
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective synthesis of α-amino ketones through palladium-catalyzed asymmetric arylation of α-keto imines†
Wei
Wen‡
 *,
Zhao-Pin
Ai‡
,
Chang-Lin
Yang
,
Chao-Xing
Li
,
Zhu-Lian
Wu
*,
Tian
Cai
and
Qi-Xiang
Guo
*
*,
Zhao-Pin
Ai‡
,
Chang-Lin
Yang
,
Chao-Xing
Li
,
Zhu-Lian
Wu
*,
Tian
Cai
and
Qi-Xiang
Guo
*
Chongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: wenwei1989@swu.edu.cn; wzl12@swu.edu.cn; qxguo@swu.edu.cn
First published on 7th March 2022
Abstract
Chiral α-amino ketones are common structural motifs in natural products and pharmaceuticals, as well as important synthons in organic synthesis. Thus, establishing efficient methods for preparing compounds with these privileged scaffolds is an important endeavor in synthetic chemistry. Herein we disclose a new catalytic asymmetric approach for the synthesis of chiral α-amino ketones through a chiral palladium-catalyzed arylation reaction of in situ generated challenging α-keto imines from previously unreported C-acyl N-sulfonyl-N,O-aminals, with arylboronic acids. The current reaction offers a straightforward approach to the asymmetric synthesis of acyclic α-amino ketones in a practical and highly stereocontrolled manner. Meanwhile, the multiple roles of the chiral Pd(II) complex catalyst in the reaction were also reported.
Introduction
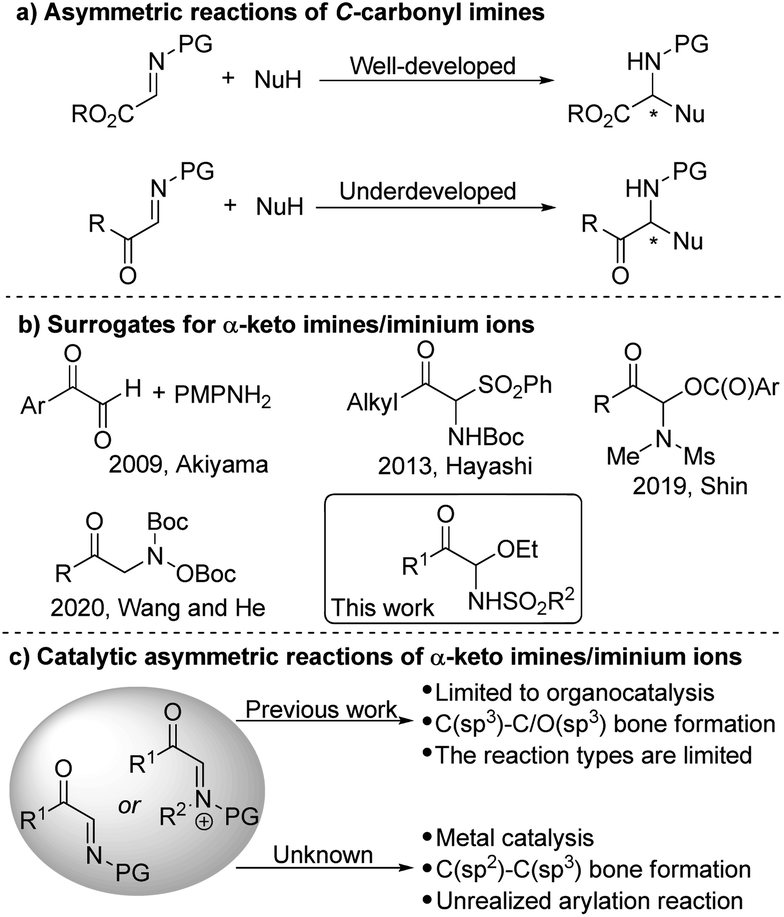
α-Amino ketones are substructures present in numerous important bioactive molecules, natural products and pharmaceuticals.1 More importantly, α-amino ketones are useful intermediates and versatile building blocks for the synthesis of polyfunctional amino derivatives due to the rich chemistry of the carbonyl groups.2 For example, they can be reduced into α-amino alcohols or oxidized into α-amino acids, and new carbon–carbon or carbon–hetero bonds can be formed by addition of various nucleophiles to the ketone groups. The design and development of new asymmetric methods and strategies for the preparation of chiral α-amino ketones is thus a critical objective in chemical synthesis. In the past decades, a few catalytic enantioselective approaches have been devised for this purpose.3–8 Such as the asymmetric electrophilic amination of ketones or enolates,4 the chiral carbene-catalyzed cross coupling of aldehydes with imines,5 the asymmetric catalyzed N–H insertion of amines with α-diazoketones or α-keto sulfonium ylides,6 the asymmetric reduction of α-keto ketimines,7 through colorful rearrangement reactions to construct enantioenriched α-amino ketones,8 and so on.9 Herein we propose a completely different approach to address these challenges.C-Carbonyl imines/iminium ions are versatile reagents and intermediates in organic synthesis.10,11 The adjacent carbonyl group not only makes the imine more reactive for electrophilic reaction, but also can undergo beneficial transformations. The metal- or organocatalytic asymmetric addition of many different types of nucleophiles to α-ester imines to synthesize various α-amino acid derivatives and heterocyclic compounds has been studied extensively (Scheme 1a).10 In contrast, so far, the application of α-keto imines/iminium ions in catalytic asymmetric synthesis is still very few.11 Recently, several organocatalytic asymmetric reactions of α-keto imines/iminium ions have been developed,12 but metal-catalyzed asymmetric conversion has not yet been achieved.
 | ||
| Scheme 1 Catalytic asymmetric reactions of C-carbonyl imines/iminium ions. | ||
One of the important reasons is that it is restricted due to the difficulty in the synthesis of α-keto imines. The classic method of preparing α-keto imines involves the condensation of glyoxal derivatives and anilines (Scheme 1b).12a However, the N-aryl α-keto imines prepared by this method were sensitive to moisture and leads to instability, which hinders its application in synthesis. In 2013, Hayashi and co-workers reported an α-N-Boc sulfone ketone derivative as a precursor of α-keto imine, but an excess of base is required.12b In addition, the potential of α-keto imines in catalytic asymmetric synthesis has not been fully exploited, the scope of applicable nucleophiles and types of chiral catalyst systems are still rather limited to date (Scheme 1c). Thus, we are interested in developing the general method for generating active α-keto imines and studying the application of α-keto imines in catalytic asymmetric synthesis.
Transition-metal-catalyzed asymmetric additions of arylboron to electron-deficient bonds are well-known for their efficient construction of C–C bonds with the simultaneous generation of stereocenters with high optical purity.13–16 Additions to imines in particular represent a powerful method for the synthesis of chiral amines.14–16 Herein, we reported C-acyl N-sulfonyl-N,O-aminals as α-keto imines surrogates. And the resulting C-acyl N-sulfonyl-N,O-aminals undergone enantioselective addition with arylboronic acids catalyzed by palladium to synthesize acyclic α-amino ketones with good yields and enantioselectivities.17
Results and discussion
Reaction condition optimization
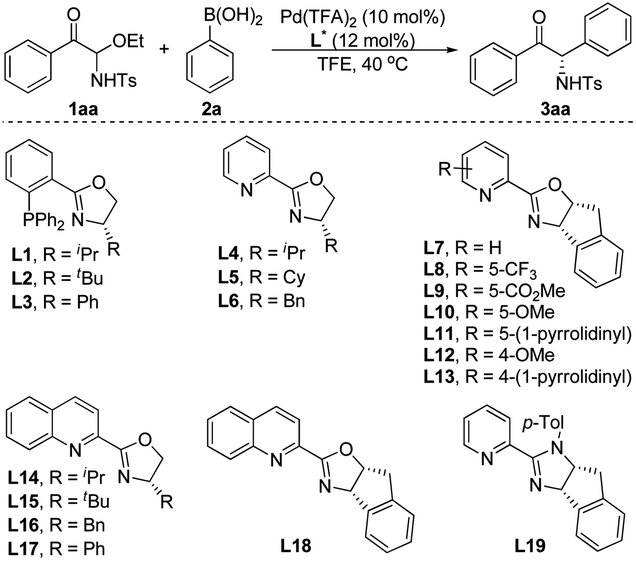
We began our study with the synthesis of active α-keto imine precursor. In the presence of tetraethoxysilane, phenylglyoxal and TsNH2 condensed to form C-acyl N-Ts N,O-aminal 1aa, and the resulting C-acyl N-Ts N,O-aminals could be recrystallized and are bench-stable for months. We envisioned that these new C-acyl N-Ts N,O-aminals could generate α-keto imines in situ and be used in asymmetric catalytic reactions to synthesize α-amino ketones and derivatives. To validate our hypothesis, we used the C-acyl N-Ts N,O-aminal 1aa as the standard substrate, and investigated the reaction conditions of the palladium-catalyzed enantioselective addition of phenylboronic acid 2a to the α-keto imines (Table 1). Phosphine–oxazoline L1 was chosen as the initial ligand and formed a complex with Pd(TFA)2 to catalyze the reaction. The reaction proceeded as expected, leading to the corresponding α-amino ketone 3aa in low yield and good ee at 40 °C using TFE as the solvent (Table 1, entry 1). Subsequently, different chiral phosphine–oxazoline ligands were screened (entries 2 and 3), but the yield of 3aa has not improved. To our delight, when pyridine–oxazoline L4 was used as the ligand, the yield was greatly improved and high enantioselectivity was obtained (entry 4). Subsequently, we studied the catalytic effects of a series of pyridine–oxazoline type ligands (entries 5–18). Most of the other ligands with substituents on oxazoline rings or pyridine rings can achieve good enantioselectivities and moderate yields. To our surprise, L6 gave racemate 3aa (entry 6). When the pyridine ring in the ligand was replaced by a quinoline ring, the enantioselectivity of 3aa was greatly reduced (entries 14–17). Interestingly, compared with the other pyridine–oxazoline ligands L4–L14, the absolute configuration of 3aa was reversed when using ligand L15 or L18 (entries 15 and 18). Another type of ligand, L19, was also tested but gave poor results (entry 19). Considering the yield and enantioselectivity of the reaction, ligand L10 was selected as the optimal ligand (entry 10).|
|
||||
|---|---|---|---|---|
| Entry | Ligand (L*) | t/h | Yieldb (%) | eec (%) |
| a Reaction conditions: 1aa (0.10 mmol), 2a (0.15 mmol), Pd(TFA)2 (10 mol%) and ligand (12 mol%) in TFE (0.5 mL), carried out in air at 40 °C. b Isolated yield. c Determined by chiral HPLC. n. d. = not determined. TFA = trifluoroacetic acetate, TFE = 2,2,2-trifluoroethanol. | ||||
| 1 | L1 | 48 | 10 | 82 |
| 2 | L2 | 48 | n. d. | n. d. |
| 3 | L3 | 48 | 3 | 73 |
| 4 | L4 | 23 | 52 | 89 |
| 5 | L5 | 17 | 53 | 90 |
| 6 | L6 | 8 | 43 | 0 |
| 7 | L7 | 17 | 59 | 93 |
| 8 | L8 | 12 | 48 | 88 |
| 9 | L9 | 7 | 50 | 96 |
| 10 | L10 | 24 | 68 | 92 |
| 11 | L11 | 24 | 53 | 89 |
| 12 | L12 | 24 | 28 | 92 |
| 13 | L13 | 25 | 38 | 90 |
| 14 | L14 | 24 | 78 | 12 |
| 15 | L15 | 48 | 38 | −59 |
| 16 | L16 | 24 | 72 | 6 |
| 17 | L17 | 48 | 58 | 1 |
| 18 | L18 | 24 | 55 | −38 |
| 19 | L19 | 10 | 39 | 74 |
The palladium catalysts, solvents and additives were screened using L10 as the ligand, but the highest yield of product 3aa was only 68%, when the enantioselectivity persisted above 90% ee (see the ESI† for details). We speculated that the poor yield of product 3aa was due to that the acyclic N-Ts α-keto imines intermediate formed in situ was easy to decompose. The bulky SO2R group would improve the stability of the α-keto imines active intermediate. Therefore, different nitrogen protecting groups (SO2R) of C-acyl N,O-aminals 1 were investigated employing Pd(TFA)2 as the palladium source and L10 as the chiral ligand (Scheme 2). The catalytic results demonstrated that substrates 1ad and 1ae were not stable under the reaction conditions, and when R = 2,4,6-trimethylphenyl, the best results were provided, and the target product 3a with 78% yield and 92% ee was obtained.
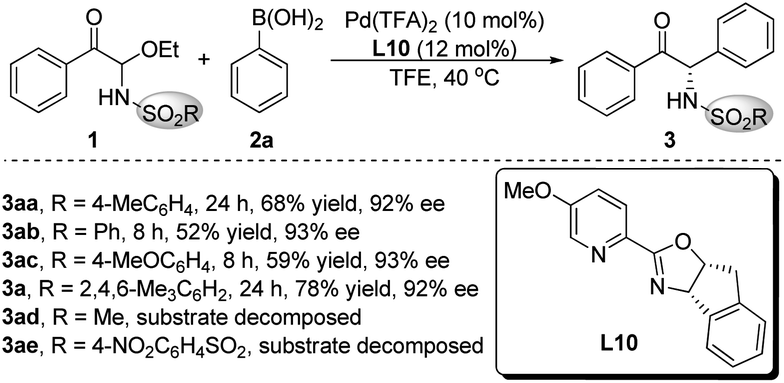 | ||
| Scheme 2 Effect of nitrogen protecting groups. | ||
Substrate scope of α-keto imines and arylboronic acids
With the optimized conditions in hand, we evaluated the substrate scope and generality. As shown in Table 2, different electron-withdrawing and electron-donating groups at different positions on the aromatic ring of the substrates 1 all offered high enantioselectivities (3b–k). However, when the para-position was substituted by N-piperidinyl, the yield of the corresponding product 3j was significantly reduced. In addition, a C-acyl N,O-aminal derived from tert-butylglyoxal was also examined, and the desired product was obtained with good yield and enantioselectivity (3l).| a Reaction conditions: 1 (0.20 mmol), 2a (0.30 mmol), Pd(TFA)2 (10 mol%) and L10 (12 mol%) in TFE (1.0 mL), carried out in air at 40 °C. Mes = 2,4,6-trimethylphenyl. |
|---|

|
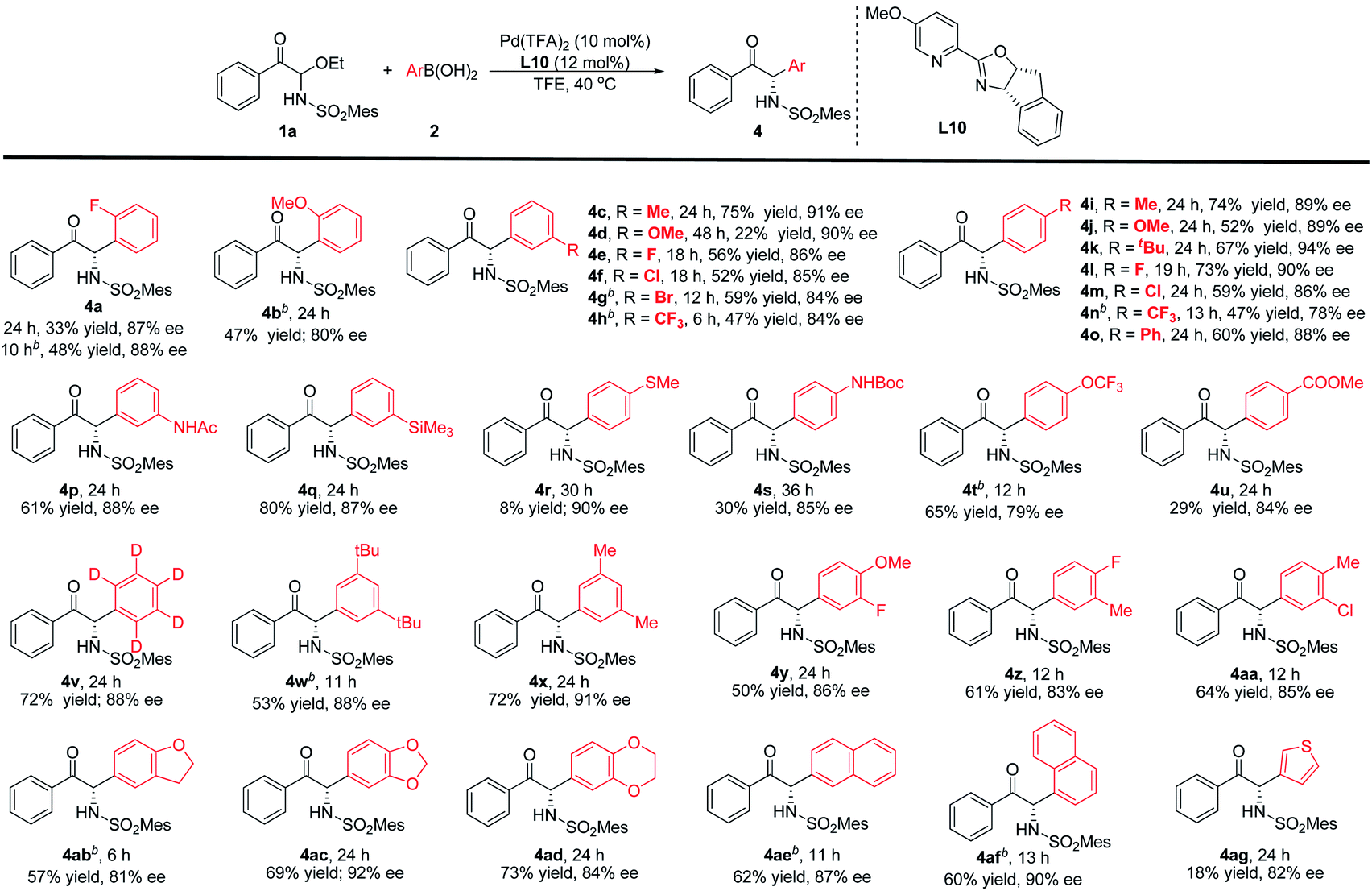
The arylboronic acid scope was also experimented (Table 3). The results suggest that reactivity could be influenced unfavourably by steric effects. For example, the ortho-position fluorine and methoxy groups greatly reduced the reactivity. Nevertheless, increasing the temperature can obtain products with moderate yields and good enantioselectivities (4a-b). The meta- and para-substituted phenylboronic acids have also been investigated. The reaction conditions were compatible with –Me, –OMe, –CF3, –tBu, –Ph and halogens, corresponding products could be obtained with moderate to good yields and good enantioselectivities (4c–o). In comparison with the arylboronic acids bearing electron-donating aryl groups, the arylboronic acids with electron-withdrawing substituent provided a slightly lower enantioselectivity. Functional group compatibility was further investigated, such as –NHPG, –TMS, –OCF3 and –CO2Me were also suitable (4p–q, 4s–u), and the corresponding arylboronic acid reacts with 1a to provide the target products with 29–80% yields and 79–88% ee. But for the 4-(methylthio)phenylboronic acid, only a small amount of the product was observed with 90% ee (4r). When phenyl-D5-boronic acid was used in this reaction, the product can be obtained with 72% yield and 88% ee (4v). Disubstituted aryl, polycyclic aryl or fused-ring arylboronic acids as nucleophiles could also be used in this reaction to attain high yields and enantioselectivities (4w–af). In addition, we also considered a heterocyclic boronic acid (4ag). 3-Thiopheneboronic acid could lead to good enantioselectivity, but the reactivity was low.
The functional group compatibility and broad substrate scope of this method can also be demonstrated by the reaction of substrate 1a with arylboronic acids with biologically active moieties. As shown in Scheme 3, when estrone- or Zetia-derived arylboronic acids were employed, the products could be obtained with good yields and diastereoselectivities. It was worth noting that when L10 or its enantiomer ent-L10 was used as ligand, diastereomeric products were produced respectively, indicating that the stereoselectivity was determined by the chiral catalyst rather than the substrates. In consequence, the system had good functional group compatibility and could be used for the late-stage modification of complex molecules.
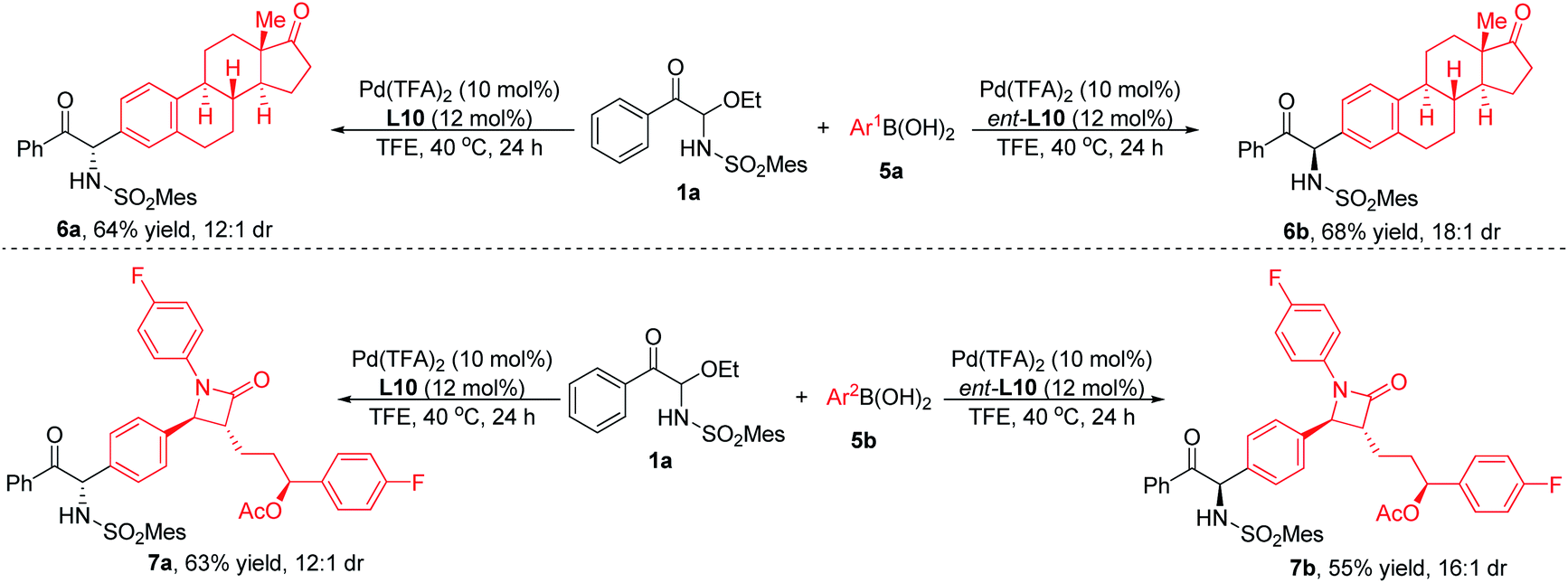 | ||
| Scheme 3 The reaction of 1a with arylboronic acids bearing biologically active moieties. | ||
Gram-scale reaction and transformations of products
A gram–scale reaction using substrate 1a and phenylboronic acid 2a was carried out, the yield and enantioselectivity of product 3a did not decrease (Scheme 4). Moreover, compound 3a could be reduced by NaBH4 to obtain 8 without a reduction in ee value. After removal of the MesSO2– group, the important 2-amino-1,2-diphenylethanol 9 in asymmetric synthesis was obtained.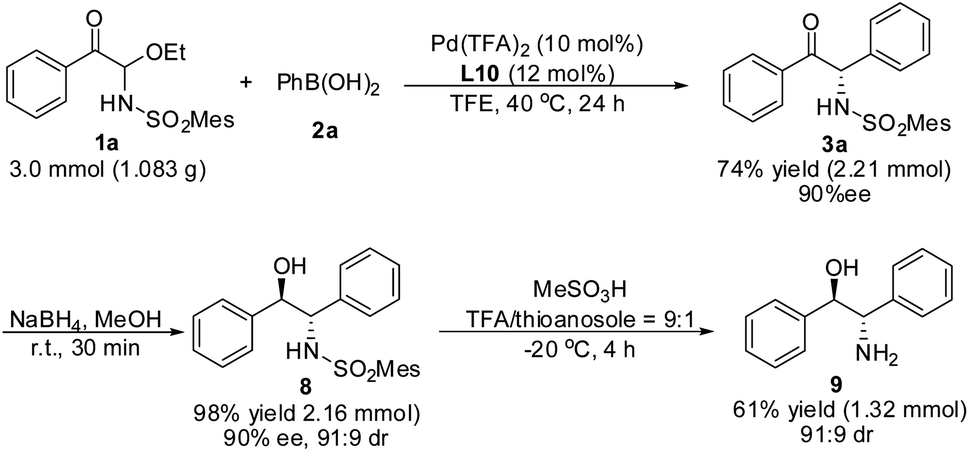 | ||
| Scheme 4 Gram-scale synthesis of (1R, 2S)-2-amino-1,2-diphenylethanol 9. | ||
Mechanism studies
To investigate the nature of C-acyl N,O-aminals as new imine precursors in situ generation of α-keto imines, the following experiments were conducted. First, we designed an experiment to capture the highly reactive α-keto imine intermediate by H2O and monitored it with 1H-NMR (Fig. 1). In the coexistence system of 1a and Pd(TFA)2, the generated phenylglyoxal after the hydrolysis reaction of α-keto imine intermediate and H2O was monitored (Fig. 1a). In the absence of Pd(TFA)2, only trace amounts of phenylglyoxal were generated (Fig. 1b). We also monitored the intermediates by in situ HRMS (Fig. 2). In the presence of Pd(TFA)2 and ligand, the α-keto imines and the compound by 1a exchanged with CF3CH2OH were monitored (Fig. 2b). These experimental results indicated that palladium effectively promoted the production of α-keto imines, and the elimination of EtOH was reversible.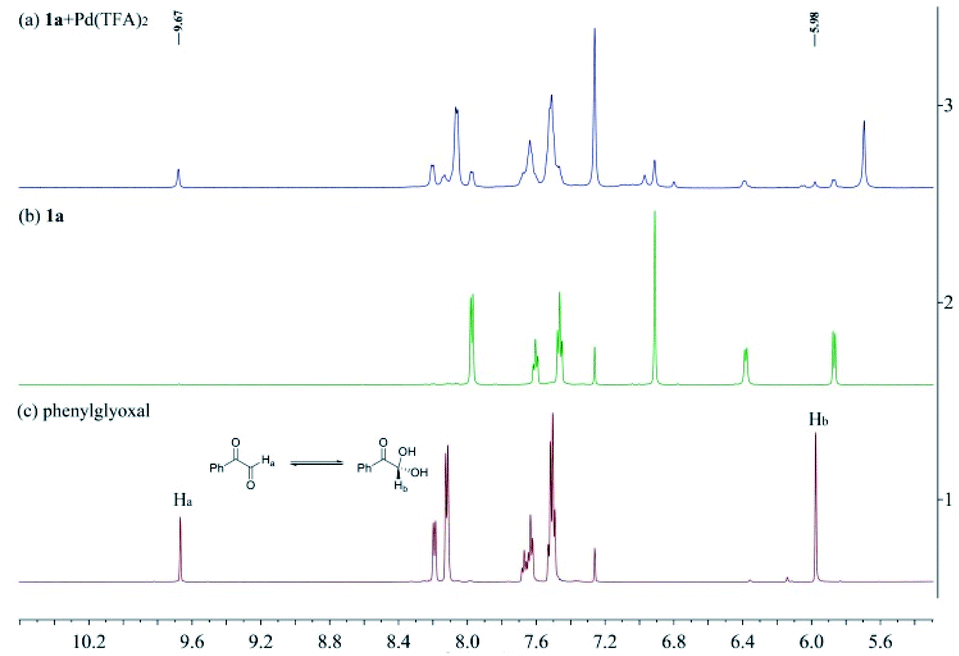 | ||
| Fig. 1 (a) 1H-NMR spectrum of 1a (0.02 mmol) and Pd(TFA)2 (0.02 mmol) in CDCl3 (0.5 mL) after stirring for 12 hours at 40 °C. (b) 1H-NMR spectrum of 1a (0.02 mmol) in CDCl3 (0.5 mL) after stirring for 12 hours at 40 °C. (c) 1H-NMR spectrum of phenylglyoxal. | ||
 | ||
| Fig. 2 (a) HRMS spectrum of 1a (0.1 mmol) in CF3CH2OH (0.5 mL) after stirring for 4 hours at 40 °C. (b) HRMS spectrum of 1a (0.1 mmol), Pd(TFA)2 (10 mol%) and L10 (12 mol%) in CF3CH2OH (0.5 mL) after stirring for 4 hours at 40 °C. | ||
Since racemic N,O-aminals 1 were employed as the precursor of the α-keto imines, we questioned whether potential kinetic resolution occurred in Pd-catalyzed arylation reactions. Accordingly, we monitored the reaction of phenylboronic acid 2a and rac-1a over different reaction times, and determined yields and enantiomeric ratios for both the generated product 3a and the recovered substrate 1a (Fig. 3). We found that the ee of the recovered starting material 1a gradually increased with the progress of the reaction. After 8 h, 1a was recovered in 10% yield and reached 48% ee (Fig. 3a). At the same time, product 3a could always be obtained with high enantioselectivity at all stages of the reaction (Fig. 3b). The results indicated that (R)-1a and (S)-1a were consumed together and the amount of one of the enantiomers decreased more rapidly. The kinetic resolution process also shed some light on the Pd catalyst and N,O-aminal substrate interactions in this catalytic system. Remarkably, the unfavorable enantiomer could still be fully converted to the products after longer reaction times. Hence, this chiral Pd-catalyzed arylation reaction could be achieved in high yield and enantioselectivity via dynamic kinetic asymmetric transformation (DyKAT).
 | ||
| Fig. 3 (a) Recovered yields and ee values of 1a at different reaction times. (b) Yields and ee values of 3a at different reaction times. | ||
Based on the above experimental results and inferred mechanism of palladium-catalyzed arylation of imines,15 a catalytic cycle was proposed (Scheme 5). First, the palladium complex F acted as a Lewis acid to promote the formation α-keto imines Ain situ from racemic C-acyl N,O-aminals 1. Simultaneously, α-keto imines A could be converted into N,O-aminals 1 and B again by the addition of ethanol and 2,2,2-trifluoroethanol, and 1 and B could also form intermediate A again under the action of Pd(II). Afterwards, α-keto imines A was coordinated with palladium Ar–Pd complex C to form intermediate D. Complex C was formed by the metal transfer of palladium complex F and arylboronic acid. Intermediate D allowed insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond into the Pd–C bond to form E. The ligand exchange of intermediate E afforded the addition product and regenerated the active Pd(II) complex F.
N bond into the Pd–C bond to form E. The ligand exchange of intermediate E afforded the addition product and regenerated the active Pd(II) complex F.
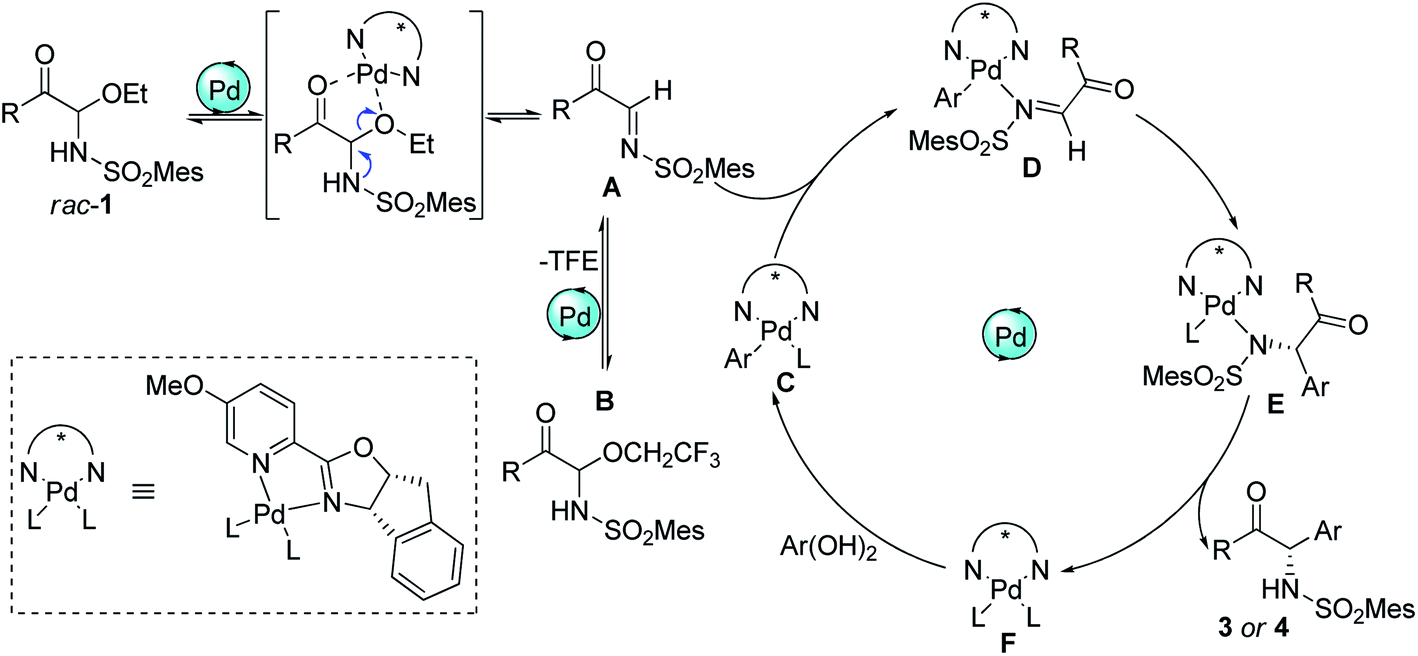 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed an efficient method for the synthesis of chiral α-amino ketones through the Pd-catalyzed asymmetric arylation of α-keto imines. The chiral Pd(II) complex catalyst played multiple roles in the reaction: (1) cooperatively activating C-acyl N,O-aminal electrophile to generate α-keto imines active intermediates, and in the process undergoes kinetic resolution of racemic 1. (2) By synergistically activating α-keto imine and arylboronic acid, the subsequent enantioselective insertion of the CN bond into Pd–C bond was promoted. The investigation of the potential applications of these C-acyl N,O-aminals as new imine precursors in other asymmetric transformations are underway in our laboratory.
Data availability
Data for this work, including optimization tables, general experimental procedures and characterization data for all new compounds are provided in the ESI.†Author contributions
W. W. and Q.-X. G. conceived and directed the project. W. W. and Z.-P. A. carried out the experiments. C.-L. Y. and C.-X. L. participated in the preparation of substrates. Z.-L. W. and T. C. performed the HRMS analysis. W. W. and Z.-L. W. wrote the paper. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Fundamental Research Funds for the Central Universities (SWU119059 and XDJK2020C034) and the Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0278).Notes and references
- (a) K. F. Foley and N. V. Cozzi, Drug Dev. Res., 2003, 60, 252–260 CrossRef CAS; (b) L.-Z. Lin, G. A. Cordell, C.-Z. Ni and J. Clardy, Phytochemistry, 1991, 30, 1311–1315 CrossRef CAS; (c) M. Kitajima, N. Kogure, K. Yamaguchi, H. Takayama and N. Aimi, Org. Lett., 2003, 5, 2075–2078 CrossRef CAS PubMed; (d) P. C. Meltzer, D. Butler, J. R. Deschamps and B. K. Madras, J. Med. Chem., 2006, 49, 1420–1432 CrossRef CAS PubMed; (e) M. C. Myers, J. Wang, J. A. Iera, J. Bang, T. Hara, S. Saito, G. P. Zambetti and D. H. Appella, J. Am. Chem. Soc., 2005, 127, 6152–6153 CrossRef CAS.
- (a) F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed; (b) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–875 CrossRef CAS PubMed.
- L. A. T. Allen, R.-C. Raclea, P. Natho and P. J. Parsons, Org. Biomol. Chem., 2021, 19, 498–513 RSC.
- (a) A. de la Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef CAS PubMed; (b) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5360–5361 CrossRef CAS PubMed; (c) M. Anada, M. Tanaka, T. Washio, M. Yamawaki, T. Abe and S. Hashimoto, Org. Lett., 2007, 9, 4559–4562 CrossRef CAS PubMed; (d) T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed; (e) X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS PubMed; (f) W. Li, X. Liu, X. Hao, X. Hu, Y. Chu, W. Cao, S. Qin, C. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2011, 133, 15268–15271 CrossRef CAS PubMed; (g) P. Phukan and A. Sudalai, Tetrahedron: Asymmetry, 1998, 9, 1001–1005 CrossRef CAS; (h) J.-L. Liang, X.-Q. Yu and C.-M. Che, Chem. Commun., 2002, 124–125 RSC.
- (a) S. M. Mennen, J. D. Gipson, Y. R. Kim and S. J. Miller, J. Am. Chem. Soc., 2005, 127, 1654–1655 CrossRef CAS PubMed; (b) D. A. DiRocco and T. Rovis, Angew. Chem., Int. Ed., 2012, 51, 5904–5906 CrossRef CAS PubMed.
- (a) B. Xu, S.-F. Zhu, X.-D. Zuo, Z.-C. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913–3916 CrossRef CAS PubMed; (b) W. Guo, Y. Luo, H. H.-Y. Sung, I. D. Williams, P. Li and J. Sun, J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed.
- (a) W. Wen, Y. Zeng, L.-Y. Peng, L.-N. Fu and Q.-X. Guo, Org. Lett., 2015, 17, 3922–3925 CrossRef CAS PubMed; (b) L. Lin, X. Bai, X. Ye, X. Zhao, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2017, 56, 13842–13846 CrossRef CAS PubMed; (c) T. Sun, G. Hou, M. Ma and X. Zhang, Adv. Synth. Catal., 2011, 353, 253–256 CrossRef CAS; (d) L. Liu, Y. Zheng, X. Hu, C. Lian, W. Yuan and X. Zhang, Chem. Res. Chin. Univ., 2014, 30, 235–241 CrossRef CAS.
- (a) X. Zhang, R. J. Staples, A. L Rheingold and W. D. Wulf, J. Am. Chem. Soc., 2014, 136, 13971–13974 CrossRef CAS PubMed; (b) T. Ooi, M. Takahashi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2002, 124, 7640–7641 CrossRef CAS PubMed; (c) A. Frongia, F. Secci, F. Capitta, P. P. Piras and M. L. Sanna, Chem. Commun., 2013, 49, 8812–8814 RSC; (d) Y. Chen, Y. Liu, Z. Li, S. Dong, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052–8056 CrossRef CAS PubMed.
- (a) X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed; (b) J. Wu, C. Zhao and J. Wang, J. Am. Chem. Soc., 2016, 138, 4706–4709 CrossRef CAS PubMed; (c) M. R. Garrett, J. C. Tarr and J. S. Johnson, J. Am. Chem. Soc., 2007, 129, 12944–12945 CrossRef CAS PubMed; (d) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636–12642 CrossRef CAS PubMed.
- (a) B. Eftekhari-Sis and M. Zirak, Chem. Rev., 2017, 117, 8326–8419 CrossRef CAS; (b) A. E. Taggi, A. M. Hafez and T. Lectka, Acc. Chem. Res., 2003, 36, 10–19 CrossRef CAS PubMed.
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed.
- (a) T. Akiyama, T. Suzuki and K. Mori, Org. Lett., 2009, 11, 2445–2447 CrossRef CAS PubMed; (b) Y. Hayashi, D. Sakamoto, H. Shomura and D. Hashizume, Chem. - Eur. J., 2013, 19, 7678–7681 CrossRef CAS PubMed; (c) N. H. Nguyen, Q. H. Nguyen, S. Biswas, D. V. Patil and S. Shin, Org. Lett., 2019, 21, 9009–9013 CrossRef CAS PubMed; (d) H. Xu, A. Nazli, C. Zou, Z.-P. Wang and Y. He, Chem. Commun., 2020, 56, 14243–14246 RSC.
- For reviews, see: (a) M. Quan, L. Wu, G. Yang and W. Zhang, Chem. Commun., 2018, 54, 10394–10404 RSC; (b) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS; (c) C. S. Marques and A. J. Burke, ChemCatChem, 2011, 3, 635–645 CrossRef CAS; (d) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (e) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093–2105 RSC; (f) A. Gutnov, Eur. J. Org. Chem., 2008, 4547–4554 CrossRef CAS; (g) N. Miyaura, Synlett, 2009, 2039–2050 CAS.
- For selected examples of Rh-catalyzed asymmetric addition of arylboron reagents to imines, see: (a) N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584–13585 CrossRef CAS PubMed; (b) M. Kuriyama, T. Soeta, X. Hao, Q. Chen and K. Tomioka, J. Am. Chem. Soc., 2004, 126, 8128–8129 CrossRef CAS PubMed; (c) R. B. C. Jagt, P. Y. Toullec, D. Geerdink, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Angew. Chem., Int. Ed., 2006, 45, 2789–2791 CrossRef CAS PubMed; (d) M. Trincado and J. A. Ellman, Angew. Chem., Int. Ed., 2008, 47, 5623–5626 CrossRef CAS PubMed; (e) R. Shintani, M. Takeda, T. Tsuji and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 13168–13169 CrossRef CAS PubMed; (f) T. Nishimura, A. Noishiki, G. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056–5059 CrossRef CAS PubMed; (g) T. Nishimura, A. Noishiki, Y. Ebe and T. Hayashi, Angew. Chem., Int. Ed., 2013, 52, 1777–1780 CrossRef CAS PubMed; (h) Z.-Q. Wang, C.-G. Feng, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336–5337 CrossRef CAS PubMed; (i) Z. Cui, H.-J. Yu, R.-F. Yang, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394–12397 CrossRef CAS PubMed; (j) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971–974 CrossRef CAS PubMed; (k) Y. Li, Y.-N. Yu and M.-H. Xu, ACS Catal., 2016, 6, 661–665 CrossRef CAS.
- For selected examples of Pd-catalyzed asymmetric addition of arylboron reagents to imines, see: (a) H. Dai, M. Yang and X. Lu, Adv. Synth. Catal., 2008, 350, 249–253 CrossRef CAS; (b) H. Dai and X. Lu, Tetrahedron Lett., 2009, 50, 3478–3481 CrossRef CAS; (c) G.-N. Ma, T. Zhang and M. Shi, Org. Lett., 2009, 11, 875–878 CrossRef CAS PubMed; (d) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540–7544 CrossRef CAS PubMed; (e) M. Quan, G. Yang, F. Xie, I. D. Gridnev and W. Zhang, Org. Chem. Front., 2015, 2, 398–402 RSC; (f) Q. He, L. Wu, X. Kou, N. Butt, G. Yang and W. Zhang, Org. Lett., 2016, 18, 288–291 CrossRef CAS PubMed; (g) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 53, 9936–9939 CrossRef CAS PubMed; (h) X. Gao, B. Wu, Z. Yan and Y.-G. Zhou, Org. Biomol. Chem., 2016, 14, 55–58 RSC; (i) Z. Yan, B. Wu, X. Gao and Y.-G. Zhou, Chem. Commun., 2016, 52, 10882–10885 RSC; (j) Z.-B. Zhao, L. Shi, F.-J. Meng, Y. Lia and Y.-G. Zhou, Org. Chem. Front., 2019, 6, 1572–1576 RSC; (k) J. Chen, X. Lu, W. Lou, Y. Ye, H. Jiang and W. Zeng, J. Org. Chem., 2012, 77, 8541–8548 CrossRef CAS PubMed; (l) T. Johnson and M. Lautens, Org. Lett., 2013, 15, 4043–4045 CrossRef CAS PubMed; (m) C. Schrapel and R. Peters, Angew. Chem., Int. Ed., 2015, 54, 10289–10293 CrossRef CAS PubMed; (n) T. Beisel and G. Manolikakes, Org. Lett., 2015, 17, 3162–3165 CrossRef CAS PubMed.
- For selected examples of Other Metal-catalyzed asymmetric addition of arylboron reagents to imines, see: (a) C. Wu, X. Qin, A. M. P. Moeljadi, H. Hirao and J. S. Zhou, Angew. Chem., Int. Ed., 2019, 58, 2705–2709 CrossRef CAS PubMed; (b) M. Quan, L. Tang, J. Shen, G. Yang and W. Zhang, Chem. Commun., 2017, 53, 609–612 RSC; (c) W. Sun, H. Gu and X. Lin, J. Org. Chem., 2018, 83, 4034–4043 CrossRef CAS PubMed.
- During the preparation of this manuscript, Song, Yang and co-workers reported a Rh-catalyzed diastereoselective addition of arylboronic acids to α-keto N-tert-butanesulfinyl aldimines: C. Wang, L. Zhou, J. Qiu, K. Yang and Q. Song, Org. Chem. Front., 2022, 9, 1016 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00386d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |