
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective formation of [Au23(SPhtBu)17]0, [Au26Pd(SPhtBu)20]0 and [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 by controlling ligand-exchange reaction†
Yuichi
Negishi
 *ab,
Hikaru
Horihata
a,
Ayano
Ebina
a,
Sayuri
Miyajima
a,
Mana
Nakamoto
a,
Ayaka
Ikeda
a,
Tokuhisa
Kawawaki
ab and
Sakiat
Hossain
a
*ab,
Hikaru
Horihata
a,
Ayano
Ebina
a,
Sayuri
Miyajima
a,
Mana
Nakamoto
a,
Ayaka
Ikeda
a,
Tokuhisa
Kawawaki
ab and
Sakiat
Hossain
a
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku–ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
bResearch Institute for Science & Technology, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
First published on 30th March 2022
Abstract
To use atomically precise metal nanoclusters (NCs) in various application fields, it is essential to establish size-selective synthesis methods for the metal NCs. Studies on thiolate (SR)-protected gold NCs (Aun(SR)m NCs) revealed that the atomically precise Aun(SR)m NC, which has a different chemical composition from the precursor, can be synthesized size-selectively by inducing transformation in the framework structure of the metal NCs by a ligand-exchange reaction. In this study, we selected the reaction of [Au25(SC2H4Ph)18]− (SC2H4Ph = 2-phenylethanethiolate) with 4-tert-butylbenzenethiol (tBuPhSH) as a model ligand-exchange reaction and attempted to obtain new metal NCs by changing the amount of thiol, the central atom of the precursor NCs, or the reaction time from previous studies. The results demonstrated that [Au23(SPhtBu)17]0, [Au26Pd(SPhtBu)20]0 (Pd = palladium) and [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (Pt = platinum) were successfully synthesized in a high proportion. To best of our knowledge, no report exists on the selective synthesis of these three metal NCs. The results of this study show that a larger variety of metal NCs could be synthesized size-selectively than at present if the ligand-exchange reaction is conducted while changing the reaction conditions and/or the central atoms of the precursor metal NCs from previous studies.
Introduction
To use atomically precise metal nanoclusters (NCs) in various application fields1–6 while taking advantage of their size-specific physicochemical properties,7–9 it is essential to establish size-selective synthesis methods for the metal NCs. Studies on thiolate (SR)-protected gold NCs (Aun(SR)m NCs),10–18 which are the most studied metal NCs, have revealed that it is extremely effective to expose the mixture of Aun(SR)m NCs with a distribution in chemical composition to harsh conditions and thereby converge them to stable NCs to selectively obtain atomically precise Aun(SR)m NC.19,20 Recent studies revealed that ligand exchange of the obtained atomically precise Aun(SR)m NC with other ligands with a different bulkiness also yields atomically precise Aun(SR)m NC with a different chemical composition.21 In the latter method, which is termed ligand-exchange-induced size/structure transformation (LEIST), new atomically precise Aun(SR)m NCs and related NCs are size-selectively synthesized by inducing transformation in the framework structure of precursor NCs by ligand exchange.22–24For such LEIST, if the reaction is carried out under different conditions from the reported condition, it may be possible to proceed with a reaction by a different pathway and/or obtain products with a different chemical composition. In the reaction between [Au25(SC2H4Ph)18]− (SC2H4Ph = 2-phenylethanethiolate, Scheme S1(a) and S2(a)†) and 4-tert-butylbenzenethiol (tBuPhSH), [Au28(SPhtBu)20]0 (SPhtBu = 4-tert-butylbenzenethiolate, Scheme S1(b) and S2(b)†) is size-selectively synthesized at 80 °C (Scheme 1(a)),25 and [Au20(SPhtBu)16]0 is also formed in ∼10% yield at 40 °C (Scheme 1(b) and S2(c)†).26 Even at 80 °C, when the amount of tBuPhSH is reduced to ∼50 times the amount of SC2H4Ph in [Au25(SC2H4Ph)18]− ([tBuPhSH]/[SC2H4Ph] ≅ 50), [Au22(SC2H4Ph)4(SPhtBu)14]0 is produced at a high proportion (Scheme 1(c) and S2(d)†).27 Based on these facts, it is expected that further modification of the reaction conditions (e.g., amount of thiol, temperature, reaction time) in LEIST will result in the size-selective synthesis of new Aun(SR)m NCs, which has not been reported previously.
 | ||
| Scheme 1 Experimental conditions for the formation of each metal NC: (a) [Au28(SPhtBu)20]0,25 (b) [Au20(SPhtBu)16]0,26 (c) [Au22(SC2H4Ph)4(SPhtBu)14]0,27 (d) [Au23(SPhtBu)17]0 (i; this work), (e) [Au26Pd(SPhtBu)20]0 (iii; this work), (f) [Au24Pt(SPhtBu)18]0,35 and (g) [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (v; this work). | ||
In addition to such changes in reaction conditions, heteroatom substitutions (Scheme S2(e) and (f)†)28–31 in the precursor Aun(SR)m NCs may also induce changes in the reaction pathway and/or chemical composition of the products.32,33 Indeed, our previous studies have shown that when [Au24Pt(SC2H4Ph)18]0 (Pt = platinum; Scheme S2(f)†),34 in which the central Au atom of [Au25(SC2H4Ph)18]− is replaced with a atom, is reacted with tBuPhSH, the ligand exchange proceeds while keeping a metal core size, unlike for [Au25(SC2H4Ph)18]−, and thereby [Au24Pt(SPhtBu)18]0, in which all ligands are exchanged with SPhtBu, can be size-selectively synthesized.35 This change in reaction pathway because of Pt substitution is related mainly to the difference in the strength of the framework between Au25(SR)18 and Au24Pt(SR)18. Because [Au24Pt(SC2H4Ph)18]0 has a stronger framework than [Au25(SC2H4Ph)18]−,36 it can maintain its framework structure even when the ligand is exchanged from SC2H4Ph to SPhtBu, and thereby [Au24Pt(SPhtBu)18]0 was synthesized in the reaction between [Au24Pt(SC2H4Ph)18]0 and tBuPhSH. Density functional theory (DFT) calculations suggest that Au24Pd(SR)18 (Pd = palladium; Scheme S2(e)†),30 in which the central Au atom of [Au25(SC2H4Ph)18]− is replaced with a Pd atom, has a framework strength between Au25(SR)18 and Au24Pt(SR)18.36 Therefore, it is expected that when [Au24Pd(SC2H4Ph)18]0 is reacted with tBuPhSH, the reaction proceeds differently from the case when [Au25(SC2H4Ph)18]− or [Au24Pt(SC2H4Ph)18]0 is used as the precursor NC, and thereby new metal NCs can be synthesized size-selectively.
The purpose of this study was to increase the variety of atomically precise metal NCs that can be synthesized size-selectively. Among the Aun(SR)m NCs, [Au25(SC2H4Ph)18]− is one of the most studied NCs,37,38 and tBuPhSH has been used frequently as an exchanged ligand in previous LEIST experiments.12,23,35 Therefore, in this study, we selected the reaction of [Au25(SC2H4Ph)18]− with tBuPhSH as a model reaction and attempted to obtain new NCs by changing the reaction conditions or the central atom of the precursor NCs. Specifically, the following changes were applied in previous reports:26,35 (1) the amount of tBuPhSH was reduced in the reaction at 40 °C (Scheme 1(d)), (2) the central Au atom of [Au25(SC2H4Ph)18]− was substituted with a Pd atom ([Au24Pd(SC2H4Ph)18]0) (Scheme 1(e)), (3) the reaction time in the reaction between [Au24Pt(SC2H4Ph)18]0 and tBuPhSH was shortened (Scheme 1(g)). These attempts led to the formation of [Au23(SPhtBu)17]0 (Scheme 1(d)), [Au26Pd(SPhtBu)20]0 (Scheme 1(e)) and [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (Scheme 1(g)) in a high proportion. To the best of our knowledge, no report exists on the selective synthesis for these three metal NCs. These results demonstrate that a larger variety of atomically precise metal NCs can be synthesized size-selectively by ligand exchange than at present if the ligand-exchange reaction is conducted while changing the reaction conditions and/or central atoms of the precursor metal NCs from previous studies.
Results and discussion
Reaction between [Au25(SC2H4Ph)18]− and tBuPhSH
Previous studies on this reaction system have shown that [Au28(SPhtBu)20]0 can be synthesized selectively when the reaction proceeds at 80 °C with a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 250,25 whereas [Au20(SPhtBu)16]0 can be formed in ∼10% yield when the reaction proceeds at 40 °C with a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 150.26 In this study, we conducted the reaction at 40 °C with a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 100 (Scheme 1(d)) to allow the reaction to proceed more slowly than in a previous report and thereby create a novel metal NC.Fig. 1A(a) and (b) show reversed-phase high-performance liquid chromatography (RP-HPLC)39–42 chromatograms of the precursor [Au25(SC2H4Ph)18]− (Fig. S1†) and the product, respectively. The chromatogram of the sample before the reaction (Fig. 1A(a)) showed a sharp peak at the retention time of [Au25(SC2H4Ph)18]− (16.0 min). In the product chromatogram (Fig. 1A(b)), the main peak (i) was observed at a retention time of 55.6 min, which differs from that of [Au25(SC2H4Ph)18]− and [Au28(SPhtBu)20]0 (59.7 min, Fig. S2†) (Table S1†).
 | ||
| Fig. 1 (A) RP-HPLC chromatogram (a) before and (b) after the reaction between [Au25(SC2H4Ph)18]− and tBuPhSH at 40 °C for 2 h. (B) Positive-ion (a) ESI and (b) MALDI mass spectra of the main product (i). (C) Positive-ion ESI mass spectrum of product ii (Fig. S3A(b)†). In the ESI-MS, Cs+ was added as a cation source. In (B) and (C), the insets compare the isotope pattern between the experimental and simulated spectra. In (B) and (C), the peak with asterisk (*) was assigned to [Au23(SC2H4Ph)(SPhtBu)16]0(Cs+)2 and [Au23(SC4H9)(SPhtBu)16]0(Cs+)2, respectively. | ||
We isolated product i and measured its electrospray ionization (ESI) mass spectrum to determine the chemical composition of product i. In the ESI mass spectrometry (MS), cesium ions (Cs+) were added to the NC solution as an ionization source.43Fig. 1B(a) shows the ESI mass spectrum of product i. In the mass spectrum, a strong peak was observed at the position (m/z = 3802.6) that was assignable to [Au23(SPhtBu)17]0(Cs+)2 (Mw = 7605.3). This result suggests that product i is neutral [Au23(SPhtBu)17]0. We also measured the matrix-assisted laser desorption/ionization (MALDI) mass spectrum of product i. A strong peak was observed at the position (m/z = 7173.4) that was assignable to [Au23(SPhtBu)16]+ (Fig. 1B(b)). In the MALDI-MS of Aun(SPhtBu)m, one SPhtBu is typically released from Aun(SPhtBu)m NCs by laser irradiation.35,44,45 The main peak was observed at a position of [Au23(SPhtBu)16]+ in the MALDI mass spectrum of [Au23(SPhtBu)17]0 because a similar release of one SPhtBu was caused by laser irradiation. We also synthesized [Au25(SC4H9)18]0 with SC4H9 (1-butanethiolate, Scheme S1(c) and Fig. S3A(a)†) as a ligand and reacted it with tBuPhSH. In the ESI (Fig. 1C) and MALDI mass spectra (Fig. S3B†) of the main product (ii, Fig. S3A(b)†), a strong peak was observed in the same positions as the product i (m/z = 3802.6 and 7173.4, respectively). Therefore, the main product contains only SPhtBu as a ligand. These results confirmed the chemical composition of the main product (i) as [Au23(SPhtBu)17]0.
Fig. 2 shows the ultraviolet-visible (UV-vis) optical absorption spectrum of the obtained [Au23(SPhtBu)17]0 (i). In the spectrum, peak structures appeared at ∼650, 545, 470, 420 and 340 nm. These peak positions are different from those in the spectrum of [Au25(SC2H4Ph)18]− (∼675, 445, 395 and 320 nm; Fig. S1(c)†),37,46 which indicates that [Au23(SPhtBu)17]0 has a different electronic structure from its precursor, [Au25(SC2H4Ph)18]−. For the geometric structure of the obtained [Au23(SPhtBu)17]0, the structure in Fig. S4† may be one of the candidates based on the DFT calculations results47 on [Au23(SCH3)17]0 (SCH3 = methanethiolate). Although we attempted to crystallize [Au23(SPhtBu)17]0 for one year to demonstrate experimentally the geometric structure by single-crystal X-ray diffraction (SC-XRD),28,48,49 we could not obtain good-quality crystals. There might be the structural isomers in [Au23(SPhtBu)17]0, leading to the difficulty in the crystallization. It is expected that the geometric structure of [Au23(SPhtBu)17]0 will be determined by DFT calculations of NC containing ligands (SPhtBu) instead of SCH3 (Fig. S5†)50–52 in future.
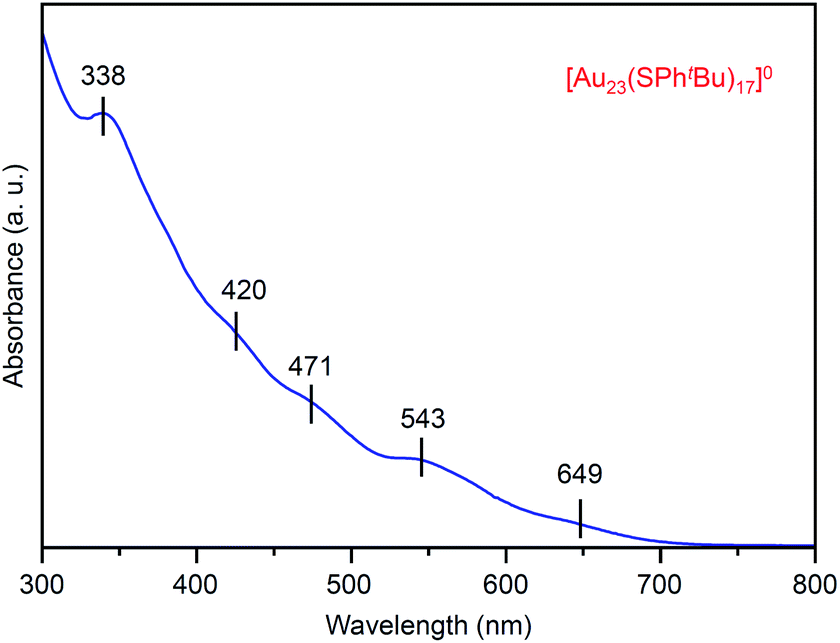 | ||
| Fig. 2 UV-vis optical absorption spectrum of product i ([Au23(SPhtBu)17]0). | ||
In this way, we obtained [Au23(SPhtBu)17]0 in a high proportion by the ligand-exchange reaction at 40 °C with a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 100 (Scheme 1(d)). The [Au23(SPhtBu)17]0 hardly decomposed even when left in a toluene solution at temperatures up to 80 °C (Fig. 3(a), (b) and S6†). Therefore, [Au23(SPhtBu)17]0 is stable in solution. Mandal et al. have also synthesized a similar NC selectively by ligand exchange using a different NC ([Au23(SC6H11)16]−, SC6H11 = cyclohexanethiolate) as a precursor.53 They assigned the product as Au23(SPhtBu)16 based on the MALDI-MS results. However, according to Fig. 1B(b) and the optical absorption spectrum of the obtained product, it is assumed that [Au23(SPhtBu)17]0 was also synthesized selectively. These results indicate that [Au23(SPhtBu)17]0 is a metal NC with a level of stability that allows for selective synthesis.
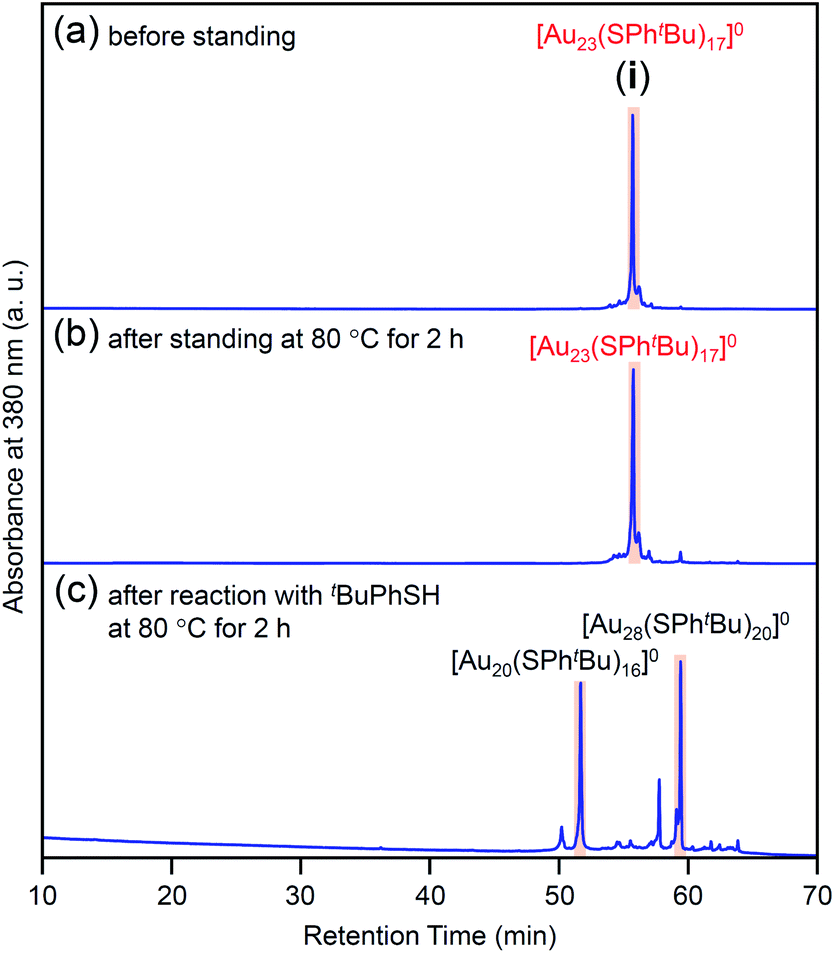 | ||
| Fig. 3 Comparison of RP-HPLC chromatogram of product i (a) before and (b) after soaking in toluene solution at 80 °C for 2 h. (c) Chromatogram of products obtained by reacting [Au23(SPhtBu)17]0 (i) with tBuPhSH in toluene solution at 80 °C (reaction time, 2 h). In (c), the main peaks at 51.6 and 59.7 min are attributed to the reported [Au20(SPhtBu)16]0 (Fig. S10†) and [Au28(SPhtBu)20]0 (Fig. S2†), respectively. The sub peaks at 50.2 and 57.6 min are attributed to [Au19(SPhtBu)15]0 and [Au29(SPhtBu)17]0, respectively (Fig. S10†). | ||
We also investigated the formation mechanism of the [Au23(SPhtBu)17]0. MALDI-27 or ESI-MS45 has been used as a reaction tracking tool in studies on product formation mechanisms. However, in this ligand-exchange reaction, the reactant ([Au25(SC2H4Ph)18]−) is a negative ion, whereas the product is neutral. Therefore, it is difficult to ionize reactants and products with the same ionization efficiency using neither negative- or positive-ion mode. In addition, in MALDI-MS, [Aun(SC2H4Ph)m−x(SPhtBu)x]0 can be ionized nondestructively,27 whereas [Aun(SPhtBu)m]0 is observed only as being dissociated (Fig. 1B(b)).35,44,45,53 Thus, it is extremely difficult to estimate the abundance distribution of the products by MALDI-MS (Fig. S7†). We found that it is also difficult to estimate the abundance distribution of the products for this reaction system using ESI-MS because the reaction intermediates ([Aun(SC2H4Ph)m−x(SPhtBu)x]0) dissociate during ESI (Fig. S8†).
In our previous studies, we separated SR-protected metal NCs by RP-HPLC (Scheme S3†) depending on the charge state and ligand combination (Scheme S4†).30,31,54,55 The charge-state distribution and abundance distribution of the products can be estimated relatively quantitatively by using such a high-resolution RP-HPLC. In addition, it is also possible to reveal the electronic structure of each separated Aun(SC2H4Ph)m−x(SPhtBu)x NC by a photodiode array (PDA) detector (Scheme S3(a)†) attached to the HPLC equipment. Therefore, we attempted to clarify the chemical composition, abundance distribution, and electronic structure of the products by the combined use of MALDI-MS and RP-HPLC.
Fig. 4 shows the RP-HPLC chromatograms of the products at each reaction time (Table S1†). These chromatograms and the MALDI mass spectra of the products at each reaction time (Fig. S7†) show the following four facts for the reaction between [Au25(SC2H4Ph)18]− and tBuPhSH (Fig. S9†): (1) first, [Au25(SC2H4Ph)18]− is oxidized to [Au25(SC2H4Ph)18]0, (2) then, SC2H4Ph is exchanged with SPhtBu, (3) these exchanges form [Au25(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–14), (4) little [Au25(SC2H4Ph)18−x(SPhtBu)x]0 (x ≥ 15) are produced. In addition, the optical absorption spectra (Fig. 5) of a series of [Aun(SC2H4Ph)m−x(SPhtBu)x]0 (x = 0–14) showed that (5) a significant change occurs in the electronic structure of [Aun(SC2H4Ph)m−x(SPhtBu)x]0 during the increase of x from 13 to 14. Based on these five facts, (1) when the number of exchanged ligands reaches 14, the framework of the product begins to distort, and (2) when the number of exchanged ligands increases above 15, the product cannot maintain its framework and changes its structure to [Au23(SPhtBu)17]0 which can include SPhtBu in a higher ratio (Scheme 2(a)). It is considered that [Au23(SPhtBu)17]0 obtained in this study was synthesized selectively by such a reaction mechanism.
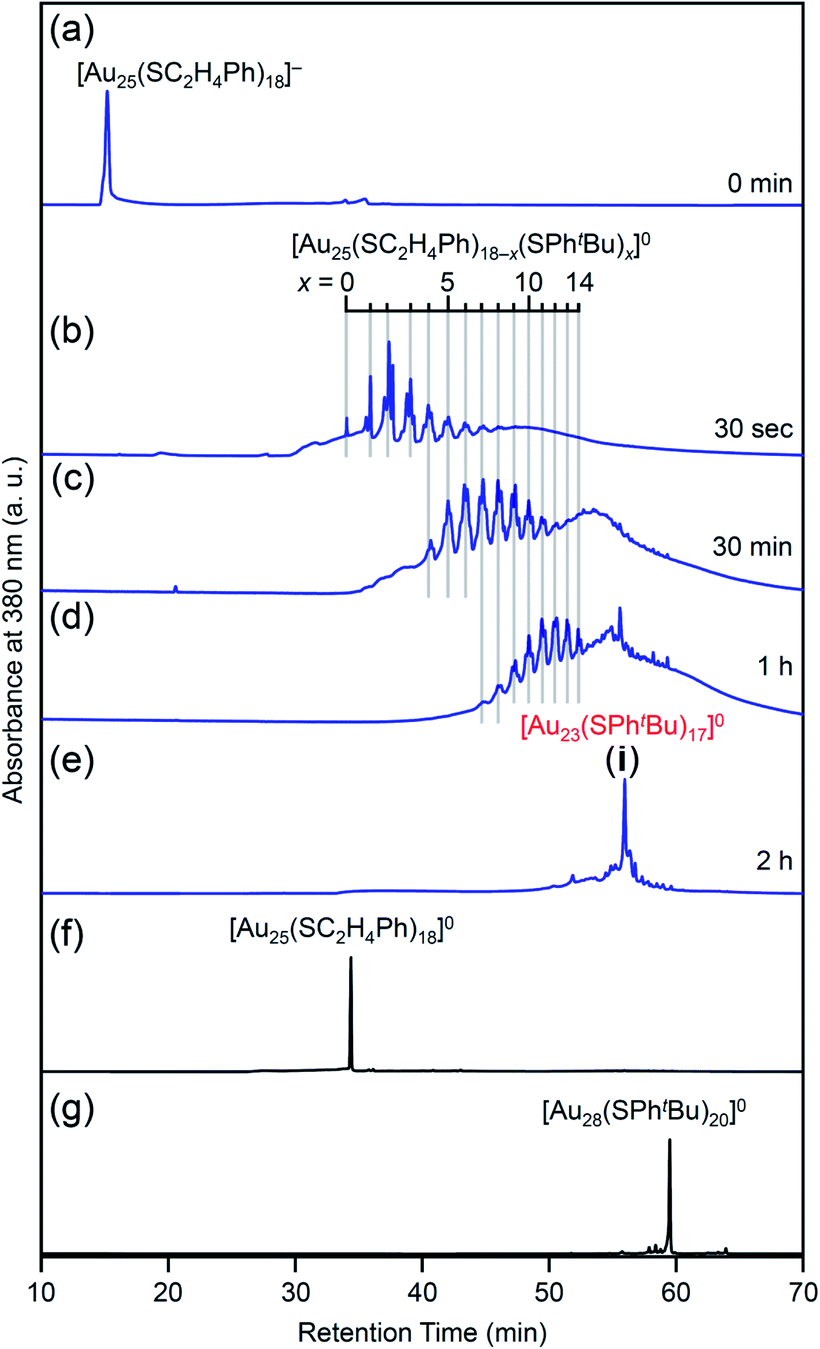 | ||
| Fig. 4 RP-HPLC chromatogram of products obtained by the reaction between [Au25(SC2H4Ph)18]− and tBuPhSH for (a) 0 min, (b) 30 s, (c) 30 min, (d) 1 h and (e) 2 h (i). RP-HPLC chromatogram of (f) [Au25(SC2H4Ph)18]0 and (g) [Au28(SPhtBu)20]0 (Fig. S2†) are shown for comparison. Ligand-exchange products eluted at longer retention times than [Au25(SC2H4Ph)18]0 because the polarity of SPhtBu is lower than that of SC2H4Ph (Scheme S4†).54 The fine peak progression in each major peak is caused by the existence of topological isomers.55 The peak attributed to [Au23(SPhtBu)17]0 was observed with a weak intensity in the MALDI mass spectra of the products (Fig. S7†) probably because of the different ionization process between [Au25(SC2H4Ph)18−x(SPhtBu)x]0 and [Au23(SPhtBu)17]0.27,35,44,45,53 This result indicates that the combined use of MALDI-MS and RP-HPLC is important to elucidate the details of the ligand-exchange mechanism while estimating the product distribution for this reaction system. | ||
 | ||
| Fig. 5 UV-vis optical absorption spectra of a series of [Au25(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–14, Fig. 4) obtained by PDA detector attached to HPLC apparatus (Scheme S3(a)†). These optical absorption spectra are not obtained for the products with a distribution in chemical composition, which has often been reported in the literature,45 but for each chemical-composition product that is separated by RP-HPLC (Fig. 4). The spectral features at x = 0–13, disappear at x = 14, which means that structural deformation starts to occur at x = 14. | ||
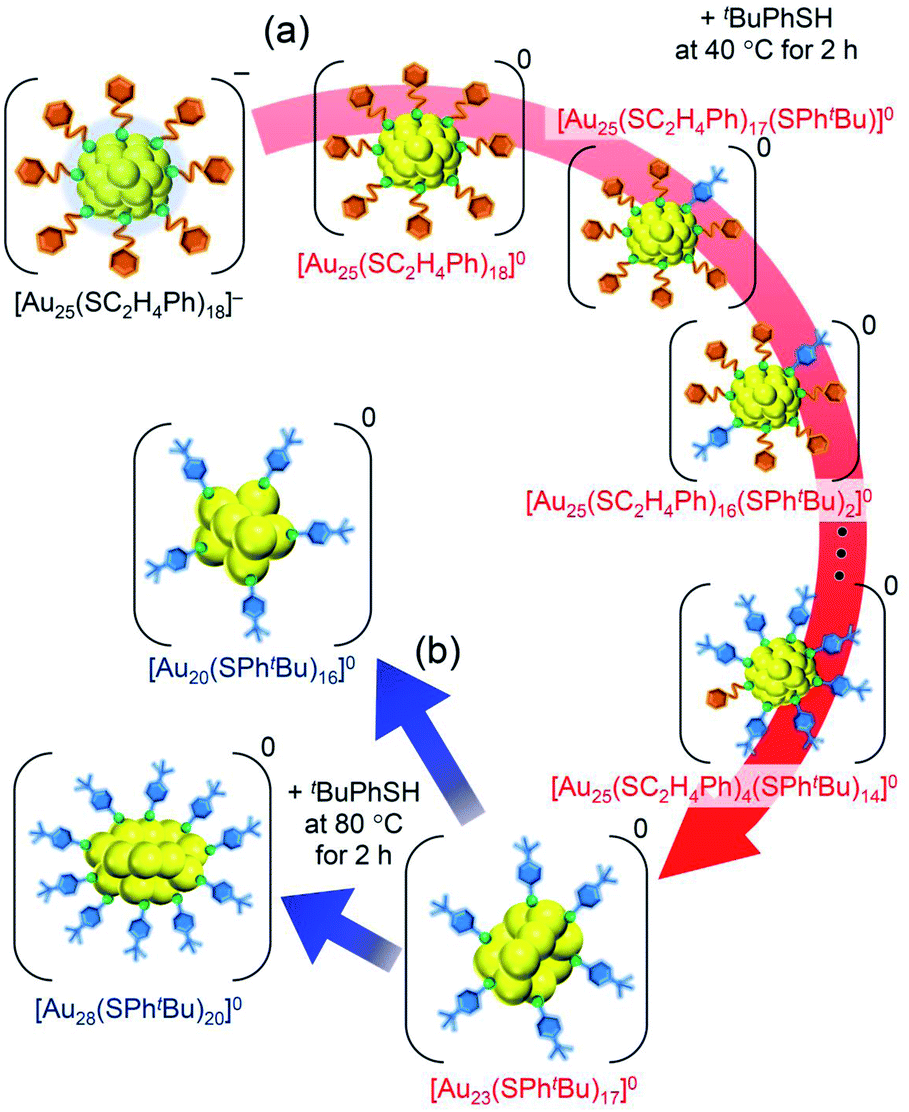 | ||
| Scheme 2 Schematic illustration of transformation from (a) [Au25(SC2H4Ph)18]− to [Au23(SPhtBu)17]0 (i) and (b) [Au23(SPhtBu)17]0 (i) to [Au20(SPhtBu)16]0 and [Au28(SPhtBu)20]0. | ||
We reacted [Au23(SPhtBu)17]0 with tBuPhSH in toluene solution at 80 °C for 2 h. [Au23(SPhtBu)17]0 disappeared and [Au20(SPhtBu)16]0 (Fig. S10†) and [Au28(SPhtBu)20]0 (Fig. S2†) were produced (Fig. 3c). Therefore, (1) [Au23(SPhtBu)17]0 reacts easily with tBuPhSH, and (2) such reactions produce [Au20(SPhtBu)16]0 and [Au28(SPhtBu)20]0 (Scheme 2(b)). It appears that [Au20(SPhtBu)16]0 and [Au28(SPhtBu)20]0 (Fig. S11†) were synthesized in previous LEIST experiments (Scheme 1(a) and (b))25,26,53 because a similar reaction occurred in the solution.
Reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH
Our previous studies56 have revealed that the reaction of [Au24Pd(SC12H25)18]0 (SC12H25 = 1-dodecanethiolate) with RSH (RSH = 1-hexanethiol, 1-octanethiol, 2-phenylethanethiol, etc.) proceeds at a faster speed than that of [Au25(SC2H4Ph)18]− with RSH. Accordingly, we conducted the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH at 25 °C to decrease the reaction speed and thereby create the novel metal NC.In the experiment, tBuPhSH was added to a toluene solution of [Au24Pd(SC2H4Ph)18]0 at a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 250, and the solution was stirred at 25 °C for 8 h (Scheme 1(e)). Fig. 6A(a) and (b) shows the RP-HPLC chromatogram of samples before (Fig. S12†) and after the reaction, respectively. The chromatogram of the sample before the reaction showed a sharp peak at the retention time of [Au24Pd(SC2H4Ph)18]0 (34.7 min). The chromatogram of the sample after the reaction (namely, the product) showed a sharp peak (iii) at a retention time of 57.4 min.
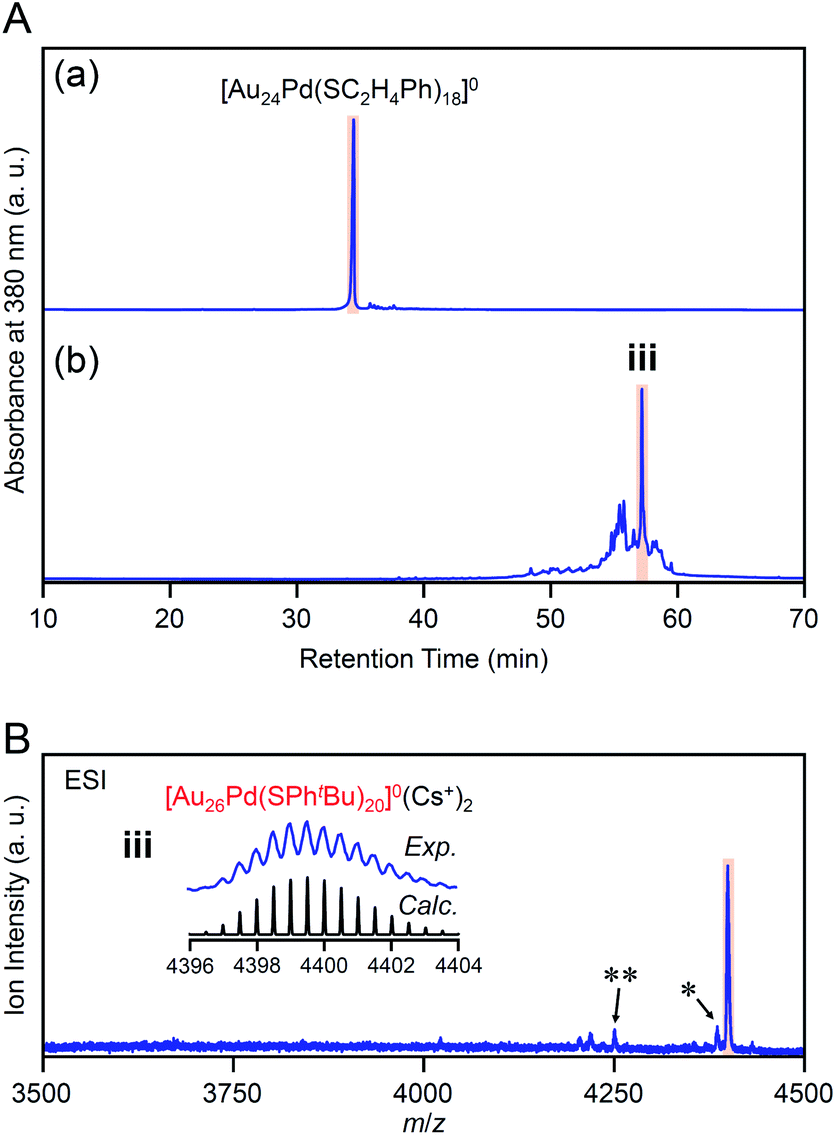 | ||
| Fig. 6 (A) RP-HPLC chromatogram of (a) before and (b) after the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH for 8 h. (B) Positive-ion ESI mass spectrum of the main products obtained by the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH (iii). The inset compares the isotope pattern between the experimental and simulated spectra. Minor peaks with (*) and (**) are assigned to [Au26Pd(SC2H4Ph)(SPhtBu)19]0(Cs+)2 and [Au27Pd(SPhtBu)17]0(Cs+)2, respectively. | ||
We isolated product iii and measured its ESI mass spectrum to determine the chemical composition of product iii. In this MS, Cs+ was also added to the NC solution as an ionization source. Fig. 6B shows the ESI mass spectrum of product iii after isolation. In the mass spectrum, a strong peak was observed at the position (m/z = 4399.16) that is attributed to [Au26Pd(SPhtBu)20]0(Cs+)2 (Mw = 8798.32). This result suggests that product iii is neutral [Au26Pd(SPhtBu)20]0. We also synthesized [Au24Pd(SC4H9)18]0 (Fig. S13(a)–(c)†), which has different ligands from [Au24Pd(SC2H4Ph)18]0, and reacted it with tBuPhSH. In the ESI mass spectrum of the main product (iv, Fig. S14†), a strong peak was observed at the same position (m/z = 4399.16) as that of product iii. These results indicate that the main product contains only SPhtBu as a ligand, which supports the above interpretation that product iii is [Au26Pd(SPhtBu)20]0. Inductively coupled plasma MS confirmed that product iii contains Pd (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 26:1.17). Similar result was also obtained by X-ray photoelectron spectroscopy (Fig. S15†).
:Pd = 26:1.17). Similar result was also obtained by X-ray photoelectron spectroscopy (Fig. S15†).
Fig. 7 shows the optical absorption spectrum of [Au26Pd(SPhtBu)20]0. In the spectrum, peaks were observed at ∼550 and 360 nm. These peak positions are different from those observed in the spectrum of [Au24Pd(SC2H4Ph)18]0 (∼950, 650, 470 and 370 nm; Fig. S12(c)†). This result indicates that the electronic structure of [Au26Pd(SPhtBu)20]0 is different from that of [Au24Pd(SC2H4Ph)18]0. The X-ray absorption fine structure analysis (Fig. S16(a)†) revealed that Au in [Au26Pd(SPhtBu)20]0 is a little reduced than Au in mono-Au [Au28(SPhtBu)20]0. As for the geometrical structure, because Pd is typically located at the center of the metal core in alloy NCs that consist of Au and Pd,30,57–61 it is reasonable to assume that Pd is also located at the center of the metal core in [Au26Pd(SPhtBu)20]0 that was obtained in this study (Fig. S17†). Unfortunately, crystallization of [Au26Pd(SPhtBu)20]0 was not achieved in this study regardless of the repeated trials for one year. However, the Fourier transform extended X-ray absorption fine structure spectrum demonstrates that Au–S and Au–Au bonds also exist in [Au26Pd(SPhtBu)20]0 as well as in [Au28(SPhtBu)20]0 (Fig. S16(b) and (c)†). The detail of the geometrical structure of [Au26Pd(SPhtBu)20]0 is expected to be elucidated by SC-XRD or DFT calculations in a future study.
 | ||
| Fig. 7 UV-vis optical absorption spectrum of product iii ([Au26Pd(SPhtBu)20]0). | ||
In this way, we have obtained [Au26Pd(SPhtBu)20]0 by the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH. The obtained [Au26Pd(SPhtBu)20]0 showed a high stability in toluene solution at 25 °C (Fig. S18†). However, [Au26Pd(SPhtBu)20]0 was not synthesized by the co-reduction of Au and Pd salts in the presence of tBuPhSH. These results indicate that it is extremely important to use a ligand-exchange reaction to synthesize [Au26Pd(SPhtBu)20]0.
We investigated the chemical composition, abundance distribution, and electronic structure of the products for this reaction system by MALDI-MS (Fig. S19†), RP-HPLC (Fig. 8 and Table S2†) and optical absorption spectroscopy (Fig. S20†). The following facts were revealed for the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH (Fig. S21†): (1) only the ligand-exchange reaction proceeds while the number of metal atoms, ligands and the electronic structure of [Au24Pd(SC2H4Ph)18]0 is maintained (Fig. S20†) until the number of exchanged ligands reaches 16, (2) [Au24Pd(SC2H4Ph)1(SPhtBu)17]0 with 17 exchanged ligands is scarcely obtained (Fig. 8 and S19†). As mentioned above, when the central atom of [Au25(SC2H4Ph)18]− was replaced by a Pt atom ([Au24Pt(SC2H4Ph)18]0), the framework of NC strengthens, and thereby [Au24Pt(SPhtBu)18]0, in which all ligands are exchanged with SPhtBu, was also obtained.35 Although the substitution of the central atom of Au25(SR)18 with a Pd atom improves the stability against decomposition in solution compared with Au25(SR)18,30 based on the interaction energy36 between the central atom (Au, Pd, or Pt) and the surrounding Au24(SR)18 structure, [Au24Pd(SC2H4Ph)18]0 is interpreted to have a framework that is not as strong as [Au24Pt(SC2H4Ph)18]0. The strongness of this structure is considered the reason why [Au24Pd(SC2H4Ph)18−x(SPhtBu)x]0 cannot maintain its framework structure when more than 16 SC2H4Ph are exchanged with SPhtBu, and changes its structure to [Au26Pd(SPhtBu)20]0, which can contain more SPhtBu (Scheme 3(a)), unlike [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0.
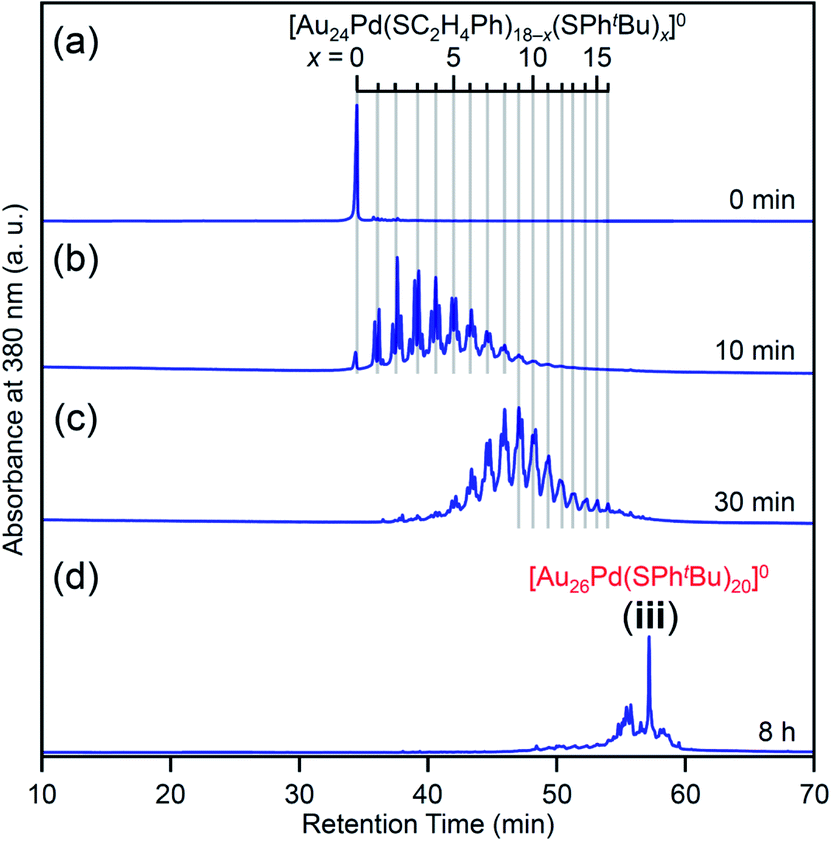 | ||
| Fig. 8 RP-HPLC chromatogram of products obtained by the reaction between [Au24Pd(SC2H4Ph)18]0 and tBuPhSH for (a) 0 min, (b) 10 min, (c) 30 min and (d) 8 h (iii). Ligand-exchange products eluted at longer retention times than [Au24Pd(SC2H4Ph)18]0 because the polarity of SPhtBu is lower than that of SC2H4Ph (Scheme S4†).54 The fine peak progression in each major peak is caused by the existence of topological isomers.55 | ||
 | ||
| Scheme 3 Schematic illustration of the transformation from (a) [Au24Pd(SC2H4Ph)18]0 to [Au26Pd(SPhtBu)20]0 (iii) and (b) [Au26Pd(SPhtBu)20]0 (iii) to [Au28(SPhtBu)20]0. | ||
We then left the obtained [Au26Pd(SPhtBu)20]0 in toluene solution at 60 °C for 14 h. As a result, [Au28(SPhtBu)20]0, which contains only Au as a metal element, was formed as the main product (Fig. S22†). Although the geometric structure of [Au28(SPhtBu)20]0 (Scheme S2(b)†) has been revealed to contain two central Au atoms,25 Au27Pd(SPhtBu)20 and Au26Pd2(SPhtBu)20, in which one or two central atoms was replaced by a Pd atom, respectively, were not produced in our experiments. In icosahedral structures, such as the metal core of [Au25(SC2H4Ph)18]− (Scheme S2(a)†), the bond length between the 12 surface atoms is extended by ∼5% compared with the distance between the central and surface atoms (Fig. S23(a)†). Thus, the central atom of the icosahedral structure is easily replaced by an atom with a smaller atomic radius.62 The metal core of [Au28(SPhtBu)20]0 has a geometric structure in which two cubic octahedra are connected. In this structure, no large difference exists between the bond lengths of the central–surface atoms and those of the surface–surface atoms (Fig. S23(b)†). For such a geometric structure, formation of a core–shell structure with Pd as the core and Au as the shell typically dose not occur. Although the chemical composition of the final product is affected by the reaction path,63,64 this geometric factor appears to be largely related to the fact that Au27Pd(SPhtBu)20 and Au26Pd2(SPhtBu)20 were not formed in our experiments.
Reaction between [Au24Pt(SC2H4Ph)18]0 and tBuPhSH
For this reaction system, our previous studies have shown that [Au24Pt(SPhtBu)18]0, in which all ligands are replaced by SPhtBu, can be synthesized selectively after reaction for 10 h (Scheme 1(f)).35 In the present study, we found that a specific reaction intermediate ([Au24Pt(SC2H4Ph)7(SPhtBu)11]0) can be obtained in a high proportion (Fig. 9 and 10) when the ligand-exchange reaction was stopped at 2 h (Scheme 4). | ||
| Fig. 9 RP-HPLC chromatogram of products obtained by the reaction between [Au24Pt(SC2H4Ph)18]0 and tBuPhSH: (a) 0 min, (b) 10 min, (c) 45 min, (d) 2 h and (e) 8 h with that of (f) [Au24Pt(SPhtBu)18]0 (Fig. S24†). Ligand-exchange products eluted at longer retention times than [Au24Pt(SC2H4Ph)18]0 because the polarity of SPhtBu is lower than that of SC2H4Ph.54 Fine peak progression in each major peak is caused by the existence of topological isomers.55 The retention time of [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–18, Table S3†) becomes irregular around x = 11, which shows the specificity of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0. | ||
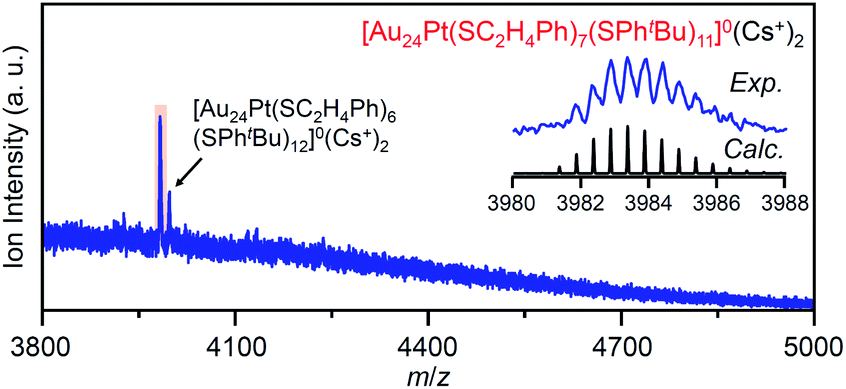 | ||
| Fig. 10 Positive-ion ESI mass spectra of main products (v) obtained by the reaction between [Au24Pt(SC2H4Ph)18]0 and tBuPhSH for 2 h. The inset compares the isotope pattern between the experimental and simulated spectra. In this sample, [Au24Pt(SC2H4Ph)6(SPhtBu)12]0 was also included as a minor product. | ||
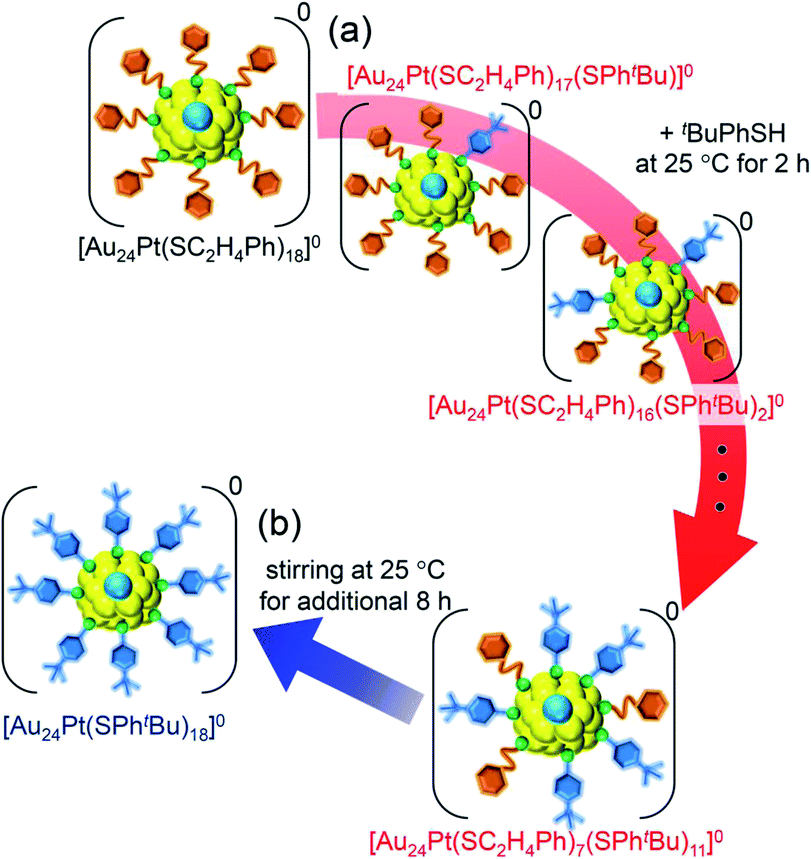 | ||
| Scheme 4 Schematic illustration of the transformation from (a) [Au24Pt(SC2H4Ph)18]0 to [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (v) and (b) [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (v) to [Au24Pt(SPhtBu)18]0. | ||
In the experiment, tBuPhSH was added to a toluene solution of [Au24Pt(SC2H4Ph)18]0 (Fig. S24†) at a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 250, and the solution was stirred at 25 °C. The chromatogram of the sample after 2 h of reaction showed a strong peak (v) at the retention time of 49.8 min (Fig. 9d and Table S3†). ESI-MS of the isolated product v revealed that product v had the chemical composition of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 (Fig. 10).
In this way, when the reaction between [Au24Pt(SPhtBu)18]0 and tBuPhSH is stopped in a short time, [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 can be obtained in a high proportion (Scheme 4(a)). It appears that [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 is thermodynamically stabilized because steric repulsion between surface ligands is lowest and/or the C–H⋯π interaction (Fig. S25†)35 is highest at this chemical composition, which leads to the formation of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 in a high proportion (Fig. S26†). In the reactions of [Au25(SC2H4Ph)18]− (Fig. 4 and S7†) or [Au24Pd(SC2H4Ph)18]0 (Fig. 8 and S17†) with tBuPhSH, [Au25(SC2H4Ph)7(SPhtBu)11]0 and [Au24Pd(SC2H4Ph)7(SPhtBu)11]0 were not synthesized in a high proportion. A comparison of these three NCs shows slight differences in the geometric structure (Fig. S27†)37,38,57 and the strength of the framework structure.36 It is assumed that these factors are related to the selective synthesis of NC with 11 exchanged ligands only in the case of the reaction between [Au24Pt(SPhtBu)18−x(SPhtBu)x]0 and tBuPhSH.
The specificity of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 was also observed in the optical absorption spectrum. Fig. 11 shows the optical absorption spectra of each [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–18) separated by RP-HPLC. The peak near 600 nm is attributed to a transition from the highest occupied molecular orbital to the lowest unoccupied molecular orbital (LUMO)+1.65 Because LUMO+1 involves orbitals that are derived from sulfur in addition to metal core orbitals,66 a replacement of the ligand in [Au24Pt(SC2H4Ph)18]0 with SPhtBu ([Au24Pt(SPhtBu)18]0) shifts this peak to the longer wavelength side.35Fig. 11 shows that (1) these shifts occur continuously with the number of exchanged ligands up to x = 10, (2) the above-mentioned peaks become less apparent when x reaches 11 and (3) the peaks appear again at x ≥ 13, and then shift continuously to the longer wavelength side with the number of exchanged ligands. This fact also implies that [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 is a special NC in a series of [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–18). The origin of this specificity of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 is expected to be elucidated by SC-XRD and DFT calculations in the future study.
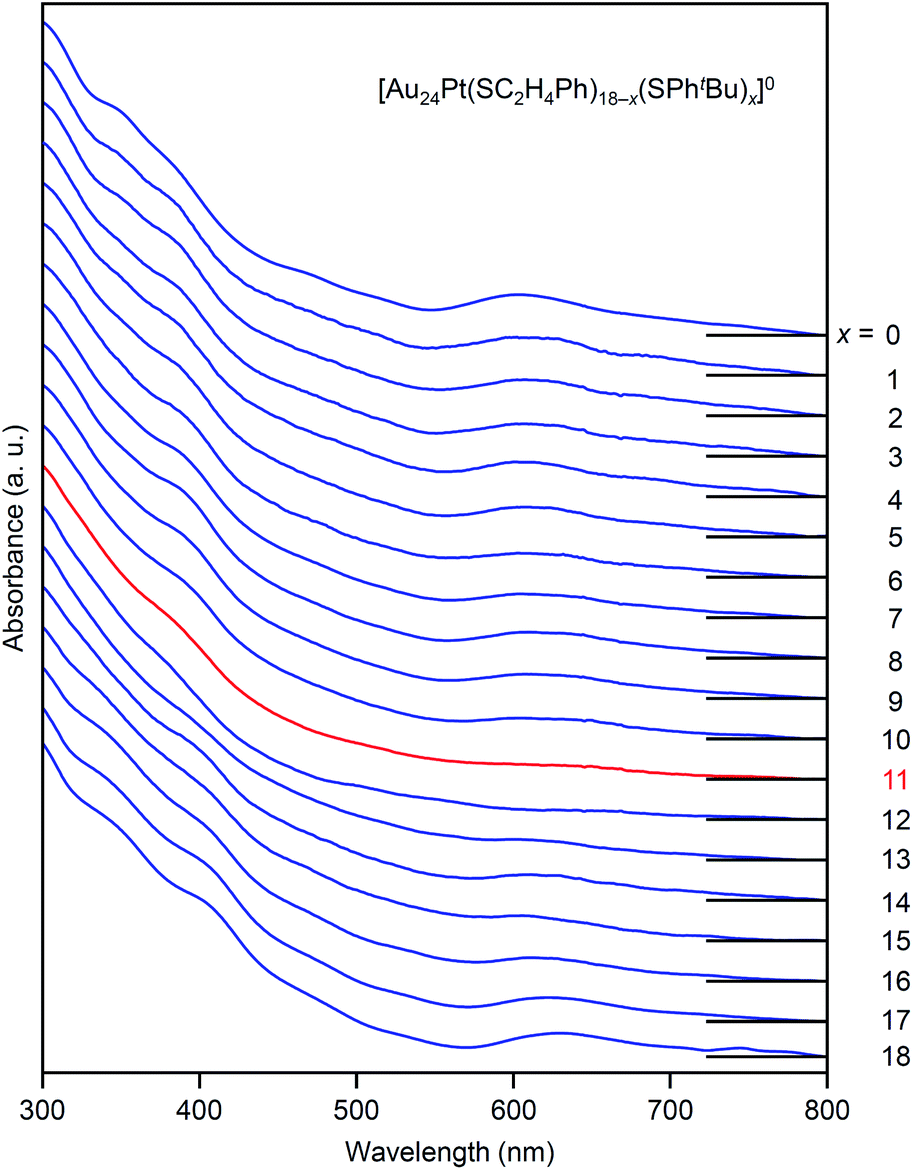 | ||
| Fig. 11 Comparison of UV-vis optical absorption spectra in a series of [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–18). These optical absorption spectra are not obtained for products with a distribution in chemical composition, which has often been reported in the literature,45 but are obtained for each chemical-composition product separated by RP-HPLC (Fig. 9). The spectral features disappear around x = 11 and then reappear at x ≥ 13, which shows the specificity of [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 in a series of [Au24Pt(SC2H4Ph)18−x(SPhtBu)x]0 (x = 0–18). | ||
Conclusions
In this study, the reaction of [Au25(SC2H4Ph)18]− with tBuPhSH was selected as a model reaction, and the amount of thiol to be reacted, the central atom of the precursor NCs, or the reaction time were changed from previous studies to create new atomically precise metal NCs. The following findings were obtained:(1) When tBuPhSH is added to a toluene solution in a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 100 and the reaction is allowed to proceed for 2 h at 40 °C, [Au23(SPhtBu)17]0 is obtained in a high proportion. In such a reaction, the framework of the product begins to distort when the number of exchanged ligands reaches 14. As the ligand exchange proceeds beyond this number, the product cannot maintain its framework structure, and its framework changes to [Au23(SPhtBu)17]0. When the obtained [Au23(SPhtBu)17]0 is reacted with tBuPhSH in toluene solution at 80 °C, [Au23(SPhtBu)17]0 changes its framework to the previously reported [Au20(SPhtBu)16]0 and [Au28(SPhtBu)20]0.
(2) When the central Au atom of [Au25(SC2H4Ph)18]− is replaced with a Pd atom ([Au24Pd(SC2H4Ph)18]0), tBuPhSH is added to the toluene solution in a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 250, and the reaction is allowed to proceed for 8 h at 25 °C, [Au26Pd(SPhtBu)20]0 is obtained in a high proportion. In such a reaction, only ligand exchange occurs with the framework structure retained until the number of the exchanged ligands reaches 16. However, as the ligand exchange proceeds beyond this number, the product is no longer able to maintain the framework, and its framework changes to [Au26Pd(SPhtBu)20]0.
(3) When the central Au atom of [Au25(SC2H4Ph)18]− is replaced with a Pt atom ([Au24Pt(SC2H4Ph)18]0), tBuPhSH is added to the toluene solution in a ratio of [tBuPhSH]/[SC2H4Ph] ≅ 250, and the reaction is allowed to proceed at 25 °C for only 2 h, [Au24Pt(SC2H4Ph)7(SPhtBu)11]0 is obtained in a high proportion.
(4) The differences in ligand-exchange reaction mechanisms of the above three reactions are caused mainly by the slight differences in framework strength (Fig. 12) and geometrical structure of the precursor metal NCs (Fig. S27†).
 | ||
| Fig. 12 Relation between the strength of the framework structure of the precursor NC and the number of changeable ligands. | ||
To the best of our knowledge, no report exists on the selective synthesis of these three metal NCs ([Au23(SPhtBu)17]0, [Au26Pd(SPhtBu)20]0 and [Au24Pt(SC2H4Ph)7(SPhtBu)11]0). These results indicate that the reaction pathway in LEIST67 and the chemical composition of the final products can be modified by controlling the amount of thiol, the type of central atom and the reaction time, which demonstrates that a variety of atomically precise metal NCs, which are synthesized size-selectively, can be increased compared with current NCs if the ligand-exchange reaction is conducted while changing the reaction conditions and/or the central atoms of the precursor metal NCs from previous studies. We have also elucidated the details of the reaction mechanism in the ligand-exchange reaction by using both MS and RP-HPLC, which was difficult using only MS often used in the literature. In future, it is expected that the details of the reaction mechanisms of various reactions that have not yet been clarified will be revealed by this combined use.
Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
Y. Negishi designed the experiments. H. Horihata. and A. Ebina conducted the measurements along with S. Miyajima, M. Nakamoto and A. Ikeda. S. Hossain and T. Kawawaki helped analyze the obtained results. Y. Negishi wrote the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ms. Kana Yoshida and Ms. Sayaka Hashimoto (Tokyo University of Science) for technical assistance in the early stage. This work was supported financially by MEXT/JSPS KAKENHI (grant number 20H02698, 20H02552 and 21J22219), Scientific Research on Innovative Areas “Coordination Asymmetry” (grant number 19H04595), “Innovations for Light-Energy Conversion” (grant number 20H05115), and “Hydrogenomics” (grant number 21H00027). Funding from Nissanken, the Yashima Environment Technology Foundation, the Yazaki Memorial Foundation for Science and Technology, TEPCO Memorial Foundation Research Grant (Basic Research), the Precise Measurement Technology Promotion Foundation, the MIKIYA Science and Technology Foundation and the Ogasawara Foundation is gratefully acknowledged.References
- T. Kawawaki, Y. Negishi and H. Kawasaki, Nanoscale Adv., 2020, 2, 17–36 RSC.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- X. Cai, W. Hu, S. Xu, D. Yang, M. Chen, M. Shu, R. Si, W. Ding and Y. Zhu, J. Am. Chem. Soc., 2020, 142, 4141–4153 CrossRef CAS PubMed.
- S. Pollitt, V. Truttmann, T. Haunold, C. Garcia, W. Olszewski, J. Llorca, N. Barrabés and G. Rupprechter, ACS Catal., 2020, 10, 6144–6148 CrossRef CAS PubMed.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- M. H. Naveen, R. Khan and J. H. Bang, Chem. Mater., 2021, 33, 7595–7612 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- Y. Li, O. Zaluzhna, B. Xu, Y. Gao, J. M. Modest and Y. J. Tong, J. Am. Chem. Soc., 2011, 133, 2092–2095 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed.
- M. S. Bootharaju, C. P. Joshi, M. J. Alhilaly and O. M. Bakr, Chem. Mater., 2016, 28, 3292–3297 CrossRef CAS.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- M. Rambukwella and A. Dass, Langmuir, 2017, 33, 10958–10964 CrossRef CAS PubMed.
- C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- C. Zeng, C. Liu, Y. Chen, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2014, 136, 11922–11925 CrossRef CAS PubMed.
- A. George, A. Sundar, A. S. Nair, M. P. Maman, B. Pathak, N. Ramanan and S. Mandal, J. Phys. Chem. Lett., 2019, 10, 4571–4576 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem. –Eur. J., 2020, 26, 16150–16193 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- X. Kang, L. Xiong, S. Wang, Y. Pei and M. Zhu, Inorg. Chem., 2018, 57, 335–342 CrossRef CAS PubMed.
- Q. Li, T.-Y. Luo, M. G. Taylor, S. Wang, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- H. Qian, D.-e. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS PubMed.
- S. Hossain, Y. Imai, D. Suzuki, W. Choi, Z. Chen, T. Suzuki, M. Yoshioka, T. Kawawaki, D. Lee and Y. Negishi, Nanoscale, 2019, 11, 22089–22098 RSC.
- D.-e. Jiang and S. Dai, Inorg. Chem., 2009, 48, 2720–2722 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS PubMed.
- V. L. Jimenez, M. C. Leopold, C. Mazzitelli, J. W. Jorgenson and R. W. Murray, Anal. Chem., 2003, 75, 199–206 CrossRef CAS PubMed.
- D. M. Black, N. Bhattarai, S. B. H. Bach and R. L. Whetten, J. Phys. Chem. Lett., 2016, 7, 3199–3205 CrossRef CAS PubMed.
- S. Knoppe and P. Vogt, Anal. Chem., 2019, 91, 1603–1609 CrossRef CAS PubMed.
- J. B. Tracy, M. C. Crowe, J. F. Parker, O. Hampe, C. A. Fields-Zinna, A. Dass and R. W. Murray, J. Am. Chem. Soc., 2007, 129, 16209–16215 CrossRef CAS PubMed.
- S. Mukherjee, D. Jayakumar and S. Mandal, J. Phys. Chem. C, 2021, 125, 12149–12154 CrossRef CAS.
- C. Zeng, C. Liu, Y. Pei and R. Jin, ACS Nano, 2013, 7, 6138–6145 CrossRef CAS PubMed.
- D.-e. Jiang, M. Kühn, Q. Tang and F. Weigend, J. Phys. Chem. Lett., 2014, 5, 3286–3289 CrossRef CAS PubMed.
- M. Waszkielewicz, J. Olesiak-Banska, C. Comby-Zerbino, F. Bertorelle, X. Dagany, A. K. Bansal, M. T. Sajjad, I. D. W. Samuel, Z. Sanader, M. Rozycka, M. Wojtas, K. Matczyszyn, V. Bonacic-Koutecky, R. Antoine, A. Ozyhar and M. Samoc, Nanoscale, 2018, 10, 11335–11341 RSC.
- C. L. Heinecke, T. W. Ni, S. Malola, V. Mäkinen, O. A. Wong, H. Häkkinen and C. J. Ackerson, J. Am. Chem. Soc., 2012, 134, 13316–13322 CrossRef CAS PubMed.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- C. M. Aikens, J. Phys. Chem. Lett., 2010, 1, 2594–2599 CrossRef CAS.
- Q. Tang, R. Ouyang, Z. Tian and D.-e. Jiang, Nanoscale, 2015, 7, 2225–2229 RSC.
- A. Tlahuice-Flores, R. L. Whetten and M. Jose-Yacaman, J. Phys. Chem. C, 2013, 117, 20867–20875 CrossRef CAS.
- M. P. Maman, A. S. Nair, H. Cheraparambil, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 1781–1788 CrossRef CAS PubMed.
- Y. Niihori, M. Matsuzaki, C. Uchida and Y. Negishi, Nanoscale, 2014, 6, 7889–7896 RSC.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- S. Tian, L. Liao, J. Yuan, C. Yao, J. Chen, J. Yang and Z. Wu, Chem. Commun., 2016, 52, 9873–9876 RSC.
- M. A. Tofanelli, T. W. Ni, B. D. Phillips and C. J. Ackerson, Inorg. Chem., 2016, 55, 999–1001 CrossRef CAS PubMed.
- K. A. Kacprzak, L. Lehtovaara, J. Akola, O. Lopez-Acevedo and H. Häkkinen, Phys. Chem. Chem. Phys., 2009, 11, 7123–7129 RSC.
- M. Laupp and J. Strähle, Angew. Chem., Int. Ed. Engl., 1994, 33, 207–209 CrossRef.
- E. Ito, S. Takano, T. Nakamura and T. Tsukuda, Angew. Chem., Int. Ed., 2021, 60, 645–649 CrossRef CAS PubMed.
- M. Akutsu, K. Koyasu, J. Atobe, N. Hosoya, K. Miyajima, M. Mitsui and A. Nakajima, J. Phys. Chem. A, 2006, 110, 12073–12076 CrossRef CAS PubMed.
- J. Peng, P. Wang, B. Wang, L. Xiong, H. Liu, Y. Pei and X. C. Zeng, Small, 2021, 17, 2000627 CrossRef CAS PubMed.
- M. P. Maman, A. S. Nair, A. M. Abdul Hakkim Nazeeja, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 10052–10059 CrossRef CAS PubMed.
- K. Kwak, Q. Tang, M. Kim, D.-e. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- M. Zhou, H. Qian, M. Y. Sfeir, K. Nobusada and R. Jin, Nanoscale, 2016, 8, 7163–7171 RSC.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experiments, table of retention time of each NC in RP-HPLC, additional mass spectra, chromatogram, analysis of geometrical structures of the precursor NCs ([Au25(SC2H4Ph)18]−, [Au24Pd(SC2H4Ph)18]0 and [Au24Pt(SC2H4Ph)18]0). See DOI: 10.1039/d2sc00423b |
| This journal is © The Royal Society of Chemistry 2022 |