
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidative N–H/P–H cross-coupling with H2 evolution towards the synthesis of tertiary phosphines†
Yong
Yuan‡
 abc,
Xue
Liu‡
a,
Jingcheng
Hu
b,
Pengjie
Wang
b,
Shengchun
Wang
b,
Hesham
Alhumade
d and
Aiwen
Lei
*abe
abc,
Xue
Liu‡
a,
Jingcheng
Hu
b,
Pengjie
Wang
b,
Shengchun
Wang
b,
Hesham
Alhumade
d and
Aiwen
Lei
*abe
aNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, P. R. China. E-mail: aiwenlei@whu.edu.cn
bCollege of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China
cCollege of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, China
dDepartment of Chemical and Materials Engineering, Center of Research Excellence in Renewable Energy and Power Systems, King Abdulaziz University, Jeddah 21589, Saudi Arabia
eKing Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 14th February 2022
Abstract
Tertiary phosphines(III) find widespread use in many aspects of synthetic organic chemistry. Herein, we developed a facile and novel electrochemical oxidative N–H/P–H cross-coupling method, leading to a series of expected tertiary phosphines(III) under mild conditions with excellent yields. It is worth noting that this electrochemical protocol features very good reaction selectivity, where only a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of amine and phosphine was required in the reaction. Moreover, this electrochemical protocol proved to be practical and scalable. Mechanistic insights suggested that the P radical was involved in this reaction.
:1 ratio of amine and phosphine was required in the reaction. Moreover, this electrochemical protocol proved to be practical and scalable. Mechanistic insights suggested that the P radical was involved in this reaction.
Introduction
Tertiary phosphines(III) find widespread use in many aspects of synthetic organic chemistry.1 For example, tertiary phosphines(III) as useful phosphorus ligands in transition metal catalyzed reactions have greatly promoted the development of organometallic chemistry (Scheme 1A).2 Furthermore, incorporation of a phosphorus(III) group into organic molecules is important for the construction of structurally diverse molecules since PIII chelation-assisted C–H functionalization can introduce various functional groups into organic molecules (Scheme 1B).3 As such, efficient and practical methods for the synthesis of tertiary phosphines are highly desirable. Among the diverse approaches for accessing tertiary phosphines(III),4 transition metal-catalyzed cross-coupling of secondary phosphines with aryl halides (or triflates) is considered one of the conceptually most reliable methods.5 However, since tertiary phosphines(III) as very good ligands can easily poison transition metal catalysts, methods for the synthesis of tertiary phosphines(III) via transition metal-catalyzed cross-coupling reactions still remain challenging. The hydrophosphination of alkenes/alkynes under transition metal catalyst-free conditions is also a known method for obtaining tertiary phosphines(III);6 however, this strategy has only realized the construction of C–P bonds. Alternatively, oxidative cross-coupling is recognized as a more ideal method for obtaining tertiary phosphines(III). However, since the P–H reagent is easily overoxidized to generate phosphine oxide and an oxidant is essential to remove surplus electrons,7 oxidative cross-coupling with the P–H reagent for accessing tertiary phosphines(III) is quite difficult.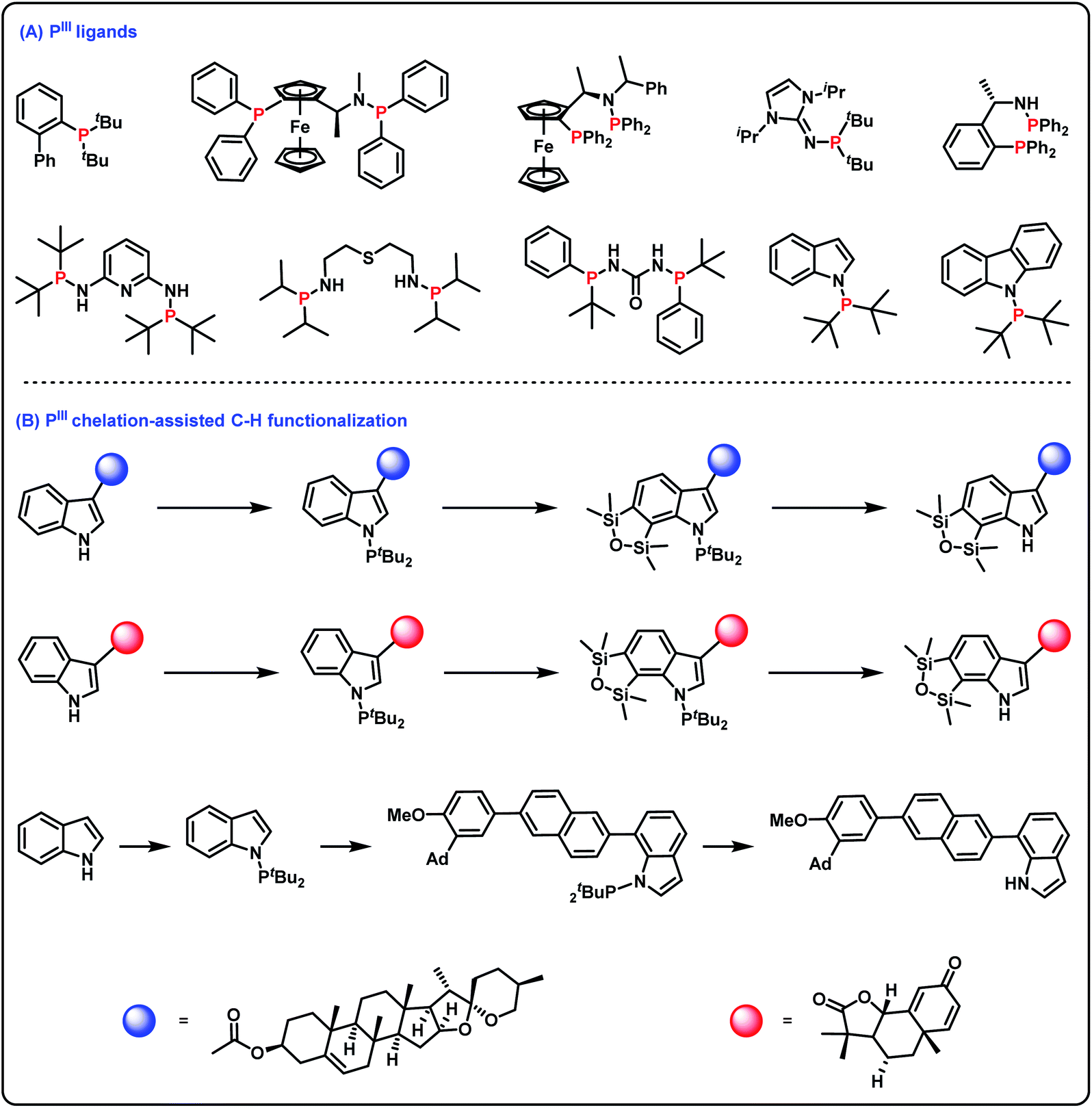 | ||
| Scheme 1 Important surrogates of PIII groups. (A) PIII ligands; (B) PIII chelation-assisted C–H functionalization. | ||
As an attractive alternative to traditional chemical oxidants, electrosynthesis achieves the function of chemical oxidants by using an anode,8 and thus can not only realize oxidative cross-coupling reactions under exogenous-oxidation-free conditions,9 but also provide a new opportunity for oxidative cross-coupling reactions that cannot occur with traditional chemical oxidants. Over the past five years, R1–H/R2–H cross-coupling reactions via electrochemical oxidation have been extensively researched.10 However, the reported electrochemical oxidative cross-coupling reactions have mainly focused on the oxidative cross-coupling of C–H with X–H (X = heteroatom), and the electrochemical oxidative X–H/Y–H (Y = heteroatom) cross-coupling for X–Y bond formation has rarely been reported.11 For example, to the best of our knowledge, the method for the synthesis of tertiary phosphines(III) via electrochemical oxidative N–H/P–H cross-coupling with H2 evolution has never been reported. As a part of our recent research interest in the field of electrochemical oxidative R1–H/R2–H cross-coupling with H2 evolution,12 we herein report a novel electrochemical oxidative N–H/P–H cross-coupling reaction under transition metal catalyst-free conditions for the first time. By using electricity as the primary energy input and KI as the mediator, oxidation side reactions were avoided and a series of important tertiary phosphines(III) were synthesized under mild reaction conditions.
Results and discussion
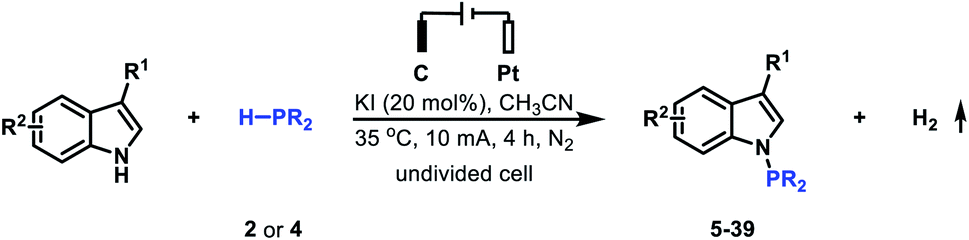
The introduction of a PtBu2 group into the N-1 position of indoles has been proved to be significant since the generated tertiary phosphine products are versatile intermediates for obtaining various indoles. To access this class of important tertiary phosphines, the cross-coupling reaction of indole 1 with di-tert-butylphosphine 2 was initially studied to evaluate the reaction conditions (Table 1). After a series of screening of reaction partners, we delightfully found that when the reaction was conducted with the use of 20 mol% KI in MeCN under a 10 mA constant current for 4 h, the electrolysis worked smoothly in an undivided cell, providing the desired N–P coupled product 3 in 95% yield (Table 1, entry 1). It is worth noting that when the reaction was finished, H2 could also be detected (see the ESI† for details). Further investigation showed that the use of electricity as the primary energy input was a key to the success of this reaction (Table 1, entry 2). KI was also important for generating N–P coupled product 3 in excellent yield. Using NaI, LiI, nBu4NCl, nBu4NBr or nBu4NI resulted in the corresponding N–H/P–H cross-coupling products in moderate to high yield (Table 1, entries 3–7); whereas when nBu4NBF4, KF, KCl, or KBr was employed in the reaction, the desired N–P coupled product 3 was obtained in 0–31% yield (Table 1, entries 8–11). The amount of KI was next explored. However, either increasing the amount of KI to 50 mol% or decreasing the amount of KI to 10 mol% furnished the N–P coupled product 3 in slightly decreased yield (Table 1, entries 12 and 13). Increasing the operating current to 15 mA nearly did not alter the reaction efficiency (Table 1, entry 14); whereas when the reaction was conducted with a 5 mA constant current, a slight loss in yield was observed (Table 1, entry 15). For the reaction, the combination of a graphite plate anode and a platinum plate cathode was found to be optimal. Either using a stainless steel plate cathode or a platinum plate anode furnished the corresponding N–H/P–H cross-coupling products in decreased yield (Table 1, entries 16 and 17).|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Reaction conditions: graphite plate (15 mm × 15 mm × 1.0 mm) as the anode, platinum plate (20 mm × 15 mm × 0.3 mm) as the cathode, undivided cell, 1 (0.5 mmol), 2 (0.5 mmol), KI (20 mol%), MeCN (10.0 mL), N2, 35 °C, 4 h. b Isolated yields. c 0.1 mmol of nBu4NBF4 was added to promote the electron transfer in solution. | ||
| 1 | None | 95 |
| 2 | No electric current | 0 |
| 3c | NaI instead of KI | 79 |
| 4c | LiI instead of KI | 65 |
| 5 | n Bu4NCl instead of KI | 35 |
| 6 | n Bu4NBr instead of KI | 66 |
| 7 | n Bu4NI instead of KI | 59 |
| 8 | n Bu4NBF4 instead of KI | 0 |
| 9c | KF instead of KI | 0 |
| 10c | KCl instead of KI | 23 |
| 11c | KBr instead of KI | 31 |
| 12 | 50 mol% KI instead of 20 mol% KI | 80 |
| 13 | 10 mol% KI instead of 20 mol% KI | 79 |
| 14 | 15 mA, 2.7 h instead of 10 mA, 4 h | 90 |
| 15 | 5 mA, 8 h instead of 10 mA, 4 h | 79 |
| 16 | Stainless steel plate instead of platinum plate | 75 |
| 17 | Platinum plate instead of graphite plate | 84 |

After establishing the optimal reaction conditions, we set out to examine the generality of this electrochemical method (Table 2). Delightfully, the electrochemical oxidative cross-coupling reactions worked well with a wide range of indole derivatives. The reaction with 3-methylindole or 3-indoleacetonitrile gave the desired N–P bond formation products in 98% and 94% yield (Table 2, 5 and 6), respectively. Tryptophol, 1-(2-(1H-indol-3-yl)ethyl)piperidin-2-one, and 3-acetylindole were converted into the corresponding cross-coupling products in 84–86% yields (Table 2, 7–9). In comparison, the C-3 substituted indoles bearing acetyloxy, acetylamino, acyl, alkenyl and phenylthio resulted in the desired products in moderate to good yields (Table 2, 10–14). The C-4, C-5, and C-6 substituted indoles were all compatible with the current electrochemical conditions. Both electron-withdrawing substituents and electron-donating groups at the C-4, C-5, or C-6 position of the indole ring did not interfere with the reaction efficiency, yielding the corresponding N–H/P–H cross-coupling products in excellent yields (Table 2, 15–25). Note that synthetically valuable halogen atoms such as F, Cl, and Br were compatible with the electrochemical conditions to give the corresponding N–H/P–H cross-coupling products in 92–95% yields (Table 2, 16–18, 20, 23). The C-7 substituted indoles, which probably because of steric hindrance were less reactive than C-3, C-4, C-5, and C-6 substituted indoles, gave the desired N–H/P–H cross-coupling products in 65–80% yields (Table 2, 26 and 27). Disubstituted indoles were also suitable cross-coupling partners, providing the corresponding N–P bond formation products in moderate to excellent yields (Table 2, 28–31). Besides substituted indoles, 4-azaindole and 7H-pyrrolo[2,3-d]pyrimidine were also compatible with the standard reaction conditions, producing the N–H/P–H cross-coupling products 32 and 33 in 75% and 41% yield, respectively. Notably, in addition to di-tert-butylphosphine, diphenylphosphine was also a good reaction partner to react with various indoles, generating the desired N–P coupled products 34–39 in moderate yields.
|
|
|---|
| a Reaction conditions: graphite plate (15 mm × 15 mm × 1.0 mm) as the anode, platinum plate (20 mm × 15 mm × 0.3 mm) as the cathode, undivided cell, N–H compounds (0.5 mmol), 2 or 4 (0.5 mmol), KI (20 mol%), MeCN (10.0 mL), N2, 35 °C, 4 h, isolated yields. b Yields were determined by 31P NMR using P(OEt)3 as an internal standard. |

|
To demonstrate the practicality of this electrochemical N–H/P–H cross-coupling method, the gram scale reaction of indole 1 with di-tert-butylphosphine 2 on a 5.0 mmol scale was conducted (Scheme 2). To our delight, when the reaction was performed with a controlled current of 20 mA for 20 h, 1.08 g cross-coupling product 3 could be obtained.
 | ||
| Scheme 2 Gram-scale synthesis. | ||
For comparative purpose, the N–H/P–H cross-coupling reaction between indole 1 and di-tert-butylphosphine 2 was performed with chemical oxidants in the absence of electrical input (Table 3). However, none of the chemical oxidants (DDQ, m-CPBA, CAN, K2S2O8, TBHP, DTBP, I2, and NIS) we examined could furnish the desired N–H/P–H cross-coupling product 3 (Table 3, entries 2–9).
|
|
||
|---|---|---|
| Entry | Conditions | Yield (%) |
| 1 | Standard conditions | 95 |
| 2 | No electric current, KI (20 mol%), CH3CN, 35 °C, DDQ (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 3 | No electric current, KI (20 mol%), CH3CN, 35 °C, m-CPBA (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 4 | No electric current, KI (20 mol%), CH3CN, 35 °C, CAN (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 5 | No electric current, KI (20 mol%), CH3CN, 35 °C, K2S208 (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 6 | No electric current, KI (20 mol%), CH3CN, 35 °C, TBHP (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 7 | No electric current, KI (20 mol%), CH3CN, 35 °C, DTBP (1.0 equiv.), 4 h, N2 | 0 (1 recovered) |
| 8 | No electric current, I2 (1.0 equiv.), CH3CN, 35 °C, 4 h, N2 | 0 (1 recovered) |
| 9 | No electric current, NIS (1.0 equiv.), CH3CN, 35 °C, 4 h, N2 | 0 (1 recovered) |

To better understand the mechanism of this electrochemical N–H/P–H cross-coupling reaction, cyclic voltammetry (CV) experiments on KI, indole 1 and di-tert-butylphosphine 2 were performed (Fig. S1,† see the ESI for details). The oxidation peaks of KI were observed at 0.70 V and 0.90 V, respectively; whereas the oxidation peaks of indole 1 and di-tert-butylphosphine 2 were both greater than 0.90 V, suggesting that the iodide ion and even the iodine radical are more easily oxidized than both indole 1 and di-tert-butylphosphine 2.
Electron paramagnetic resonance (EPR) experiments were also carried out to investigate the details of this electrochemical N–H/P–H cross-coupling reaction (Fig. 1). Electrolyzing indole 1 under standard conditions for 15 min, a mixed signal of the 3,4-dihydro-2,2-dimethyl-2H-pyrrole 1-oxide (DMPO) trapping nitrogen radical (AN = 13.8, AH = 17.8, and AN = 3.0) and oxidized DMPO (AN = 14.1) was identified (Fig. 1A). Electrolyzing di-tert-butylphosphine 2 under standard conditions for 30 min, a mixed signal of the DMPO trapping phosphorus radical (AN = 15.4, AH = 20.6, and AP = 25.7) and DMPO trapping tert-butyl radical (AN = 14.6, AH = 20.7) was identified (Fig. 1C). The adduct of the phosphorus radical to DMPO was also detected by high resolution mass spectrometry (see the ESI† for details). By contrast, a relatively weak radical signal was observed under the conditions of absence of KI (Fig. 1D), whereas when indole 1 was electrolyzed in the absence of KI, no radical signal was observed (Fig. 1B). These results indicated that KI not only played the role of the electrolyte, but also acted as a mediator to access the phosphorus radical and nitrogen radical.
 | ||
| Fig. 1 Electron paramagnetic resonance (EPR) spectra. | ||
Control experiments were performed to clarify whether di-tert-butyliodophosphine 40 is the key intermediate for generating the N–P coupled product (Scheme 3). Replacing di-tert-butylphosphine 2 with di-tert-butyliodophosphine 40 produces the desired N–H/P–H cross-coupling product 3 in 72% yield; whereas when the reaction was conducted in the absence of electrical input or KI, no or only 13% yield of N–P coupled product 3 was formed. These results indicated that di-tert-butyliodophosphine 40 might be a key reaction intermediate for generating product 3 and both electric current and KI are important for oxidizing indole 1 to nitrogen radical.
 | ||
| Scheme 3 Control experiments. | ||
Based on the experiments described above and previous reports,13,14 a plausible reaction mechanism between indole 1 and di-tert-butylphosphine 2 is presented in Scheme 4. The anodic oxidation of the iodide ion leads to the formation of the iodine radical. The iodine radical reacts with di-tert-butylphosphine 2 to furnish the phosphorus radical and then to access di-tert-butyliodophosphine 40. At the same time, indole 1 reacts with in situ generated I+ to form an unstable N-iodo intermediate. The homolysis of the unstable N–I bond provides the nitrogen radical and iodine radical. Finally, radical–radical cross-coupling between the phosphorus radical and nitrogen radical gives the desired N–P coupled product 3.
 | ||
| Scheme 4 Proposed mechanism. | ||
Conclusions
In summary, we have developed a facile and novel electrochemical oxidative N–H/P–H cross-coupling reaction for the synthesis of tertiary phosphines. The reaction was performed in a simple undivided cell with excellent yields. Good reaction selectivity was achieved with a 1:1 ratio of amine and phosphine. This electrochemical protocol provides a new way for the synthesis of tertiary phosphines. We anticipate that this work will stimulate the research interest of chemists in the P radical.
Data availability
Data for this work, including experimental procedures and characterization data for all new compounds are provided in the ESI.†Author contributions
A. L. conceived the project. A. L. and Y. Y. designed the experiments. Y. Y., X. L., J. H., P. W. and S. W. performed and analyzed experiments. Y. Y., A. L. and H. A. wrote the manuscript. X. L. wrote the ESI† and contributed other related materials. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2021YFA1500100), the National Natural Science Foundation of China (22031008) and the Science Foundation of Wuhan (2020010601012192). This research work was also funded by Institution Fund Projects under grant no. (IFPIP: 438-135-1442). The authors gratefully acknowledge technical and financial support from the Ministry of Education and the King Abdulaziz University, DSR, Jeddah, Saudi Arabia.Notes and references
- (a) N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS PubMed; (b) M. E. Dmitriev and V. V. Ragulin, Russ. J. Gen. Chem., 2011, 81, 1786 CrossRef CAS; (c) A. Hassanabadi, M. H. Mosslemin, E. Abyar and M. Taleb-Malamiri, J. Chem. Res., 2012, 36, 497–499 CrossRef CAS.
- (a) D.-Y. Wang, X.-P. Hu, J. Deng, S.-B. Yu, Z.-C. Duan and Z. Zheng, J. Org. Chem., 2008, 73, 2011–2014 CrossRef CAS PubMed; (b) T. Gross, S. Chou, A. Dyke, B. Dominguez, M. Groarke, J. Medlock, M. Ouellette, J. P. Reddy, A. Seger, S. Zook and A. Zanotti-Gerosa, Tetrahedron Lett., 2012, 53, 1025–1028 CrossRef CAS; (c) P. A. Donets and N. Cramer, J. Am. Chem. Soc., 2013, 135, 11772–11775 CrossRef CAS PubMed; (d) T. Schlatzer and R. Breinbauer, Adv. Synth. Catal., 2021, 363, 668–687 CrossRef CAS PubMed; (e) C. You, S. Li, X. Li, H. Lv and X. Zhang, ACS Catal., 2019, 9, 8529–8533 CrossRef CAS; (f) N. W. Boaz, E. B. Mackenzie, S. D. Debenham, S. E. Large and J. A. Ponasik Jr, J. Org. Chem., 2005, 70, 1872–1880 CrossRef CAS PubMed.
- (a) D. Wang, X. Chen, J. J. Wong, L. Jin, M. Li, Y. Zhao, K. N. Houk and Z. Shi, Nat. Commun., 2021, 12, 524 CrossRef CAS PubMed; (b) A. J. Borah and Z. Shi, J. Am. Chem. Soc., 2018, 140, 6062–6066 CrossRef CAS PubMed; (c) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504–1508 CrossRef CAS PubMed; (d) J. Wen, D. Wang, J. Qian, D. Wang, C. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 2078–2082 CrossRef CAS PubMed.
- (a) L. M. Broomfield, Y. Wu, E. Martin and A. Shafir, Adv. Synth. Catal., 2015, 357, 3538–3548 CrossRef CAS; (b) N. P. Kenny, K. V. Rajendran, E. V. Jennings and D. G. Gilheany, Chem.–Eur. J., 2013, 19, 14210–14214 CrossRef CAS PubMed.
- E. G. N. Yu, A. Veits and O. S. Vinogradova, Russ. J. Gen. Chem., 2005, 75, 1060–31068 CrossRef.
- (a) D. Bissessar, J. Egly, T. Achard, P. Steffanut and S. Bellemin-Laponnaz, RSC Adv., 2019, 9, 27250–27256 RSC; (b) Y. Moglie, M. J. González-Soria, I. Martín-García, G. Radivoy and F. Alonso, Green Chem., 2016, 18, 4896–4907 RSC; (c) A. N. Barrett, H. J. Sanderson, M. F. Mahon and R. L. Webster, Chem. Commun., 2020, 56, 13623–13626 RSC; (d) T. Bunlaksananusorn and P. Knochel, Tetrahedron Lett., 2002, 43, 5817–5819 CrossRef CAS; (e) H. Hu and C. Cui, Organometallics, 2012, 31, 1208–1211 CrossRef CAS.
- (a) T. L. Gianetti, R. E. Rodríguez-Lugo, J. R. Harmer, M. Trincado, M. Vogt, G. Santiso-Quinones and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 15323–15328 CrossRef CAS PubMed; (b) Y. Huang, Y. Li, P. H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865–4868 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (b) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (c) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (d) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (e) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- (a) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058–10086 RSC; (b) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed.
- (a) L.-Y. Xie, S. Peng, F. Liu, J.-Y. Yi, M. Wang, Z. Tang, X. Xu and W.-M. He, Adv. Synth. Catal., 2018, 360, 4259–4264 CrossRef CAS; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (c) Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS PubMed; (d) Y.-Z. Yang, Y.-C. Wu, R.-J. Song and J.-H. Li, Chem. Commun., 2020, 56, 7585–7588 RSC; (e) Y. Wang, P. Qian, J.-H. Su, Y. Li, M. Bi, Z. Zha and Z. Wang, Green Chem., 2017, 19, 4769–4773 RSC; (f) S. Herold, D. Bafaluy and K. Muñiz, Green Chem., 2018, 20, 3191–3196 RSC; (g) L. Zhang, Z. Zhang, J. Zhang, K. Li and F. Mo, Green Chem., 2018, 20, 3916–3920 RSC; (h) S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252–255 CrossRef CAS PubMed.
- (a) H. Liang, L.-J. Wang, Y.-X. Ji, H. Wang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1839–1844 CrossRef CAS PubMed; (b) Z. Yang, J. Zhang, L. Hu, L. Li, K. Liu, T. Yang and C. Zhou, J. Org. Chem., 2020, 85, 3358–3363 CrossRef CAS PubMed; (c) R. Wang, X. Dong, Y. Zhang, B. Wang, Y. Xia, A. Abdukader, F. Xue, W. Jin and C. Liu, Chem.–Eur. J., 2021, 27, 14931–14935 CrossRef CAS PubMed; (d) N. Sbei, G. M. Martins, B. Shirinfar and N. Ahmed, Chem. Rec., 2020, 20, 1530–1552 CrossRef CAS PubMed; (e) Y. Deng, S. You, M. Ruan, Y. Wang, Z. Chen, G. Yang and M. Gao, Adv. Synth. Catal., 2020, 363, 464–469 CrossRef.
- (a) H. Wang, M. He, Y. Li, H. Zhang, D. Yang, M. Nagasaka, Z. Lv, Z. Guan, Y. Cao, F. Gong, Z. Zhou, J. Zhu, S. Samanta, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2021, 143, 3628–3637 CrossRef CAS PubMed; (b) Y. Yuan, Y. Cao, J. Qiao, Y. Lin, X. Jiang, Y. Weng, S. Tang and A. Lei, Chin. J. Chem., 2019, 37, 49–52 CrossRef CAS; (c) Q. Wang, X. Zhang, P. Wang, X. Gao, H. Zhang and A. Lei, Chin. J. Chem., 2021, 39, 143–148 CrossRef CAS.
- S. Wang, S. Tang and A. Lei, Sci. Bull., 2018, 63, 1006–1009 CrossRef CAS.
- (a) P. C. L. Hermosilla, J. M. García de la Vega and C. Sieiro, J. Phys. Chem. A, 2005, 109, 1114–1124 CrossRef PubMed; (b) K. Schwedtmann, S. Schulz, F. Hennersdorf, T. Strassner, E. Dmitrieva and J. J. Weigand, Angew. Chem., Int. Ed., 2015, 54, 11054–11058 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07248j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |