
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hypoxia-activated NO donor for the treatment of myocardial hypoxia injury†
Wen
Zhou
ab,
Wanxiang
Yang
ab,
Keyu
Fan
ab,
Wuyang
Hua
ab and
Shaohua
Gou
 *ab
*ab
aJiangsu Province Hi-Tech Key Laboratory for Biomedical Research, Southeast University, Nanjing, 211189, China. E-mail: 2219265800@qq.com
bPharmaceutical Research Center and School of Chemistry and Chemical Engineering, Southeast University, Nanjing, 211189, China
First published on 28th February 2022
Abstract
As present NO donor drugs cannot localize to release NO at the hypoxic site, along with the short half-life and bidirectional regulation of NO, they are unable to overcome low bioavailability and side effects in the treatment of myocardial hypoxia injury. In this study, we designed and prepared a novel hypoxia-activated NO donor (Hano) by hybridization of a known NO donor compound (Nno) with a hypoxia-activated group. Hano and isosorbide dinitrate were compared in terms of NO release and anti-myocardial hypoxia injury. Furthermore, the effects of Hano and Nno on releasing NO, dilating blood vessels, and preventing myocardial hypoxia injury were studied and compared in smooth muscle cells, cardiomyocytes and mice. The results showed that the NO release by Hano increased either in smooth muscle cells or in myocardial cells under hypoxia conditions. Significantly, Hano was found capable of dilating blood vessels and attenuating hypoxia injury both in vitro and in vivo, and has great potential as a hypoxia-activated NO donor drug to treat hypoxic heart diseases.
Introduction
Nitric oxide (NO) donors can release exogenous NO molecules in the body, which are commonly used for the treatment of coronary heart disease caused by ischemia and hypoxia.1–3 However, NO has a bidirectional regulatory effect in vivo. It can dilate blood vessels and restore blood supply in hypoxia sites, but also causes oxidative damage to normal tissues. Available NO donor drugs cannot locally release NO at the hypoxic injury site and increase side effects to normal tissues, while the short half-life of NO itself reduces the bioavailability of NO donors. Therefore, we designed a NO donor with moderate half-life and local release of NO.4–6In detail, myocardial hypoxia injury is closely related to NO deficiency.1,7 Clinical studies have found that coronary heart disease patients have a low intra-coronary NO level.8,9 As NO is a kind of endothelial relaxing factor, it can activate soluble guanylate cyclase (sGC) and catalyze the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP) in smooth muscle cells. It is known that cGMP can activate protein kinase G (PKG), a primary effector molecule of NO, while PKG is involved in angiotensin converting enzyme (ACE) related signaling pathways to regulate the contraction of smooth muscles and cause smooth muscle vasodilatation to restore blood flow.10–12 Therefore, the vasodilation effect of NO donors could be studied via ACE and related pathways.
In myocardial cells, under the hypoxia conditions, the electron transport chain in the mitochondria is blocked due to insufficient oxygen supply. Research indicates that some electrons leak out of the respiratory chain and combine with oxygen to produce a large number of ROS, causing oxidative injury and resulting in cell apoptosis.13,14 However, it has been evidenced that, by dilating blood vessels to restore oxygen supply, NO can regulate excessive ROS. Moreover, mechanistic target of rapamycin complex-1 (mTORC1) activation is closely related to diseases in main organs including the heart. PKG downregulates the protein expression of mTORC1 and attenuates myocardial hypoxia injury.15 In addition, tuberin (TSC2) is a GTPase-activating protein and prominent intrinsic regulator of mTORC1. And protein kinase G (PKG) phosphorylates these TSC2 sites and constitutively inhibits mTORC1. Thus, suppression of hypertrophy and stimulation of autophagy in cardiomyocytes by PKG require TSC2 phosphorylation.16 In short, TSC2 phosphorylation and mTORC1 expression variations and ROS detection can be used as reliable basis to analyze the efficacy of NO donors.
Hypoxia-activated prodrugs (HAPs) have been studied for years to treat hypoxia related diseases. HAPs enhance the release of active substances to hypoxic sites by increased enzyme activity under the hypoxic conditions. Nitroarene is widely used in the design of HAPs.17,18 It has been reported that intracellular nitroreductase (NTR) selectively reduces nitro groups and converts HAPs into bioactive molecules.19 And NTR, highly expressed in hypoxic cells, promotes the efficient release of active molecules in a hypoxic microenvironment, showing up hypoxia selectivity.20 Since hypoxia occurs in many human diseases such as tumor and coronary heart diseases, promotion of molecular hypoxic selectivity is an effective strategy to improve drug efficacy, decrease drug dosage and reduce side effects on normal tissues.21,22 Such a molecular design of related drugs has been applied to anti-tumor drugs so far, which releases active groups in the hypoxic microenvironment of tumors. It is also used in fluorescent probe design for diagnosing hypoxia, but hypoxia-activated drug design hasn't been used to optimize anti-myocardial hypoxia injury drugs.
Hypoxia is not only the main phenotype of coronary heart disease, myocardial infarction and other cardiovascular diseases, but also an important cause of myocardial injury.23,24 Therefore, in this study, we attempted to introduce a hypoxia-activated unit, nitroarene, into a NO donor moiety so as to construct a novel compound (Hano), which is presumed to release NO in a hypoxic microenvironment. To demonstrate the efficacy of the resulting compound under hypoxic conditions, NO release quantity and the activity of Hano at both cellular and animal levels were detected and analyzed. Hypoxia activated optimization for NO donors to treat coronary heart disease was originally studied.
Results and discussion
Compound Hano was synthesized and characterized by 1H-NMR, 13C-NMR and ESI/TOF MS. All results verify that the structure of the compound coincides with what we designed (Scheme 1, Fig. S1–S5, Table S1–S2†).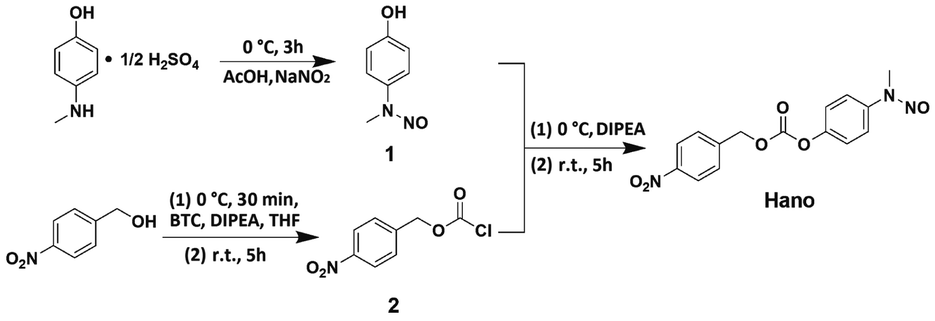 | ||
| Scheme 1 Preparation of compound Hano. | ||
The efficacy of Hano to attenuate hypoxia injury was first compared with isosorbide dinitrate (ISDN), a common clinical nitrate ester NO donor drug. The results of CCK8 assay showed that under normoxia, both Hano and ISDN had no impact on H9c2 cell viability (Fig. 1A). Under hypoxic conditions, as shown in Fig. 1B, cell viability was reduced to 74% of that under normoxia. Hano and ISDN recovered cell viabilities to different degrees. Within the concentration range of 0.001–10 μM, ISDN has the best drug effect at 0.1 μM, and the cell viability was restored to 92% of normal cells. The effect of Hano was concentration-dependent obviously. Cell viability could be recovered to 99% of normal cells when the concentration was 10 μM, which was significantly better compared with ISDN. In Fig. 1C, the Griess method was used to compare the NO release rates of two compounds. The results showed that ISDN had higher release rates under normoxia and hypoxia, 36% and 20%, respectively. The release rate of Hano under normoxia and hypoxia was 1% and 25%, respectively, which indicated that the NO release of Hano had obvious hypoxia selectivity. We further compared the effects of two compounds against myocardial hypoxia injury by confocal microscopy. As shown in Fig. 1D, in the double staining experiment of Hoechst 33258 and propidium iodide (PI), the intensity of blue fluorescence was proportional to the degree of early apoptosis, and that of red fluorescence was proportional to the degree of late apoptosis and necrosis. The fluorescence intensity was very weak in normal cells. Both the blue and red fluorescence intensities of the hypoxic modeling cells were significantly increased. The fluorescence intensities of cells cultured with 1 μM Hano or ISDN administration were weaker than that of the modeling group. The red fluorescence intensity of the Hano group almost disappeared. At the cellular level, it showed that Hano was better than ISDN in inhibiting apoptosis and necrosis of myocardial cells.
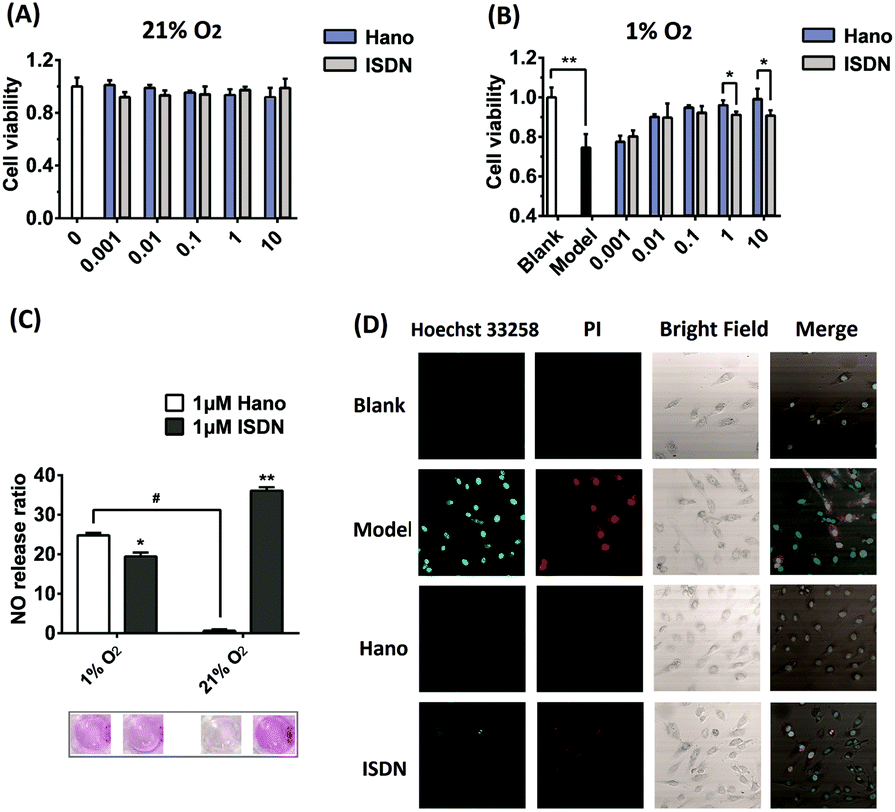 | ||
| Fig. 1 H9c2 cell viabilities influenced by Hano or isosorbide dinitrate (ISDN) (0.001–10 μM), respectively, under 21% (A) or 1% oxygen conditions (B), and data were expressed as the mean ± SD from three independent experiments. *p <0.05, **<0.01; (C) NO releases and the corresponding histograms in Hano or ISDN (1 μM) treated H9c2 cells under normoxia or hypoxia conditions with Griess reagent; (D) Hoechst 33258 and PI apoptosis detection of the blank group, model group (1% O2) and medicated groups (10 μmol L−1 Hano or ISDN under 1% O2, respectively) detected by confocal microscopy. | ||
In view of Hano's superior anti-myocardial hypoxia injury effect to the marketed drugs under hypoxia, and the preliminary discovery that Hano achieved hypoxia-induced release of NO, the efficacy and mechanism of its hypoxia-induced release of NO were further studied.
After normoxic or hypoxic treatment, CCK8 detection showed that cell viabilities changed under different circumstances. Under normoxia conditions (21% O2), H9c2 cells dosed with Hano or Nno (0.001–10 μM) had almost the same cell viability as that of the blank group, suggesting that the two compounds have no toxic effects on cardiomyocytes. However under hypoxia conditions (1% O2), cell viability of the modelling group decreased to 70% compared with that of the blank group. Intriguingly, cell viability of the Hano group gradually increased depending on the concentration increase of Hano from 0.001 to 10 μM. In contrast, cell viability of the Nno group remained almost the same as that of the modeling group. Different from Nno, Hano has an obvious advantage to promote cell viability in a hypoxic microenvironment in comparison with Nno (Fig. 2A).
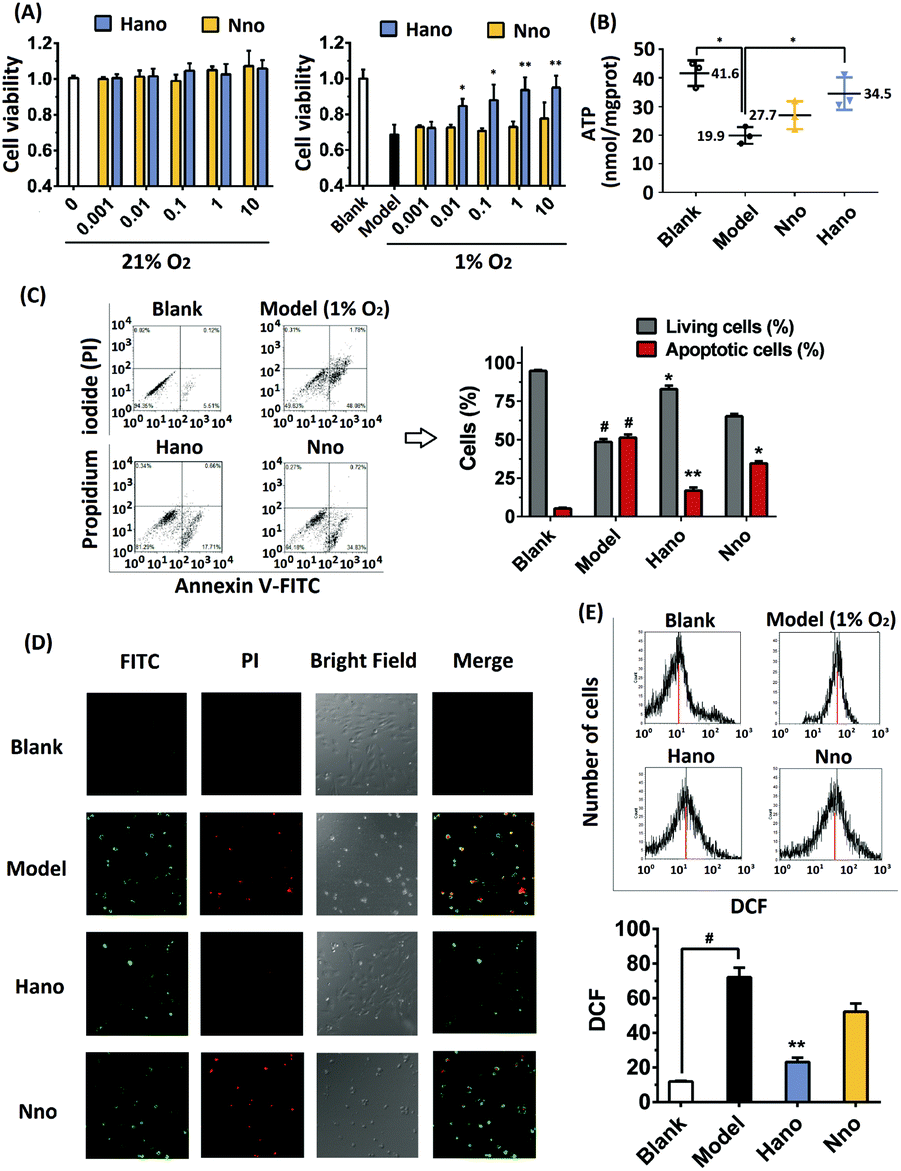 | ||
| Fig. 2 (A) Cell viabilities influenced by Hano or Nno (0.001–10 μM), respectively, under 21% or 1% oxygen conditions. Data were expressed as the mean ± SD from three independent experiments. *p <0.05, **p <0.01 versus the model group; (B) ATP regulation effect of Hano or Nno under hypoxia treated with H9c2 cells. Data were expressed as the mean ± SD from three independent experiments. *p <0.05; (C) H9c2 cell membrane integrities of blank, model (1% O2) and medicated groups (10 μmol L−1 Hano or Nno under 1% O2, respectively) detected with FITC and PI staining by flow cytometry; (D) Annexin V-FITC apoptosis detection of blank, model (1% O2) and medicated groups (10 μmol L−1 Hano or Nno under 1% O2, respectively) detected by confocal microscopy; (E) cell distribution of H9c2 cells under different fluorescence intensities of DCF to detect ROS levels in blank, model (1% O2) and medicated groups (1% O2 and 10 μM Hano or Nno, respectively). | ||
Under normoxic conditions, the ATP value in the blank group was 41.6 nmol mgprot−1, while its value decreased to 19.9 nmol mgprot−1 under hypoxic conditions. As observed in Hano and Nno treated groups, the ATP levels were restored at 34.5 and 27.7 nmol mgprot−1, respectively. The ATP levels in different groups indicate that hypoxia treatment in a tri-gas incubator can reduce the ATP level, while Hano is capable of restoring the ATP level greater than Nno (Fig. 2B).
Cell membrane integrities of different groups were examined by flow cytometry using Annexin V-FITC and propidium iodide (PI) double staining. Cells were distinguished as normal cells (FITC–, PI–), early apoptotic cells (FITC+, PI–), and late apoptotic and necrotic cells (FITC+, PI+).25 As shown in Fig. 2C, the proportion of normal cells in the blank group was as high as 94.35%. In the hypoxia model group, normal cells reduced to 49.83% while the sum of apoptotic and necrotic cells increased to 49.86%. Significantly, in the medicated group, apoptotic and necrotic cells reduced to 18.37% and 35.55% in Hano and Nno treated groups, respectively.
Cell apoptosis and necrosis were also detected by confocal microscopy with Annexin V-FITC and propidium iodide (PI) double-staining. In Fig. 2D, the intensity of green fluorescence is proportional to the degree of early apoptosis, and that of red fluorescence is proportional to the degree of late apoptosis and necrosis. As observed, the green and red fluorescence in the blank group was weak. In the model group, two fluorescence intensities were significantly enhanced, and a large number of apoptotic and necrotic cells were stained with either red or green fluorescence. In the Hano group, the intensity of red fluorescence significantly diminished or even disappeared, and the green fluorescence was rather weakened compared with that in the model group. Notably, the cell morphology was significantly improved, indicating that Hano had a strong effect on anti-apoptosis and necrosis. Different from that in the Hano group, both the green and red fluorescence intensities in the Nno group were similar to that in the model group, demonstrating that Nno had a weaker effect on anti-apoptosis and necrosis. The above experimental results indicate that hypoxia stimulation can lead to cell apoptosis and necrosis. Among the test groups, Hano can most effectively resist cell apoptosis and necrosis caused by hypoxia.
Under hypoxic conditions, perturbation in electron transport is associated with leakage of electrons from the respiratory chain, resulting in increased ROS that could lead to myocardial hypoxia oxidative injury.26 From the flow cytometry experiments, it was observed that the specific abscissa value related to the DCF peak values increased from 12.23 to 68.18 under hypoxia treatment. Hano reduced the ROS level to 21.47 while Nno to 55.60 (Fig. 2E). The results indicate that Hano can much more effectively reduce the production of ROS than Nno.
Since Hano exhibited much potent anti-apoptotic and necrosis activity when compared with a normal NO donor compound (Nno), we further studied the mechanism of Hano to attenuate hypoxia injury. In smooth muscle cells HCASMC, NTR activity increased under hypoxia (Fig. 3A), which facilitated the reduction of the nitro group in Hano to release NO a little more easily than that in the Nno group.
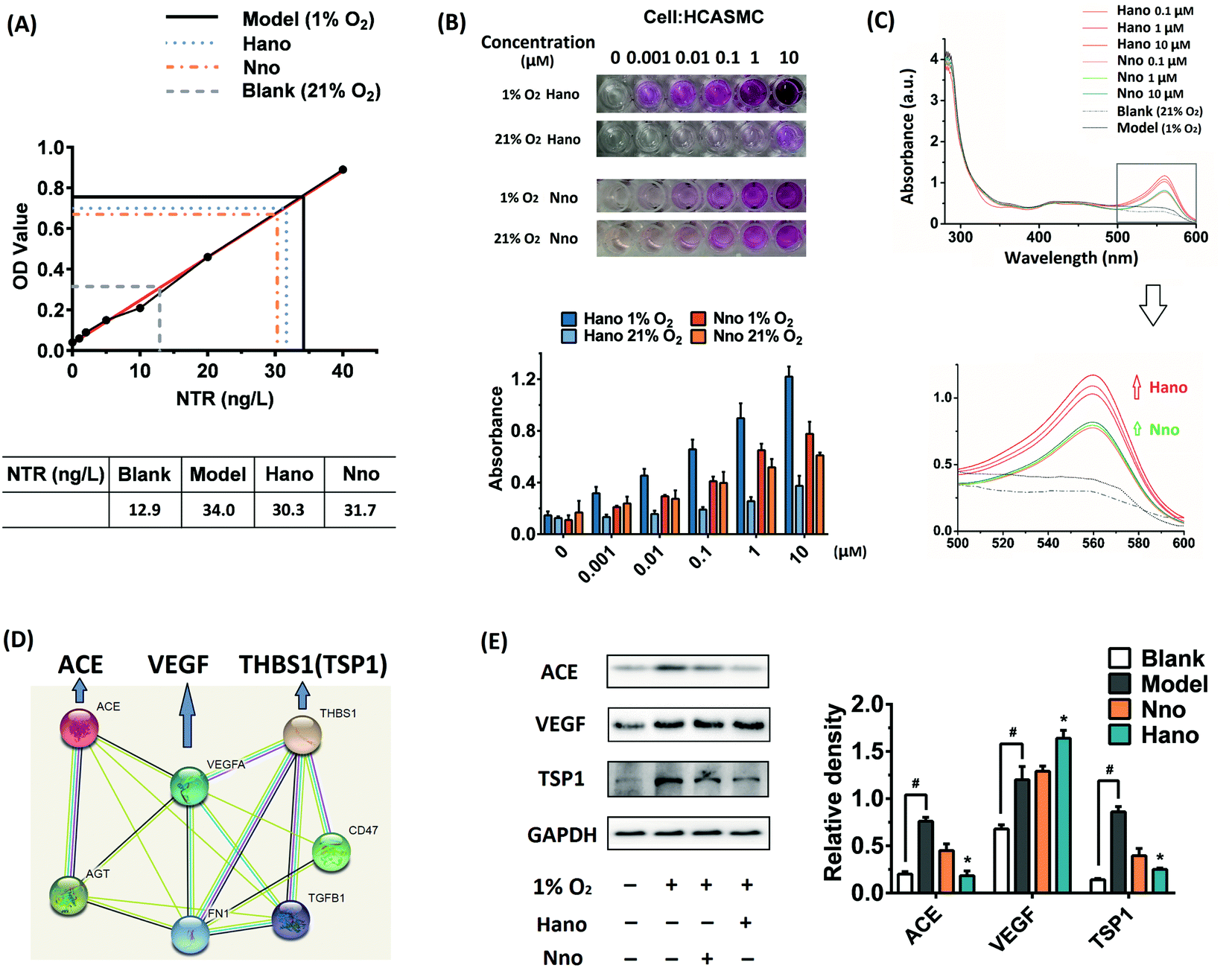 | ||
| Fig. 3 (A) Nitroreductase (NTR) standard curve and NTR activities of blank, model and drug (Hano or Nno) treated groups under hypoxia; (B) NO releases and the corresponding histograms in Hano or Nno (0–10 μM) treated HCASMC cells under normoxia or hypoxia conditions with Griess reagent; (C) UV spectra of blank, model and drug (Hano or Nno) treated groups with Griess reagent, and the amplified absorption peaks at about 560 nm; (D) protein–protein interactions among ACE, VEGF and THBS1 (TSP1) from the STRING database; (E) ACE, VEGF and TSP1 protein expressions in HCASMC cells treated with or without 10 μM Hano or Nno. | ||
In HCASMC cells, detected with the Griess method, NO levels of cells cultured with Hano under hypoxic or normoxic conditions showed significant differences. As shown in Fig. 3B, under normoxic conditions, the NO concentration in the Hano group (0–10 μM) was low, while under hypoxic conditions, with the increase of the Hano concentration, the release of NO increased significantly. The NO amount in Nno cultured cells under normoxia or hypoxia was similar, and the NO level was higher than that of Hano under normoxic conditions but lower than that of Hano under hypoxic conditions. The results indicated that Hano had obvious hypoxia selectivity. UV spectral comparison in Fig. 3C further showed that under hypoxic conditions, with the increase of the Hano concentration, the characteristic absorption peak of Griess reagent at 560 nm increased more significantly than that of the Nno group, which also indicates that the NO level of the Hano group was higher than that of Nno under hypoxia.
In the analysis of STRING protein interaction, ACE interacts with VEGF and THBS1 (TSP1) (Fig. 3D). As an upstream gene, angiotensin converting enzyme (ACE) overexpression is an important factor leading to vasoconstriction and myocardial hypoxia. Therefore, ACE inhibitors were used to treat heart failure and other cardiovascular diseases.27 The vascular endothelial growth factor (VEGF) is involved in angiogenic cell differentiation, which is closely related to the regulation of angiogenesis and vascular permeability.28 As thrombospondin1 (TSP1) inhibits angiogenesis, the elevated expression of TSP1 increases vascular endothelial adhesion and platelet aggregation, causing myocardial ischemia and hypoxia.29
In the western blot assay under hypoxia conditions (1% O2), protein expressions of ACE and TSP1 were obviously upregulated. It was found that Hano downregulated ACE and TSP1 overexpression more significantly than Nno (Fig. 3E). As VEGF expression was upregulated under hypoxia, it was thought to be an adaptive response of cells to hypoxia. Notably, Hano and Nno could upregulate VEGF expression at different levels. These results hinted that the improved NO releasing level by Hano under hypoxic conditions could relax smooth muscles more effectively to expand blood vessels than that by Nno.
NO levels in H9c2 cells were examined through the fluorescence intensity of NO probe DAF-FM DA. As shown in Fig. 4A, the NO level in the Hano group increased more visibly under hypoxic conditions than that in the Nno group. Besides, the fluorescence emission spectrum of DAF-FM was also detected by using a fluorescence spectrophotometer (Fig. 4B), in which the peak intensity variation trends were consistent with the fluorograph.
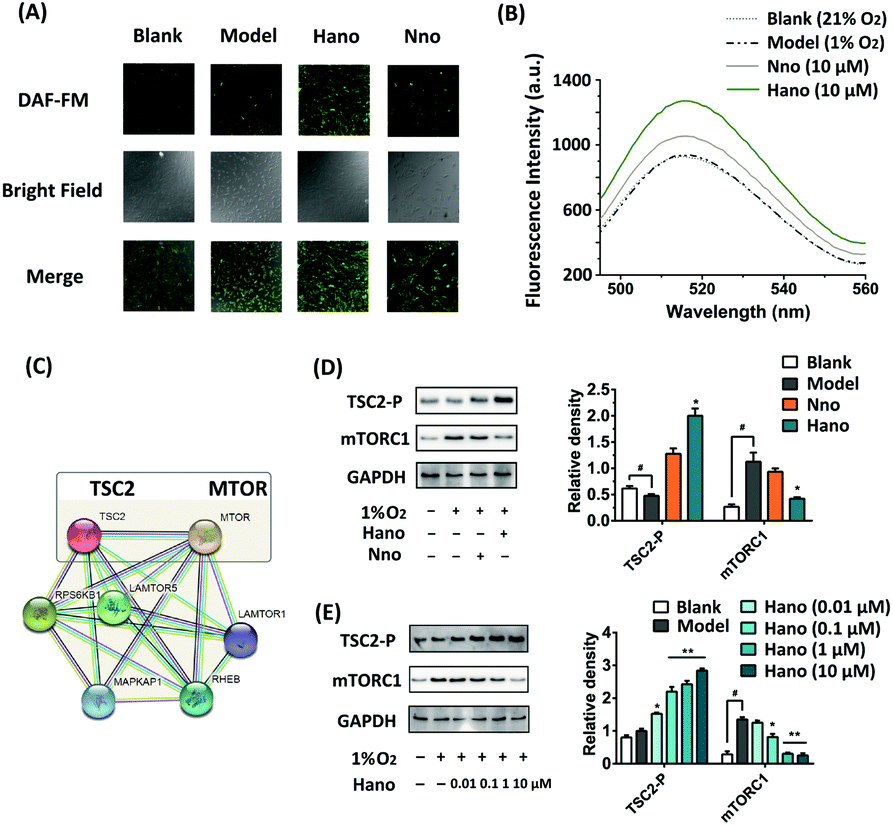 | ||
| Fig. 4 (A) NO levels of blank, model and 10 μM Hano/Nno treated groups in situ detected with NO fluorescent probe DAF-FM DA (Ex: 495 nm, Em, 515 nm); (B) NO levels of Hano or Nno (10 μM) treated H9c2 cells under hypoxia (DAF-FM, Ex: 495 nm, Em, 515 nm); (C) protein–protein interactions between TSC2 and MTOR (mTORC1) from the STRING database; (D) TSC2-P and mTORC1 protein expressions in H9c2 cells treated with or without 10 μM Hano or Nno; (E) TSC2-P and mTORC1 protein expressions in H9c2 cells treated with Hano (0–10 μM). | ||
In the analysis of STRING protein interactions, the protein–protein interaction between TSC2 and mTORC1 could be searched (Fig. 4C). It has been proved that turberin (TSC2) negatively regulates mTORC1 signaling in the pathway associated with myocardial injury. Under hypoxic conditions, TSC2 could be activated to inhibit the target of rapamycin complex-1 (mTORC1) signaling. As an important regulatory factor of cell proliferation, TSC2 can inhibit the expression of mTORC1 when it is phosphorylated.16 In our western blot assay (Fig. 4D), the TSC2-P expression was upregulated in the Hano treated group, and the expression of mTORC1 was inhibited as expected to achieve anti-hypoxia injury. Subsequent concentration gradient western blot assay showed that the protein expression regulated by Hano was concentration-dependent (Fig. 4E).
It could be proved, upon the above studies on two types of NO donors, that Hano released NO with significant hypoxia selectivity, so we also explored this feature from the perspective of the structure–activity relationship. The electron cloud distribution of Hano and Nno was simulated, and it could be seen that after Hano was reduced to Hano–NH2 by nitroreductase (NTR), the electron cloud of Hano was shifted from the –NO2 end to the –NO end. The increased electronegativity of the NO site is conducive to the combination with the heme iron ion in the active center of Cyt-P450 and the release of NO, thus illustrating hypoxia selectivity. However, Nno did not undergo such a structural variation and electron cloud migration that it didn't have hypoxia selectivity (Fig. 5A, Table 1). After being kept at r.t. for 6 h, the aqueous solution of Hano was found stable with a purity of 97.82%, which was comparable to the purity before the placement (98.21%, Fig. S5†). The in vitro NTR enzymatic reaction was tracked by HPLC, in which Hano (18.0 min) was completely transformed into Hano-NH2 (10.4 min) within 1 h (Fig. 5B). LC-MS detection verified the molecular weights of two compounds (Fig. S6 and S7†).
 | ||
| Fig. 5 (A) Proposed NO release mechanism of Hano illustrated by the relevant electron cloud distribution simulation of Nno and Hano; (B) HPLC diagrams and the corresponding curve graph of Hano and Hano-NH2 variations after standing at r.t. for 6 h (1) and during the NTR catalyzed reaction (2–4). | ||
| Hano | Hano-NH2 | Nno | |
|---|---|---|---|
| Electric charge (NNO) | 1.03 | 0.11 | 0.13 |
| Electric charge (ONO) | 0.17 | −0.65 | −0.63 |
In animal experiments, macroscopic enzyme mapping assay (triphenyltetrazolium chloride, TTC test) was applied to detect the myocardial ischemia degrees in the animal heart of different groups. Since isoproterenol (ISO) could induce coronary microvasospasm and vasoconstriction, which in turn causes myocardial hypoxia, it was used as a modeling agent to construct a myocardial ischemia model in mice. An increased percentage of ischemia infarct size was observed in isoproterenol (ISO) administered mice (the white area, note the difference between the reflective area and the white area). The mouse pretreated with Hano (20 mg kg−1) showed a moderately low infarct size in contrast to the mouse model, whereas the mouse pretreated with Nno (20 mg kg−1) had no conspicuous effect (Fig. 6A). Fig. 6B shows the hematoxylin–eosin (HE) staining images of cardiac tissue slices from each group. The morphology of cardiomyocytes in the blank group was normal spindle shape, and the cells were arranged in an orderly manner. The myocardial tissue of the ISO modeling mouse in the model group showed obvious edema with disordered cell arrangements and blurred boundaries. The myocardial pathological injuries in Hano and Nno groups were alleviated at certain degrees. The Hano group had clear cell boundaries, and the disorder of cell arrangements was significantly improved. In contrast, the severity of edema in the Nno group was attenuated compared to that in the model group, but the disorder of cell arrangements was not improved.
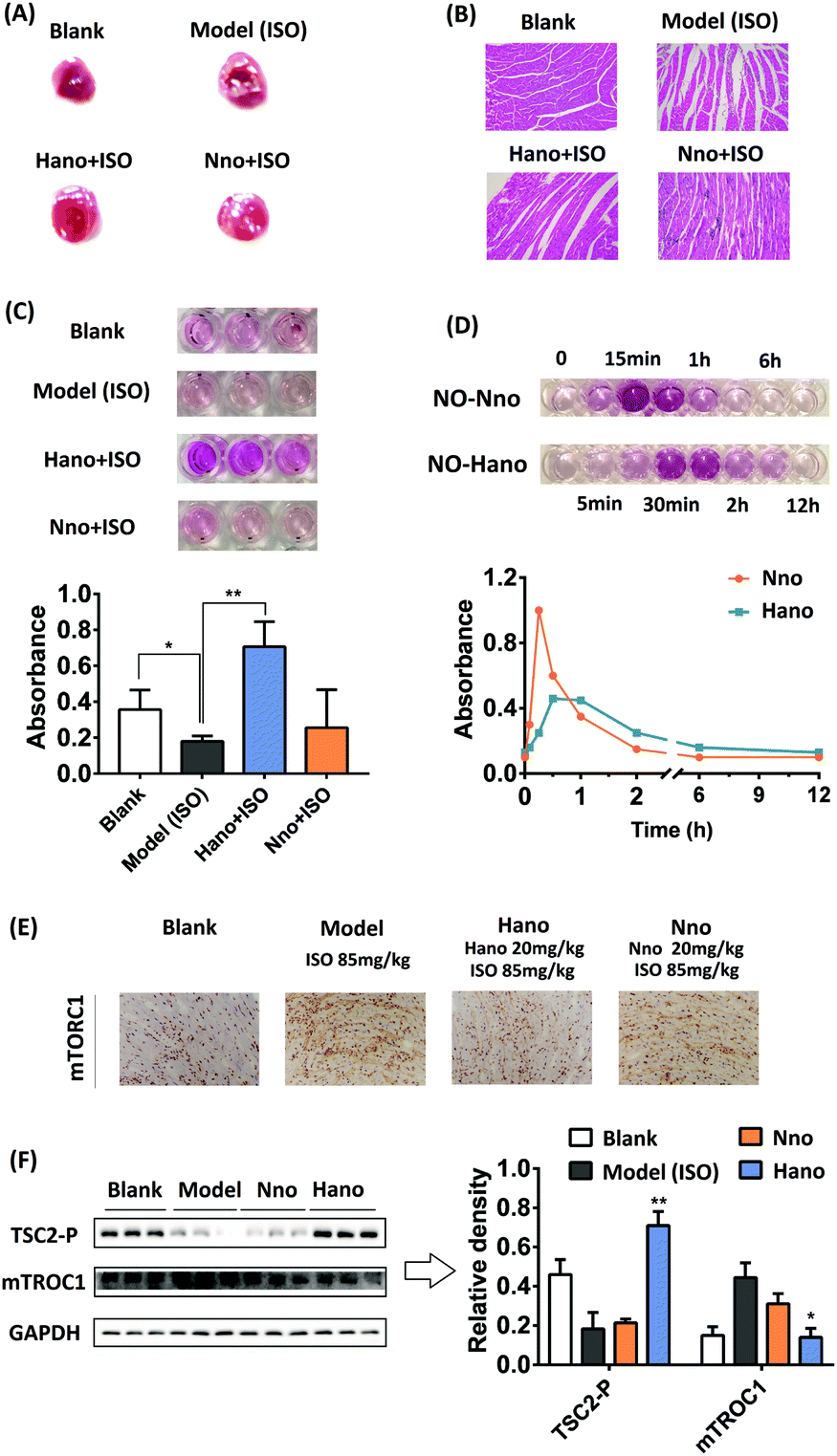 | ||
| Fig. 6 (A) TTC test of mouse heart tissues in blank, model (85 mg kg−1 ISO) and medicated groups (20 mg kg−1 Hano or Nno+85 mg kg−1 ISO); (B) effect of pretreatment with Hano or Nno on pathological changes in the ISO induced myocardial ischemia injury mouse model by HE staining; (C) NO release of 10 μM Hano or Nno in heart tissues compared with blank and model groups, and the corresponding histograms, *p <0.05, **p <0.01; (D) NO level at 0–12 h in mouse serum treated with compound Nno or Hano (10 mg kg−1); (E) immunohistochemistry examinations of mTORC1 accumulation in mouse heart; (F) TSC2-P and mTORC1 protein expressions in myocardial tissue in response to ISO treatment with or without Hano/Nno, respectively, *p <0.05, **p <0.01 compared with the model group. | ||
Griess experimental results showed different NO levels in the heart tissue homogenate, in which the NO level in the Hano group was the highest among the tested groups (Fig. 6C, each group has 3 parallel samples). Fig. 6D shows the difference in the variation trend of NO levels in the mouse serum treated with Hano or Nno at 10 mg kg−1. The NO level in the Nno-treated serum reached a peak at about 15 min and quickly decreased within 1 h, while that in the Hano-treated serum delayed its peak at about 30 min, with the half-life from about 1 h in the former group to about 2 h in the latter one. Significantly, the peak value in the Hano group was much lower than that in the Nno group, indicating that Hano released NO smoothly under normoxic conditions.
The mTORC1 protein expression in myocardial tissues of different groups was measured using immunohistochemistry assay (IHC). Compared with the blank group, the mTORC1 level in the ISO induced model group was enhanced obviously. The overexpression of mTORC1 was suppressed more substantially in the Hano group when compared with the Nno group (Fig. 6E). In the western blot assay (Fig. 6F), TSC2-P was activated by Hano while mTORC1 expression was inhibited, which was consistent with the results of IHC assay. Creatine kinase MB isoenzyme (CK-MB) and cardiac troponin I (cTNI) are two important indicators to diagnose myocardial injury through detecting enzyme levels in serum. The effect of Hano on reducing CK-MB activity and downregulating the cTNI level was greater than that of Nno (Table 2).
| CK-MB (U L−1) | cTNI (pg mL−1) | |
|---|---|---|
| Blank | 54.7 ± 21.1 | 20.2 ± 3.5 |
| Model | 251.5 ± 49.3 | 797.0 ± 102.6 |
| Hano | 118.2 ± 35.4 | 264.1 ± 41.5 |
| Nno | 203.3 ± 56.9 | 583.3 ± 77.3 |
Conclusions
Conclusively, in order to enhance the bioavailability of NO donor drugs in a hypoxic microenvironment, a compound Hano was designed and prepared by conjugating a hypoxia activated group with a NO donor molecule. By releasing NO intensively under hypoxic conditions, Hano could effectively overcome the shortcomings of NO caused by its short half-life and bidirectional regulation. Besides, it could greatly improve the NO activity of expanding blood vessels and anti-hypoxia injury. Our results reveal that construction of hypoxia activated NO donors is feasible for myocardial injury protection.Data availability
All experimental procedures and data associated with this article was contained in the ESI.†Author contributions
Dr Wen Zhou was involved in most of the molecular biology experiments, data analysis and manuscript writing; Wanxiang Yang participated in the in vitro experiments and the supplementary HPLC and LC-MS detection during the revision stage; Keyu Fan carried out the in vivo experiments and prepared the relevant figures and tables; Wuyang Hua synthesized the compounds; Prof. Shaohua Gou was the group leader, responsible for the whole proposal organization, compound and experimental design, and manuscript improvement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Natural Science Foundation of Jiangsu Province (No. BK20180570) and the Jiangsu Province Hi-Tech Key Laboratory for Biomedical Research for financial support to this work.References
- S. I. Bibli, A. Papapetropoulos, E. K. Iliodromitis, A. Daiber, V. Randriamboavonjy, S. Steven, P. Brouckaert, A. Chatzianastasiou, K. E. Kypreos, D. J. Hausenloy, I. Fleming and I. Andreadou, Cardiovasc. Res., 2019, 115, 625–636 CrossRef CAS PubMed.
- Y. Q. Yang, Z. J. Huang and L. L. Li, Nanoscale, 2021, 13, 444–459 RSC.
- A. Daiber and T. Munzel, Antioxid. Redox Signaling, 2015, 23, 899–942 CrossRef CAS PubMed.
- T. Yang, A. N. Zelikin and R. Chandrawati, Adv. Sci., 2018, 5, 1701043 CrossRef PubMed.
- T. Yang, A. N. Zelikin and R. Chandrawati, Small, 2020, 16, 1907635 CrossRef CAS PubMed.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- U. Forstermann and W. C. Sessa, Eur. Heart J., 2012, 33, 829–837 CrossRef PubMed.
- C. Farah, L. Y. M. Michel and J. L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS PubMed.
- B. L. Yu, F. Ichinose, D. B. Bloch and W. M. Zapol, Br. J. Pharmacol., 2019, 176, 246–255 CrossRef CAS PubMed.
- T. Kessler, J. Wobst, B. Wolf, J. Eckhold, B. Vilne, R. Hollstein, S. von Ameln, T. A. Dang, H. B. Sager, P. M. Rumpf, R. Aherrahrou, A. Kastrati, J. L. M. Bjorkegren, J. Erdmann, A. J. Lusis, M. Civelek, F. J. Kaiser and H. Schunkert, Circulation, 2017, 136, 476–489 CrossRef CAS PubMed.
- J. Scotcher, O. Prysyazhna, A. Boguslavskyi, K. Kistamas, N. Hadgraft, E. D. Martin, J. Worthington, O. Rudyk, P. R. Cutillas, F. Cuello, M. J. Shattock, M. S. Marber, M. R. Conte, A. Greenstein, D. J. Greensmith, L. Venetucci, J. F. Timms and P. Eaton, Nat. Commun., 2016, 7, 13187 CrossRef CAS PubMed.
- A. Velagic, C. Qin, O. L. Woodman, J. D. Horowitz, R. H. Ritchie and B. K. Kemp-Harper, Front. Pharmacol., 2020, 11, 727 CrossRef CAS PubMed.
- U. Forstermann, N. Xia and H. G. Li, Circ. Res., 2017, 120, 713–735 CrossRef PubMed.
- J. Tejero, S. Shiva and M. T. Gladwin, Physiol. Rev., 2019, 99, 311–379 CrossRef CAS PubMed.
- C. Farah, L. Y. M. Michel and J. L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS PubMed.
- M. J. Ranek, K. M. Kokkonen-Simon, A. Chen, B. L. Dunkerly-Eyring, M. P. Vera, C. U. Oeing, C. H. Patel, T. Nakamura, G. S. Zhu, D. Bedja, M. Sasaki, R. J. Holewinski, J. E. Van Eyk, J. D. Powell, D. I. Lee and D. A. Kass, Nature, 2019, 566, 264–269 CrossRef CAS PubMed.
- L. J. O'Connor, C. Cazares-Korner, J. Saha, C. N. G. Evans, M. R. L. Stratford, E. M. Hammond and S. J. Conway, Nat. Protoc., 2016, 11, 781–794 CrossRef PubMed.
- N. Baran and M. Konopleva, Clin. Cancer Res., 2017, 23, 2382–2390 CrossRef CAS PubMed.
- B. Brennecke, Q. H. Wang, Q. Y. Zhang, H. Y. Hu and M. Nazare, Angew. Chem., Int. Ed., 2020, 59, 8512–8516 CrossRef CAS PubMed.
- X. Q. Meng, J. L. Zhang, Z. H. Sun, L. H. Zhou, G. J. Deng, S. P. Li, W. J. Li, P. Gong and L. T. Cai, Theranostics, 2018, 8, 6025–6034 CrossRef CAS PubMed.
- C. Jin, Q. M. Zhang and W. Lu, Eur. J. Med. Chem., 2017, 132, 135–141 CrossRef CAS PubMed.
- A. Hajime, T. Norihiko, I. Takayuki, S. Hiroaki and N. Satoshi, Nat. Commun., 2019, 10, 2824 CrossRef PubMed.
- L. W. Andersen, M. J. Holmberg, K. M. Berg, M. W. Donnino and A. Granfeldt, JAMA, J. Am. Med. Assoc., 2019, 321, 1200–1210 CrossRef PubMed.
- H. K. Eltzschig and T. Eckle, Nat. Med., 2011, 17, 1391–1401 CrossRef CAS PubMed.
- Z. Liu, M. Wang, H. Wang, L. Fang and S. Gou, J. Med. Chem., 2020, 63, 186–204 CrossRef CAS PubMed.
- S. Cadenas, Free Radicals Biol. Med., 2018, 117, 76–89 CrossRef CAS PubMed.
- M. Molitor, W. S. Rudi, V. Garlapati, S. Finger, R. Schuler, S. Kossmann, J. Lagrange, T. S. Nguyen, J. Wild, T. Knopp, S. H. Karbach, M. Knorr, W. Ruf, T. Munzel and P. Wenzel, Cardiovasc. Res., 2021, 117, 162–177 CrossRef CAS PubMed.
- Y. Liu, M. Paterson, S. L. Baumgardt, M. G. Irwin, Z. Xia, Z. J. Bosnjak and Z. D. Ge, Cardiovasc. Res., 2019, 115, 168–178 CrossRef CAS PubMed.
- N. M. Rogers, M. Sharifi-Sanjani, M. Y. Yao, K. Ghimire, R. Bienes-Martinez, S. M. Mutchler, H. E. Knupp, J. Baust, E. M. Novelli, M. Ross, C. St Croix, J. C. Kutten, C. A. Czajka, J. C. Sembrat, M. Rojas, D. Labrousse-Arias, T. N. Bachman, R. R. Vanderpool, B. S. Zuckerbraun, H. C. Champion, A. L. Mora, A. C. Straub, R. A. Bilonick, M. J. Calzada and J. S. Isenberg, Cardiovasc. Res., 2017, 113, 15–29 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, results and figures. See DOI: 10.1039/d2sc00048b |
| This journal is © The Royal Society of Chemistry 2022 |