the fibers in a 1:100 ratio with TasA.189In vitro TapA contributes to the polymerization of TasA into fibers. More specifically, a sequence of 8 amino acids in the N-terminal of TapA has been identified as being crucial in the initiation of the self-assembly of TasA.190 Additionally, the 5 conserved cysteine residues in TapA play a minor role in the formation of a robust biofilm. The presence of TapA has furthermore been shown to be required in order to anchor TasA fibers to the cell surface.63,189 Incorporation of D-amino acids into the peptidoglycan results in release of TasA fibers from the cell surface and disassembly of the biofilm.191 Besides TapA, TasA has also been found to interact with BslA during fiber formation in the biofilm. BslA acts synergistically with other matrix components to facilitate the assembly of the biofilm matrix.70In vivo, overexpression of TasA was not sufficient to overcome the effects of deletion of BslA. Besides aiding in the self-assembly of TasA, BslA also self-assembles on its own at interfaces to form a hydrophobic surface layer around the biofilm.72
Although no specific nucleator protein has been identified for the PSM peptides from S. aureus, interactions between the individual peptides have been investigated though cross-seeding experiments. No specific pattern in cross-seeding was seen between αPSMs and βPSMs but PSMα1 was found to cross-seed all the other PSM peptides.192 Based on the kinetics of the aggregation of PSM peptides along with the cross-seeding it was speculated that PSMα3 that aggregates extremely fast is able to kick-start the functional amyloid formation in biofilm. The PSMα3 aggregates are then able to cross-seed PSMα1 that normally aggregates much slower in the absence of PSMα3 aggregates. The PSMα1 aggregates can then proceed to cross-seed all the other remaining PSM peptides to ensure that even the more slowly aggregating peptides aggregate on a timescale relevant for the bacteria during biofilm formation. This ensures that βPSM peptides which have been found to have the most pronounced impact on S. aureus biofilm structuring60 are able to aggregate in a time scale relevant for the bacteria.
This mechanism of less amyloidogenic peptides/proteins being seeded by more highly amyloidogenic peptides/proteins has also been observed for other functional amyloids not involved in biofilm formation. Different members of the CRES-family which comprise part of the complex extracellular amyloid matrix in mouse spermatozoa have been found to cross-seed each other in a manner similar to the PSM peptides and these are also seeding of CsgA by CsgB.193 The cross-seeding between the CRES subgroup members suggests that it may be an evolutionarily conserved mechanism to control the assembly of some functional amyloids.
Cross-seeding between functional amyloids and pathological amyloids
Cross-seeding between functional amyloids and pathological amyloids is an emerging interesting research area due to recent discoveries of a link between functional amyloids in the gut and progression in neurodegenerative diseases. During the progression of PD it has been observed that αSyn aggregates in the form of an αSyn-immunoreactive Lewy body and Lewy neurite pathology spreads through the brain in a progressive manner from the brainstem to the telencephalon.194 In the earliest stages aggregates are detected in the olfactory bulb, as well as in the dorsal motor nucleus of the vagus nerve in the medulla oblongata. In later stages αSyn aggregate pathology is found more widespread in the brainstem via the pons and midbrain, in the basal forebrain and, ultimately, in the neocortex. The spreading of aggregated species of αSyn in mice has been demonstrated to happen through cell-to-cell transmission of pathologic αSyn aggregates in anatomically interconnected regions upon injections of synthetic αSyn fibers leading to nigrostriatal degeneration.195 Furthermore, the injections of αSyn aggregates in rats and the subsequent pattern of αSyn accumulation observed suggests that the cell-to-cell spreading occurs only through retrograde transmission.196 In rodent models the cell-to-cell spreading of αSyn aggregates has also been used to demonstrate a spreading and propagation from neurons in the gut to the brain through the vagus nerve and/or the spinal cord.197–199 Furthermore, the bacterial composition of the gut microbiome has been observed to lead to increased neuroinflammation and αSyn aggregate deposits in the brain of αSyn expressing mice. As a link emerges between neurons in the gut and the brain and the possible spread of pathological amyloid species, the interactions between functional amyloids formed from bacteria and pathological amyloids prove to be an interesting topic highly related to bacterial biofilm forming functional amyloids.200In mice it was found that monomeric CsgA was able to accelerate the aggregation of αSyn even under sub-stoichiometric conditions.200 The accelerated aggregation occurs through transient interactions between CsgA and αSyn. In the same mouse model Tau aggregation was not found to be accelerated by CsgA monomers. When αSyn is overexpressed in mice it was seen that simultaneous colonization with curli-producing E. coli in the gut promotes αSyn pathology in both the gut and the brain. Furthermore, curli expression is required for E. coli to intensify αSyn-induced behavioral deficits, including intestinal and motor impairments in the mice. Interestingly CsgA is not the only component of the curli apparatus found to interact with αSyn. CsgC and CsgE, both of which act as periplasmic chaperones and prevent aggregation of CsgA intracellularly,4,38 also interact with αSyn although in different ways. CsgC inhibits the aggregation of αSyn while CsgE surprisingly accelerates the aggregation of αSyn.201
PD is not the only neurodegenerative disease to be linked to functional bacterial amyloids, as CsgA pre-formed seeds have been found to also accelerate the aggregation of Aβ, and fibrillation inhibitors are shared between CsgA and Aβ.202 Curli cross-seeding exerted a complicated concentration dependent effect on Aβ1–40 fibrillogenesis kinetics.203
In human semen fragments of prostatic acid phosphatase (PAP248–286) increase HIV infection efficiency through increased virus adhesion to target cells. This enhancement of infection only occurs when PAP248–286 is in the form of amyloid aggregates termed SEVI (Semen Enhancer of Viral Infection), however monomeric PAP248–286 aggregates very slowly and exogenous factors have been suggested as a promoter of SEVI fiber formation in vivo.203 Indeed it has been demonstrated that curli amyloids of both CsgA and CsgB can act as a catalyst for SEVI formation from PAP248–286. At low concentrations in vitro, cross-seeding with curli fibers results in fibers of PAP248–286 that retain the ability to enhance HIV infection. Kinetic analysis of the cross-seeding of PAP248–286 with curli moderately affects the nucleation rate while significantly enhancing the growth of fibers from existing nuclei and hence the elongation rate. During the study it was also shown that curli amyloids of both CsgA and CsgB were able to cross-seed IAPP leading to a decreased lag-time but simultaneously a strong inhibition is observed in IAPP elongation.203
FapC has also been found to interact with proteins associated with neurodegenerative diseases, specifically αSyn and Aβ. In vitro FapC can seed Aβ while templating the structure of the resulting Aβ amyloid fibers to resemble that of FapC fibers.204In vivo in zebrafish larvae FapC was able to induce accelerated Aβ aggregation and Aβ pathology. In adult zebrafish FapC was found to accelerate Aβ-induced cognitive pathology. To extend the results to the bacterial biofilm it was found that co-injection of Aβ and the protein part of P. aeruginosa biofilm samples into adult zebrafish elevated Aβ pathology. This elevated pathology was not observed in zebrafish injected with only the biofilm protein sample. Based on these results Aβ can also interact with the fibers when they are present in a complex setting such as a biofilm despite the morphology of the biofilm protein sample being different from that of FapC. The changes in morphology were attributed to the presence of other structural components of the biofilm in the sample.204
FapC is also capable of interacting with αSyn. FapC binds in vitro to αSyn monomers, oligomers, and aggregates.205 Full-length FapC protein slightly accelerates αSyn aggregation while the FapC deletion mutant where all three repeat regions have been deleted and hence only the loop regions remain (ΔR1R2R3) increases the lag-time of αSyn aggregation. The fibrillation inhibition was found to be due to the formation of hetero-oligomers possibly enhanced by disulfide bond formation in FapC ΔR1R2R3. Other deletion mutants where only one or two repeat regions had been deleted did not increase the lag-time of αSyn. Interestingly, only the monomeric protein is responsible for the interactions between FapC and αSyn as no seeding effects were seen for FapC aggregates on αSyn aggregation.
Although several links have been found between functional bacterial amyloids and neurodegenerative diseases, it cannot be concluded that biofilm formation is involved in the causative effects of such neurodegenerative diseases. For example, it has also been shown that biofilm can actually protect against protein aggregation. In C. elegans the probiotic B. subtilis has been found to inhibit αSyn aggregation by altering the host sphingolipid metabolism and thereby reduce αSyn aggregation through biofilm formation in the gut.206
In summary, functional amyloids are found to interact with a wide variety of other proteins both from bacteria and from humans. They are even emerging as contributing factors involved in the development of the pathology of various human diseases with pathology linked to protein aggregation.
Concluding remarks
It is becoming increasingly clear that functional bacterial amyloids involved in biofilm formation are not solitary inert structural components in the biofilm. Cross-talk between different aggregated species is emerging as a common theme and not only a phenomenon observed between various functional amyloids and pathological amyloids. Self-assembled structures of various metabolites have also been found to induce aggregation of pathological amyloid species. Fibrillar aggregates of homocysteine have been found to display cross-talk with Aβ,207 and amyloid-like aggregates of quinolinic acid have been observed to cross-seed αSyn aggregation.208 Despite the recent focus and many new insights gained in the field, the molecular mechanisms underlining the interactions between different amyloid species are very limited. This lack of molecular insight limits possibilities to develop strategies to better combat biofilm related bacterial infections. We hope that this review will make a contribution towards a more complete understanding.Our fight against AMR and biofilm related infections relies strongly on our knowledge of the molecular details of biofilm forming functional amyloids. With the recent fast pace and exponential growth in biofilm, amyloid fibril and functional amyloid related research we believe that the future is very bright. As of today, structural and mechanistic models are predominantly still missing, along with high-resolution structures of the key elements of this complex network in biofilms. Recent developments in the field of structural biology by cryo-EM and solid-state NMR will pave the way towards our atomic level understanding of proteins in vitro and in vivo. All in all, we will be able to understand, manipulate and fight persistent infections, AMR, biofilms and amyloids forming them.
Author contributions
The manuscript was conceived, designed and written by UA and MA with equal contributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Roberto Kolter for the permission to reuse the B. subtilis biofilm pictures, and the inspiration for our biofilm illustration. We thank Daniel Lopez for additional biofilm pictures. Frans Mulder and Daniel Otzen are acknowledged for supplying the solution NMR spectrum of monomeric FapC and helpful discussion. Robert Tycko is acknowledged for supplying the solid-state NMR spectrum of CsgA. We are thankful to Kresten Lindorff-Larsen for the PDB-like structure file of CsgA and to Morten S. Dueholm for discussions on FapC structure model. Meytal Landau is acknowledged for providing the assembly crystal structure file for CsgA 6G9G fragment. UA acknowledges Anne Diehl, Madhu Nagaraj, Kürsat Turgay, Matthias Hiller and Hartmut Oschkinat for making the initial stages of functional amyloid work possible. UA acknowledges financial support from the DFF Mobilex and Aarhus University AIAS Cofund grants. MA gratefully acknowledges financial support from Aarhus University Research Foundation AUFF-E-2017-7-16 and L'oreal and Unesco For Women in Science National Award (Denmark).References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed.
- J. Oh, J. G. Kim, E. Jeon, C. H. Yoo, J. S. Moon, S. Rhee and I. Hwang, J. Biol. Chem., 2007, 282, 13601–13609 CrossRef CAS.
- M. R. Chapman, L. S. Robinson, J. S. Pinkner, R. Roth, J. Heuser, M. Hammar, S. Normark and S. J. Hultgren, Science, 2002, 295, 851–855 CrossRef CAS PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS.
- J. F. Berson, A. C. Theos, D. C. Harper, D. Tenza, G. Raposo and M. S. Marks, J. Cell Biol., 2003, 161, 521–533 CrossRef CAS PubMed.
- K. Si, S. Lindquist and E. R. Kandel, Cell, 2003, 115, 879–891 CrossRef CAS.
- V. Coustou, C. Deleu, S. Saupe and J. Begueret, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9773–9778 CrossRef CAS PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- M. Malone, T. Bjarnsholt, A. J. McBain, G. A. James, P. Stoodley, D. Leaper, M. Tachi, G. Schultz, T. Swanson and R. D. Wolcott, J. Wound Care, 2017, 26, 20–25 CrossRef CAS PubMed.
- B. C. Wu, E. F. Haney, N. Akhoundsadegh, D. Pletzer, M. J. Trimble, A. E. Adriaans, P. H. Nibbering and R. E. Hancock, npj Biofilms Microbiomes, 2021, 7, 1–13 CrossRef.
- T. F. Mah and G. A. O'Toole, Trends Microbiol., 2001, 9, 34–39 CrossRef CAS.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, Lancet, 2022, 399, 629–655 CrossRef CAS.
- R. Gallardo, N. A. Ranson and S. E. Radford, Curr. Opin. Struct. Biol., 2020, 60, 7–16 CrossRef CAS PubMed.
- S. M. Ulamec, D. J. Brockwell and S. E. Radford, Front. Neurosci., 2020, 14, 611285 CrossRef PubMed.
- M. R. Sawaya, M. P. Hughes, J. A. Rodriguez, R. Riek and D. S. Eisenberg, Cell, 2021, 184, 4857–4873 CrossRef CAS PubMed.
- N. Salinas, T. L. Povolotsky, M. Landau and I. Kolodkin-Gal, Microbiol. Mol. Biol. Rev., 2020, 85, e00062–00020 Search PubMed.
- P. Ragonis-Bachar and M. Landau, Curr. Opin. Struct. Biol., 2021, 68, 184–193 CrossRef CAS.
- S. A. Levkovich, E. Gazit and D. Laor Bar-Yosef, Trends Microbiol., 2021, 29, 251–265 CrossRef CAS.
- P. C. Ke, R. Zhou, L. C. Serpell, R. Riek, T. P. Knowles, H. A. Lashuel, E. Gazit, I. W. Hamley, T. P. Davis and M. Fändrich, Chem. Soc. Rev., 2020, 49, 5473–5509 RSC.
- V. A. Sergeeva, N. V. Zakharova, A. E. Bugrova, N. L. Starodubtseva, M. I. Indeykina, A. S. Kononikhin, V. E. Frankevich and E. N. Nikolaev, Eur. J. Mass Spectrom., 2020, 26, 158–161 CrossRef CAS PubMed.
- M. S. Rubel, S. A. Fedotov, A. V. Grizel, J. V. Sopova, O. A. Malikova, Y. O. Chernoff and A. A. Rubel, Life, 2020, 10, 156 CrossRef CAS.
- M. Bokhove, D. Claessen, W. de Jong, L. Dijkhuizen, E. J. Boekema and G. T. Oostergetel, J. Struct. Biol., 2013, 184, 301–309 CrossRef CAS PubMed.
- W. De Jong, H. A. Wösten, L. Dijkhuizen and D. Claessen, Mol. Microbiol., 2009, 73, 1128–1140 CrossRef CAS PubMed.
- C. J. Alteri, J. Xicohtencatl-Cortes, S. Hess, G. Caballero-Olin, J. A. Giron and R. L. Friedman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5145–5150 CrossRef CAS PubMed.
- G. G. Sgro, F. A. Ficarra, G. Dunger, T. E. Scarpeci, E. M. Valle, A. Cortadi, E. G. Orellano, N. Gottig and J. Ottado, Mol. Plant Pathol., 2012, 13, 1047–1059 CrossRef CAS PubMed.
- A. Taglialegna, L. Matilla-Cuenca, P. Dorado-Morales, S. Navarro, S. Ventura, J. A. Garnett, I. Lasa and J. Valle, npj Biofilms Microbiomes, 2020, 6, 15 CrossRef CAS PubMed.
- D. Romero and R. Kolter, Int. Microbiol., 2014, 17, 65–73 CAS.
- C. Jeter and A. G. Matthysse, Mol. Plant-Microbe Interact., 2005, 18, 1235–1242 CrossRef CAS PubMed.
- J. H. Ryu and L. R. Beuchat, Appl. Environ. Microbiol., 2005, 71, 247–254 CrossRef CAS PubMed.
- J. H. Ryu, H. Kim, J. F. Frank and L. R. Beuchat, Lett. Appl. Microbiol., 2004, 39, 359–362 CrossRef PubMed.
- C. Prigent-Combaret, G. Prensier, T. T. Le Thi, O. Vidal, P. Lejeune and C. Dorel, Environ. Microbiol., 2000, 2, 450–464 CrossRef CAS PubMed.
- D. Otzen and P. H. Nielsen, Cell. Mol. Life Sci., 2008, 65, 910–927 CrossRef CAS PubMed.
- M. F. Gebbink, D. Claessen, B. Bouma, L. Dijkhuizen and H. A. Wosten, Nat. Rev. Microbiol., 2005, 3, 333–341 CrossRef CAS PubMed.
- M. M. Barnhart and M. R. Chapman, Annu. Rev. Microbiol., 2006, 60, 131–147 CrossRef CAS PubMed.
- S. Kanamaru, H. Kurazono, A. Terai, K. Monden, H. Kumon, Y. Mizunoe, O. Ogawa and S. Yamamoto, Int. J. Antimicrob. Agents, 2006, 28(Suppl 1), S21–S25 CrossRef CAS.
- N. D. Hammer, J. C. Schmidt and M. R. Chapman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12494–12499 CrossRef CAS PubMed.
- M. L. Evans, E. Chorell, J. D. Taylor, J. Aden, A. Gotheson, F. Li, M. Koch, L. Sefer, S. J. Matthews, P. Wittung-Stafshede, F. Almqvist and M. R. Chapman, Mol. Cell, 2015, 57, 445–455 CrossRef CAS PubMed.
- A. A. Nenninger, L. S. Robinson, N. D. Hammer, E. A. Epstein, M. P. Badtke, S. J. Hultgren and M. R. Chapman, Mol. Microbiol., 2011, 81, 486–499 CrossRef CAS PubMed.
- X. Wang, N. D. Hammer and M. R. Chapman, J. Biol. Chem., 2008, 283, 21530–21539 CrossRef CAS PubMed.
- X. Wang and M. R. Chapman, J. Mol. Biol., 2008, 380, 570–580 CrossRef CAS PubMed.
- D. Otzen and R. Riek, Cold Spring Harbor Perspect. Biol., 2019, 11, a033860 CrossRef CAS PubMed.
- A. Olsen, H. Herwald, M. Wikstrom, K. Persson, E. Mattsson and L. Bjorck, J. Biol. Chem., 2002, 277, 34568–34572 CrossRef CAS PubMed.
- X. Wang, D. R. Smith, J. W. Jones and M. R. Chapman, J. Biol. Chem., 2007, 282, 3713–3719 CrossRef CAS PubMed.
- M. S. Dueholm, S. V. Petersen, M. Sonderkaer, P. Larsen, G. Christiansen, K. L. Hein, J. J. Enghild, J. L. Nielsen, K. L. Nielsen, P. H. Nielsen and D. E. Otzen, Mol. Microbiol., 2010, 77, 1009–1020 CAS.
- M. S. Dueholm, M. T. Sondergaard, M. Nilsson, G. Christiansen, A. Stensballe, M. T. Overgaard, M. Givskov, T. Tolker-Nielsen, D. E. Otzen and P. H. Nielsen, MicrobiologyOpen, 2013, 2, 365–382 CrossRef CAS PubMed.
- K. L. Palmer, L. M. Mashburn, P. K. Singh and M. Whiteley, J. Bacteriol., 2005, 187, 5267–5277 CrossRef CAS PubMed.
- M. S. Dueholm, D. Otzen and P. H. Nielsen, PLoS One, 2013, 8, e76630 CrossRef CAS PubMed.
- P. H. Nielsen, M. S. Dueholm, T. R. Thomsen, J. L. Nielsen and D. Otzen, in Biofilm highlights, Springer, 2011, pp. 41–62 Search PubMed.
- S. L. Rouse, W. J. Hawthorne, J. L. Berry, D. S. Chorev, S. A. Ionescu, S. Lambert, F. Stylianou, W. Ewert, U. Mackie, R. M. L. Morgan, D. Otzen, F. A. Herbst, P. H. Nielsen, M. Dueholm, H. Bayley, C. V. Robinson, S. Hare and S. Matthews, Nat. Commun., 2017, 8, 263 CrossRef PubMed.
- S. L. Rouse, W. J. Hawthorne, S. Lambert, M. L. Morgan, S. A. Hare and S. Matthews, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2016, 72, 892–896 CrossRef CAS PubMed.
- L. F. B. Christensen, J. S. Nowak, T. V. Sonderby, S. A. Frank and D. E. Otzen, J. Biol. Chem., 2020, 295, 13031–13046 CrossRef CAS PubMed.
- C. B. Rasmussen, G. Christiansen, B. S. Vad, C. Lynggaard, J. J. Enghild, M. Andreasen and D. Otzen, Protein Sci., 2019, 28, 633–642 CrossRef CAS PubMed.
- T. V. Sonderby, H. O. Rasmussen, S. A. Frank, J. Skov Pedersen and D. E. Otzen, J. Mol. Biol., 2021, 434, 167337 CrossRef PubMed.
- K. Schwartz, A. K. Syed, R. E. Stephenson, A. H. Rickard and B. R. Boles, PLoS Pathog., 2012, 8, e1002744 CrossRef CAS PubMed.
- A. Peschel and M. Otto, Nat. Rev. Microbiol., 2013, 11, 667–673 CrossRef CAS PubMed.
- A. Bleem, R. Francisco, J. D. Bryers and V. Daggett, npj Biofilms Microbiomes, 2017, 3, 16 CrossRef PubMed.
- R. Wang, K. R. Braughton, D. Kretschmer, T. H. Bach, S. Y. Queck, M. Li, A. D. Kennedy, D. W. Dorward, S. J. Klebanoff, A. Peschel, F. R. DeLeo and M. Otto, Nat. Med., 2007, 13, 1510–1514 CrossRef CAS PubMed.
- E. Tayeb-Fligelman, O. Tabachnikov, A. Moshe, O. Goldshmidt-Tran, M. R. Sawaya, N. Coquelle, J. P. Colletier and M. Landau, Science, 2017, 355, 831–833 CrossRef PubMed.
- S. Periasamy, H. S. Joo, A. C. Duong, T. H. Bach, V. Y. Tan, S. S. Chatterjee, G. Y. Cheung and M. Otto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1281–1286 CrossRef CAS PubMed.
- N. Salinas, J. P. Colletier, A. Moshe and M. Landau, Nat. Commun., 2018, 9, 3512 CrossRef PubMed.
- S. S. Branda, F. Chu, D. B. Kearns, R. Losick and R. Kolter, Mol. Microbiol., 2006, 59, 1229–1238 CrossRef CAS PubMed.
- D. Romero, C. Aguilar, R. Losick and R. Kolter, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2230–2234 CrossRef CAS PubMed.
- A. Driks, Mol. Microbiol., 2011, 80, 1133–1136 CrossRef CAS PubMed.
- N. R. Stanley and B. A. Lazazzera, Mol. Microbiol., 2004, 52, 917–924 CrossRef CAS PubMed.
- F. Chu, D. B. Kearns, S. S. Branda, R. Kolter and R. Losick, Mol. Microbiol., 2006, 59, 1216–1228 CrossRef CAS PubMed.
- A. G. Stover and A. Driks, J. Bacteriol., 1999, 181, 1664–1672 CrossRef CAS PubMed.
- A. G. Stover and A. Driks, J. Bacteriol., 1999, 181, 7065–7069 CrossRef CAS PubMed.
- R. Terra, N. R. Stanley-Wall, G. Cao and B. A. Lazazzera, J. Bacteriol., 2012, 194, 2781–2790 CrossRef CAS PubMed.
- A. Ostrowski, A. Mehert, A. Prescott, T. B. Kiley and N. R. Stanley-Wall, J. Bacteriol., 2011, 193, 4821–4831 CrossRef CAS PubMed.
- K. Kobayashi and M. Iwano, Mol. Microbiol., 2012, 85, 51–66 CrossRef CAS PubMed.
- L. Hobley, A. Ostrowski, F. V. Rao, K. M. Bromley, M. Porter, A. R. Prescott, C. E. MacPhee, D. M. van Aalten and N. R. Stanley-Wall, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13600–13605 CrossRef CAS PubMed.
- J. W. Costerton, P. S. Stewart and E. P. Greenberg, Science, 1999, 284, 1318–1322 CrossRef CAS PubMed.
- H. C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS PubMed.
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS.
- A. Berry and S. E. Radford, Biochem. Soc. Symp., 2001, 68, 1–26 CrossRef.
- D. M. Fowler, A. V. Koulov, C. Alory-Jost, M. S. Marks, W. E. Balch and J. W. Kelly, PLoS Biol., 2006, 4, e6 CrossRef PubMed.
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS PubMed.
- L. Giehm, N. Lorenzen and D. E. Otzen, Methods, 2011, 53, 295–305 CrossRef CAS.
- J. D. Sipe, M. D. Benson, J. N. Buxbaum, S. Ikeda, G. Merlini, M. J. M. Saraiva and P. Westermark, Amyloid, 2014, 21, 221–224 CrossRef PubMed.
- A. Diehl, Y. Roske, L. Ball, A. Chowdhury, M. Hiller, N. Moliere, R. Kramer, D. Stoppler, C. L. Worth, B. Schlegel, M. Leidert, N. Cremer, N. Erdmann, D. Lopez, H. Stephanowitz, E. Krause, B. J. van Rossum, P. Schmieder, U. Heinemann, K. Turgay, U. Akbey and H. Oschkinat, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3237–3242 CrossRef CAS PubMed.
- M. Torok, S. Milton, R. Kayed, P. Wu, T. McIntire, C. G. Glabe and R. Langen, J. Biol. Chem., 2002, 277, 40810–40815 CrossRef PubMed.
- V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1698, 131–153 CrossRef CAS PubMed.
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS PubMed.
- W. T. Astbury, S. Dickinson and K. Bailey, Biochem. J., 1935, 29, 2351–2360 CrossRef CAS PubMed.
- A. J. Geddes, K. D. Parker, E. D. T. Atkins and E. Beighton, J. Mol. Biol., 1968, 32, 343–344 CrossRef CAS.
- M. Fandrich and C. M. Dobson, EMBO J., 2002, 21, 5682–5690 CrossRef PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405–1412 CrossRef CAS PubMed.
- M. Groenning, J. Chem. Biol., 2010, 3, 1–18 CrossRef PubMed.
- H. LeVine, Amyloid, 1995, 2, 1–6 CrossRef CAS.
- A. M. Morris, M. A. Watzky and R. G. Finke, Biochim. Biophys. Acta, 2009, 1794, 375–397 CrossRef CAS PubMed.
- C. C. F. Blake, L. C. Serpell, M. Sunde, O. Sandgren and E. Lundgren, Nature and Origin of Amyloid Fibrils, 1996, vol. 199, pp. 6–21 Search PubMed.
- M. Sunde and C. Blake, in Advances in Protein Chemistry, ed. R. Wetzel, 1997, vol. 50, Protein Misassembly, pp. 123–159 Search PubMed.
- L. C. Serpell, C. C. F. Blake and P. E. Fraser, Biochemistry, 2000, 39, 13269–13275 CrossRef CAS PubMed.
- N. P. Reynolds, J. Adamcik, J. T. Berryman, S. Handschin, A. A. H. Zanjani, W. Li, K. Liu, A. Zhang and R. Mezzenga, Nat. Commun., 2017, 8, 1338 CrossRef PubMed.
- C. P. Jaroniec, C. E. MacPhee, V. S. Bajaj, M. T. McMahon, C. M. Dobson and R. G. Griffin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 711–716 CrossRef CAS.
- R. Tycko and R. B. Wickner, Acc. Chem. Res., 2013, 46, 1487–1496 CrossRef CAS PubMed.
- F. Shewmaker, R. P. McGlinchey and R. B. Wickner, J. Biol. Chem., 2011, 286, 16533–16540 CrossRef CAS PubMed.
- C. Wasmer, A. Lange, H. Van Melckebeke, A. B. Siemer, R. Riek and B. H. Meier, Science, 2008, 319, 1523–1526 CrossRef CAS PubMed.
- R. Guerrero-Ferreira, N. M. Taylor, D. Mona, P. Ringler, M. E. Lauer, R. Riek, M. Britschgi and H. Stahlberg, eLife, 2018, 7, e36402 CrossRef PubMed.
- M. A. Walti, F. Ravotti, H. Arai, C. G. Glabe, J. S. Wall, A. Bockmann, P. Guntert, B. H. Meier and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4976–E4984 CrossRef CAS PubMed.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed.
- P. G. Wolynes, Biochimie, 2015, 119, 218–230 CrossRef CAS PubMed.
- H. Van Melckebeke, C. Wasmer, A. Lange, E. Ab, A. Loquet, A. Bockmann and B. H. Meier, J. Am. Chem. Soc., 2010, 132, 13765–13775 CrossRef CAS PubMed.
- D. Li and C. Liu, Nat. Chem. Biol., 2021, 17, 237–245 CrossRef CAS PubMed.
- M. Fandrich, S. Nystrom, K. P. R. Nilsson, A. Bockmann, H. LeVine and P. Hammarstrom, J. Intern. Med., 2018, 283, 218–237 CrossRef CAS PubMed.
- C. Seuring, J. Verasdonck, P. Ringler, R. Cadalbert, H. Stahlberg, A. Bockmann, B. H. Meier and R. Riek, J. Phys. Chem. B, 2021, 125, 484 CrossRef CAS PubMed.
- D. T. Murray, M. Kato, Y. Lin, K. R. Thurber, I. Hung, S. L. McKnight and R. Tycko, Cell, 2017, 171, 615–627 CrossRef CAS PubMed.
- M. P. Hughes, M. R. Sawaya, D. R. Boyer, L. Goldschmidt, J. A. Rodriguez, D. Cascio, L. Chong, T. Gonen and D. S. Eisenberg, Science, 2018, 359, 698–701 CrossRef CAS PubMed.
- R. Hervas, M. J. Rau, Y. Park, W. J. Zhang, A. G. Murzin, J. A. J. Fitzpatrick, S. H. W. Scheres and K. Si, Science, 2020, 367, 1230 CrossRef CAS PubMed.
- M. Andreasen, G. Meisl, J. D. Taylor, T. C. T. Michaels, A. Levin, D. E. Otzen, M. R. Chapman, C. M. Dobson, S. J. Matthews and T. P. J. Knowles, mBio, 2019, 10, e02279-18 CrossRef PubMed.
- S. H. Alijanvand, A. Peduzzo and A. K. Buell, Front. Mol. Biosci., 2021, 8, 669994 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS PubMed.
- D. Willbold, B. Strodel, G. F. Schroder, W. Hoyer and H. Heise, Chem. Rev., 2021, 121, 8285–8307 CrossRef CAS PubMed.
- E. Nogales and S. H. W. Scheres, Mol. Cell, 2015, 58, 677–689 CrossRef CAS PubMed.
- E. Callaway, Nature, 2015, 525, 172–174 CrossRef CAS PubMed.
- W. Kuhlbrandt, Science, 2014, 343, 1443–1444 CrossRef PubMed.
- K. M. Towle, C. T. Lohans, M. Miskolzie, J. Z. Acedo, M. J. van Belkum and J. C. Vederas, Biochemistry, 2016, 55, 4798–4806 CrossRef CAS PubMed.
- N. E. Mammeri, J. Hierrezuelo, J. Tolchard, J. Cámara-Almirón, J. Caro-Astorga, A. Álvarez-Mena, A. Dutour, M. Berbon, J. Shenoy and E. Morvan, FASEB J., 2019, 33, 12146–12163 CrossRef PubMed.
- L. Sewell, F. Stylianou, Y. Xu, J. Taylor, L. Sefer and S. Matthews, Sci. Rep., 2020, 10, 7896 CrossRef CAS PubMed.
- T. Konuma, E. Chatani, M. Yagi, K. Sakurai, T. Ikegami, H. Naiki and Y. Goto, J. Mol. Biol., 2011, 405, 851–862 CrossRef CAS PubMed.
- N. A. Whittemore, R. Mishra, I. Kheterpal, A. D. Williams, R. Wetzel and E. H. Serpersu, Biochemistry, 2005, 44, 4434–4441 CrossRef CAS PubMed.
- R. Tycko, in Annual Review of Physical Chemistry, ed. S. R. Leone, P. S. Cremer, J. T. Groves and M. A. Johnson, 2011, vol. 62, pp. 279–299 Search PubMed.
- B. H. Meier, R. Riek and A. Bockmann, Trends Biochem. Sci., 2017, 42, 777–787 CrossRef CAS PubMed.
- L. Bousset, L. Pieri, G. Ruiz-Arlandis, J. Gath, P. H. Jensen, B. Habenstein, K. Madiona, V. Olieric, A. Bockmann, B. H. Meier and R. Melki, Nat. Commun., 2013, 4, 2575 CrossRef PubMed.
- R. Guerrero-Ferreira, N. M. I. Taylor, A. A. Arteni, P. Kumari, D. Mona, P. Ringler, M. Britschgi, M. E. Lauer, A. Makky, J. Verasdonck, R. Riek, R. Melki, B. H. Meier, A. Bockmann, L. Bousset and H. Stahlberg, Elife, 2019, 8, e48907 CrossRef CAS PubMed.
- F. Castellani, B. van Rossum, A. Diehl, M. Schubert, K. Rehbein and H. Oschkinat, Nature, 2002, 420, 98–102 CrossRef CAS PubMed.
- A. K. Schutz, T. Vagt, M. Huber, O. Y. Ovchinnikova, R. Cadalbert, J. Wall, P. Guntert, A. Bockmann, R. Glockshuber and B. H. Meier, Angew. Chem., Int. Ed., 2015, 54, 331–335 CrossRef PubMed.
- M. D. Tuttle, G. Comellas, A. J. Nieuwkoop, D. J. Covell, D. A. Berthold, K. D. Kloepper, J. M. Courtney, J. K. Kim, A. M. Barclay, A. Kendall, W. Wan, G. Stubbs, C. D. Schwieters, V. M. Y. Lee, J. M. George and C. M. Rienstra, Nat. Struct. Mol. Biol., 2016, 23, 409–415 CrossRef CAS PubMed.
- V. Chevelkov, K. Rehbein, A. Diehl and B. Reif, Angew. Chem., Int. Ed., 2006, 45, 3878–3881 CrossRef CAS PubMed.
- U. Akbey, S. Lange, W. T. Franks, R. Linser, K. Rehbein, A. Diehl, B. J. van Rossum, B. Reif and H. Oschkinat, J. Biomol. NMR, 2010, 46, 67–73 CrossRef CAS PubMed.
- J. R. Lewandowski, J. N. Dumez, U. Akbey, S. Lange, L. Emsley and H. Oschkinat, J. Phys. Chem. Lett., 2011, 2, 2205–2211 CrossRef CAS.
- V. Agarwal, S. Penzel, K. Szekely, R. Cadalbert, E. Testori, A. Oss, J. Past, A. Samoson, M. Ernst, A. Bockmann and B. H. Meier, Angew. Chem., Int. Ed., 2014, 53, 12253–12256 CrossRef CAS PubMed.
- S. K. Vasa, P. Rovo and R. Linser, Acc. Chem. Res., 2018, 51, 1386–1395 CrossRef CAS PubMed.
- E. Barbet-Massin, A. J. Pell, J. S. Retel, L. B. Andreas, K. Jaudzems, W. T. Franks, A. J. Nieuwkoop, M. Hiller, V. Higman, P. Guerry, A. Bertarello, M. J. Knight, M. Felletti, T. Le Marchand, S. Kotelovica, I. Akopjana, K. Tars, M. Stoppini, V. Bellotti, M. Bolognesi, S. Ricagno, J. J. Chou, R. G. Griffin, H. Oschkinat, A. Lesage, L. Emsley, T. Herrmann and G. Pintacuda, J. Am. Chem. Soc., 2014, 136, 12489–12497 CrossRef CAS PubMed.
- K. Xue, R. Sarkar, C. Motz, S. Asami, D. C. R. Camargo, V. Decker, S. Wegner, Z. Tosner and B. Reif, Sci. Rep., 2017, 7, 7444 CrossRef PubMed.
- C. M. Quinn, M. Z. Wang and T. Polenova, in Protein NMR: Methods and Protocols, ed. R. Ghose, 2018, vol. 1688, pp. 1–35 Search PubMed.
- B. Reif, S. E. Ashbrook, L. Emsley and M. Hong, Nat. Rev. Methods Primers, 2021, 1, 2 CrossRef CAS PubMed.
- A. Konig, D. Scholzel, B. Uluca, T. Viennet, U. Akbey and H. Heise, Solid State Nucl. Magn. Reson., 2019, 98, 1–11 CrossRef PubMed.
- U. Akbey and H. Oschkinat, J. Magn. Reson., 2016, 269, 213–224 CrossRef CAS PubMed.
- K. K. Frederick, Biophys. J., 2019, 116, 203A CrossRef.
- K. Jaudzems, T. Polenova, G. Pintacuda, H. Oschkinat and A. Lesage, J. Struct. Biol., 2019, 206, 90–98 CrossRef CAS PubMed.
- U. Akbey, W. T. Franks, A. Linden, S. Lange, R. G. Griffin, B. J. van Rossum and H. Oschkinat, Angew. Chem., Int. Ed., 2010, 49, 7803–7806 CrossRef CAS PubMed.
- A. L. Cogen, K. Yamasaki, J. Muto, K. M. Sanchez, L. C. Alexander, J. Tanios, Y. P. Lai, J. E. Kim, V. Nizet and R. L. Gallo, PLoS One, 2010, 5, e8557 CrossRef PubMed.
- E. Tayeb-Fligelman, N. Salinas, O. Tabachnikov and M. Landau, Structure, 2020, 28, 301–313 CrossRef CAS PubMed.
- P. Marinelli, I. Pallares, S. Navarro and S. Ventura, Sci. Rep., 2016, 6, 34552 CrossRef CAS PubMed.
- M. Zaman and M. Andreasen, Microorganisms, 2021, 9, 117 CrossRef CAS PubMed.
- F. Shewmaker, R. P. McGlinchey, K. R. Thurber, P. McPhie, F. Dyda, R. Tycko and R. B. Wickner, J. Biol. Chem., 2009, 284, 25065–25076 CrossRef CAS PubMed.
- T. Schubeis, P. Yuan, M. Ahmed, M. Nagaraj, B. J. van Rossum and C. Ritter, Angew. Chem., Int. Ed. Engl., 2015, 54, 14669–14672 CrossRef CAS.
- D. S. Wishart, B. D. Sykes and F. M. Richards, J. Mol. Biol., 1991, 222, 311–333 CrossRef CAS.
- D. S. Wishart, C. G. Bigam, A. Holm, R. S. Hodges and B. D. Sykes, J. Biomol. NMR, 1995, 5, 67–81 CrossRef CAS PubMed.
- P. Tian, W. Boomsma, Y. Wang, D. E. Otzen, M. H. Jensen and K. Lindorff-Larsen, J. Am. Chem. Soc., 2015, 137, 22–25 CrossRef CAS PubMed.
- A. Daskalov, N. El Mammeri, A. Lends, J. Shenoy, G. Lamon, Y. Fichou, A. Saad, D. Martinez, E. Morvan, M. Berbon, A. Grelard, B. Kauffmann, M. Ferber, B. Bardiaux, B. Habenstein, S. J. Saupe and A. Loquet, Front. Mol. Neurosci., 2021, 14, 670513 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- N. Ferreira, H. Gram, Z. A. Sorrentino, E. Gregersen, S. I. Schmidt, L. Reimer, C. Betzer, C. Perez-Gozalbo, M. Beltoja, M. Nagaraj, J. Wang, J. S. Nowak, M. D. Dong, K. Willen, E. Cholak, K. Bjerregaard-Andersen, N. Mendez, P. Rabadia, M. Shahnawaz, C. Soto, D. E. Otzen, U. Akbey, M. Meyer, B. I. Giasson, M. Romero-Ramos and P. H. Jensen, Acta Neuropathol., 2021, 142, 87–115 CrossRef CAS PubMed.
- T. Schubeis, M. Nagaraj and C. Ritter, in Split Inteins: Methods and Protocols, ed. H. D. Mootz, 2017, vol. 1495, pp. 147–160 Search PubMed.
- M. Nagaraj, M. Ahmed, J. Lyngso, B. S. Vad, A. Boggild, A. Fillipsen, J. S. Pedersen, D. E. Otzen and U. Akbey, J. Mol. Biol., 2020, 432, 2232–2252 CrossRef CAS PubMed.
- S. L. Rouse, S. J. Matthews and M. S. Dueholm, J. Mol. Biol., 2018, 430, 3685–3695 CrossRef CAS PubMed.
- H. O. Rasmussen, D. E. Otzen and J. S. Pedersen, Biophys. J., 2021, 120, 2262–2275 CrossRef CAS PubMed.
- N. El Mammeri, J. Hierrezuelo, J. Tolchard, J. Camara-Almiron, J. Caro-Astorga, A. Alvarez-Mena, A. Dutour, M. Berbon, J. Shenoy, E. Morvan, A. Grelard, B. Kauffmann, S. Lecomte, A. de Vicente, B. Habenstein, D. Romero and A. Loquet, FASEB J., 2019, 33, 12146–12163 CrossRef CAS PubMed.
- M. Ghrayeb, S. Hayet, N. Lester-Zer, Y. Levi-Kalisman and L. Chai, Microorganisms, 2021, 9, 529 CrossRef CAS PubMed.
- N. Lorenzen, S. B. Nielsen, A. K. Buell, J. D. Kaspersen, P. Arosio, B. S. Vad, W. Paslawski, G. Christiansen, Z. Valnickova-Hansen, M. Andreasen, J. J. Enghild, J. S. Pedersen, C. M. Dobson, T. P. Knowles and D. E. Otzen, J. Am. Chem. Soc., 2014, 136, 3859–3868 CrossRef CAS PubMed.
- Z. Najarzadeh, H. Mohammad-Beigi, J. Nedergaard Pedersen, G. Christiansen, T. V. Sonderby, S. A. Shojaosadati, D. Morshedi, K. Stromgaard, G. Meisl, D. Sutherland, J. Skov Pedersen and D. E. Otzen, Biomolecules, 2019, 9, 659 CrossRef CAS.
- Z. Najarzadeh, J. N. Pedersen, G. Christiansen, S. A. Shojaosadati, J. S. Pedersen and D. E. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 140263 CrossRef CAS PubMed.
- H. M. Swasthi and S. Mukhopadhyay, J. Biol. Chem., 2017, 292, 19861–19872 CrossRef CAS PubMed.
- R. Malishev, R. Abbasi, R. Jelinek and L. Chai, Biochemistry, 2018, 57, 5230–5238 CrossRef CAS PubMed.
- G. Y. Cheung, K. Rigby, R. Wang, S. Y. Queck, K. R. Braughton, A. R. Whitney, M. Teintze, F. R. DeLeo and M. Otto, PLoS Pathog., 2010, 6, e1001133 CrossRef PubMed.
- J. P. Rasigade, S. Trouillet-Assant, T. Ferry, B. A. Diep, A. Sapin, Y. Lhoste, J. Ranfaing, C. Badiou, Y. Benito, M. Bes, F. Couzon, S. Tigaud, G. Lina, J. Etienne, F. Vandenesch and F. Laurent, PLoS One, 2013, 8, e63176 CrossRef CAS PubMed.
- M. Laabei, W. D. Jamieson, Y. Yang, J. van den Elsen and A. T. Jenkins, Biochim. Biophys. Acta, 2014, 1838, 3153–3161 CrossRef CAS PubMed.
- B. G. Surewaard, R. Nijland, A. N. Spaan, J. A. Kruijtzer, C. J. de Haas and J. A. van Strijp, PLoS Pathog., 2012, 8, e1002606 CrossRef CAS PubMed.
- C. Galvagnion, A. K. Buell, G. Meisl, T. C. Michaels, M. Vendruscolo, T. P. Knowles and C. M. Dobson, Nat. Chem. Biol., 2015, 11, 229–234 CrossRef CAS PubMed.
- D. Campoccia, L. Montanaro and C. R. Arciola, Int. J. Mol. Sci., 2021, 22, 9100 CrossRef CAS PubMed.
- J. T. Blakeman, A. L. Morales-García, J. Mukherjee, K. Gori, A. S. Hayward, N. J. Lant and M. Geoghegan, Langmuir, 2019, 35, 6468–6475 CrossRef CAS PubMed.
- J. L. Lister and A. R. Horswill, Front. Cell. Infect. Microbiol., 2014, 4, 178 Search PubMed.
- J. A. Jurcisek and L. O. Bakaletz, J. Bacteriol., 2007, 189, 3868–3875 CrossRef CAS PubMed.
- E. E. Mann, K. C. Rice, B. R. Boles, J. L. Endres, D. Ranjit, L. Chandramohan, L. H. Tsang, M. S. Smeltzer, A. R. Horswill and K. W. Bayles, PLoS One, 2009, 4, e5822 CrossRef PubMed.
- M. R. Kiedrowski, J. S. Kavanaugh, C. L. Malone, J. M. Mootz, J. M. Voyich, M. S. Smeltzer, K. W. Bayles and A. R. Horswill, PLoS One, 2011, 6, e26714 CrossRef CAS PubMed.
- P. M. Gallo, G. J. Rapsinski, R. P. Wilson, G. O. Oppong, U. Sriram, M. Goulian, B. Buttaro, R. Caricchio, S. Gallucci and Ç. Tükel, Immunity, 2015, 42, 1171–1184 CrossRef CAS PubMed.
- K. Schwartz, M. Ganesan, D. E. Payne, M. J. Solomon and B. R. Boles, Mol. Microbiol., 2016, 99, 123–134 CrossRef CAS PubMed.
- R. M. Shanks, N. P. Donegan, M. L. Graber, S. E. Buckingham, M. E. Zegans, A. L. Cheung and G. A. O'Toole, Infect. Immun., 2005, 73, 4596–4606 CrossRef CAS PubMed.
- C. B. Ibberson, C. P. Parlet, J. Kwiecinski, H. A. Crosby, D. K. Meyerholz and A. R. Horswill, Infect. Immun., 2016, 84, 1917–1929 CrossRef CAS PubMed.
- S. Mishra and A. R. Horswill, mSphere, 2017, 2, e00135-17 CrossRef PubMed.
- Z. Najarzadeh, M. Zaman, V. Sereikaite, K. Stromgaard, M. Andreasen and D. E. Otzen, J. Biol. Chem., 2021, 297, 100953 CrossRef CAS PubMed.
- M. Hammar, Z. Bian and S. Normark, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6562–6566 CrossRef CAS PubMed.
- M. Hammar, A. Arnqvist, Z. Bian, A. Olsen and S. Normark, Mol. Microbiol., 1995, 18, 661–670 CrossRef CAS PubMed.
- Z. Bian and S. Normark, EMBO J., 1997, 16, 5827–5836 CrossRef CAS PubMed.
- Q. Shu, S. L. Crick, J. S. Pinkner, B. Ford, S. J. Hultgren and C. Frieden, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6502–6507 CrossRef CAS PubMed.
- N. D. Hammer, B. A. McGuffie, Y. Zhou, M. P. Badtke, A. A. Reinke, K. Brannstrom, J. E. Gestwicki, A. Olofsson, F. Almqvist and M. R. Chapman, J. Mol. Biol., 2012, 422, 376–389 CrossRef CAS.
- D. Romero, H. Vlamakis, R. Losick and R. Kolter, Mol. Microbiol., 2011, 80, 1155–1168 CrossRef CAS.
- D. Romero, H. Vlamakis, R. Losick and R. Kolter, J. Bacteriol., 2014, 196, 1505–1513 CrossRef PubMed.
- I. Kolodkin-Gal, D. Romero, S. Cao, J. Clardy, R. Kolter and R. Losick, Science, 2010, 328, 627–629 CrossRef CAS PubMed.
- M. Zaman and M. Andreasen, eLife, 2020, 9, e59776 CrossRef CAS PubMed.
- H. Q. Do, A. Hewetson, C. G. Borcik, M. C. Hastert, S. Whelly, B. J. Wylie, R. B. Sutton and G. A. Cornwall, J. Biol. Chem., 2021, 296, 100250 CrossRef CAS.
- H. Braak, K. Del Tredici, U. Rub, R. A. de Vos, E. N. Jansen Steur and E. Braak, Neurobiol. Aging, 2003, 24, 197–211 CrossRef.
- K. C. Luk, V. Kehm, J. Carroll, B. Zhang, P. O'Brien, J. Q. Trojanowski and V. M. Lee, Science, 2012, 338, 949–953 CrossRef CAS PubMed.
- K. L. Paumier, K. C. Luk, F. P. Manfredsson, N. M. Kanaan, J. W. Lipton, T. J. Collier, K. Steece-Collier, C. J. Kemp, S. Celano, E. Schulz, I. M. Sandoval, S. Fleming, E. Dirr, N. K. Polinski, J. Q. Trojanowski, V. M. Lee and C. E. Sortwell, Neurobiol. Dis., 2015, 82, 185–199 CrossRef CAS PubMed.
- S. Holmqvist, O. Chutna, L. Bousset, P. Aldrin-Kirk, W. Li, T. Bjorklund, Z. Y. Wang, L. Roybon, R. Melki and J. Y. Li, Acta Neuropathol., 2014, 128, 805–820 CrossRef PubMed.
- N. Uemura, H. Yagi, M. T. Uemura, Y. Hatanaka, H. Yamakado and R. Takahashi, Mol. Neurodegener., 2018, 13, 21 CrossRef PubMed.
- N. Van Den Berge, N. Ferreira, H. Gram, T. W. Mikkelsen, A. K. O. Alstrup, N. Casadei, P. Tsung-Pin, O. Riess, J. R. Nyengaard, G. Tamguney, P. H. Jensen and P. Borghammer, Acta Neuropathol., 2019, 138, 535–550 CrossRef CAS PubMed.
- T. R. Sampson, C. Challis, N. Jain, A. Moiseyenko, M. S. Ladinsky, G. G. Shastri, T. Thron, B. D. Needham, I. Horvath, J. W. Debelius, S. Janssen, R. Knight, P. Wittung-Stafshede, V. Gradinaru, M. Chapman and S. K. Mazmanian, eLife, 2020, 9, e53111 CrossRef CAS PubMed.
- E. Chorell, E. Andersson, M. L. Evans, N. Jain, A. Gotheson, J. Aden, M. R. Chapman, F. Almqvist and P. Wittung-Stafshede, PLoS One, 2015, 10, e0140194 CrossRef PubMed.
- S. Perov, O. Lidor, N. Salinas, N. Golan, E. Tayeb-Fligelman, M. Deshmukh, D. Willbold and M. Landau, PLoS Pathog., 2019, 15, e1007978 CrossRef CAS PubMed.
- K. Hartman, J. R. Brender, K. Monde, A. Ono, M. L. Evans, N. Popovych, M. R. Chapman and A. Ramamoorthy, PeerJ, 2013, 1, e5 CrossRef PubMed.
- I. Javed, Z. Zhang, J. Adamcik, N. Andrikopoulos, Y. Li, D. E. Otzen, S. Lin, R. Mezzenga, T. P. Davis, F. Ding and P. C. Ke, Adv. Sci., 2020, 7, 2001299 CrossRef CAS PubMed.
- L. F. B. Christensen, K. F. Jensen, J. Nielsen, B. S. Vad, G. Christiansen and D. E. Otzen, ACS Omega, 2019, 4, 4029–4039 CrossRef CAS PubMed.
- M. E. Goya, F. Xue, C. Sampedro-Torres-Quevedo, S. Arnaouteli, L. Riquelme-Dominguez, A. Romanowski, J. Brydon, K. L. Ball, N. R. Stanley-Wall and M. Doitsidou, Cell Rep., 2020, 30, 367–380 CrossRef CAS PubMed e367..
- D. Sade Yazdi, D. Laor Bar-Yosef, H. Adsi, T. Kreiser, S. Sigal, S. Bera, D. Zaguri, S. Shaham-Niv, D. S. Oluwatoba, D. Levy, M. Gartner, T. D. Do, D. Frenkel and E. Gazit, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017575118 CrossRef PubMed.
- O. Tavassoly, D. Sade, S. Bera, S. Shaham-Niv, D. J. Vocadlo and E. Gazit, J. Mol. Biol., 2018, 430, 3847–3862 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |