
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric dipicolylamine based mass tags for mass cytometry†
Yefeng
Zhang
a,
Peng
Liu
 b,
Daniel
Majonis
b and
Mitchell A.
Winnik
*ac
b,
Daniel
Majonis
b and
Mitchell A.
Winnik
*ac
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada. E-mail: m.winnik@utoronto.ca
bFluidigm Canada Inc., 1380 Rodick Road, Suite 400, Markham, ON L3R 4G5, Canada
cDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, ON M5S 3E5, Canada
First published on 23rd February 2022
Abstract
Mass cytometry is an emerging powerful bioanalytical technique for high-dimensional single-cell analysis. In this technique, cells are stained with metal-isotope-tagged antibodies and are analyzed by an inductively coupled plasma time-of-flight mass spectrometer. While there are more than 100 stable isotopes available in the m/z 75 to 209 detection range of the instrument, only about 50 parameters can be measured per cell because current reagents are metal-chelating polymers with pendant aminocarboxylate chelators that only bind hard metal ions such as the rare earths and Bi3+. Here we describe the synthesis and characterization of a new type of metal-chelating polymer with pendant dipicolylamine chelators suited to binding intermediate to soft metals such as rhenium and platinum. We introduce two different conjugation strategies, a thiol–maleimide reaction that works well for rhenium, and a DBCO-azide click reaction designed to avoid potential complications of Pt and other heavy metals interacting with thiol groups. We show that these polymers can serve as new elemental mass tags for mass cytometry. Antibody-polymer conjugates of CD20 and CD8a prepared by both coupling reactions were employed in conjunction with commercial metal-conjugated antibodies for multi-parameter single-cell immunoassays.
Introduction
Modern medical research requires highly sensitive, multiplexed assays of cellular biomarkers to interrogate the complex biology of underlying diseases.1–3 Biomarkers are defined as characteristic proteins, genes, or small molecules that can be measured and evaluated as indicators of normal biological or pathological states of a cell. Mass cytometry (MC) was designed to overcome the multiplexing limitations of flow cytometry by employing heavy metal isotopes as tags combined with cytometric injection of cells into an inductively coupled plasma time-of-flight mass spectrometer.4 Reagents for biomarker detection are antibodies (Abs) labeled with metal-chelating polymers.5–12 Mass channels are used not just to detect cell surface proteins for cell type identification, but also to detect transcription factors, intracellular cytokines, and phosphorylation states of intracellular proteins (cell signaling).13 Additional mass channels may be used to detect cellular metabolism, hypoxia and enzymatic activity.14–16Current MC instrumentation can detect atomic masses in the range of amu 75 to 209. There are theoretically ∼130 isotopes in this mass range, but those that can be attached to Abs for protein detection are currently limited by the types of metal chelating pendant groups that can be attached to the polymer backbone. Current polymer reagents carry polyaminocarboxylates, typically diethylenetriaminepentaacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), with multiple oxygen and nitrogen donors that can form strong complexes with hard metal ions such as the lanthanide ions. A recent review identified 49 stable isotopes available for protein detection (38 lanthanides, 89Y, In(2), 209Bi, and Cd(7)), as well as 14 additional isotopes available for cell identification.17 While this extent of multiplexing is impressive, it falls short of the full capability of MC as a technique. A recent review points out that researchers are often faced with a dilemma of trying to cast a broad net and obtain as much information as possible at a particular level of cellular behavior or take a highly-targeted approach to reveal a more limited number of cellular features with higher resolution.13
The most effective way to expand the parameter space accessible to mass cytometry would be the development of metal-chelating polymers with pendant groups able to carry soft to intermediate metals. A partial list of soft to intermediate heavy metal ions and their isotopes would include Pd(6), Pt(6), Ag(2), Hg(5), Re(2). Previous experiments have shown that polymers bearing polyaminocarboxylate pendant groups were able to bind Pd2+ and Pt2+.11 However, after attachment to Abs, the conjugates were unable to detect the target antigens and served only to label dead cells in a cell suspension. A likely explanation is that intracellular thiols were able to displace these metals from the polyaminocarboxylate ligands. This result emphasizes the need for polymers with new pendant ligands designed to bind soft metal ions.
The dipicolylamine (DPA) ligand has been employed in the field of radiopharmaceuticals for chelating radioactive 99mTc tricarbonyl core (fac-[99mTc(CO)3]+) and 188Re tricarbonyl core (fac-[188Re(CO)3]+).18–21 The resulting chelates exhibit great stability towards ligand substitution and decomposition due to the d6 low-spin electron configuration of Tc(I) and Re(I).20,22,23 The element rhenium has two naturally occurring isotopes, both 185Re (37.4%) and 187Re (62.6%), are within the mass range of the current CyTOF® mass cytometer (75–209 amu). The DPA ligand has also been employed in the field of bioinorganic chemistry for the development of new platinum(II)-based anticancer drugs.24–26 The element platinum has six naturally occurring isotopes (m/z 190, 192, 194, 195, 196, 198), of which four are commercially available at high enrichment levels (>95%). While the small molecule-based chemotherapy drug, cisplatin, has been used for cell viability staining, cellular barcoding and immunophenotyping in mass cytometry, a polymeric platinum mass tag has yet to be developed.27–30
Polymers for mass cytometry applications have to meet several important criteria. First, the polymer should have a relatively narrow distribution of chain lengths such that each labeled antibody carries a similar number of metal ions. Second, the metals must be bound in a way that they do not undergo exchange during storage or applications. Third, the polymer must contain functional groups for antibody conjugation. Finally, the polymer must be water-soluble since bioassays are performed in aqueous media. While metal-chelating polymers bearing polyaminocarboxylate chelators designed for binding lanthanides lend themselves to high water solubility, some chelators for metal ions beyond lanthanides are rather hydrophobic. Metal-chelating polymers with hydrophobic chelators require additional solubilizing moieties to render them water-soluble. In 2019, we reported a polymer with a terminal biotin, pendant desferrioxamine (DFO) groups designed to bind Zr ions, and multiple pendant PEG24 chains to promote water solubility.6 Zr would contribute four stable isotopes (m/z 90, 91, 92, 94) for detection. While we could detect Zr with polymers bound to streptavidin-coated polystyrene microbeads, the polymer was too short and carried too few Zr atoms to be useful on its own as a MC reagent. Longer polymers had solubility problems, likely associated with the hydrophobic nature of the benzyl-DFO chelator. It would not be possible to include the Zr polymer in multiparameter experiments on PBMC samples. Nevertheless, it proved useful for demonstrating that Zr could in fact be detected by mass cytometry. We note that a more successful Zr mass tag based upon metal–organic framework nanoparticles has recently been reported.31
In this contribution, we describe the synthesis of a new type of metal-chelating polymer with pendant dipicolylamine (DPA) chelators that can complex with the rhenium tricarbonyl core (fac-[Re(CO)3]+) or with the platinum(II) ion. For the rhenium mass tag, the initial polymer synthesized, with a polyacrylamide backbone, a lysine-based spacer, DPA pendant groups and a maleimide functional end group, was soluble in water, but its fac-[Re(CO)3]+ complex caused the polymer to precipitate. We overcame this problem by introducing an oligoethylene glycol (methoxyPEG6) to the pendant groups. For the platinum mass tag, we employed a similar polymer structure to that of rhenium mass tag but with multiple additional azide functional groups being introduced to the pendant groups. These polymers could be attached to primary antibodies, and these conjugates showed comparable performance to their Maxpar® counterparts in suspension MC applications.
Results and discussion
Rhenium mass tag synthesis
The rhenium-chelating polymer was synthesized as shown in Scheme 1. We began with the synthesis of the activated ester polymer 1, poly(pentafluorophenyl acrylate) (PPFPA) by reversible addition–fragmentation chain transfer (RAFT) polymerization.32,33 The polymerization was carried out at 70 °C using 2-(dodecylthiocarbonothioylthio)-2-methylpropanoic acid (DDMAT) as the chain transfer agent (CTA) and 2,2′-azobis(2-methylpropionitrile) (AIBN) as the thermal initiator in 1,4-dioxane. A monomer-to-CTA to initiator molar ratio ([M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CTA]:[I]) of 30:1:0.1 was chosen to tailor the molecular weight of the resulting polymer 1, with an apparent number average molecular weight (MnGPC) of 9400 g mol−1 and a relatively narrow molecular weight distribution (Đ = 1.25) being obtained at ca. 73% conversion (Fig. S1, ESI†). The degree of polymerization (DP) was determined using 1H NMR spectroscopy by comparing the integration of the PPFPA backbone peaks (δ = 3.11 ppm) with that of the methyl unit for the dodecyl chain end (δ = 0.88 ppm) (ESI, Fig. S2†). In this way, we found the DP of polymer 1 was ca. 20. Additionally, the 19F NMR spectrum of polymer 1 displayed three broad peaks at −153.2, −156.8, and −162.3 ppm with an integration ratio of 2:1:2 corresponding to the pentafluorophenyl groups along the polymer backbone (Fig. S2†).
:[CTA]:[I]) of 30:1:0.1 was chosen to tailor the molecular weight of the resulting polymer 1, with an apparent number average molecular weight (MnGPC) of 9400 g mol−1 and a relatively narrow molecular weight distribution (Đ = 1.25) being obtained at ca. 73% conversion (Fig. S1, ESI†). The degree of polymerization (DP) was determined using 1H NMR spectroscopy by comparing the integration of the PPFPA backbone peaks (δ = 3.11 ppm) with that of the methyl unit for the dodecyl chain end (δ = 0.88 ppm) (ESI, Fig. S2†). In this way, we found the DP of polymer 1 was ca. 20. Additionally, the 19F NMR spectrum of polymer 1 displayed three broad peaks at −153.2, −156.8, and −162.3 ppm with an integration ratio of 2:1:2 corresponding to the pentafluorophenyl groups along the polymer backbone (Fig. S2†).
 | ||
| Scheme 1 Synthesis of rhenium-chelating polymer (m = 20). (a) (1) DPA ligand 5, RT, 13 h, DMF; (2) ethanolamine, RT, 3 h; (b) (1) DMTMM, RT, 5 min, PB buffer (0.2 M, pH 8.0); (2) mPEG6-NH2, RT, 15 h; (c) (1) TCEP (50 mM), RT, 1 h, H2O; (2) Bis-Mal-PEG6, RT, 90 min, DMF/PB buffer (0.2 M, pH 7.0). | ||
Next, polymer 1 was treated with a slight excess of a lysine-derived dipicolylamine (DPA) ligand 5, (1.6 equivalents with respect to PFPA repeating units) at room temperature to obtain polymer 2. Ligand 5 was prepared in three steps, as shown in Scheme S1 (ESI),† by (i) direct reductive alkylation of a Boc protected lysine precursor,23 (ii) Boc deprotection by hydrochloric acid (HCl in dioxane), followed by (iii) conversion of the amine hydrochloride salt to free base with NaOH (ESI, Fig. S3†). The aminolysis reaction was monitored by 19F NMR. The reaction was rapid, as shown by the NMR spectra in Fig. S4 (ESI),† in which the broad signals corresponding to PFPA units along the backbone disappeared, while sharp signals corresponding to the released pentafluorophenol appeared. After purification, the 1H NMR spectrum of polymer 2 showed a set of resonances corresponding to the introduction of ligand 5 while the 19F spectrum displayed no signal (ESI, Fig. S5†). These NMR studies suggest complete modification of the PFPA units with near-quantitative incorporation of ligand 5 along the polymer backbone. In addition, the UV-vis spectrum of polymer 2 in methanol showed negligible absorbance at 309 nm, suggesting the cleavage of the trithiocarbonate group during aminolysis (ESI, Fig. S6†). A sharp absorbance peak located at 262 nm was observed, corresponding to the absorption from the 2-pyridyl groups of the chelator.
The carboxylic acid (COOH) moiety of ligand 5 was initially designed with two goals in mind. First, we anticipated that the carboxylate ion would improve the water solubility of the rhenium-loaded polymer since the [Re(CO)3(DPA)]+ complex itself is rather hydrophobic. Second, this complex also has a net positive charge. Polymers with a positive charge in each pendant group can interact non-specifically with cells that commonly have a negatively charged outer membrane. The carboxylate provides a potential counterion, so that each pendant group is zwitterionic. Zwitterionic pendant groups have been beneficial in different radioimmunotherapy applications of metal-chelating polymers.34 Disappointedly, we found that rhenium-loaded polymer 2 had low solubility in water or in PB buffer. To try to address this aqueous solubility problem, polymer 2 was further modified with a short methoxy polyethylene glycol (mPEG6-NH2) as shown in Scheme 1, step b. The goal here was not only to enhance the water solubility of the polymer, but also to provide a PEG corona to shield the positively charged complex from interaction with cells.
Specifically, polymer 2 was first treated with excess (4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride) (DMTMM, ca. 8 molar equivalents to each carboxylic acid group) in PB buffer (pH 8.0) for a brief time (5 min) to activate the carboxylic acid functional groups. Following this, excess mPEG6-NH2 (ca. 7 molar equivalents to each carboxylic acid group) was added and the reaction solution was stirred overnight. The PEGylated version of polymer 2 was subsequently obtained after purification by spin-filtration, which we refer to as polymer 3. The 1H NMR spectrum of polymer 3 confirms PEGylation, with the appearance of new peaks at 3.32 ppm and 3.59 ppm corresponding to the methoxy group and the ethylene glycol repeating units of the mPEG6, respectively (ESI, Fig. S7†). A rhenium loading experiment indicated that metal-loaded polymer 3 was soluble in both water and PB buffer.
Polymer 3 was further modified to install a maleimide functional group that enables subsequent bioconjugation (Scheme 1, step c). First, any disulfide bonds that may have formed in prior steps were reduced with tris(2-carboxyethyl)phosphine (TCEP). The newly reduced thiol groups were then reacted with an excess of a bismaleimide (Bis-Mal-PEG6) to afford the maleimide-functionalized polymer 4. The 1H NMR spectrum of polymer 4 showed the appearance of a new peak at 6.89 ppm corresponding to the maleimide (Fig. 1). The ratio of integration of this peak to that of the chelator protons at 8.27 ppm is 1:20, which suggests nearly quantitative incorporation of the maleimide functional group into the polymer.
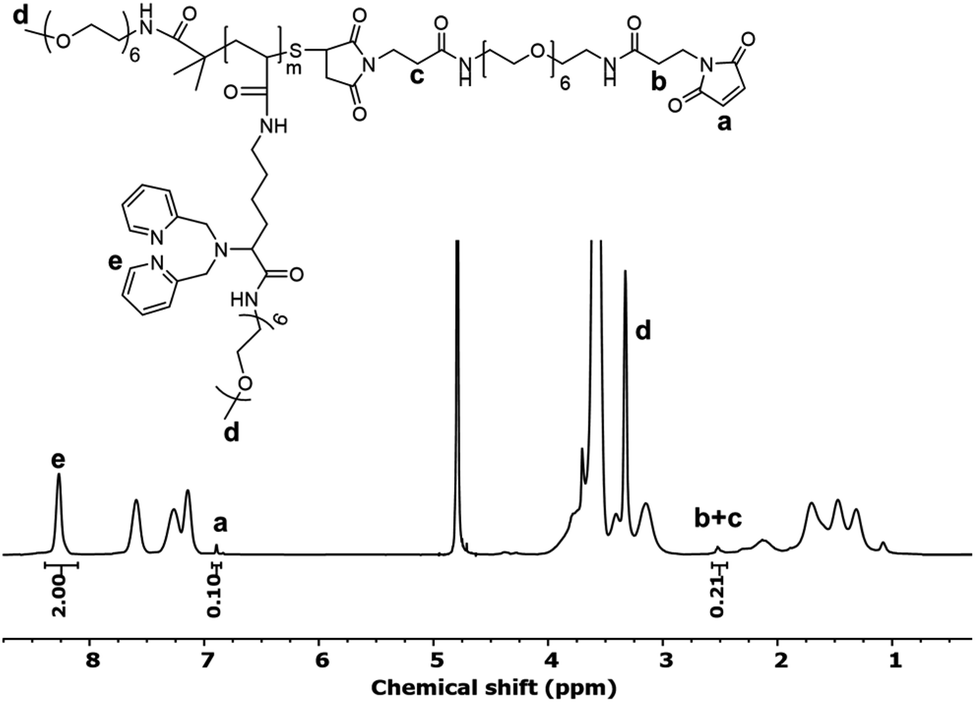 | ||
| Fig. 1 1H NMR spectrum of maleimide functionalized polymer 4 in deuterated water (D2O). | ||
Platinum mass tag synthesis
As mentioned above, one requirement for the metal-chelating polymer to be used in mass cytometry is the presence of functional groups for antibody attachment. One concern in designing a platinum-chelating polymer is the compatibility of functional groups in the polymer structure with the platinum(II) ion. For example, Pt(II) can interact with both amino and carboxylate groups. Many platinum-based anti-cancer drugs are complexes of ligands containing these two functional groups.35 Pt(II) can also interact with the thiol group. Mei et al. prepared platinum-conjugated CD45 Ab by reacting cisplatin with a TCEP reduced Ab.30 Moreover, Pt(II)-alkene (e.g. Zeise's salt) and alkyne complexes have also been reported in literature.36 We speculated that the presence of the aforementioned functional groups along with the DPA ligands in the polymer structure might interfere with the platinum loading process. On the other hand, DeRose's group has reported a series of azide-containing Pt(II) derivatives for target analysis by click chemistry, suggesting minimal interactions between Pt(II) and azide functional groups.37,38 Taking all these factors into consideration, we decided to introduce pendant azide functional groups to the polymer structure for antibody conjugation via click reactions.The synthesis of the clickable platinum-chelating polymer consists of two major steps as shown in Scheme 2. Starting from the same polymer 1, we first attached the DPA ligand through the aminolysis reaction as described above. The resulting polymer was further treated with TCEP followed by a large excess of maleimide to cap the thiol end group of the polymer. The end-capped polymer 6 was then reacted with an excess of DMTMM (ca. 8-fold to each COOH) followed by a mixture of mPEG6-NH2 and azide-PEG6-NH2. The molar ratio of the two PEGs was 1:1. Assuming the same reactivity of the two PEGs, we anticipated that there were ca. 10 randomly distributed azide groups along every polymer backbone. Fig. 2a presents the 1H NMR spectrum of the PEGylated polymer 7. Similarly, we observed new peaks at 3.0–4.0 ppm, suggesting successful PEGylation. In addition, compared to the mPEG6-only modified polymer 4 (Fig. 1), there is a decrease in the peak a (methoxy group of mPEG6) integral and the appearance of a new peak b at 3.43 ppm (methylene group next to the azide group). A careful analysis of the integrations of the two peaks suggested that the ratio of the azide-PEG6 to the mPEG6 was 1.1:1. Details of the analysis can be found in the ESI (Fig. S8).† These 1H NMR results suggested the presence of both PEGs in polymer 7. We further confirmed the presence of azide groups in polymer 7 by Fourier transform infrared (FTIR) spectroscopy as shown in Fig. 2b. Compared to the rhenium chelating polymer 4 (Fig. 3b), polymer 7 showed a new absorption peak located at 2100 cm−1, which corresponds to the N3 asymmetric vibration.39
 | ||
| Scheme 2 Synthesis of platinum-chelating polymer (m = 10, n = 10). (a) (1) DPA ligand 5, RT, 13 h, DMF; (2) ethanolamine, RT, 3 h; (3) TCEP, maleimide; (b) (1) DMTMM, RT, PB buffer (0.2 M, pH 8.0); (2) mPEG6-NH2 and azide-PEG6-NH2, RT, 12 h. | ||
 | ||
| Fig. 2 (a) 1H NMR spectrum of the clickable platinum-chelating polymer 7 in deuterated water (D2O). (b) FTIR spectrum of the platinum chelating polymer 7. | ||
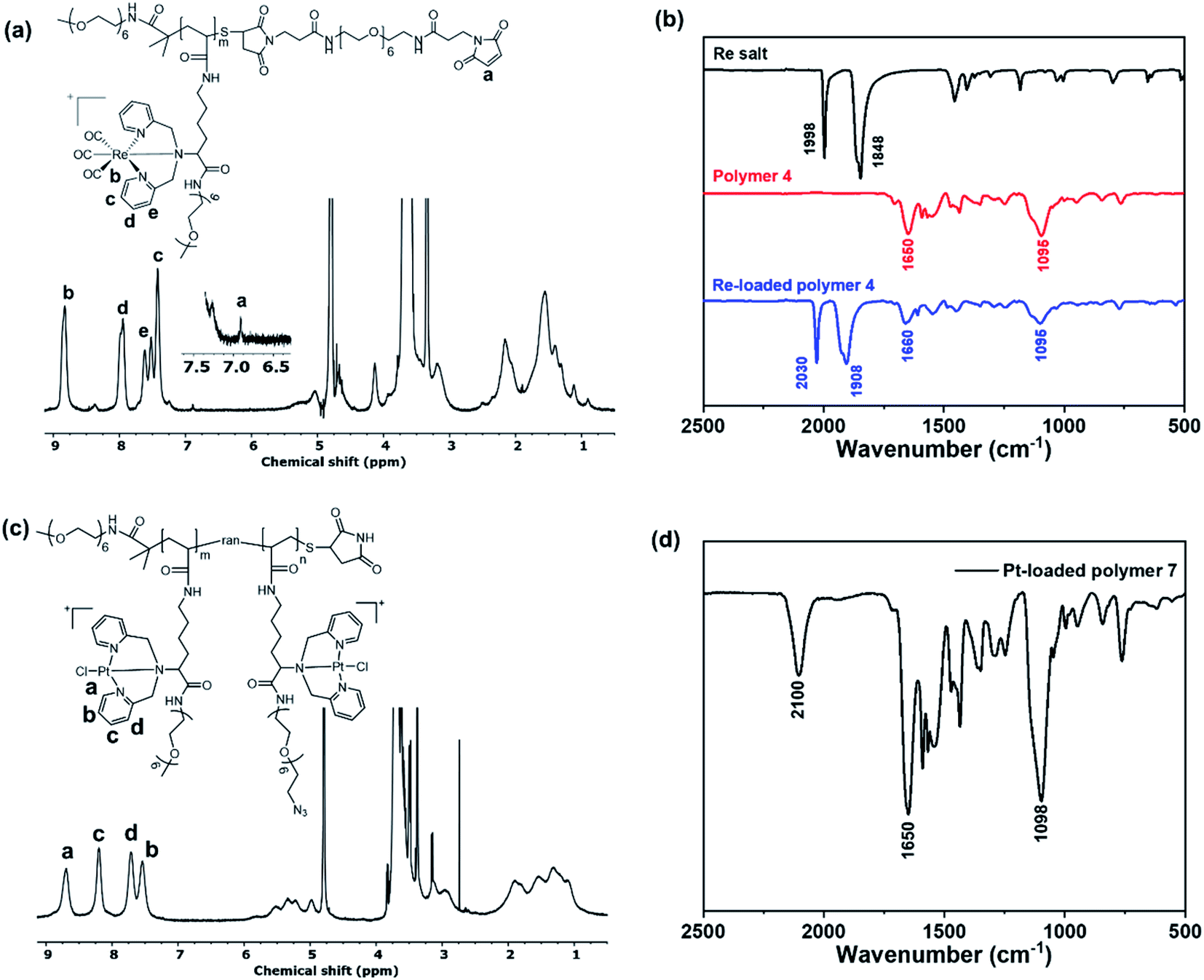 | ||
| Fig. 3 (a) Structure of Re-loaded polymer 4 and its 1H NMR spectrum in deuterated water (D2O). (b) FTIR spectra of the Re salt, polymer 4 and Re-loaded polymer 4. (c) Structure of Pt-loaded polymer 7 and its 1H NMR spectrum in deuterated water (D2O). (d) FTIR spectrum of Pt-loaded polymer 7. | ||
Metal loading
Rhenium loading was achieved by incubating polymer 4 with a slight excess of rhenium salt (1.2-fold to each chelator), [NEt4]2[Re(CO)3Br3],40 in anhydrous methanol at 37 °C for 2 h. Fig. 3a presents the 1H NMR spectrum of polymer 4 after loading with rhenium (denoted PolyRe). Compared to the unloaded polymer 4, all pyridyl proton signals were shifted downfield due to the electron withdrawing inductive effects of Re(I). Importantly, the maleimide group survived under these reaction conditions (ESI, Fig. S9†). Successful loading was further confirmed by FTIR as shown in Fig. 3b. The rhenium salt showed two strong carbonyl absorptions at 1848 and 1998 cm−1. Polymer 4 showed absorption bands at 1650 and 1095 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of amide group and vibrations of ether C–O–C bonds in PEG, respectively.41 After loading, two new absorption bands corresponding to carbonyl groups appeared at 1908 and 2030 cm−1, suggesting the presence of the fac-[Re(CO)3]+ core. PolyRe can be lyophilized for long-term storage and redissolved in PB buffer before bioconjugation (ESI, Fig. S10†).
O stretching of amide group and vibrations of ether C–O–C bonds in PEG, respectively.41 After loading, two new absorption bands corresponding to carbonyl groups appeared at 1908 and 2030 cm−1, suggesting the presence of the fac-[Re(CO)3]+ core. PolyRe can be lyophilized for long-term storage and redissolved in PB buffer before bioconjugation (ESI, Fig. S10†).
Platinum loading was achieved by incubating polymer 7 with potassium tetrachloroplatinate (K2PtCl4, 1.2-fold to each chelator, dissolved in DMSO) in anhydrous methanol at 45 °C for 2 h. Fig. 3c shows the 1H NMR of the purified Pt-loaded polymer 7 (denoted PolyPt). Compared to the metal-free polymer 7, all pyridyl proton signals were shifted downfield, suggesting the formation of the metal complex. In addition, the azide groups survived the metal loading process as indicated by the FTIR spectrum of PolyPt, where the characteristic absorption band of the azide group remains unchanged (Fig. 3d). PolyPt can also be lyophilized for long-term storage and redissolved in PB buffers before bioconjugation (ESI, Fig. S11†).
Antibody conjugation
For the rhenium mass tag, antibody-polymer conjugates (natRe-CD20 and natRe-CD8a) were prepared following standard Maxpar® antibody labeling protocol. Briefly, the antibody was partially reduced by TCEP, washed in a spin filter, and then mixed with an excess of polymer tag, and the mixture was incubated at 37 °C for 1 h. The goal of this protocol is to selectively reduce disulfide bonds in the hinge region of the Ab, while otherwise maintaining the intact IgG structure. To remove, we initially employed a spin filter device (Amicon, Ultra-0.5, 50 kDa) but found significant residual polymer even after multiple washes. Therefore, the antibody-polymer conjugate was further purified by fast-protein liquid chromatography to remove most of the free polymer (ESI, Fig. S12 and S13†). Note that it is important to remove the unconjugated polymers, because we found that these free polymers could interact non-specifically with major cell populations in human PBMCs (ESI, Fig. S14†). Fortunately, as will be shown below, the Ab-polymer conjugate is much less susceptible to this non-specific binding.For the platinum mass tag, the antibody-polymer conjugate (natPt-CD20) was prepared via the DBCO-azide click reaction. Specifically, the anti-CD20 Ab was reacted with DBCO-PEG4-TFP to introduce the DBCO groups. The tetrafluorophenyl (TFP) ester group reacts with lysine amino groups on the Ab. The number of DBCO per Ab was ca. 3 determined by comparing the DBCO absorbance at 309 nm to the Ab absorbance at 280 nm (ESI, Fig. S15†). The DBCO modified Ab was then reacted with an excess of PolyPt (ca. 13-fold molar excess to each Ab). Excess polymer was removed by spin filtration (Amicon, Ultra-0.5, 100 kDa).
Mass cytometry immunoassays with the rhenium mass tag
The performance of our rhenium mass tag in mass cytometry immunoassays was first evaluated with a natRe-CD20 conjugate. CD20 is a high expression cell surface maker expressed exclusively on B-lymphocytes. The natRe-CD20 conjugate was tested in a 4-plex assay for the identification of B-lymphocytes in human PBMC samples. In these experiments, human PBMCs were stained with a 4-plex antibody panel (Fig. 4a), including Maxpar® reagents 154Sm-CD45, 160Gd-CD14, 170Er-CD3 as well as natRe-CD20. A separate staining panel, where natRe-CD20 was replaced with 147Sm-CD20, was used as a positive control. The natRe-CD20 conjugate was titrated at concentrations of 0.1, 0.3, 0.5 and 1.0 μg mL−1. As shown in Fig. 4c–f, the natRe-CD20 conjugate allowed distinct separation of B cell subsets (CD3−CD20+) from the rest of cell subsets within PBMCs at all titers. The median 187Re signal intensities generated by the natRe-CD20 conjugate at titers of 0.1, 0.3, 0.5 and 1.0 μg mL−1 were 98, 312, 442 and 827, respectively (Fig. 4h). In comparison, the median signal intensity generated by the 147Sm-CD20 conjugate was 528 (Fig. 4g). In addition, CD20 staining using the natRe-CD20 conjugate yielded similar CD3−CD20+ B-cell frequencies as the 147Sm-CD20 conjugate, confirming the utility of our rhenium mass tag in the mass cytometry immunoassays (Fig. 4b and d).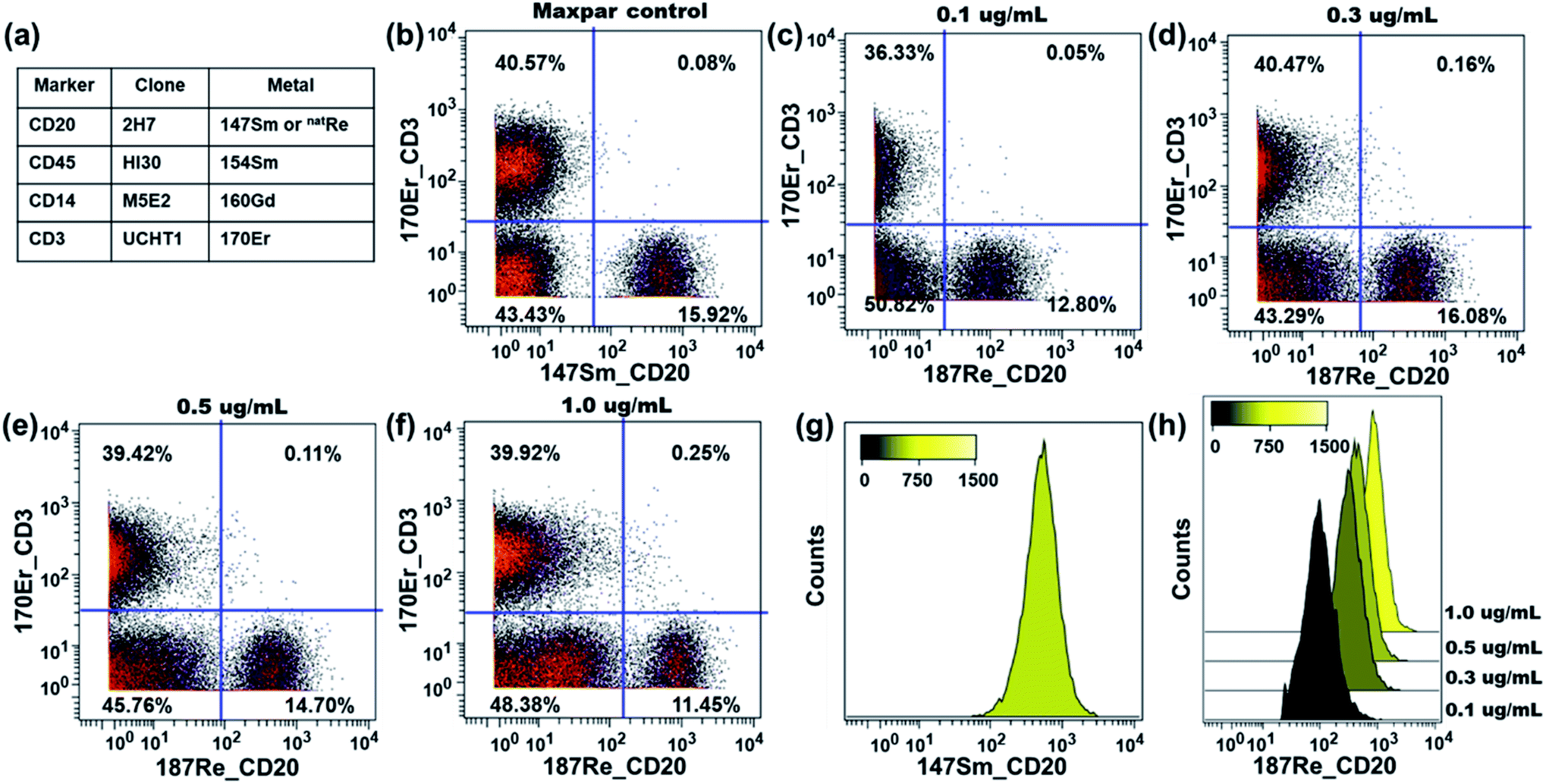 | ||
| Fig. 4 Functionality assay of the natRe-CD20 conjugate with human PBMC samples. The rhenium 187 channel was used for data analysis. (a) Antibody staining panel employed in the assay. (b) Biaxial scatter plot of 170Er-CD3 vs.147Sm-CD20 within human PBMCs at the optimal titer. Biaxial scatter plots of 170Er-CD3 vs.187Re-CD20 within human PBMCs at different titers of 187Re-CD20: (c) 0.1 μg mL−1; (d) 0.3 μg mL−1; (e) 0.5 μg mL−1; (f) 1.0 μg mL−1. (g) Signal distribution histogram for the CD45+CD3−CD20+ B-cells detected by the 147Sm-CD20 conjugate at the optimal titer. (h) Signal distribution histograms for the CD45+CD3−CD20+ B-cells detected by the natRe-CD20 conjugate at different titers. | ||
The performance of the rhenium mass tag was further evaluated in a 7-plex assay with a natRe-CD8a conjugate (Fig. 5a). The CD8 antigen is a cell surface glycoprotein found on most cytotoxic T lymphocytes. Here the natRe-CD8a conjugate was used to identify the CD8+ T-cell population within PBMCs. We titrated the natRe-CD8a conjugate at concentrations of 0.05, 0.1 and 0.2 μg mL−1. A Maxpar® 162Dy-CD8a conjugate was used as a positive control. As shown in Fig. 5c–e, the CD8+ T-cell population within PBMCs could be easily identified with the natRe-CD8a conjugate at all titers. The median 187Re signal intensities generated by the natRe-CD8a conjugate at titers of 0.05, 0.1 and 0.2 μg mL−1 were 364, 374 and 636, respectively (Fig. 5g). In contrast, the 162Dy-CD8a conjugate gave a median signal intensity of 1288 (Fig. 5f). While the natRe-CD8a conjugate showed a lower staining intensity compared to the positive control, the frequencies of the CD8+ T-cell population determined from the natRe-CD8a conjugate were nevertheless comparable to that of the 162Dy-CD8a conjugate at optimal titer (Fig. 5b). In addition, we observed minimal non-specific binding of the natRe-CD8a conjugate to CD8a negative cells such as B-cells (CD45+CD3−CD20+) (Fig. S16†).
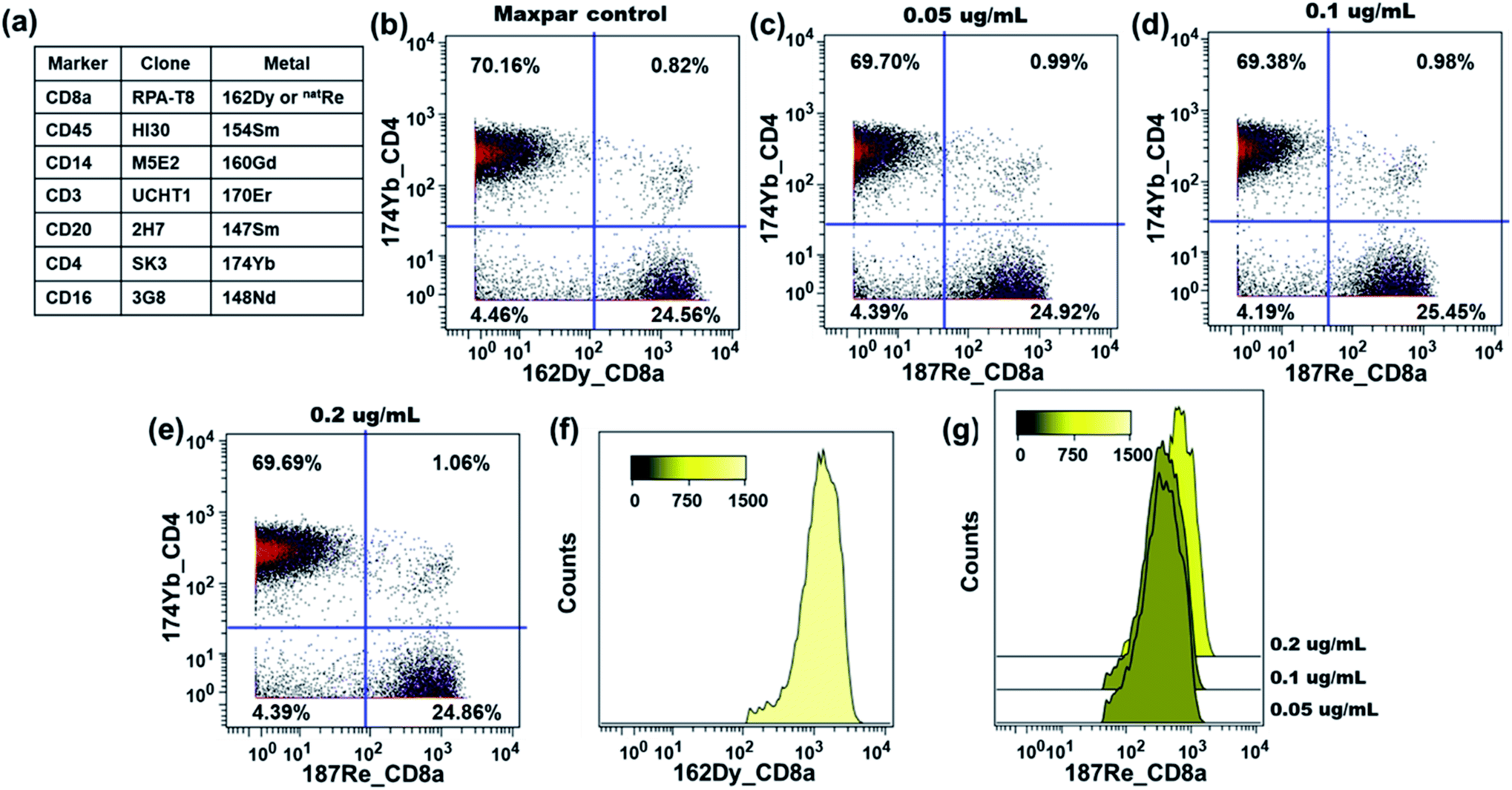 | ||
| Fig. 5 Functionality assay of the natRe-CD8a conjugate with human PBMC samples. The rhenium 187 channel was used for data analysis. (a) Antibody staining panel employed in the assay. (b) Biaxial scatter plot of 174Yb-CD4 vs.162Dy-CD8a within human PBMCs at the optimal titer. Biaxial scatter plots of 174Yb-CD4 vs.187Re-CD8a within human PBMCs at different titers of 187Re-CD8a: (c) 0.05 μg mL−1; (d) 0.1 μg mL−1; (e) 0.2 μg mL−1. (f) Signal distribution histogram for the CD45+CD3+CD4−CD8+ T-cells detected by the 162Dy-CD8a conjugate at the optimal titer. (g) Signal distribution histograms for the CD45+CD3+CD4−CD8+ T-cells detected by the natRe-CD8a conjugate at different titers. | ||
These results demonstrate that our rhenium mass tag can be conjugated to antibodies via the thiol–maleimide reaction for the detection of cellular biomarkers in mass cytometry immunoassays. These rhenium-tagged antibodies provide accurate quantification for single-cell immunophenotyping experiments that is comparable to the commercial Maxpar® conjugates.
Mass cytometry immunoassays with the platinum mass tag
The performance of our platinum mass tag in mass cytometry immunoassays was evaluated with a natPt-CD20 conjugate. A similar 4-plex antibody staining panel was employed for the functionality tests including Maxpar® reagents 154Sm-CD45, 160Gd-CD14, 170Er-CD3 as well as natPt-CD20. The natPt-CD20 conjugate was titrated at concentrations of 0.1, 0.3, 0.5, 1.0 and 2.5 μg mL−1. As shown in Fig. 6b, at the titer of 0.1 μg mL−1, the natPt-CD20 conjugate could not separate the B-cell population (CD45+CD3−CD20+) from the non-T/B cells (CD45+CD3−CD20−). As the titer increased from 0.3 μg mL−1 to 1.0 μg mL−1, the B-cell population in the PBMCs could be clearly identified with the natPt-CD20 conjugate. At the highest titer, however, we observed a significant increase of the 195Pt signal in the non-T/B cells, resulting a poor separation between the two populations. This is likely caused by the non-specific binding of the natPt-CD20 conjugate to the non-T/B cells. Nevertheless, in the medium titer range, the natPt-CD20 conjugate allowed distinct separation of the CD45+CD3−CD20+ B-cells from PBMCs. These results clearly emphasize the importance of antibody titration to optimize the staining in the mass cytometry experiments as a lower antibody concentration may not be able to pull out the target cell population while a very high antibody concentration may result in significant non-specific binding. | ||
| Fig. 6 Functionality assay of the natPt-CD20 conjugate with human PBMC samples. The platinum 195 channel was used for data analysis. (a) Biaxial scatter plot of 170Er-CD3 vs.147Sm-CD20 within human PBMCs at the optimal titer. Biaxial scatter plots of 170Er-CD3 vs.195Pt-CD20 within human PBMCs at different titers of 195Pt-CD20: (b) 0.1 μg mL−1; (c) 0.3 μg mL−1; (d) 0.5 μg mL−1; (e) 1.0 μg mL−1; (f) 2.5 μg mL−1. | ||
Non-specific interactions between polymers and cells
In the previous section, we showed that the rhenium mass tag itself could interact non-specifically with PBMCs. Here we also examined the interaction of the platinum mass tag with PBMCs. For this experiment, an antibody cocktail containing Maxpar® conjugates (Fig. 5a) as well as PolyPt was used to stain the PBMCs. As shown in Fig. 7, the platinum mass tag itself could also interact with the major cell subsets of the PBMCs. In addition, we observed that the CD45+CD3+ T-cells had less binding of PolyPt compared to the CD45+CD3−CD20+ B-cells while the non-T/B cells (CD45+CD3−CD20−) had the highest binding level. Interestingly, the same trend was also observed for that of the rhenium mass tag (Fig. S14†).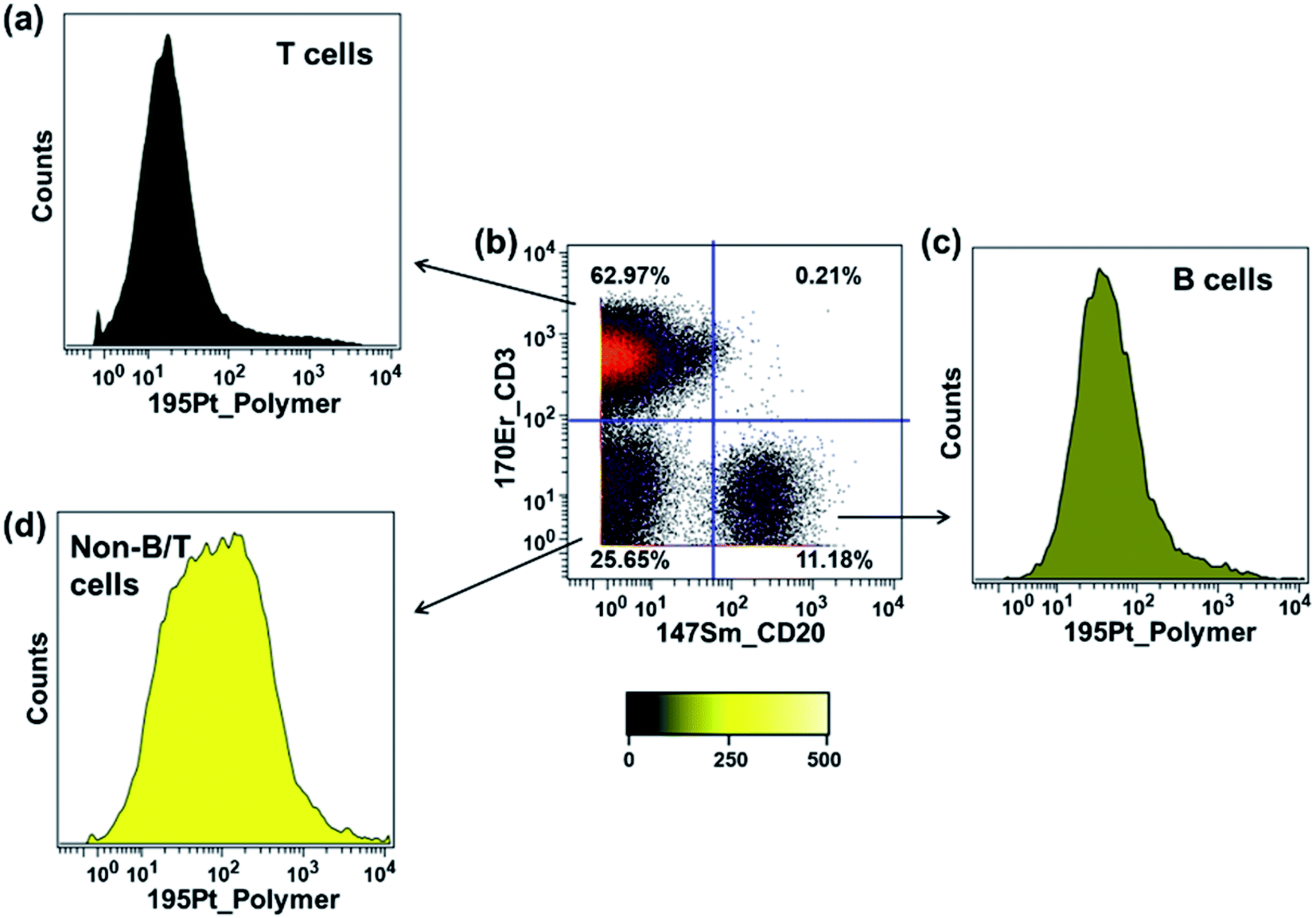 | ||
| Fig. 7 Non-specific binding test of PolyPt with human PBMC samples. The polymer staining concentration was 5 μg mL−1. The platinum 195 channel was used for data analysis. (a) 195Pt signal distribution histogram for the CD45+CD3+ T-cells. Median 195Pt signal intensity: 18. (b) Biaxial scatter plot of 170Er-CD3 vs.147Sm-CD20 within human PBMCs at the optimal titer. (c) 195Pt signal distribution histogram for the CD45+CD3−CD20+ B-cells. Median 195Pt signal intensity: 44. (d) 195Pt signal distribution histogram for the non-B/T cells (CD45+CD3−CD20−). Median 195Pt signal intensity: 89. | ||
In general, polymers can bind to cell membranes via hydrophobic, electrostatic, and hydrogen bonding interactions.42 However, in the case of a polymer carrying multiple metal complexes, transchelation, for example via an exchange reaction with ligands present on the cell surface, can lead to metal attachment to a cell. This would be also detected as non-specific binding. To examine this possibility, we carried out a series of ligand challenge experiments, in which we challenged both polymer tags with a 1000-fold excess (i.e. 50 molar excess to each complex) of either glycine, cysteine, or histidine in PBS buffer (pH 7.4) for 3 days at room temperature. We chose cysteine and histidine as our model biological chelating ligands because they are reported to interact with soft metal ions such as palladium and platinum via the thiol or histidyl group.43–45 Tridentate chelators containing thiol or histidyl groups for the rhenium tricarbonyl core have also been reported.46,47 Glycine was included to mimic the N-terminal amino group or the C-terminal carboxylic group of the peptides.
Following incubation, excess ligands were removed by washing the polymer three times with PBS using a spin filter (Amicon, 3 kDa). The UV-vis absorption of the purified polymer was recorded and compared to that of the original polymer. As shown in Fig. 8a for the PolyRe mass tag, no significant difference in the absorption peak shape was observed after exposure to the challenging ligand, and the ratio of the absorbance at 265 and 300 nm was also unchanged. In addition, we measured the absorbance of the filtrate and observed no significant UV-absorbance between 260 and 350 nm, suggesting no rhenium tricarbonyl core was displaced from its DPA complex in the ligand challenge experiments. These results are consistent with previous reports showing that the dipicolylamine rhenium tricarbonyl complex was stable against ligand exchange.20,23
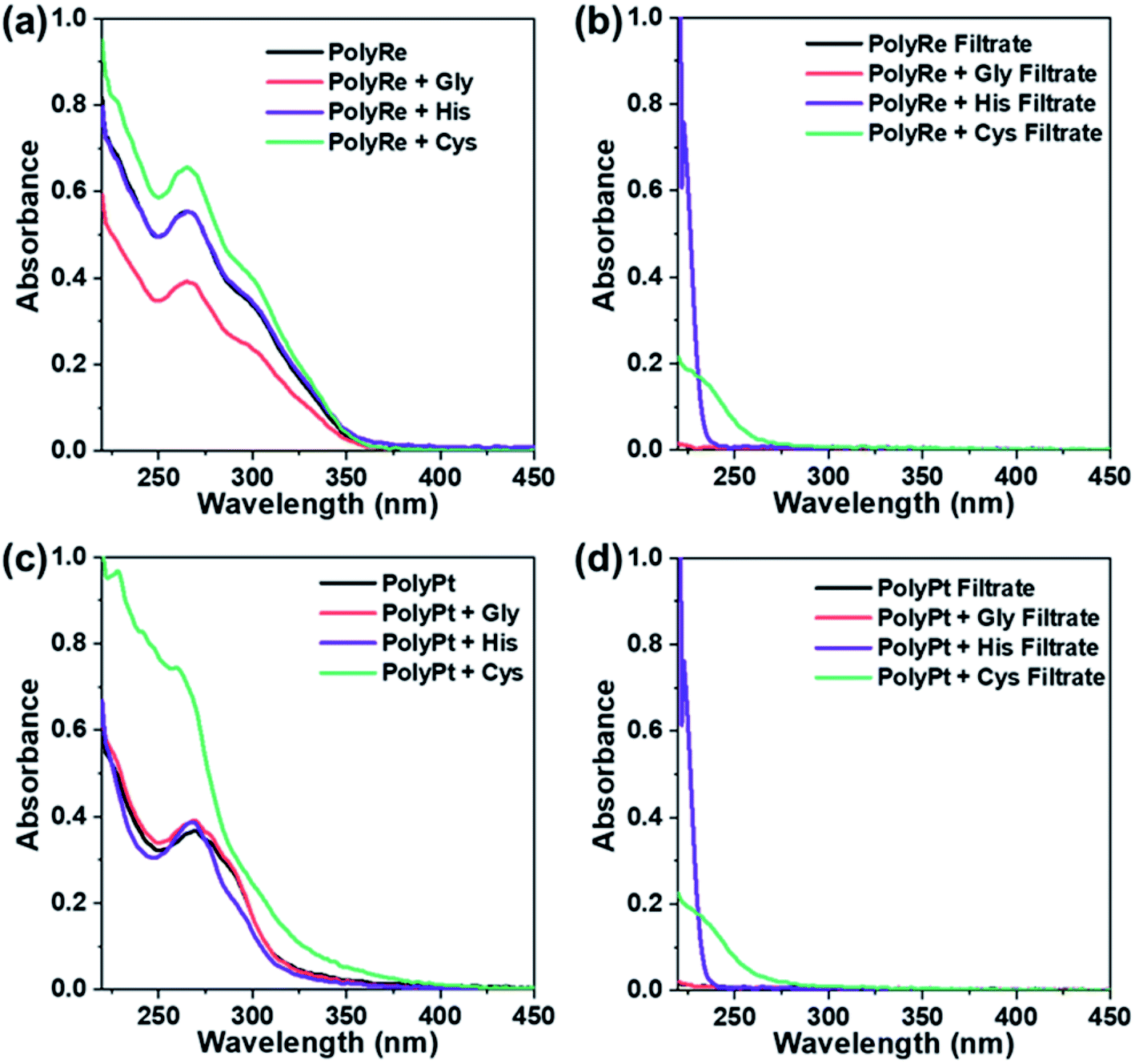 | ||
| Fig. 8 Ligand challenge experiments with PolyRe and PolyPt mass tags. (a) UV-vis spectra of PolyRe mass tag (black) and purified PolyRe mass tag after challenging with glycine (Gly, red), histidine (His, purple) and cysteine (Cys, green). (b) UV-vis spectra of the first filtrate during the polymer purification. (c) UV-vis spectra of PolyPt mass tag (black) and purified PolyPt mass tag after challenging with glycine (Gly, red), histidine (His, purple) and cysteine (Cys, green). (d) UV-vis spectra of the first filtrate during the polymer purification. | ||
For the PolyPt mass tag, the shape of the absorption peak did not change after being challenged with glycine (Fig. 8c). With histidine, we observed a slight decrease in the absorbance between 280 and 300 nm. With cysteine, however, we observed a significant change in the absorption spectrum. In these experiments, we also measured the absorbance of the filtrate and observed no significant UV-absorbance between 260 and 350 nm, suggesting no displacement of the Pt out of the complex. These results indicate that both histidine and cysteine can interact with the PolyPt mass tag via the histidyl or thiol group. Sarkar et al. reported a cyclometalated trinuclear Ir(III)/Pt(II) complex and used it as a luminescent probe for histidine-rich proteins. In their complex, they also employed the dipicolylamine ligand to bind Pt(II) and they showed that the chloride ion in the fourth coordination site of the Pt metal center can be replaced with the imidazole nitrogen or the sulphur atom of histidine or cysteine.48 Therefore, in our case, it is also likely that the chloride ligand in the Pt complex undergoes ligand substitution with histidine or cysteine.
Results from the experiments described above teach us two lessons: first, the PolyRe mass tag is kinetically stable towards ligand substitution. Therefore, the observed non-specific binding is likely due to hydrophobic and/or electrostatic interactions with the cells. Second, the PolyPt mass tag can interact with histidine and cysteine, but the Pt ions are not extracted from the polymer. However, the PolyPt mass tag may also bind to exposed cell surface thiol and histidyl groups, generating non-specific binding signals. In addition, we note that the commercial Maxpar® conjugates are often supplied in BSA-containing buffers. BSA has a free thiol group. The results above also indicate that BSA-containing buffers may not be suitable for long-term storage of the PolyPt-Ab conjugates.
Conclusions
In summary, we have developed a new generation of metal-chelating polymer with pendant dipicolylamine groups suitable for binding intermediate to soft metals such as rhenium and platinum. We introduced two different conjugation strategies, a thiol–maleimide reaction that works well for the rhenium-chelating polymer, and a DBCO-azide click reaction designed to avoid potential complications of platinum interacting with thiol groups. Both polymers were employed in multiplexed immunoassays of PBMCs and shown to be able to quantify cell populations as effectively as commercial Maxpar® conjugates. Despite being modified with a PEG corona, both polymers still showed some level of non-specific binding to PBMCs. In addition, although the Pt mass tag appeared to perform satisfactorily in the immunoassays, we showed that the Pt–Cl bond in the Pt mass tag is susceptible to ligand substitution reactions and could be a potential source of interference in MC applications. These are topics that are currently under study in our laboratories and will be reported in future publications.While we have demonstrate that this new type of polymer can bind rhenium and platinum ions for application to mass cytometry, we note that DPA is also an effective metal chelator for a number of other soft metal ions, such as Pd, Ag and Hg.25,49 Thus, the polymer with DPA pendant groups opens the door to the preparation of other mass tags based upon soft to intermediate metals. We consider the results reported here to be important first steps in the development of soft-metal–metal chelating polymer-conjugates as reagents for mass cytometry.‡
Data availability
Data for this paper, including NMR spectra and mass cytometry data are available at https://doi:10.5683/SP3/WADUHE.Author contributions
Y. Z., P. L. and M. A. W. conceived the project. Y. Z. designed the polymer and carried out all the ligand synthesis, polymer synthesis, characterizations and bioconjugation. P. L. and D. M. designed the mass cytometry experiments. P. L. carried out all the mass cytometry experiments and FPLC purification. Y. Z. and P. L. analyzed the mass cytometry data. Y. Z. and M. A. W. wrote the manuscript. M. A. W. supervised the project.Conflicts of interest
Drs P. Liu and D. Majonis are employees of Fluidigm Canada. A US patent application has been filed on these polymers.Acknowledgements
We thank NSERC Canada and Fluidigm Canada for their financial support of this research. We also thank Mr Edmond Wong for his helpful input.Notes and references
- Q. Baca, A. Cosma, G. Nolan and B. Gaudilliere, Cytometry, Part B, 2017, 92, 10–11 CrossRef PubMed.
- G. K. Behbehani, Clin. Lab. Med., 2017, 37, 945–964 CrossRef PubMed.
- T. Konry, I. Smolina, J. M. Yarmush, D. Irimia and M. L. Yarmush, Small, 2011, 7, 395–400 CrossRef CAS PubMed.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. D. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed.
- B. Allo, X. D. Lou, A. Bouzekri and O. Ornatsky, Bioconjugate Chem., 2018, 29, 2028–2038 CrossRef CAS PubMed.
- H. J. Cho, P. Liu, J. Pichaandi, T. L. L. Closson, D. Majonis, P. L. A. Leighton, E. Swanson, O. Ornatsky, V. Baranov and M. A. Winnik, Eur. Polym. J., 2019, 120, 109175 CrossRef CAS.
- G. Han, S. Y. Chen, V. D. Gonzalez, E. R. Zunder, W. J. Fantl and G. P. Nolan, Cytometry, Part A, 2017, 91, 1150–1163 CrossRef CAS PubMed.
- N. Illy, D. Majonis, I. Herrera, O. Omatsky and M. A. Winnik, Biomacromolecules, 2012, 13, 2359–2369 CrossRef CAS PubMed.
- X. D. Lou, G. H. Zhang, I. Herrera, R. Kinach, O. Ornatsky, V. Baranov, M. Nitz and M. A. Winnik, Angew. Chem., Int. Ed., 2007, 46, 6111–6114 CrossRef CAS PubMed.
- D. Majonis, I. Herrera, O. Ornatsky, M. Schulze, X. D. Lou, M. Soleimani, M. Nitz and M. A. Winnik, Anal. Chem., 2010, 82, 8961–8969 CrossRef CAS PubMed.
- D. Majonis, O. Ornatsky, R. Kinach and M. A. Winnik, Biomacromolecules, 2011, 12, 3997–4010 CrossRef CAS PubMed.
- D. Majonis, O. Ornatsky, D. Weinrich and M. A. Winnik, Biomacromolecules, 2013, 14, 1503–1513 CrossRef CAS PubMed.
- M. H. Spitzer and G. P. Nolan, Cell, 2016, 165, 780–791 CrossRef CAS PubMed.
- J. Bassan, L. M. Willis, R. N. Vellanki, A. Nguyen, L. J. Edgar, B. G. Wouters and M. Nitz, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8155–8160 CrossRef CAS PubMed.
- L. J. Edgar, R. N. Vellanki, A. Halupa, D. Hedley, B. G. Wouters and M. Nitz, Angew. Chem., Int. Ed., 2014, 53, 11473–11477 CrossRef CAS PubMed.
- L. J. Edgar, R. N. Vellanki, T. D. McKee, D. Hedley, B. G. Wouters and M. Nitz, Angew. Chem., Int. Ed., 2016, 55, 13159–13163 CrossRef CAS PubMed.
- G. J. Han, M. H. Spitzer, S. C. Bendall, W. J. Fantl and G. P. Nolan, Nat. Protoc., 2018, 13, 2121–2148 CrossRef CAS PubMed.
- R. T. M. de Rosales, C. Finucane, J. Foster, S. J. Mather and P. J. Blower, Bioconjugate Chem., 2010, 21, 811–815 CrossRef PubMed.
- A. Duatti, Nucl. Med. Biol., 2021, 92, 202–216 CrossRef CAS PubMed.
- K. Ranasinghe, S. Handunnetti, I. C. Perera and T. Perera, Chem. Cent. J., 2016, 10, 71 CrossRef PubMed.
- T. Storr, C. L. Fisher, Y. Mikata, S. Yano, M. J. Adam and C. Orvig, Dalton Trans., 2005, 654–655 RSC.
- V. Fernandez-Moreira, F. L. Thorp-Greenwood and M. P. Coogan, Chem. Commun., 2010, 46, 186–202 RSC.
- M. K. Levadala, S. R. Banerjee, K. P. Maresca, J. W. Babich and J. Zubieta, Synthesis, 2004, 1759–1766 CAS.
- Z. Chen, Y. Wu, Q. Zhang and Y. Zhang, J. Coord. Chem., 2020, 73, 1817–1832 CrossRef CAS.
- X. Xue, C. Qian, H. Fang, H.-K. Liu, H. Yuan, Z. Guo, Y. Bai and W. He, Angew. Chem., Int. Ed., 2019, 58, 12661–12666 CrossRef CAS PubMed.
- Q. P. Qin, B. Q. Zou, Z. F. Wang, X. L. Huang, Y. Zhang, M. X. Tan, S. L. Wang and H. Liang, Eur. J. Med. Chem., 2019, 183, 111727 CrossRef CAS PubMed.
- H. G. Fienberg, E. F. Simonds, W. J. Fantl, G. P. Nolan and B. Bodenmiller, Cytometry, Part A, 2012, 81A, 467–475 CrossRef CAS PubMed.
- F. J. Hartmann, E. F. Simonds and S. C. Bendall, Sci. Rep., 2018, 8, 10 CrossRef PubMed.
- R. L. McCarthy, D. H. Mak, J. K. Burks and M. C. Barton, Sci. Rep., 2017, 7, 6 CrossRef PubMed.
- H. E. Mei, M. D. Leipold and H. T. Maecker, Cytometry, Part A, 2016, 89, 292–300 CrossRef CAS PubMed.
- J. Dang, H. Li, L. Zhang, S. Li, T. Zhang, S. Huang, Y. Li, C. Huang, Y. Ke, G. Shen, X. Zhi and X. Ding, Adv. Mater., 2021, 33, 2008297 CrossRef CAS PubMed.
- R. Bou Zerdan, Z. Geng, B. Narupai, Y. J. Diaz, M. W. Bates, D. S. Laitar, B. Souvagya, A. K. Van Dyk and C. J. Hawker, J. Polym. Sci., 2020, 58, 1989–1997 CrossRef CAS.
- F. D. Jochum and P. Theato, Macromolecules, 2009, 42, 5941–5945 CrossRef CAS.
- A. J. Boyle, P. Liu, Y. Lu, D. Weinrich, D. A. Scollard, G. N. N. Mbong, M. A. Winnik and R. M. Reilly, Pharm. Res., 2013, 30, 104–116 CrossRef CAS PubMed.
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS PubMed.
- D. Balcells, O. Eisenstein, M. Tilset and A. Nova, Dalton Trans., 2016, 45, 5504–5513 RSC.
- J. D. White, M. F. Osborn, A. D. Moghaddam, L. E. Guzman, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2013, 135, 11680–11683 CrossRef CAS PubMed.
- R. Wirth, J. D. White, A. D. Moghaddam, A. L. Ginzburg, L. N. Zakharov, M. M. Haley and V. J. DeRose, J. Am. Chem. Soc., 2015, 137, 15169–15175 CrossRef CAS PubMed.
- E. Lieber, C. N. R. Rao, T. S. Chao and C. W. W. Hoffman, Anal. Chem., 1957, 29, 916–918 CrossRef CAS.
- R. Alberto, A. Egli, U. Abram, K. Hegetschweiler, V. Gramlich and P. A. Schubiger, J. Chem. Soc., Dalton Trans., 1994, 2815–2820 RSC.
- I. M. Deygen and E. V. Kudryashova, Colloids Surf., B, 2016, 141, 36–43 CrossRef CAS PubMed.
- E. Marie, S. Sagan, S. Cribier and C. Tribet, J. Membr. Biol., 2014, 247, 861–881 CrossRef CAS PubMed.
- D. Cocic, S. Jovanovic, S. Radisavljevic, J. Korzekwa, A. Scheurer, R. Puchta, D. Baskic, D. Todorovic, S. Popovic, S. Matic and B. Petrovic, J. Inorg. Biochem., 2018, 189, 91–102 CrossRef CAS PubMed.
- N. Hadjiliadis, N. Ferderigos, J. L. Butour, H. Marzarguil, G. Gasmi and J. P. Laussac, Inorg. Chem., 1994, 33, 5057–5064 CrossRef CAS.
- X. M. Luo, W. Huang, Y. H. Mei, S. Z. Zhou and L. G. Zhu, Inorg. Chem., 1999, 38, 1474–1480 CrossRef CAS.
- D. Donghi, D. Maggioni, G. D'Alfonso, F. Amigoni, E. Ranucci, P. Ferruti, A. Manfredi, F. Fenili, A. Bisazza and R. Cavalli, Biomacromolecules, 2009, 10, 3273–3282 CrossRef CAS PubMed.
- G. Makris, O. Karagiorgou, D. Papagiannopoulou, A. Panagiotopoulou, C. P. Raptopoulou, A. Terzis, V. Psycharis, M. Pelecanou, I. Pirmettis and M. S. Papadopoulos, Eur. J. Inorg. Chem., 2012, 3132–3139 CrossRef CAS.
- A. Sarkar, R. Kumar, B. Das, P. S. Ray and P. Gupta, Dalton Trans., 2020, 49, 1864–1872 RSC.
- K. Seubert, D. Boehme, J. Koesters, W.-Z. Shen, E. Freisinger and J. Z. Mueller, Z. Anorg. Allg. Chem., 2012, 638, 1761–1767 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures, scheme, and experimental details. See DOI: 10.1039/d2sc00595f |
| ‡ Fluidigm, CyTOF, EQ, Helios and Maxpar are trademarks and/or registered trademarks of Fluidigm Corporation in the United States and/or other countries. For research use only. Not for use in diagnostic procedures. |
| This journal is © The Royal Society of Chemistry 2022 |