
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemo- and regio-divergent access to fluorinated 1-alkyl and 1-acyl triazenes from alkynyl triazenes†
Jin-Fay
Tan
a,
Carl Thomas
Bormann
b,
Kay
Severin
 b and
Nicolai
Cramer
*a
b and
Nicolai
Cramer
*a
aLaboratory of Asymmetric Catalysis and Synthesis, EPFL SB ISIC LCSA, BCH 4305, CH-1015 Lausanne, Switzerland. E-mail: nicolai.cramer@epfl.ch
bLaboratory of Supramolecular Chemistry, EPFL SB ISIC LCS, BCH 3307, CH-1015 Lausanne, Switzerland
First published on 9th February 2022
Abstract
The 1,1,2,2-tetrafluoroethylene unit is prevalent in bioactive molecules and functional materials. Despite being in principle a straightforward strategy to access this motif, the direct tetrafluorination of alkynes involves very hazardous or inconvenient reagents. Therefore, safer and convenient alternatives are sought after. We developed a mild and operationally simple perfluorination method converting 1-alkynyl triazenes into 1,1,2,2-tetrafluoro alkyl triazenes, employing cheap and readily accessible reagents. Moreover, a judicious tuning of the reaction conditions enables access to α-difluoro triazenyl ketones. Complementary, electrophilic fluorination of alkynyl triazenes gives rise to the regioisomeric α-difluoro acyl triazenes. These three chemo- and regio-divergent protocols enable access to elusive fluorinated 1-alkyl and 1-acyl triazenes, thus expanding the chemical space for these unusual entities. Furthermore, several reaction intermediates and side products revealed insights on the reaction pathways that may be useful for further fluorination chemistry of alkynes.
Introduction
The incorporation of fluorine atoms or fluoroalkyl groups in organic molecules can alter various physical and chemical properties such as metabolic stabilities, lipophilicities, electron distributions, acid–base equilibria and pharmacological activities.1 These unique characteristics render fluorine-containing functionalities highly appreciated in the design and development of pharmaceuticals, agrochemical candidates and materials.2 As a consequence, the introductions or formations of fluorinated moieties have been a topic of intensive investigations.3 With respect to the fluoroalkyl functionality, 1,1,2,2-tetrafluoroethylene and difluoromethylene moieties are frequently encountered in bioactive molecules and advanced materials (Fig. 1).4 Well-established approaches allow for the transfer of difluoromethylene and (CF2)n (n > 2) groups.3a,5 In contrast, the chemistry of isolated 1,1,2,2-tetrafluoroethylene (–CF2CF2–) units6 is less common. It remains underdeveloped compared to the structurally similar but widely-employed trifluoromethyl (–CF3) and perfluoroethyl (–C2F5) groups. To date, there is only a handful of methods for the constructions of tetrafluoroethylene linkages, namely addition reactions across tetrafluoroethenes,7 the use of halotetrafluoroalkane as a CF2CF2 transfer group,8 exhaustive deoxyfluorination of 1,2-diketones using organosulfur reagents9 and lastly, direct fluorinations of internal alkynes.10 Among them, direct fluorinations of alkynes might be the most straightforward and attractive strategy, due to the prompt accessibility and prevalence of carbon–carbon triple bonds in organic molecules (Scheme 1a). However, the number of reports on this seemingly simple procedure is very limited. Moreover, the reported procedures primarily involve harsh conditions using hazardous or inconvenient reagents, such as fluorine gas,10a hydrogen fluoride,10b xenon difluoride10d and iodine monofluoride or bromine monofluoride.10c Additions of fluoride across different activated alkynes have been investigated.11 Most transformation resulted either in the transfer of a single fluoride or yielding fluoro alkenes. Therefore, a mild and practical fluorination procedure of alkynes would be a very valuable addition to a chemist's toolbox for the expedient synthesis of 1,1,2,2-tetrafluoroethylene units. | ||
| Fig. 1 Selected examples of functional molecules containing 1,1,2,2-tetrafluoroethylene and difluoromethylene linkages. | ||
 | ||
| Scheme 1 Fluorinative transformations of alkynes and 1-alkynyl triazenes. | ||
The triazene group12 is a highly useful functionality in a broad variety of molecules having applications in anticancer therapy,13 materials,14 total synthesis15 and solid phase synthesis.16 Triazenes with alkyl or acyl substituents at their N3 atom have been widely studied in medicinal chemistry and organic synthesis (Scheme 1b). Specifically, 3,3-dialkyl triazenes are among the most commonly synthesized triazene compounds due to their easy access via the corresponding dialkyl amines. Moreover, 3-methyl-substituted triazenyl units are essential for cytotoxicity and tumor inhibitory activities in anti-cancer drugs such as dacarbazine13b and temozolomide.13c 3-Acyl triazenes have been reported in numerous bioactive compounds,17 as synthetic precursors for aminyl radicals,18 acylating agents19 and chemo-dosimeters for cyanide.20 In contrast, there are much less reports dealing with the synthesis and use of 1-alkyl21 and 1-acyl triazenes.22 Therefore, a rapid and modular access to 1-alkyl and 1-acyl triazenes would open up opportunities for investigations of their underexplored chemical and biological properties. Over the past years, the use of 1-alkynyl triazenes in organic synthesis has been receiving a growing interest.12a,23,24 Leveraging on their ynamide-like reactivity profile,25 we herein report three sets of convenient protocols that allow the fully chemo- and regio-divergent formations of tetrafluoro-alkyl triazenes, difluoro-alkyl triazenes and α-difluoro acyl triazenes from 1-alkynyl triazenes (Scheme 1c). Such fluorinated 1-alkyl and 1-acyl triazenes expand the chemical space for medicinal chemistry studies involving triazene molecules. Furthermore, these transformations provide potentially useful mechanistic insights for related chemistry with other alkynes.
Results and discussion
We initiated our perfluorination studies by exposing 1-naphthyl-substituted alkynyl triazene 1a to various electrophilic halogen sources and fluorides (Table 1). Reaction of 1a with HF·py and 5,5-dimethyl-1,3-diiodo hydantoin (DIH) caused a strong decomposition of the starting material. However, formation of tetrafluoro alkyl triazene 2a in 7% yield was observed as well (entry 1). We postulated that formation of 2a arises from a double iodofluorination of 1a followed by a subsequent substitution of the two iodides by fluoride anions. Such formation of a tetrafluoroethylene unit from an alkynyl triazene is not reported and encouraged us to further optimize the transformation. Switching from HF·py to AgF as fluoride source significantly improved the yield of 2a to 38% while as well giving small amounts of α-difluoro triazenyl ketone 3a and 1,2-diketone 4a (entry 2). Both compounds possibly arise from a displacement of the iodides by a water molecule from remaining residual traces of water in the solvent. A screening of electrophilic halogen sources X+ did not provide an improvement in yield or selectivity, but additionally gave iodofluoro alkenyl triazene 5a, α-difluoro acyl triazene 6a and diiodo alkenyl triazene 7a (entries 3–5). In most instances, these products were present only in trace amounts. The use of I2 increased the yield of 5a, while IBr produced more compound 3a (entries 4 and 5).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X+ | F− | % Conv.b | 2a [%] | 3a [%] | 4a [%] | 5a [%] |
| a Conditions: 0.10 mmol 1a, 2.5 equiv. X+, 10.0 equiv. F−, 0.1 M in CHCl3, 23 °C for 2 h in the dark. b Conversion and yields determined by 1H-NMR with an internal standard. c 11% NMR yield of 6a. d 1.2 equiv. DIH, 4.0 equiv. DAST. e With ambient light. f 1.2 equiv. DIH, 2.0 equiv. DAST. | |||||||
| 1 | DIH | HF·py | 69 | 7 | — | — | — |
| 2 | DIH | AgF | 100 | 38 | 4 | 4 | — |
| 3 | NIS | AgF | 100 | 30 | 15 | 9 | <2 |
| 4c | I2 | AgF | 100 | 26 | 15 | 14 | <2 |
| 5 | IBr | AgF | 100 | 8 | 30 | 17 | <2 |
| 6 | DIH | TBAF | 100 | 0 | 0 | <2 | 44 |
| 7 | DIH | TASF | 100 | 0 | 0 | <2 | 31 |
| 8 | DIH | CsF | 100 | 0 | 0 | <2 | — |
| 9 | DIH | DAST | 100 | 92 | 0 | 0 | — |
| 10d | DIH | DAST | 100 | 87 | 0 | 6 | — |
| 11d,e | DIH | DAST | 100 | 43 | 31 | 10 | — |
| 12 | DIH | DAST | 100 | 0 | 83 | 16 | — |

With DIH as best electrophilic source X+, we proceeded to screen other fluorides. Tetrabutylammonium fluoride (TBAF) and tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) favored clearly the formation of 2a (entries 6 and 7), while CsF caused substrate decomposition (entry 8). Gratifyingly, the use of DAST delivered desired product 2a in 92% yield (entry 9). Decreasing the amount of DIH to 1.2 equivalents did not result in a major drop in yield, indicating that both iodine atoms on the hydantoin are consumed in the transformation. The amount of DAST was lowered to four equivalents without compromising the yield and selectivity for 2a (entry 10). However, any further decrease had a negative impact on the product selectivity. Interestingly, the absence of light is essential for selective and efficient formation of 2a (entry 11). Next, we investigated the possibility of shifting the equilibrium towards α-difluoro triazenyl ketone 3a formation. Since only two fluorine atoms are incorporated in 3a, reducing the amount of DAST would be a logical step. The optimal stoichiometry was indeed found to be two equivalents of DAST, giving 3a in 83% yield (entry 12).
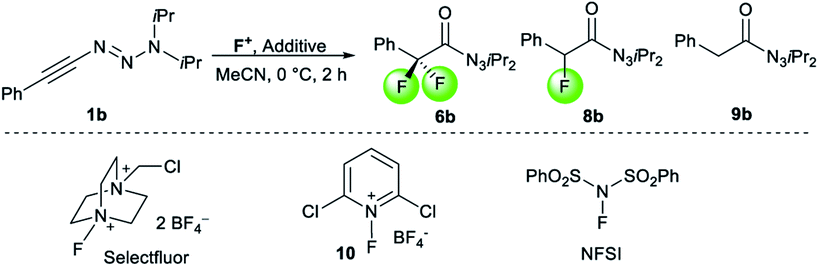
The observation of α-difluoro acyl triazene 6 prompted us to investigate its selective formation from 1. Due to the electronic properties and reactivity profile of alkynyl triazenes analogous to that of ynamides,25,26 we envisioned that an electrophilic fluorination might lead to 6 (Table 2). To investigate the selective formation of 6b, we initially exposed alkynyl triazene 1b to 2.5 equivalents of Selectfluor as an electrophilic fluorine source (F+) and water as an additive (entry 1). Full decomposition of starting material 1b occurred at room temperature. Reasoning that Selectfluor might act as an activator cleaving the triazene group, milder conditions were tested. Indeed, conducting the reaction at a lower temperature of 0 °C provided a mixture of acyl triazene 6b, mono fluoro acyl triazene 8b and the hydrated side product 9b (entry 2). An increased amount of Selectfluor did not improve the outcome (entry 3). Other electrophilic fluorine sources such as N-fluorobenzenesulfonimide (NFSI) or N-fluoropyridinium salt 10 resulted in inferior yields (entries 4 and 5). Notably, the presence of water was found to be essential for the formation of product 6 (entry 6). To suppress 8b formation, which possibly results from the competition of H+ with F+, we turned to more basic additives. Aqueous sodium hydroxide gave no improvement (entry 7). To our delight, a major improvement occurred with alkylammonium hydroxide hydrates, significantly increasing both yield and selectivity for 6b (entries 8–11). The optimal stoichiometry of the superior tetramethylammonium hydroxide was found to be 2.5 equivalents (entry 10), providing 6b in 85% yield. Finally, a methanolic solution of the hydroxide salt majorly caused decomposition, suggesting that methanol is not able to substitute water as a nucleophile (entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | F+ | Additive (equiv.) | 6b [%] | 8b [%] | 9b [%] |
| a Conditions: 0.1 mmol 1b, F+, additive, in 0.4 mL MeCN at 0 °C for 2 h; conversion and yields determined by 1H-NMR with an internal standard. b 23 °C. c 3.0 equiv. Selectfluor. d Reaction was conducted in the dark. e Isolated yield. | |||||
| 1b | Selectfluor | H2O (3) | 0 | 0 | 1 |
| 2 | Selectfluor | H2O (3) | 44 | 38 | 2 |
| 3c | Selectfluor | H2O (3) | 43 | 40 | 3 |
| 4d | NFSI | H2O (3) | 0 | 37 | 5 |
| 5 | 10 | H2O (3) | 0 | 0 | 6 |
| 6 | Selectfluor | — | 0 | 0 | 7 |
| 7 | Selectfluor | 2 M NaOH (3) | 45 | 36 | 9 |
| 8 | Selectfluor | (Bu4NOH)·30H2O (3) | 60 | 12 | 11 |
| 9 | Selectfluor | (Me4N)OH·5H2O (3) | 83 | 9 | 12 |
| 10 | Selectfluor | (Me 4 N)OH·5H 2 O (2.5) | 85 | 6 | 13 |
| 11 | Selectfluor | (Me4N)OH·5H2O (1.5) | 66 | 7 | 14 |
| 12 | Selectfluor | (Me4N)OH in MeOH (2.5) | <5 | 0 | 15 |
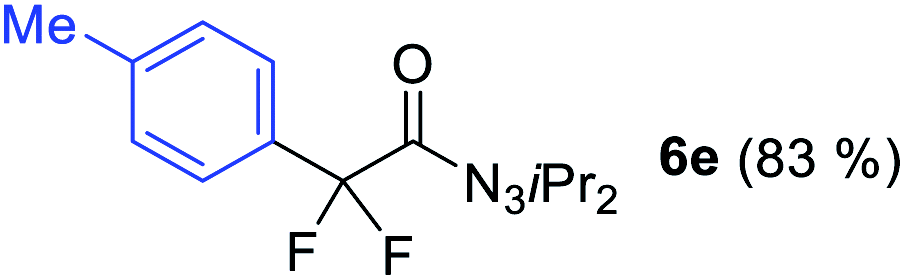
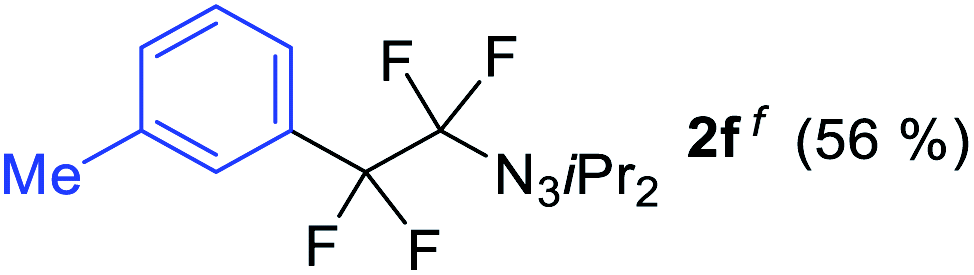
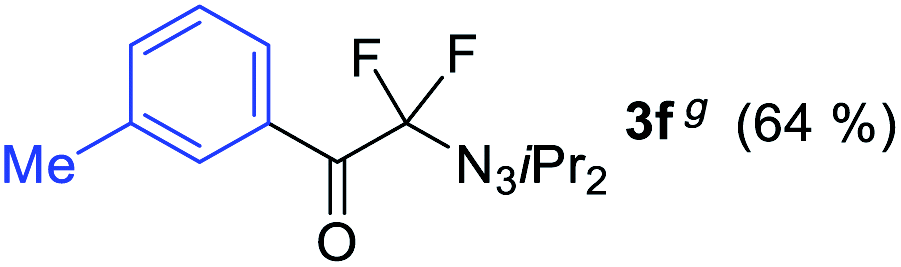
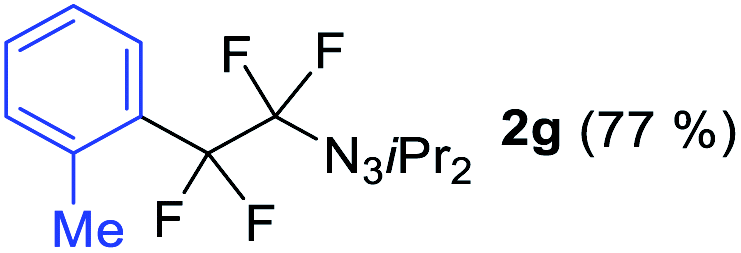
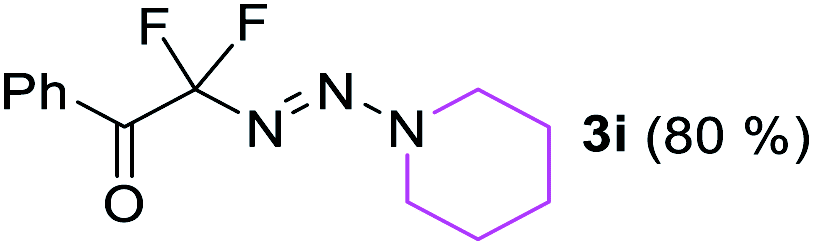
With the optimized three selective conditions (A, B and C), the substrate scope for the perfluorination and regio-divergent oxyfluorinations of 1-alkynyl triazenes was investigated (Table 3). The perfluorinative condition A reliably delivered 1,1,2,2-tetrafluorinated alkyl triazenes 2b–2g with electron-rich and electron-poor aryl groups substituents R in good to excellent yields (entries 2–7). The same substrate set underwent both regioselective oxyfluorinations delivering α-difluoro triazenyl ketones 3b–3g (condition B) and α-difluoro acyl triazenes 6b–6g (condition C) with moderate to excellent yields. The structures of 2a and 6e were unambiguously confirmed by X-ray crystallographic analyses (see ESI†).27 Noteworthy, the transformations also accommodate electron-rich heterocyclic substrates. Subjecting thienyl-substituted alkynyl triazene 1h to conditions A and B, a concomitant iodination at 2-position of the thiophene by DIH occurred forming 2h and 3h (entry 8). Condition C delivered expected thienyl product 6h. A piperidinyl group on the R′ of N3 (1i) was also well accepted (entry 9). Alkyl groups like cyclopropyl (1j), cyclopentyl (1k), tert-butyl (1l) and methoxymethyl (1m) all consistently afforded acyl triazenes 6j–6m in moderate to good yields (entries 10–13) under condition C. These substrates did not provide fluorinated products 2 and 3 under the conditions A or B, with the products being mono iodo acyl triazene 10 (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | 1 | 2 (Condition A)a | 3 (Condition B)b | 6 (Condition C)c |
| a Condition A: 0.1 mmol 1, 0.12 mmol DIH, 0.4 mmol DAST, 0.1 M in CHCl3 at 23 °C for 2 h in the dark. b Condition B: 0.1 mmol 1, 0.12 mmol DIH, 0.2 mmol DAST, 0.1 M in CHCl3 at 23 °C for 2 h in the dark. c Condition C: 0.1 mmol 1, 0.25 mmol Selectfluor, 0.25 mmol (Me4N)OH·5H2O, 0.25 M in MeCN at 0 °C for 2 h under ambient light. d With 5.0 equiv. DAST. e With 0.3 equiv. DAST. f With 10.0 equiv. DAST. g For 5 h. h With 2.4 equiv. DIH. | ||||
| 1 | 1a |
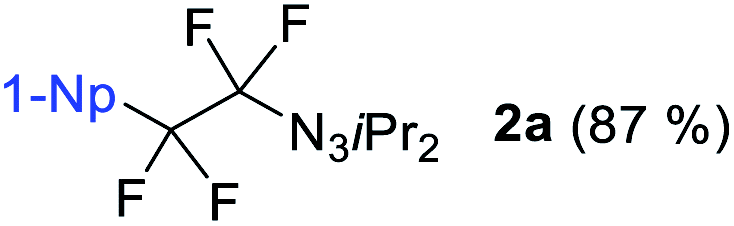
|
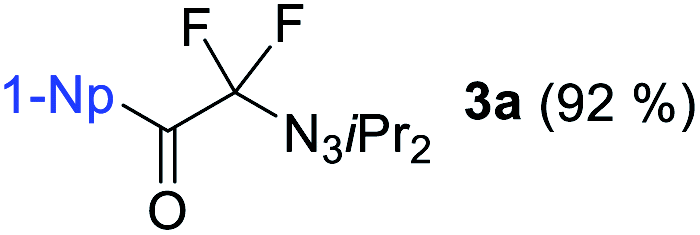
|
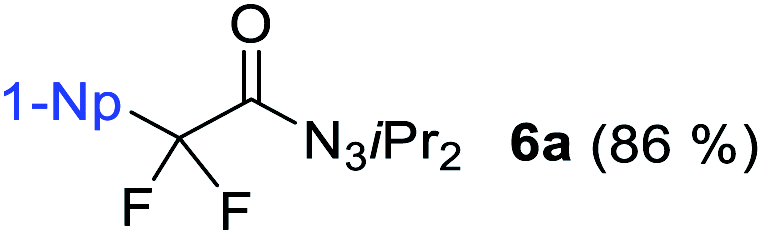
|
| 2 | 1b |
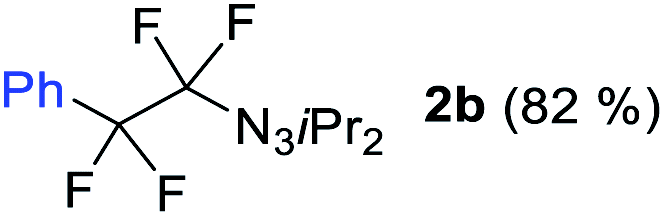
|

|
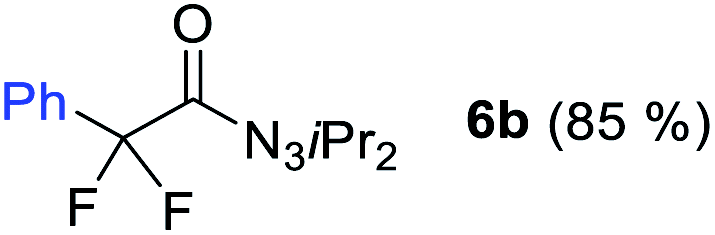
|
| 3 | 1c |
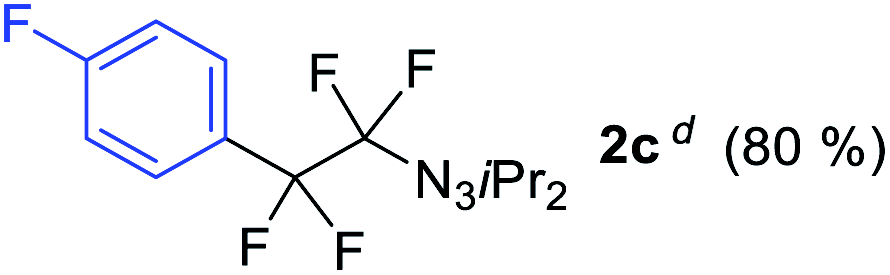
|
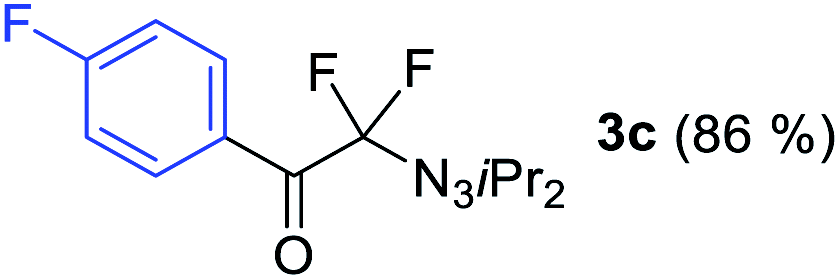
|
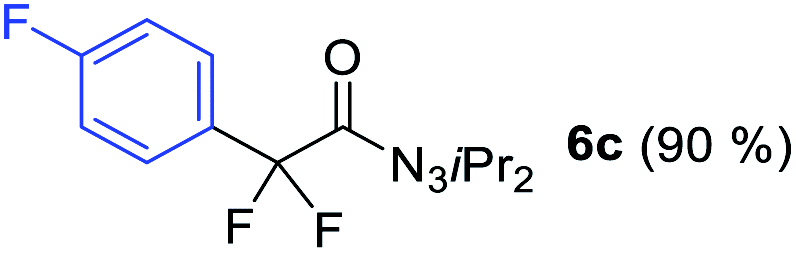
|
| 4 | 1d |
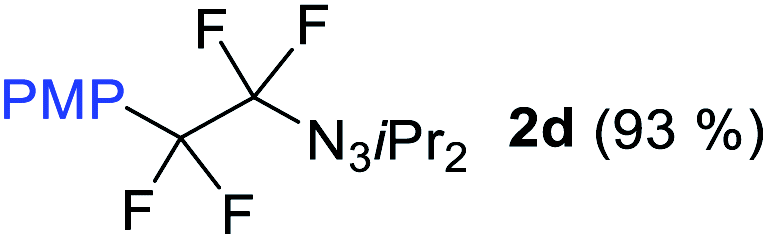
|
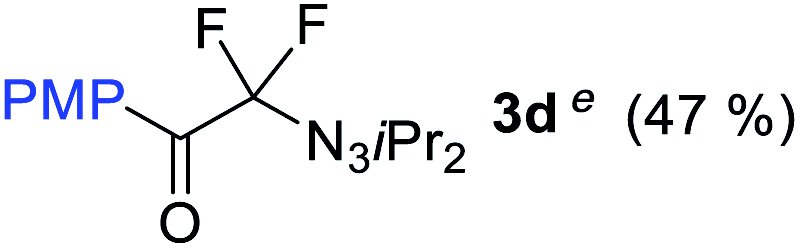
|
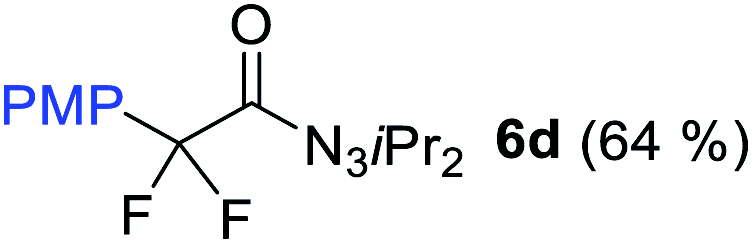
|
| 5 | 1e |

|

|
|
| 6 | 1f |
|
|

|
| 7 | 1g |
|
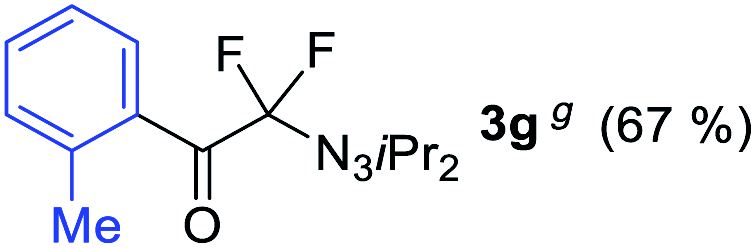
|
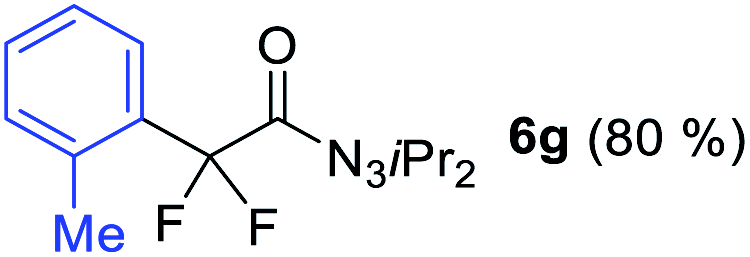
|
| 8 | 1h |

|

|
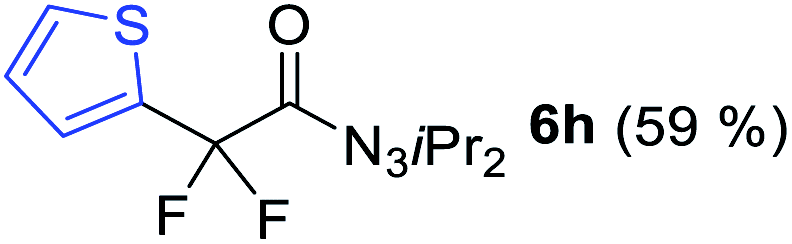
|
| 9 | 1i |
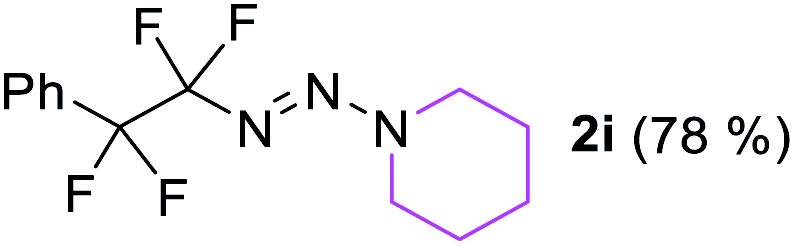
|
|
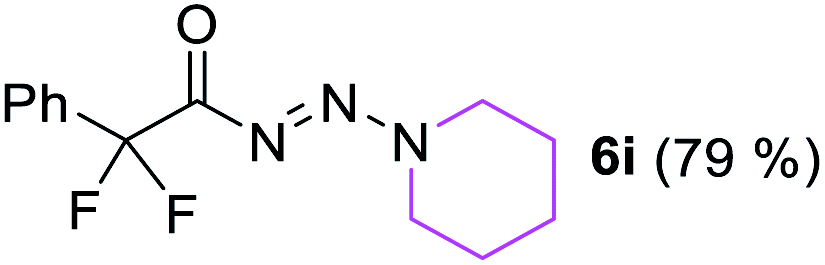
|
| 10 | 1j |

|

|
|
| 11 | 1k |
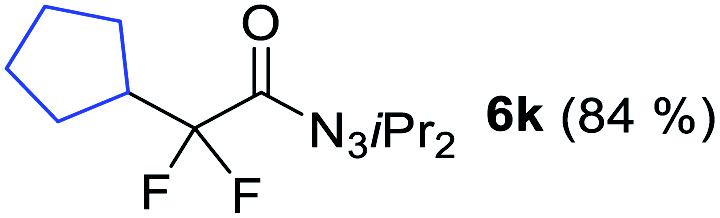
|
||
| 12 | 1l |

|
||
| 13 | 1m |

|
||

Acyl triazenes are behaving as activated carboxylic acid derivatives21a and could be synthetically leveraged as such. Indeed, exposure of naphthyl-substituted product 6a to BF3·OEt2 in methanol smoothly provided corresponding methyl ester 11a (Scheme 2). Notably, Lewis-acid activation in a suitable acceptor environment such as mesitylene as a solvent initiated a clean Friedel–Crafts acylation leading to ketone 12b with an excellent yield. Attempts to replace the triazene moiety of compounds 2 and 3 by other nucleophiles largely failed, supporting their unusual electronic nature (see as well Scheme 3).
 | ||
| Scheme 2 Functionalization of acyl triazene 6a. | ||
 | ||
| Scheme 3 Reactivity and mechanistic studies. | ||
The detection and isolation of fluoro iodo alkene 5a as well as diiodo alkene 7a during the optimization study raised questions regarding their roles in the reaction. To shed some light on the mechanism, we studied the behavior of these species. When compound 5a was subjected to the standard condition of generating 2a, productive full conversion was observed leading to 2a in 79% and trace amount of 3a (Scheme 3a). This result suggests that 5a acts as intermediate species during the conversion of 1-alkynyl triazene 1a to either 2a or 3a. Moreover, product 2a can be formed exposing 3a to DAST only (Scheme 3b). This corroborates the observations that excess DAST shifts the ratio towards formation of 2a over 3a. Next, we tested the formation of diiodo alkene 7a. By combining substrate 1a with 0.6 equivalents of DIH and 1.2 equivalents of tetrabutylammonium iodide, diiodo alkene 7a was formed in 92% yield (Scheme 3c). No product at all was observed in the absence of the iodide source, proving that iodide anions are required for formation of 7a. Isolated 7a was subsequently subjected to the fluorination condition with excess DIH. Even after prolonged reaction times, no fluorinated product was observed and the starting material was fully recovered (Scheme 3d). This result is evidence that 7a is not part of the productive reaction pathway. During the course of our functionalization attempts of products 2a and 3a, we found that hydrolysis products 6a and 4a were formed under strongly acidic conditions (Scheme 3e). This observation may be explained by α-oxydefluorinations of these α-difluoro triazenes, which appear analogous to α-difluoro amino derivatives. α-Oxydefluorinations of α-difluoro amino derivatives under acidic conditions into amides are well-reported phenomena.28 Noteworthy, theses fluorinated triazenes display a much higher stability compared to acid-lability of normal 1-alkyl triazenes.29
Taking these studies into account, we propose a plausible pathway for the formations of fluorinated products 2, 3 and 6 (Scheme 4). The first iodofluorination of 1-alkynyl triazene 1 by an electrophilic iodine source and a fluoride anion leads to fluoro iodo alkenyl triazene 5, which is stable enough for isolation. A second iodofluorination could deliver transient diiodo difluoro intermediate 13. It is a rather unstable species depending on the conditions rapidly collapsing into either 2 or 3. When excess fluoride anions are present, the two iodides on 13 get displaced by fluorides furnishing tetrafluorinated alkyl triazene 2. In a low-fluoride environment, the iodides are instead substituted by the oxygen atom of a molecule of water from residual moisture traces present in the solvent. In combination with an electrophilic iodine source, the liberated iodides could react with 1 to yield diiodo alkene 7, a side product that does not react further under the conditions. Essentially, the formation of 7 leads to a dead end compromising the yield of 2 and 3. Lastly, the α-oxydefluorinations of 2 and 3 accounts for the observations of 6 and 4 respectively.
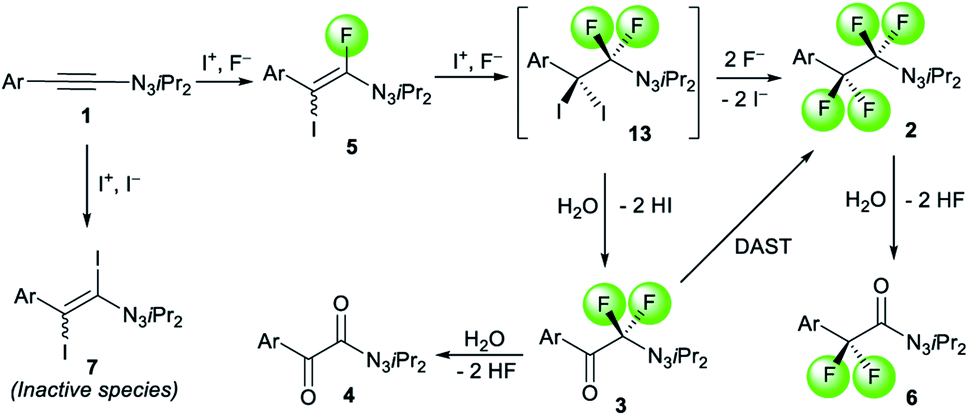 | ||
| Scheme 4 Suggested pathway map of the formation of fluorinated products and intermediates. | ||
Conclusions
In summary, we have developed a mild and operationally simple tetrafluorination procedure of alkynyl triazenes employing cheap and readily accessible reagents. The transformation provides access to alkyl triazenes containing the valuable 1,1,2,2-tetrafluoro ethylene motif. Moreover, a judicious tuning of the reaction conditions promotes an oxydifluorination pathway leading to the selective formation of α-difluoro triazenyl ketones. Complementary, an electrophilic fluorination of the alkynyl triazenes using Selectfluor enables access to α-difluoro acyl triazenes, which can be subsequently elaborated into α-difluoro esters or α-difluoro ketones. These three distinct fluorinative transformations represent a valuable addition to a chemist's toolbox for the expedient synthesis of the 1,1,2,2-tetrafluoroethylene and the α-difluoro carbonyl motifs.Author contributions
JFT and NC conceived, designed and directed the project. JFT conducted the experiments. CTB synthesized the alkynyl triazenes. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by EPFL. We thank Dr R. Scopelliti and Dr F. Fadaei-Tirani for X-ray crystallographic analysis of compounds 2a and 6e.Notes and references
- (a) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, ed. V. Gouverneur and K. Müller, Imperial College Press, London, UK, 2012, vol. 6, pp. 1–558 Search PubMed; (b) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Chichester, UK, 2009, pp. 1–640 Search PubMed; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (d) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (e) D. B. Harper and D. O'Hagan, Nat. Prod. Rep., 1994, 11, 123–133 RSC.
- (a) Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, ed. P. Kirsch, Wiley-VCH, Weinheim, 2006, pp. 1–320 Search PubMed; (b) Fluorine in Organic Chemistry, ed. R. D. Chambers, Blackwell Publishing Ltd, Oxford, 2004, pp. 1–406 Search PubMed.
- (a) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS; (b) F. Giornal, S. Pazenok, L. Rodefeld, N. Lui, J. P. Vors and F. R. Leroux, J. Fluorine Chem., 2013, 152, 2–11 CrossRef CAS; (c) J. Boyer, E. Arnoult, M. Médebielle, J. Guillemont, J. Unge and D. Jochmans, J. Med. Chem., 2011, 54, 7974–7985 CrossRef CAS PubMed; (d) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed; (e) M. E. Christy, C. D. Colton, M. Mackay, W. H. Staas, J. B. Wong, E. L. Engelhardt, M. L. Torchiana and C. A. Stone, J. Med. Chem., 1977, 20, 421–430 CrossRef CAS PubMed.
- (a) R. Jia, X. Wang and J. Hu, Tetrahedron Lett., 2021, 75, 153182 CrossRef CAS; (b) M. Miele and V. Pace, Aust. J. Chem., 2021, 74, 623–625 CrossRef CAS; (c) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139–152 CrossRef CAS; (d) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC; (e) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecouke, Chem.–Eur. J., 2015, 21, 12836–12865 CrossRef CAS PubMed; (f) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- Review: J. Václavík, I. Klimánková, A. Budinská and P. Beier, Eur. J. Org. Chem., 2018, 3554–3593 CrossRef.
- (a) K. Jiang, S. Han, M. Ma, L. Zhang, Y. Zhao and M. Chen, J. Am. Chem. Soc., 2020, 142, 7108–7115 CrossRef CAS PubMed; (b) H. Sakaguchi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2018, 57, 328–332 CrossRef CAS PubMed; (c) T. Kawashima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2018, 140, 17423–17427 CrossRef CAS PubMed; (d) T. Kawashima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2017, 139, 17795–17798 CrossRef CAS PubMed; (e) H. Sakaguchi, Y. Uetake, M. Ohashi, T. Niwa, S. Ogoshi and T. Hosoya, J. Am. Chem. Soc., 2017, 139, 12855–12862 CrossRef CAS PubMed; (f) L. Li, C. Ni, Q. Xie, M. Hu, F. Wang and J. Hu, Angew. Chem., Int. Ed., 2017, 56, 9971–9975 CrossRef CAS PubMed; (g) M. Ohashi, H. Shirataki, K. Kikushima and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 6496–6499 CrossRef CAS PubMed; (h) M. Ohashi, T. Kambara, T. Hatanaka, H. Saijo, R. Doi and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 3256–3259 CrossRef CAS PubMed.
- Review: V. G. Nenajdenko, V. M. Muzalevskiy and A. V. Shastin, Chem. Rev., 2015, 115, 973–1050 CrossRef CAS PubMed.
- (a) Y. Chang, A. Tewari, A. I. Adi and C. Bae, Tetrahedron, 2008, 64, 9837–9842 CrossRef CAS; (b) R. P. Singh, U. Majumder and J. M. Shreeve, J. Org. Chem., 2001, 66, 6263–6267 CrossRef CAS PubMed; (c) W. R. Hasek, W. C. Smith and V. A. Engelhardt, J. Am. Chem. Soc., 1960, 82, 543–551 CrossRef CAS.
- (a) J. Gatenyo and S. Rozen, J. Fluorine Chem., 2009, 130, 332–335 CrossRef CAS; (b) C. York, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1994, 59, 6493–6494 CrossRef CAS; (c) S. Rozen and M. Brand, J. Org. Chem., 1986, 51, 222–225 CrossRef CAS; (d) A. Gregorcic and M. Zupan, J. Org. Chem., 1979, 44, 4120–4122 CrossRef CAS.
- Reaction with ynamides: (a) X. Zeng, J. Li, C. K. Ng, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 2924–2928 CrossRef CAS PubMed; (b) Y. Xi, G. Zhu, L. Tang, S. Ma, D. Zhang, R. Zhang, G. He and H. Zhu, Org. Biomol. Chem., 2017, 15, 7218–7226 Search PubMed; (c) G. Zhu, S. Qiu, Y. Xi, D. Zhang, R. Zhang, G. He and H. Zhu, Org. Biomol. Chem., 2016, 14, 7746–7753 RSC; (d) G. He, S. Qui, H. Huang, G. Zhu, D. Zhang, R. Zhang and H. Zhu, Org. Lett., 2016, 18, 1856–1859 CrossRef CAS PubMed; (e) B. Metayer, G. Compain, K. Jouvin, A. Martin-Mingot, C. Bachmann, J. Marrot, G. Evano and S. Thibaudeau, J. Org. Chem., 2015, 80, 3397–3410 CrossRef CAS PubMed; (f) G. Compain, K. Jouvin, A. Martin-Mingot, G. Evano, J. Marrot and S. Thibaudeau, Chem. Commun., 2012, 48, 5196–5198 RSC , with alkyoxy alkynes; (g) L. Hu, C. Che, Z. Tan and G. Zhu, Chem. Commun., 2015, 51, 16641–16644 RSC , with electron-deficient alkynes; (h) T. J. O'Connor and F. Dean Toste, ACS Catal., 2018, 8, 5947–5951 CrossRef PubMed; (i) X. Zeng, Z. Lu, S. Liu, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2017, 359, 4062–4066 CrossRef CAS PubMed; (j) X. Zeng, S. Liu, G. B. Hammond and B. Xu, Chem.–Eur. J., 2017, 23, 11977–11981 CrossRef CAS PubMed.
- For reviews, see: (a) A. A. Suleymanov and K. Severin, Angew. Chem., Int. Ed., 2020, 60, 6879–6889 CrossRef PubMed; (b) D. K. Kölmel, N. Jung and S. Bräse, Aust. J. Chem., 2014, 67, 328–336 CrossRef; (c) S. Bräse, Acc. Chem. Res., 2004, 37, 805–816 CrossRef PubMed; (d) D. B. Kimball and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3338–3498 CrossRef CAS.
- (a) A. P. Francisco, E. Mendes, A. R. Santos and M. J. Perry, Curr. Pharm. Des., 2019, 1623–1642 CrossRef CAS PubMed; (b) L. Serrone, M. Zeuli, F. M. Sega and F. J. Cognetti, J. Exp. Clin. Cancer Res., 2000, 19, 21–34 CAS; (c) E. S. Newlands, M. F. G. Stevens, S. R. Wedge, R. T. Wheelhouse and C. Brock, Cancer Treat. Rev., 1997, 23, 35–61 CrossRef CAS PubMed.
- (a) A. K. Flatt, B. Chen and J. M. Tour, J. Am. Chem. Soc., 2005, 127, 8918–8919 CrossRef CAS PubMed; (b) J. L. Delgado, P. de la Cruz, V. López-Arza, F. Langa, D. B. Kimball, M. M. Haley, Y. Araki and O. Ito, J. Org. Chem., 2004, 69, 2661–2668 CrossRef CAS PubMed; (c) L. Jones, J. S. Schumm and J. M. Tour, J. Org. Chem., 1997, 62, 1388–1410 CrossRef CAS; (d) J. S. Moore, Acc. Chem. Res., 1997, 30, 402–413 CrossRef CAS.
- (a) C. Gil and S. Bräse, J. Comb. Chem., 2009, 11, 175–197 CrossRef CAS PubMed; (b) S. Dahmen and S. Bräse, Angew. Chem., Int. Ed., 2000, 39, 3681–3683 CrossRef CAS; (c) S. Bräse, S. Dahmen and M. Pfefferkorn, J. Comb. Chem., 2000, 2, 710–715 CrossRef PubMed.
- (a) C. Torres-García, D. Pulido, F. Albericio, M. Royo and E. Nicolás, J. Org. Chem., 2014, 79, 11409–11415 CrossRef PubMed; (b) K. C. Nicolaou and C. N. C. Boddy, J. Am. Chem. Soc., 2002, 124, 10451–10455 CrossRef CAS PubMed; (c) K. C. Nicolaou, C. N. C. Boddy, H. Li, A. E. Koumbis, R. Hughes, S. Natarajan, N. F. Jain, J. M. Ramanjulu, S. Bräse and M. E. Solomon, Chem.–Eur. J., 1999, 5, 2602–2621 CrossRef CAS.
- (a) J. Vajs, C. Proud, A. Brozovic, M. Gazvoda, A. Lloyd, D. I. Roper, M. Osmak, J. Košmrlj and C. G. Dowson, Eur. J. Med. Chem., 2017, 127, 223–234 CrossRef CAS PubMed; (b) A. Sousa, F. Santos, M. M. Gaspar, S. Calado, J. D. Pereira, E. Mendes, A. P. Francisco and M. J. Perry, Bioorg. Med. Chem., 2017, 25, 3900–3910 CrossRef CAS PubMed; (c) Z. S. Mouhri, E. Goodfellow, S. P. Kelley, R. S. Stein, R. D. Rogers and B. J. JeanClaude, Molecules, 2017, 22, 1183 CrossRef PubMed; (d) D. Cappoen, J. Vajs, C. Uythethofken, A. Virag, V. Mathys, M. Kočevar, L. Verschaeve, M. Gazvoda, S. Polanc, K. Huygen and J. Košmrlj, Eur. J. Med. Chem., 2014, 77, 193–203 CrossRef CAS PubMed; (e) A. Monteiro, S. J. Almeida, G. Cabral, P. Severino, P. A. Videira, A. Sousa, R. Nunes, J. D. Pereira, A. P. Francisco, M. J. Perry and E. Mendes, Eur. J. Med. Chem., 2013, 70, 1–9 CrossRef CAS PubMed; (f) M. J. Perry, E. Mendes, A. L. Simplício, A. Coelho, R. V. Soares, J. Iley, R. Moreira and A. P. Francisco, Eur. J. Med. Chem., 2009, 44, 3228–3234 CrossRef CAS PubMed.
- H. Lu and C. Li, Tetrahedron Lett., 2005, 46, 5983–5985 CrossRef CAS.
- B. Štefane, U. Černigoj, M. Kočevar and S. Polanc, Tetrahedron Lett., 2001, 42, 6659–6662 CrossRef.
- Y. Chung, H. Lee and K. H. Ahn, J. Org. Chem., 2006, 71, 9470–9474 CrossRef CAS PubMed.
- (a) A. A. Suleymanov, E. Le Du, Z. Dong, B. Muriel, R. Scopelliti, F. Fadaei-Tirani, J. Waser and K. Severin, Org. Lett., 2020, 22, 4517–4522 CrossRef CAS PubMed; (b) R. H. Smith Jr and C. J. Michejda, Synthesis, 1983, 476–477 CrossRef; (c) D. H. Sieh, D. J. Wilbur and C. J. Michejda, J. Am. Chem. Soc., 1980, 102, 3883 CrossRef CAS; (d) R. J. LeBlanc and K. Vaughn, Can. J. Chem., 1972, 50, 2544–2551 CrossRef CAS; (e) W. Kirmse and U. Seipp, Chem. Ber., 1974, 107, 745–758 CrossRef CAS; (f) D. M. Gale, W. J. Middleton and C. G. Krespan, J. Am. Chem. Soc., 1965, 87, 657–658 CrossRef CAS.
- (a) I. R. Landman, E. Acuña-Bolomey, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Org. Lett., 2019, 21, 6408–6412 CrossRef CAS PubMed; (b) A. Glowacki, V. Jeux, G. Gasnier, L. Joucla, G. Jacob and E. Lacôte, Synlett, 2018, 29, 566–570 CrossRef CAS; (c) W.-M. Shi, X.-P. Ma, C.-X. Pan, G.-F. Su and D.-L. Mo, J. Org. Chem., 2015, 80, 11175–11183 CrossRef CAS PubMed; (d) I. R. Dunkin, M. A. Lynch, R. Withnall, A. J. Boulton and N. Henderson, J. Chem. Soc., Chem. Commun., 1989, 1777–1778 RSC; (e) A. J. Boulton and P. J. Devi, J. Chem. Soc., Chem. Commun., 1988, 631–632 RSC; (f) N. Egger, L. Hoesch and A. S. Dreiding, Helv. Chim. Acta, 1983, 66, 1416–1426 CrossRef CAS.
- G. Kiefer, T. Riedel, P. J. Dyson, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 302–305 CrossRef CAS PubMed.
- (a) J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, Chem. Sci., 2021, 12, 9140–9145 RSC; (b) C. T. Bormann, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Org. Biomol. Chem., 2021, 19, 8113–8117 RSC; (c) J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, ACS Catal., 2020, 10, 3790–3796 CrossRef CAS; (d) C. T. Bormann, F. G. Abela, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Eur. J. Org. Chem., 2020, 2130 CrossRef CAS; (e) J.-F. Tan, C. T. Bormann, F. G. Perrin, F. M. Chadwick, K. Severin and N. Cramer, J. Am. Chem. Soc., 2019, 141, 10372–10383 CrossRef CAS PubMed; (f) T. Wezeman, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Adv. Synth. Catal., 2019, 361, 1383–1388 CrossRef CAS; (g) A. A. Suleymanov, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Adv. Synth. Catal., 2018, 360, 4178–4183 CrossRef CAS; (h) D. Kossler, F. G. Perrin, A. A. Suleymanov, G. Kiefer, R. Scopelliti, K. Severin and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 11490–11493 CrossRef CAS PubMed; (i) L. N. Jeanbourquin, R. Scopelliti, F. Tirani-Fadaei and K. Severin, Helv. Chim. Acta, 2017, 100, e1700186 CrossRef.
- F. Perrin, G. Kiefer, L. Jeanbourquin, S. Racine, D. Perrotta, J. Waser, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 13393–13396 CrossRef CAS PubMed.
- J.-L. Li, E. Lin, X.-L. Han, Q. Li and H. Wang, Org. Lett., 2019, 21, 4255–4258 CrossRef CAS PubMed.
- Supplementary crystallographic data of the following compounds can be found from The Cambridge Crystallographic Data Centre: 2141776 (2a) and 2100359 (6e)†.
- (a) P. L. Coe and J. H. Sleigh, J. Fluorine Chem., 1981, 17, 403–407 CrossRef CAS; (b) R. Dannley and R. Taborsky, J. Org. Chem., 1957, 22, 77–78 CrossRef CAS.
- (a) R. H. Smith, C. L. Denlinger, R. Kupper, S. R. Koepke and C. J. Michejda, J. Am. Chem. Soc., 1984, 106, 1056–1059 CrossRef; (b) D. H. Sieh and C. J. Michejda, J. Am. Chem. Soc., 1981, 103, 442–445 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of all new compounds. CCDC 2141776 and 2100359. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00294a |
| This journal is © The Royal Society of Chemistry 2022 |