
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolation of the elusive bisbenzimidazole Bbim3−˙ radical anion and its employment in a metal complex†
Florian
Benner
 and
Selvan
Demir
*
and
Selvan
Demir
*
Department of Chemistry, Michigan State University, 578 South Shaw Lane, East Lansing, Michigan 48824, USA. E-mail: sdemir@chemistry.msu.edu
First published on 14th April 2022
Abstract
The discovery of singular organic radical ligands is a formidable challenge due to high reactivity arising from the unpaired electron. Matching radical ligands with metal ions to engender magnetic coupling is crucial for eliciting preeminent physical properties such as conductivity and magnetism that are crucial for future technologies. The metal-radical approach is especially important for the lanthanide ions exhibiting deeply buried 4f-orbitals. The radicals must possess a high spin density on the donor atoms to promote strong coupling. Combining diamagnetic 89Y (I = 1/2) with organic radicals allows for invaluable insight into the electronic structure and spin-density distribution. This approach is hitherto underutilized, possibly owing to the challenging synthesis and purification of such molecules. Herein, evidence of an unprecedented bisbenzimidazole radical anion (Bbim3−˙) along with its metalation in the form of an yttrium complex, [K(crypt-222)][(Cp*2Y)2(μ-Bbim˙)] is provided. Access of Bbim3−˙ was feasible through double-coordination to the Lewis acidic metal ion and subsequent one-electron reduction, which is remarkable as Bbim2− was explicitly stated to be redox-inactive in closed-shell complexes. Two molecules containing Bbim2− (1) and Bbim3−˙ (2), respectively, were thoroughly investigated by X-ray crystallography, NMR and UV/Vis spectroscopy. Electrochemical studies unfolded a quasi-reversible feature and emphasize the role of the metal centre for the Bbim redox-activity as neither the free ligand nor the Bbim2− complex led to analogous CV results. Excitingly, a strong delocalization of the electron density through the Bbim3−˙ ligand was revealed via temperature-dependent EPR spectroscopy and confirmed through DFT calculations and magnetometry, rendering Bbim3−˙ an ideal candidate for single-molecule magnet design.
Introduction
The stabilization of organic radicals and molecules comprising an open shell ligand, respectively, has always been exceptionally challenging which can be ascribed to the high reactivity arising from the unpaired electron that is able to initiate various side reactions such as dimerization or electron transfer reactions.1–3 Notably, specific nitrogen-containing heteroaromatic compounds can house an unpaired electron and coordinate to metal ions while preserving their open shell nature which led to complexes with unprecedented chemical and physical properties.4,5 For instance devised transition metal catalysts containing radical ligands show boosted chemical reactivity due to ligand-based redox processes and as such are a real alternative to heavier d-block catalysts comprising expensive metals.6 Importantly, the implementation of radical units engenders unique physical properties, which is reflected most notably in conductivity7,8 and magnetism. Specifically regarding the latter and in stark contrast to the effects of closed-shell ligands, diffuse radical spin orbitals provide an efficient platform for direct magnetic exchange coupling.9,10 Here, the spin density on the coordinating nitrogen donor bridges needs to be sufficiently large to allow for strong coupling which together with high magnetic anisotropy may result in paramount magnetic materials whether it be solid-state of higher nuclearity or of molecular nature.9,11 A low spin density on the donors of the open shell ligands, as is often observed for nitroxide-based radicals,10 usually leads in combination with paramagnetic lanthanide ions innate to contracted 4f orbitals to weak magnetic exchange. This diminishes the excellence of multi-metallic single-molecule magnets (SMMs) which is a class of molecules that feature magnetic memory and have potential applications in high-density information storage and spin-based electronics.12–14 Specifically, weak magnetic coupling typically results in fast magnetic relaxation with a lack of remanent magnetization, where the single-molecule magnet behaviour originates primarily from the single-ion effect.10,15–18Notably, over the past decade, only a handful of new radical ligands, with higher spin densities than seen for nitronyl nitroxides, could be successfully generated encased between lanthanide ions. In fact, the combination of the highly anisotropic tripositive dysprosium and terbium ions with the radical anions of bipyrimidine,11,19 tetra-2-pyridinylpyrazine,20 indigo,17 and hexaazatrinaphthalene,21 gave rise to powerful single-molecule magnets with substantial coercivities and remanence, Fig. 1. The largest coercivity for any molecule was attained with the compact and highly anionic N23−˙ radical bridge.9 Although impressive, this radical unit is highly reactive precluding both synthetic modifications and access of larger molecular clusters. Since the retention of magnetization aka open magnetic hysteresis at high temperatures is of utmost importance for realizing the applications of single-molecule magnets, new controllable radical ligands are in high demand that are able to engender strong magnetic coupling to fulfil the aspiration for multinuclear single-molecules with large overall ground states. Intriguingly, none of the probed organic radicals, Fig. 1, has been studied in a complex containing diamagnetic rare earth metal ions. In particular, the use of yttrium would be beneficial as it possesses a stable isotope (89Y) with a natural abundance of 100% and a nuclear spin of ½ enabling a profound insight into the electronic structure though subjecting the corresponding yttrium molecules to invaluable characterisation methods such as EPR spectroscopy and computations, respectively. Despite a clear path, transferring the analogous synthetic path for the lanthanide molecules to the respective yttrium congeners is greatly challenging possibly due to the ionic size difference, albeit small, and to the greater electropositivity of the rare earth. Remarkably, only two tetradentate radical ligand-containing yttrium metallocene complexes were hitherto isolated,22,23 where neither were employed in the development of single-molecule magnets. Hence, our synthetic strategy relies on generating new radical bridging ligands and capturing them in between trivalent yttrium ions to access invaluable spin density distribution by EPR spectroscopy24 which will provide a handle to assess the suitability of radical ligands for strong magnetic exchange coupling when combined with paramagnetic metals. Our approach includes in-depth computational studies to gain insight into the singly occupied molecular orbital (SOMO). Here, we show the isolation of an unprecedented radical ligand trapped between two yttrium centres and provide full investigation encompassing single crystal X-ray diffraction, EPR spectroscopy, cyclic voltammetry, and DFT studies. The quest for strongly coupled multinuclear single-molecule magnets with large overall spin ground states necessitates the evolution of new radical ligands that foster efficient exchange pathways between metal ions. Hence, we focused on the development of new nitrogen-based radicals by probing synthetically and electrochemically the accessibility of numerous oxidation states.
 | ||
| Fig. 1 Radical anions of the organic ligands 5,6,11,12,17,18-hexaazatrinaphthylene (han), 2,3,5,6-tetrakis(2-pyridyl)pyrazine (tppz), indigo (ind), and 2,2′-bipyrimidine (bpym) employed for bi- and trimetallic single-molecule magnets, and the discovered radical anion of 2,2′-bisbenzimidazole (Bbim). | ||
The organic molecule 2,2-bisbenzimidazole (Bbim) as a potentially redox-active ligand sparked our interest since it possesses coordinative ability where all four nitrogen donor atoms bind to metal centres as seen in transition metal complexes,25–27 and an aromatic nature inherent to π* orbitals which are required for the uptake of an electron. Although innate to these paramount characteristics, to the best of our knowledge, neither a Bbim nor a Bbim-based radical have been hitherto isolated, crystallographically characterised and spectroscopically detected, respectively. Noteworthy, even electrochemical investigations on the bare Bbim ligand are unknown. Strikingly, multiple studies on Bbim-bridged complexes indeed highlight the redox-inactivity of Bbim.25,26,28,29 Recognizing the synthetic yet inspiring challenge, we formulated synthetic routes to isolate the first Bbim radical captured in between sterically saturated YIII ions.
Experimental methods
General information
All manipulations were performed under inert conditions using either standard Schlenk techniques or nitrogen-filled glovebox. House nitrogen was purified through a MBraun HP-500-MO-OX gas purifier. nHexane, toluene and THF were purified by refluxing over potassium using benzophenone as an indicator and distilled prior to use. The chemicals pentamethylcyclopentadiene (Cp*H), allylmagnesium chloride (2.0 M in THF), potassium bistrimethylsilylamide (KN(Si(CH3)3)2), anhydrous YCl3, and 2.2.2-cryptand (crypt-222) were purchased from Sigma-Aldrich. 2.2.2-Cryptand (crypt-222) was recrystallized from nhexane prior to use. KCp*,30 (HNEt3)(BPh4),31 Cp*2Y(BPh4),19 H2Bbim,32 (Li(TMEDA))2Bbim33 and KC8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 were synthesized according to literature procedures.
34 were synthesized according to literature procedures.
Synthesis of K2Bbim
A solution of potassium bistrimethylsilylamide (KN(Si(CH3)3)2) (0.5386 g, 2.288 mmol, in THF) was added dropwise to a yellow suspension of H2Bbim (1.0551 g, 5.0292 mmol in 10 mL THF) while stirring. An immediate colour change to blue was observed. After completed addition, the mixture was stirred for 21 h at room temperature, upon which a colourless solid formed. The mixture was centrifuged, and the yellow supernatant was removed. The remaining yellowish solid was thoroughly washed with ∼10 mL THF and centrifuged again. After removal of the supernatant the solid was washed twice with THF. The obtained off-white solid was dried under vacuum and, after removal of the majority of solvent, dried under vacuum at 45 °C for 40 min to obtain a fluffy colourless solid (yield: 0.6428 g, 2.071 mmol, 90%). Anal. calcd for C14H8K2N4: C, 54.17; H, 2.60; N, 18.05. Found: C, 54.50; H, 2.94; N, 17.61.Synthesis of (Cp*2Y)2(μ-Bbim), 1
To a THF solution (13 mL) of the yttrium tetraphenylborate complex Cp*2Y(BPh4) (0.2005 g, 0.2955 mmol) the off-white bisbenzimidazole ligand K2Bbim (0.0458 g, 0.148 mmol) was added as a solid. An immediate colour change to yellow and the formation of colourless solids could be observed. The mixture was stirred for 3.5 h, briefly left standing for the solids to settle, and filtrated through a Celite plug. The clear yellow solutions were evaporated to dryness to afford yellow and colourless solids which were extracted three times with toluene. Colourless insoluble solids were removed via filtration through Celite to give rise to a clear, yellow filtrate which was evaporated to dryness. The resulting yellow solids were redissolved in a minimum amount of toluene and stored at −30 °C for crystallization. Crystalline yield of 1: 0.0804 g (pale yellow block-shaped crystals, 0.0889 mmol, 60%). 1H NMR (THF-d6, 500 MHz) δ (ppm) = 7.47 (dd, 3JH–H = 3.40 Hz, 6.09 Hz, 2H, Ha/Hb), 7.26 (dd, 3JH–H = 3.40 Hz, 6.09 Hz, 1H, Ha/Hb), 1.82 (s, 15H, HMe). Additional signals: δ (ppm) = 3.58 (s, THF-d8), 1.73 (s, THF-d8). 13C NMR (THF-d8, 125 MHz) δ (ppm) = 144.9 (Cc/Cd), 123.1 (s, Ca/Cb), 119.3 (s, CCp), 118.4 (s, Ca/Cb), 11.7 (CMe). Additional signals: δ (ppm) = 67.6 (q, THF-d8), 25.5 (q, THF-d8). Anal. calcd for C54H68N4Y2: C, 68.20; H, 7.21; N 5.89. Found: C, 68.24; H, 7.18; N 5.76. IR (ATR, cm−1): 3050 (w), 2904 (m), 2856 (m), 2723 (vw), 1604 (vw), 1572 (vw), 1442 (m), 1362 (vs), 1346 (vs), 1279 (s), 1251 (m), 1165 (vw), 1140 (vw), 1120 (w), 1060 (vw), 1020 (m), 1008 (m), 978 (w), 913 (m), 801 (vw), 777 (m), 741 (vs), 689 (w).Synthesis of [K(crypt-222)][(Cp*2Y)2(μ-Bbim˙)], 2
To a THF solution of the yttrium complex, 1, (0.0541 g, 0.0569 mmol, in 5–8 mL THF), 2.2.2-cryptand (0.0215 g, 0.0571 mmol, in 1 mL THF) was added and stirred for 5 min. Addition of KC8 (0.0077 g, 0.057 mmol) resulted in an immediate colour change to dark green and the formation of insoluble grey materials, presumably graphite. After stirring for 20 min, the mixture was filtered to remove insoluble solids and the clear, dark green filtrate was stored at −30 °C for crystallization. Crystalline yield of 2: 0.0254 mg (dark green block-shaped crystals, 0.0186 mmol, 33%). The low solubility and paramagnetic nature of 2 precluded an assignment of the NMR signals. The 13C NMR shifts were obtained from a 1H/13C-HSQC-NMR spectrum while no signals were found in a standard 13C NMR spectrum. 1H NMR (THF-d8, 500 MHz) δ (ppm) = 3.66 (s, 2.2.2-crypt), 3.53 (s, 2H, 2.2.2-crypt), 2.54 (s, 2H, 2.2.2-crypt), 2.05–0.64 (br s). Additional signals: δ (ppm) = 3.58 (s, THF-d8), 1.78, (s, THF) 1.73 (s, THF-d8). 13C NMR (THF-d8, 125 MHz) δ (ppm) = 67.36 (crypt-222), 54.9 (s, crypt-222). Additional signals: δ (ppm) = 67.4 (q, THF-d8), 25.4 (q, THF-d8). Anal. calcd for C72H104N4KN6O6Y2: C, 63.28; H, 7.67; N, 6.15. Found: C, 62.99; H, 7.87; N, 5.87. IR (ATR, cm−1): 3042 (vw), 2960 (vw), 2884 (w), 2851 (w), 2723 (vw), 1715 (vw), 1553 (m), 1478 (vw), 1439 (m), 1353 (w), 1294 (vw), 1258 (s), 1248 (s), 1221 (m), 1174 (vw), 1133 (m), 1102 (s), 1077 (s), 1025 (w), 1009 (m), 950 (m), 931 (m), 919 (m), 902 (m), 862 (w), 831 (w), 820 (w), 777 (vw), 764 (w), 752 (w), 718 (s), 670 (vw).Single-crystal X-ray diffraction
Data of complex 1 were collected on a Bruker CCD (charge coupled device) based diffractometer using MoKα radiation. The instrument was equipped with an Oxford Cryostream low-temperature apparatus operating at 173 K. Data were measured using omega and phi scans of 1.0° per frame for 30 s. The total number of images was based on results from the program COSMO35 where redundancy was expected to be 4.0 and completeness of 100% out to 0.83 Å. Cell parameters were retrieved using APEX II software36 and refined using SAINT on all observed reflections. Data reduction was performed using the SAINT software37 which corrects for Lp. Scaling and absorption corrections were applied using SADABS38 multi-scan technique.Data on 2 were collected on a XtaLAB Synergy DualflexHyPix four-circle diffractometer, equipped with a HyPix Hybrid Pixel Array Detector. The crystal was kept at 100 K during data collection. Data were measured using MoKα radiation. The maximum resolution that was achieved was Θ = 71.296° (0.81 Å). The CrysAlisPro software package39 was used to retrieve and refine the cell parameters, as well as for data reduction. Correction for absorption effects was done using a numerical correction based on Gaussian integration over a multifaceted crystal model and an empirical correction using spherical harmonics, implemented in SCALE3 ABSPACK40 scaling algorithm (spherical harmonics and frame scaling).
Using Olex2,41 the structures were solved with the ShelXT42 structure solution program using intrinsic phasing and refined with version 2018/3 of ShelXL43 using least squares minimization. All non-hydrogen atoms are refined anisotropically. Hydrogen atoms were calculated by geometrical methods and refined as a riding model. The crystals used for the diffraction study showed no decomposition during data collection. Crystal data and structure refinement for all compounds are shown in Table S1.†
NMR spectroscopy
NMR spectra were recorded on a 500 MHz Agilent DirectDrive2 and calibrated to the residual solvent signals (benzene-d6: δH = 7.16 ppm, THF-d8: δH = 3.58 ppm, δC = 67.6 ppm). Signal multiplicities are abbreviated as: s (singlet), d (doublet), m (multiplet), br (broad). Air-sensitive samples were prepared in a nitrogen-filled glovebox using either standard NMR tubes air-tight sealed or by using Young NMR tubes. NMR solvents (benzene-d6, THF-d8) were dried by storing over Na/K alloy for several days and filtered prior to use.EPR spectroscopy
Solution state EPR spectra were recorded on a Bruker Elexsys E-680X in Young quartz EPR tubes using a 1 mmol L−1 solution of 2 in thoroughly dried THF. The spectra were simulated by using the EasySpin software package44 for MATLAB. Variable temperature EPR spectra of the same concentration were recorded ascending from 200 to 280 K and the obtained derivative spectra subsequently basline corrected and doubly integrated for intensity analysis (Fig. S13 and Table S2†).IR spectroscopy
IR spectra were recorded with an Agilent Cary 630 ATR spectrometer in an argon-filled glovebox.UV/vis spectroscopy
UV/vis spectra were recorded in an argon-filled glovebox with an Agilent Cary 60 spectrometer, equipped with QP600-1-SR fiber optics and a Square One cuvette holder from Ocean Insight.SQUID magnetometry
Temperature-dependent magnetic susceptibility (χMT vs. T) and field-dependent magnetization (M vs. T) data were collected with a Quantum Design MPMPS3 SQUID magnetometer. Crystalline material of 2 was washed with cold THF and dried under vacuum for 1 h prior to sample preparation. Molten eicosane (66.5 mg (0.2329 mmol)) was added at 60 °C to the sample (29.5 mg (0.0216 mmol)) to immobilize the crystallites and to ensure good thermal contact between sample and the bath. The sample was sealed airtight and transferred to the SQUID magnetometer.The diamagnetic correction was done by measuring the susceptibility data of a blank sample, comprising the same amount of eicosane as the sample of 2 in an identical setup, and subtracting the obtained susceptibility data of the blank from the radical.
Cyclic voltammetry measurements
All cyclic voltammetry experiments were conducted under inert atmosphere in either an argon- or nitrogen-filled glovebox. Data of complex 1 and the free ligand (Li(TMEDA))2Bbim were recorded on a Metrohm μAutolabIII/FRA2 in dichloromethane with (nBu4N)(PF6) as supporting electrolyte (0.1 mol L−1) with a setup of Pt working electrode, Ag/AgNO3 reference electrode and Pt mesh counter electrode. Complex 2 was measured using a PGSTAT204 from Metrohm with 1.3 mmol L−1 sample solution in THF with (nBu4N)(PF6) as supporting electrolyte (0.25 mol L−1) in conjunction with a glassy carbon working electrode, a Pt spring counter electrode and a Pt wire pseudo reference electrode. All voltammograms were externally referenced to a ferrocene solution with identical supporting electrolyte concentration.Elemental analysis
Elemental analysis were obtained at Michigan State University. In a nitrogen-filled glovebox, solid samples (∼1–3 mg) were weighed into tin sample holders and folded multiple times to ensure proper sealing from surrounding atmosphere. Samples were transferred to the instrument under exclusion of air.Computational methods
DFT calculations were carried out on a truncated model of the Bbim radical-bridged complex 2 using the Gaussian program suite (revision B01).45 Starting coordinates for the geometry optimizations were taken from the crystallographically obtained molecule structure where all Cp* methyl groups were substituted by H atoms in order to reduce computational costs (herein called 2′). In order to evaluate the impact of the exchange correlation functional the geometry optimizations were carried out using the uB3LYP46–49 and uTPSS50 functionals. Geometry optimizations were carried out on the def2-SV(P)51,71 level on all atoms with the ECP28MDF52 pseudo potential on the Y atoms and grimme's dispersion correction GD353,54 using tight convergence criteria (obtained energies for 2′: −1607.537621 H (uTPSS) and −1607.22118 H (uB3LYP), Fig. S20 and S21,† Tables S6 and S7).† The obtained structures were confirmed to be minimum structures from frequency calculations on the same level of theory. Both functionals predict slightly shorter Y–N distances relative to the parameters extracted from X-ray analysis (0.0152–0.0218 Å) and thus, marginally overestimate these bonding interaction, while the central C2–C2′ bond is found to be longer by 0.0099–0.0154 Å. Single point calculations with the NLMO55 keyword as implemented in NBO656 on the optimized structures were carried out on the (def2-SV(P)51,71 (C, H atoms)/def2-TZVP51 (N atoms)/ECP28MDF_VDZ52 (Y atoms)) level. The obtained parameters describing the bonding in 2′ are given in Tables S8–S14.†
Results and discussion
Synthesis and structural characterisation
The devised two-step synthetic route consists of a salt elimination reaction followed by a chemical reduction reaction, Fig. 2. The initial salt metathesis reaction employs the organometallic yttrium tetraphenylborate complex Cp*2Y(BPh4), (where Cp* = pentamethylcyclopentadienyl) which is accessible through a multi-step synthetic path.9,57 The weakly interacting BPh4− anion allows facile insertion of neutral donor ligands, metathesis reactions with salts, and in situ reductions with a strong reducing reagent.11,17,20 The N-heterocyclic ligand H2Bbim was prepared from commercially available o-phenylenediamine and 2,2,2,-trichloroacetimidate,32 and subsequently deprotonated with KN(Si(CH3)3)2 in THF to give K2Bbim in 90% yield. Treatment of Cp*2Y(BPh4) with K2Bbim afforded the first bisbenzimidazolyl-bridged yttrium metallocene complex (Cp*2Y)2(μ-Bbim) (1). This molecule represents the first example of a homometallic lanthanide complex containing a bisbenzimidazole as a bridge. Pale yellow crystals of 1 suitable for X-ray analysis were obtained from a concentrated toluene solution at −35 °C in 52% yield. Compound 1 exhibits a diyttrium complex that resides on a crystallographic inversion centre, such the two yttrium ions are equivalent by symmetry. Each YIII centre is eight-coordinate composed of two η5-binding Cp* rings and two ligating N atoms of the bridging dianionic imidazole ligand Bbim2−. Selected interatomic distances (Å) and angles (°) for 1 are given in the caption of Fig. 2.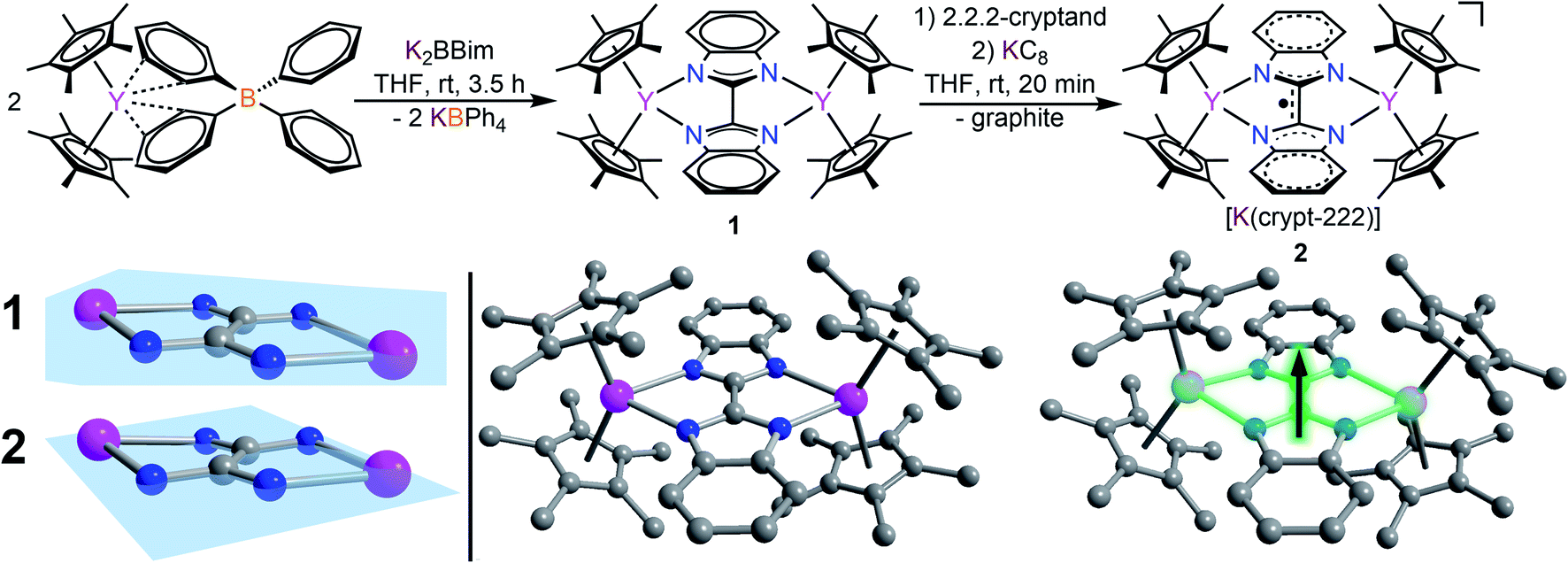 | ||
| Fig. 2 Top: synthesis of (Cp*2Y)2(μ-Bbim) (1) through salt metathesis reaction of Cp*2Y(BPh4) with K2Bbim and subsequent reduction of 1 with KC8 to [K(crypt-222)][(Cp*2Y)2(μ-Bbim˙)] (2). Bottom left: magnification of the ligand core, signifying increased tilting of the Bbim ligand upon chemical reduction from 1 to 2. Bottom right: structures of 1 and 2. Pink, blue, and gray, spheres represent yttrium, nitrogen, and carbon atoms, respectively. All hydrogen atoms, solvent molecules and the counter ion ([K(crypt-222)]+ for (2) are omitted for clarity. Selected interatomic distances [Å] and angles [°] for 1: C2–C2′: 1.445(6), avg. Y–N: 2.412(3), avg. Y–C: 2.655(4), Y–Y: 6.214(1), avg. Y–Cpcent: 2.366(1), Cpcent–Y–Cpcent: 138.6(1), Y–N–N–Y: 11.8(5), Bimplane1–Bimplane2: 0.1(1). For 2: C2–C2′: 1.402(3), avg. Y–N: 2.363(2), avg. Y–C: 2.677(2), Y–Y: 6.030(1), avg. Y–Cpcent: 2.390(1), Cpcent–Y–Cpcent: 135.8(1), Y1–N1–N3–Y2: 23.3(2), Bimplane1–Bimplane2: 3.1(1). | ||
The average Cpcent–Y (where Cpcent = centroid) and Y–N distances of 2.366(1) Å and 2.412(3) Å, and Cpcent–Y–Cpcent angle of 138.6(1)° are of similar magnitude to values in yttrocene complexes with N-donor ligands in the equatorial plane such as Cp*2Yphen (where phen = 1,10-phenanthroline) (2.343/2.354 Å, 2.403(2)/2.413(2) and 144.2°, respectively).22 The Bbim ligand is planar and symmetrically coordinated to both metal centres, featuring a slightly distorted Y1–N1–N3–Y2 angle of 11.8(5)°, possibly due to the steric demand of the Cp* rings in close proximity.
The chemical reduction of 1 with KC8 in the presence of 2.2.2-cryptand (crypt-222) caused an instantaneous distinct colour change from yellow to dark green and formed [K(crypt-222)][(Cp*2Y)2(μ-Bbim˙)] (2) containing the first evidence for a Bbim˙ radical, Fig. 2. Compound 2 constitutes simultaneously the first Bbim radical-containing complex for any metal ion. Dark green crystals of 2 suitable for single-crystal X-ray diffraction were grown from concentrated THF solutions at −35 °C. The diyttrium complex 2 exhibits two crystallographically inequivalent yttrium centres that are ligated by two η5-Cp* units and two N atoms of the bridging Bbim3−˙ radical trianion. The dinuclear yttrium moiety of 2 composes the anionic part, whereas the cationic unit consists of the [K(crypt-222)]+ counterion. Selected interatomic distances (Å) and angles (°) for 2 are given in the caption of Fig. 2. The structure of the [(Cp*2Y)2(μ-Bbim˙)]− anion features noticeable changes, albeit subtle, owing to the uptake of an electron, Fig. 2 bottom left. The central C–C and Y–N distances are decreased by 0.0488 Å (2.363(2) Å) and 0.0429 Å (1.402(3) Å), whereas the Y–Cpcent distances (0.0246 Å) are slightly elongated and the Cpcent–Y–Cpcent bending angle reduced by −2.8°. Structural changes such as the C–C bond reduction in heteroaromatic ligands have been frequently observed and indicate the population of the vacant ligand π* orbital. A prime example constitutes 4,4′-bipyridine, where the central C–C bond is shortened by 0.060 Å.19 Furthermore, the one-electron reduction of the Bbim ligand afforded more than doubled Y–Bbim3–˙–Y dihedral angle of 26.4(2)° resulting in a distinct displacement of the Y ions above and below the Bbim˙3− plane, respectively. Notably, the Y–Y distance is with 6.030(1) Å also decreased by 0.185 Å compared to 1.
Spectroscopic characterisation
UV-vis spectra collected on compounds 1 and 2 differ substantially (Fig. 3): 1 exhibits mainly transitions in the UV region that can be ascribed to the π–π* transitions of the Cp* ligands, whereas 2 features broad absorption signals in the visible region (403, 423, 434, 701 nm) and close to the NIR region (784 and 884 nm), respectively. In comparison, similar near IR transitions have been detected for other organic radicals.58 For instance, the trans-indigo (Ind) and the corresponding radical ind−˙ variant, exhibited transitions at 790 and 880 nm, respectively, which were assigned to π → π* transitions from the SOMO to the vacant LUMO + 1.59 By contrast, the dinuclear ruthenium complex meso-[(Ru(bpy)2)2(μ-Ind−˙)](ClO4)3 (where Ind = Indigo) featured the corresponding transitions considerably shifted towards the near IR region (>1000 nm), while mixed transitions including MLCT and LMCT occurred in the visible region.60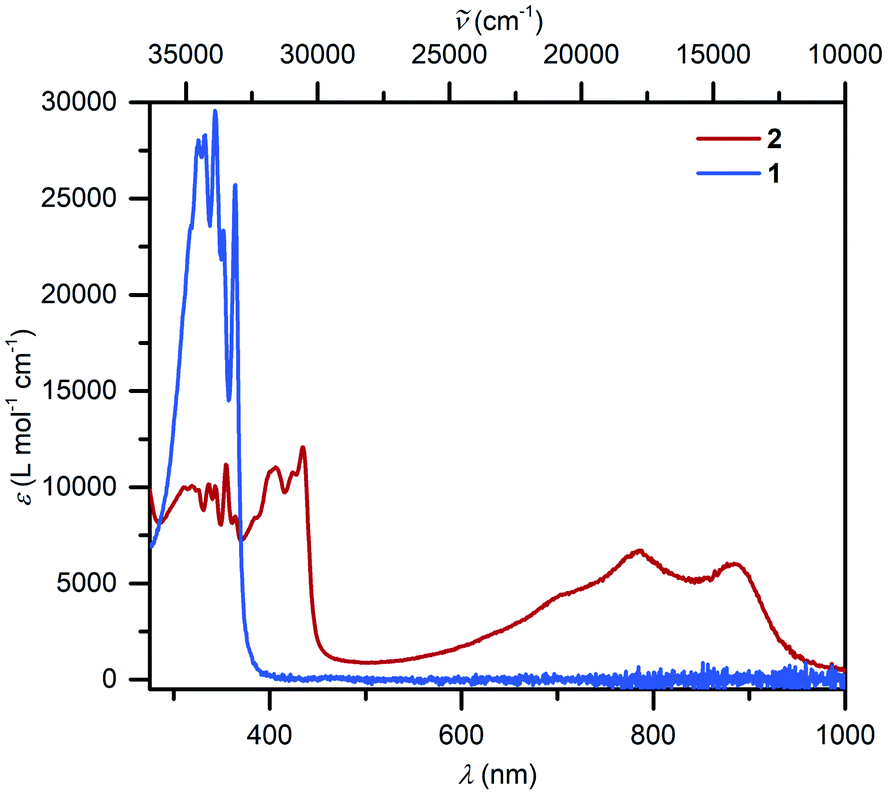 | ||
| Fig. 3 UV/vis spectra of (Cp*2Y)2(μ-Bbim), 1 (8.054 × 10−6 mol L−1, blue) and [K(crypt-222)][(Cp*2Y)2(μ-Bbim˙)], 2, (5.091 × 10−5 mol L−1, red), taken in THF. | ||
The 1H NMR spectra taken for 1 and 2 in thf-d8 differ from one another which originate from the presence of a paramagnetic organic radical in 2. For 1, the two paramount sets of resonances at 7.56 and 7.28 ppm with 3JH–H = 3.05, 5.90 Hz, are allocated to the aromatic protons of the Bbim2− ligand. These signals are absent in 2 accompanied by the emergence of new peaks at 3.66, 3.53 and 2.54 ppm, which are attributed to 2.2.2-cryptand (Fig. S8–S10†). The very broad signal between 2.05–0.64 ppm likely arises from the paramagnetic radical. Compound 2 features in THF-d8 a low solubility, preventing more elaborate NMR experiments where NMR spectra can only be regarded as phenomenological proof for the presence of 2.
To gain further insight into the electronic structure of the Bbim3−˙-bridged complex 2, variable-temperature solution X-band EPR spectra were recorded between 190 K and 298 K, and subsequently simulated by using the EasySpin software package (Fig. 4, 5, S11 and S12†).44 Each spectrum features one symmetric signal innate to well-resolved hyperfine couplings that gradually shrinks with decreasing temperatures. All spectra consist of 19 main lines, however, a strong temperature dependence of the signal shape was monitored. At room temperature, further splitting by superhyperfine couplings of the 1H-nuclei of the aromatic moiety occurred, where such couplings were gradually less resolved with decreasing temperatures, while at 190 K the fine structure essentially vanished (Fig. S11†). This signal splitting suggests that the radical interacts strongly with the four 14N nuclei (S = 1) and the eight 1H nuclei (S = ½). The g-values obtained from simulating the EPR spectra (g = 2.00554 (294 K), 2.00557 (280–200 K)) are close to the value of a free electron (g = 2.0023), indicating negligible interaction with the diamagnetic yttrium ion (89Y: S = ½). This is also reflected in the hyperfine coupling constants for the 14N-, the two 1H- and the 89Y species attesting a significant delocalization of the spin density onto the outer phenyl rings. Such a predominance of the 15N-/1H-over 89Y hyperfine coupling constants in organic radical-containing yttrium complexes is reminiscent of observations for other yttrium radical complexes such as [Y2((μ-bpytz−˙)2)(THMD)4] (where bpytz = 3,6-bis(3,5-dimethyl-pyrazolyl)-1,2,4,5-tetrazine, and TMHD = 2,2,6,6-tetramethyl-3,5-heptanedionate).61
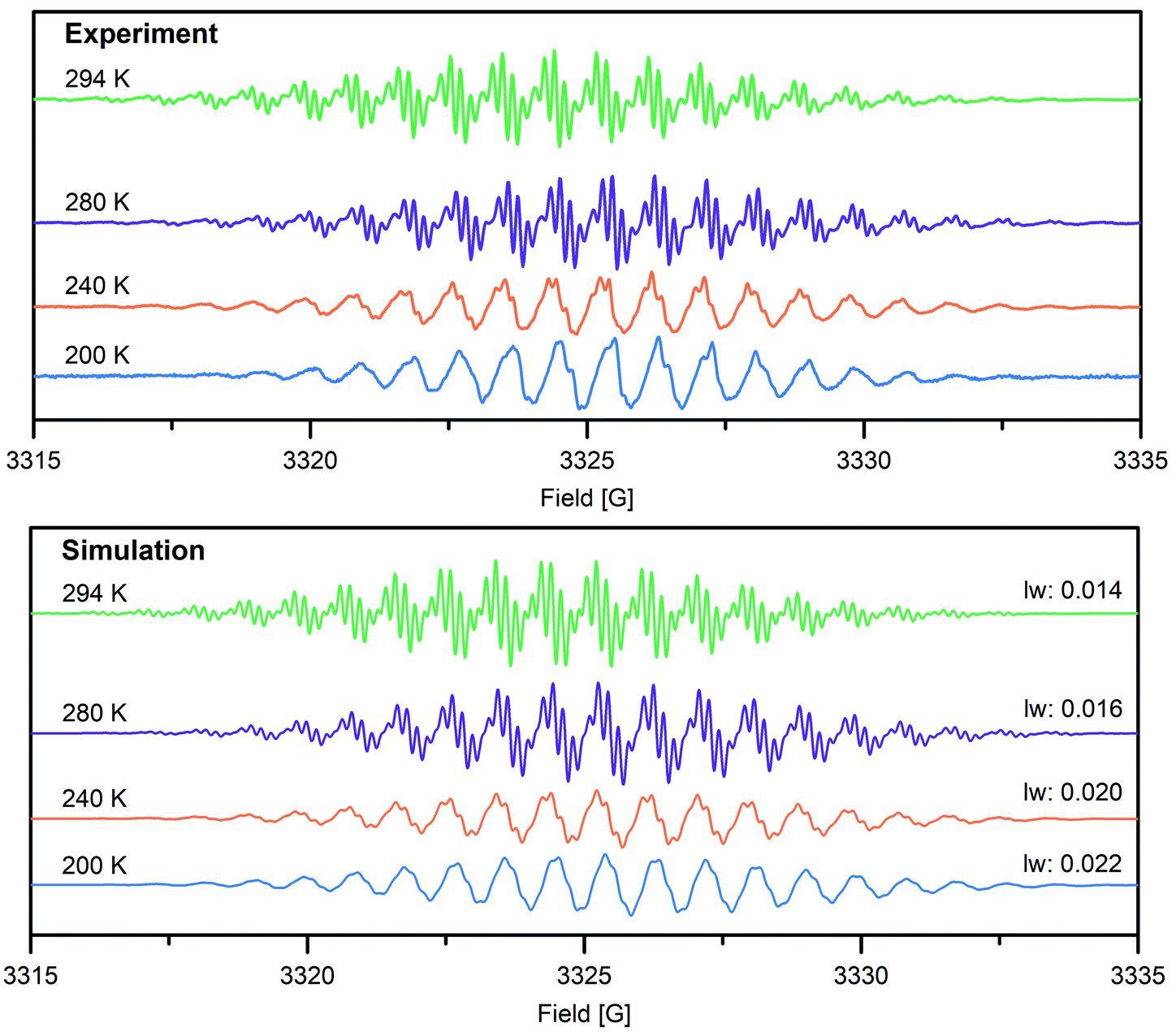 | ||
| Fig. 4 Top: variable-temperature solution X-band EPR spectra of 2 in THF (1 mmol L−1) at 294 K (green trace), 280 K (purple trace), 240 K (orange trace) and 200 K (blue trace). Bottom: simulated EPR spectra using a spin system of 4 14N, 4 1H1, 4 1H2 and 2 89Y nuclei, A(14N) = 5.1 MHz, A1(1H) = 2.30 MHz, A2(1H) = 0.54 MHz, A(89Y) = 0.42 MHz), g = 2.00554 (294 K), 2.00557 (280–200 K). Different linewidths (lw) were chosen for each temperature to account for increasingly less resolved hyperfine couplings (given in mT). | ||
 | ||
| Fig. 5 Top: solution X-band EPR spectrum of 2 in THF (1 mmol L−1) at 298 K (orange trace) at 9.769 GHz. Bottom: simulated EPR spectrum using a spin system of 4 14N, 4 1H1, 4 1H2 and 2 89Y nuclei with A(14N) = 5.10 MHz, A1(1H) = 2.30 MHz, A2(1H) = 0.54 MHz, A(89Y) = 0.42 MHz, as hyperfine coupling constants and g = 2.00525 (blue trace). The linewidth was set to 0.015 mT. | ||
Dimerization of organic radicals62 can occur for organic radicals such as phenalenyl-63 and olympicenyl64 radicals, giving rise to EPR silent π-stacked structures in solution. Among those, solid π-stacked radicals (“pancake bonds”)65 are particularly intriguing owing to singular properties such as electric conductivity and magnetic ordering. Notably, the intensity of VT EPR spectra was used to determine dimerization constants. Upon lowering the temperature, for a monomeric complex an increase in intensity is expected according to Boltzmann's law, whereas an incremental decrease in intensity originates from diamagnetic π-stacked dimers as a result of dimerization. To probe potential π-dimerization in 2, the integrated intensities of the VT EPR spectra between 200 K and 280 K were meticulously scrutinized (Fig. S13 and Table S2†). With decreasing temperature, the derivative (Fig. 4) and integrated (Fig. S13†) VT EPR spectra, feature a loss in hyperfine coupling resolution, concomitant with a progressive disappearance of hyperfine splittings. This may be a consequence of a gradual decreased molecular motion of 2 and the associated rotational correlation time, ultimately leading to incomplete averaging of the anisotropic hyperfine coupling tensor. This precludes an assignment of the EPR intensity decrease to dimerization and is rather attributed to originate from poorly-resolved hyperfine coupling especially considering that a dimerization of 2 is substantially hampered by the two coordinating Cp* ligands to each metal center.
Electrochemical study
The electrochemistry of 2 was probed by performing cyclic voltammetry (CV) measurements in THF solution with (nBu4N)PF6 as supporting electrolyte, Fig. 6 and S19.† A reproducible quasi-reversible feature was observed at −1.30(7) V vs. Fc/Fc+ on the time scale of the electrochemical experiment, suggesting that dinuclear yttrium complexes with Bbim in two oxidation states (Bbim3−˙/4−) are accessible. This result is remarkable given that electrochemical approaches on most organometallic rare earth complexes bearing redox-active ligands lead to insufficient information regarding reduction potentials. By contrast, electrochemical measurements conducted on 1 and ((TMEDA)Li)2Bbim33 lack (quasi)reversible features (Fig. S18†), which is in line with the alleged redox-inactivity of complexes containing H2BBim or BBim2−.25,26 Albeit non-trivial comparison as the challenges are different in transition metal chemistry and other ligands are involved, electrochemical experiments on mono- and dinuclear complexes of molybendum66,67 and ruthenium68 with redox-active bridging ligands other than Bbim have shown shifts towards less negative redox potentials upon complexation of the given ligand relative to the free variant. Taken this into consideration along with our experimental and electrochemical results, we conclude that the redox potential for the free Bbim ligand is too negative to be readily accessible with a strong chemical reducing agent. Therefore, we propose that for the successful one-electron reduction of Bbim2− to its trianionic radical state Bbim3−˙, its coordination to a highly Lewis acidic metal centre such as YIII is required as this lowers the LUMO orbitals of Bbim, rendering them accessible for the population with an additional electron. | ||
| Fig. 6 Cyclic voltammogram of 2 measured in THF at 300 K (0.25 M (nBu4N)PF6, 1.3 mmol L−1 analyte solution, υ = 50 mV s−1). | ||
Magnetic characterisation
The solid-state magnetic properties of 2 were probed via SQUID magnetometry (Fig. 7 and S14–S16†). Temperature-dependent magnetic susceptibility measurements (χMT vs. T) were performed between 2 K and 300 K at 0.5 T and 1.0 T. The room temperature χMT value is in good agreement with the expected value for an isolated radical (0.419 cm3 K mol−1 (0.5 T) and 0.403 cm3 K mol−1 (1.0 T), calculated: 0.375 cm3 K mol−1). The value at 1 T is in better accordance with the calculated value than the lower field which may account for overcoming a potentially apparent weak inter radical magnetic coupling. Indeed, the χMT vs. T was fitted with the program Phi69 and the best fits were obtained by consideration of small antiferromagnetic intermolecular coupling terms (zJ′, −0.088(12) cm−1 (0.5 T), −0.098(8) cm−1 (1.0 T)), Fig. S15.† The fitted g-values are smaller (2.0035(28) (0.5 T), 1.9927(17) (1.0 T)) compared to the g-values obtained from EPR spectroscopy, hinting at additional intermolecular interactions in the solution state. Fitting the temperature-dependent inverse susceptibility curves (1/χMvs. T) to a Curie–Weiss law gave excellent fits to small Weiss constants (−1.897 K (0.5 T), −1.492 K (1.0 T)), indicative of antiferromagnetic coupling, Fig. S14.† | ||
| Fig. 7 Variable-temperature dc magnetic susceptibility data (χMT vs. T) for 2, collected from 2 to 300 K under a 1.0 T applied dc field (A). Field-dependent magnetization data (M vs. T) for 2, collected from 2 K to 10 K in 2 K increments between 0 T and 7 T (B). The black lines represent fits to a Brillouin law (Table S5†). | ||
In addition, field-dependent magnetization measurements (M vs. T) were conducted from 2 K to 10 K in 2 K increments and with dc fields ranging from 0 to 7 T. The saturation magnetization measured at 2 K is with MS = 0.9 μB very close to the calculated value of Ms = 1 μB for an organic radical. For an ideal paramagnet, the temperature-dependent magnetization can be described by the Brillouin law which was employed to fit the experimental M vs. T curves of 2, Fig. 7B and S16.† The resulting fits are in excellent agreement with the experimental data and reveal that approximately 90% of radical spins contribute to the Curie–Weiss susceptibility. This high value suggests further that the paramagnetism observed for 2 is indeed of molecular origin. The reduced magnetization plot reveals that all measured magnetization curves superimpose to one single curve as expected for an organic radical with negligible magnetic anisotropy.
Quantum chemical study of yttrium–Bbim bonding
The electronic structure and nature of bonding in 2 were further examined by density functional theory (DFT) calculations and natural localized molecular orbital (NLMO)55 analysis using the Gaussian software suite.45 Due to the size of the system, a hypothetical model complex was employed for all calculations, where all methyl groups on the Cp rings were replaced by H atoms [(Cp2Y)2(μ-Bbim˙)]−, 2′). To shed light on the orbital contributions and spin densities in 2′, NLMO analyses were performed on the optimized geometries using the B3LYP46–49 functional and basis sets of triple zeta on the bonding N51 and double zeta quality on Y52 atoms (Fig. S20 and S21, Tables S6 and S7†). The structural changes accompanied by the chemical reduction of complex 1 to 2 can be well-elucidated considering the difference of the computed SOMO and SOMO-1 orbitals (Fig. S22†): the SOMO is populated with one electron and contains bonding regions on the bridging C2–C2′ and the Y–N bonds of opposite phase, while the SOMO-1 features opposing phases along C2 and C2′ and N1/N1′ bonds. Hence, upon chemical reduction of 1 these bonds experience a shortening whereas the N1–C2/C2–N3 bonds undergo an elongation (0.0313 Å).The computational results evince a highly ionic bonding between the yttrium and nitrogen atoms leading to the absence of bonding NLMOs between these atoms with strong donations of the nitrogen lone pairs (LPs) into yttrium lone valences found in the second order perturbation analysis (13.66–19.49 kcal mol−1 in the α- and 13.04–19.40 kcal mol−1 in the β-spin-manifold, Fig. S23 and Tables S8–S11†). The NLMO analysis further reveals equally small atomic hybrid contributions of the yttrium atom to nitrogen LPs for the α-spin manifold: each two hybridized nitrogen LPs with 7.7% yttrium contribution and two purely π nitrogen LPs with smaller donation (2.4–2.2%). By contrast, the β-manifold contains exclusively NLMOs with higher yttrium contribution (7.4%). This further corroborates the assignment of a strongly ionic bond in the Bbim3−˙ radical-bridged complex 2′ and accordingly in 2. In sum, the calculated Mulliken spin densities (Fig. 8 and Table S12†) confirmed the trend suggested by the hyperfine coupling constants obtained via EPR spectroscopy: the majority of the spin density is accumulated on the central N4C2 moiety and merely a small amount is delocalized onto the arene system. In comparison to other radical-bridged yttrium complexes, such as [{(Me3Si)2N)2Y(THF)}2(μ-η2:η2-N2˙)][K(THF)6], where the spin density is massively confined to the N23−˙ radical bridging ligand,70 the delocalization of the unpaired electron is greatly augmented in 2/2′.
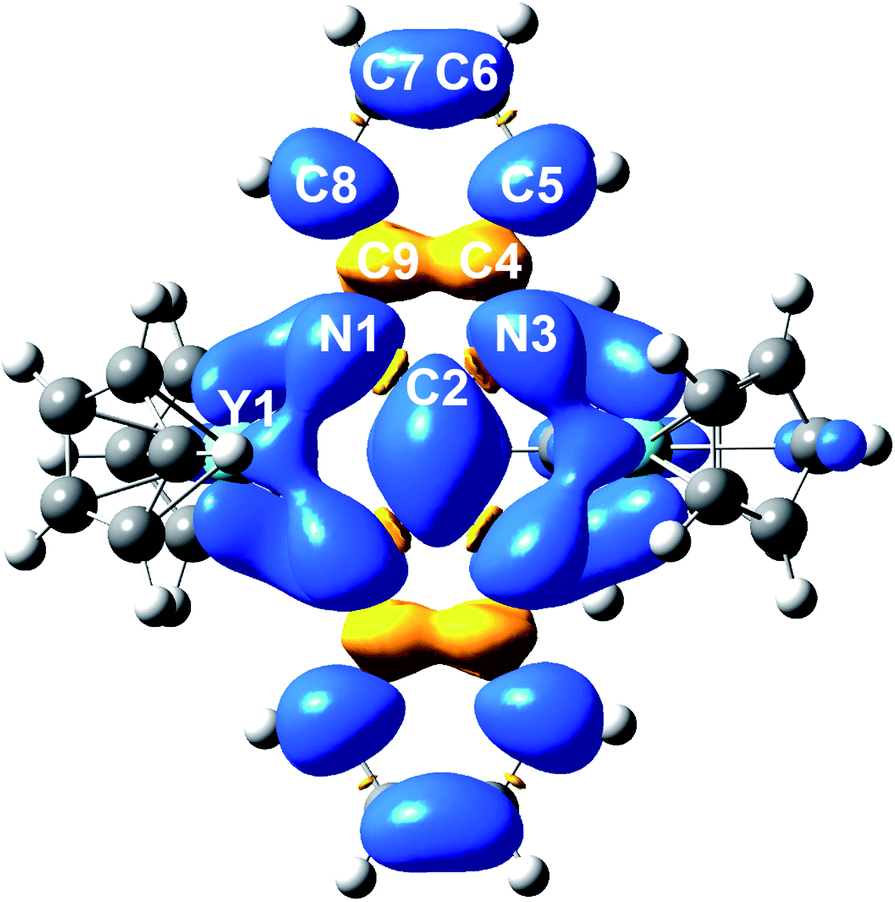 | ||
| Fig. 8 DFT calculated spin density map of 2′ (iso values: 0.02). Average calculated Mulliken spin densities: Y: 0.0395, N: 0.0900, C2: 0.158, C4/C9: −0.0279, C5/C8: 0.0679, C6/C7: 0.0163. | ||
Conclusion
Taken together, the foregoing findings provide evidence of the generation and isolation of a hitherto unknown radical ligand upon complexation to rare earth metal ions. This work portrays the first unambiguous report on an isolable Bbim3−˙ radical bridging ligand for any metal ion. In particular, the unprecedented Bbim3−˙ radical, stabilized through the coordination to two yttrocene moieties, was proven by single-crystal X-ray diffraction, variable-temperature EPR spectroscopy, SQUID magnetometry, electrochemistry, and computations. Additional characterisation by UV-vis- and NMR spectroscopy gave further insight into the electronic structure. The combined experimental and theoretical approach unfolded a mainly ionic coordination with a substantial delocalized spin density on the central atoms of the bridging ligand. The parent combined EPR and DFT study represents a rare instance of an organic radical-bridged dinuclear yttrium metallocene complex and will have huge ramifications for thorough understanding of future lanthanide-radical materials. The outlined synthetic procedure for Bbim3−˙ generation provides a platform to novel functional conductive and magnetic materials containing rare earth metals. Future research will be geared towards the synthesis of Bbim3−˙-bridged complexes containing heavy lanthanide ions with high magnetic anisotropy. The isolation of such compounds will pave the way for studying the utility of Bbim3−˙ radicals in promoting strong magnetic exchange coupling and tied to that realizing new advances in the forefront of radical-bridged single-molecule magnets.Data availability
All computational data, spectroscopic data, supplementary figures and tables, and detailed crystallographic information can be found in the ESI.† (CCDC): 2123273 (1), 2123274 (2).Author contributions
F. B. prepared and characterised the compounds via crystallography, spectroscopy, magnetometry, and computations. S. D. assisted with data analysis, formulated, and directed the research, and wrote the manuscript with input of F. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. D. is grateful to the Department of Chemistry at Michigan State University (MSU) for generous start-up funds. We are grateful to Dr Rui Huang (MSU) for assistance with CHN analyses. This work was supported in part through computational resources and services provided by the Institute for Cyber-Enabled Research at MSU. Funding for the Single Crystal X-ray diffractometer was provided by the MRI program by the National Science Foundation under Grant No. CHE-1919565.Notes and references
- B. Tang, J. Zhao, J. F. Xu and X. Zhang, Tuning the stability of organic radicals: From covalent approaches to non-covalent approaches, Chem. Sci., 2020, 11, 1192–1204 RSC.
- R. G. Hicks, What's new in stable radical chemistry?, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- Z. X. Chen, Y. Li and F. Huang, Persistent and Stable Organic Radicals: Design, Synthesis, and Applications, Chem, 2021, 7, 288–332 CAS.
- J. O. Moilanen, N. F. Chilton, B. M. Day, T. Pugh and R. A. Layfield, Strong Exchange Coupling in a Trimetallic Radical-Bridged Cobalt(II)-Hexaazatrinaphthylene Complex, Angew. Chem., Int. Ed., 2016, 128, 5611–5615 CrossRef.
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, A N23− radical-bridged terbium complex exhibiting magnetic hysteresis at 14 K, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed.
- P. J. Chirik and K. Wieghardt, Radical ligands confer nobility on base-metal catalysts, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- M. Saha and S. Bandyopadhyay, Stimuli Responsive Stable Radical Anion for Conductance Switching, J. Phys. Chem. C, 2021, 125, 6427–6432 CrossRef CAS.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Air-Stable Organic Radicals: New-Generation Materials for Flexible Electronics?, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Giant coercivity and high magnetic blocking temperatures for N23− radical-bridged dilanthanide complexes upon ligand dissociation, Nat. Commun., 2017, 8, 2144 CrossRef PubMed.
- S. Demir, I. R. Jeon, J. R. Long and T. D. Harris, Radical ligand-containing single-molecule magnets, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS.
- C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, Substituent Effects on Exchange Coupling and Magnetic Relaxation in 2,2′-Bipyrimidine Radical-Bridged Dilanthanide Complexes, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS PubMed.
- S. Sanvito, Molecular spintronics, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- E. Coronado, Molecular magnetism: from chemical design to spin control in molecules, materials and devices, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- M. Urdampilleta, S. Klayatskaya, M. Ruben and W. Wernsdorfer, Magnetic interaction between a radical spin and a single-molecule magnet in a molecular spin-valve, ACS Nano, 2015, 9, 4458–4464 CrossRef CAS PubMed.
- A. J. Hutchings, F. Habib, R. J. Holmberg, I. Korobkov and M. Murugesu, Structural rearrangement through lanthanide contraction in dinuclear complexes, Inorg. Chem., 2014, 53, 2102–2112 CrossRef CAS PubMed.
- P. Evans, D. Reta, C. A. P. Goodwin, F. Ortu, N. F. Chilton and D. P. Mills, A double-dysprosocenium single-molecule magnet bound together with neutral ligands, Chem. Commun., 2020, 56, 5677–5680 RSC.
- F.-S. Guo and R. A. Layfield, Strong direct exchange coupling and single-molecule magnetism in indigo-bridged lanthanide dimers, Chem. Commun., 2017, 53, 3130–3133 RSC.
- D. N. Woodru, R. E. P. Winpenny and R. A. Layfield, Lanthanide Single-Molecule Magnets, Chem. Rev., 2013, 113, 5110–5148 CrossRef PubMed.
- S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, Exchange coupling and magnetic blocking in bipyrimidyl radical-bridged dilanthanide complexes, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS PubMed.
- S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Exchange coupling and magnetic blocking in dilanthanide complexes bridged by the multi-electron redox-active ligand 2,3,5,6-tetra(2-pyridyl)pyrazine, Chem. Sci., 2014, 5, 4702–4711 RSC.
- C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, A Trinuclear Radical-Bridged Lanthanide Single-Molecule Magnet, Angew. Chem., Int. Ed., 2017, 56, 10103–10107 CrossRef CAS PubMed.
- M. R. MacDonald, J. W. Ziller and W. J. Evans, Coordination and Reductive Chemistry of Tetraphenylborate Complexes of Trivalent Rare Earth Metallocene Cations, [(C5Me5)2Ln][(μ-Ph)2BPh2], Inorg. Chem., 2011, 50, 4092–4106 CrossRef CAS PubMed.
- F. Delano IV, E. Castellanos, J. McCracken and S. Demir, A rare earth metallocene containing a 2,2′-azopyridyl radical anion, Chem. Sci., 2021, 12, 15219–15228 RSC.
- J. R. Bolton, in Radical Ions, ed. E. T. Kaiser and L. Kevan, John Wiley & Sons, New York, London, Sydney, 1st edn, 1968, pp. 1–33 Search PubMed.
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Multimetallic Ruthenium(II) Complexes Based on Biimidazole and Bibenzimidazole: Effect of Dianionic Bridging Ligands on Redox and Spectral Properties, Inorg. Chem., 1990, 29, 167–175 CrossRef CAS.
- M. Haga and A. M. Bond, Electrochemistry of Symmetrical and Asymmetrical Dinuclear Ruthenium, Osmium, and Mixed-Metal 2,2′-Bipyridine Complexes Bridged by 2,2′-Bibenzimidazolate, Inorg. Chem., 1991, 30, 475–480 CrossRef CAS.
- S. Y. Yao, Y. L. Ou and B. H. Ye, Asymmetric Synthesis of Enantiomerically Pure Mono-and Binuclear Bis(cyclometalated) Iridium(III) Complexes, Inorg. Chem., 2016, 55, 6018–6026 CrossRef CAS PubMed.
- P. H. Dinolfo, M. E. Williams, C. L. Stern and J. T. Hupp, Rhenium-based molecular rectangles as frameworks for ligand-centered mixed valency and optical electron transfer, J. Am. Chem. Soc., 2004, 126, 12989–13001 CrossRef CAS PubMed.
- M. Haga, T. Matsumura-Inoue and S. Yamabe, Binuclear Complexes of Ruthenium and Osmium Bridged by 2,2′-Bibenzimidazolate: Preparation, Electrochemistry, and Formation of Mixed-Valence Complexes, Inorg. Chem., 1987, 26, 4148–4154 CrossRef CAS.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, Structure, reactivity, and density functional theory analysis of the six-electron reductant, [(C5Me5)2U]2(μ-η6:η6-C6H6), synthesized via a new mode of (C5Me5)3M reactivity, J. Am. Chem. Soc., 2004, 126, 14533–14547 CrossRef CAS PubMed.
- B. J. Barker and P. G. Sears, Conductance behaviour of some ammonium and partially substituted ammonium tetraphenylborates in oxazolidones, J. Phys. Chem., 1974, 78, 2687–2688 CrossRef CAS.
- A. F. Henwood, D. Antón-García, M. Morin, D. Rota Martir, D. B. Cordes, C. Casey, A. M. Z. Slawin, T. Lebl, M. Bühl and E. Zysman-Colman, Conjugated, rigidified bibenzimidazole ancillary ligands for enhanced photoluminescence quantum yields of orange/red-emitting iridium(III) complexes, Dalton Trans., 2019, 48, 9639–9653 RSC.
- B. F. Fieselmann, D. N. Hendrickson and G. D. Stucky, Synthesis, Electron Paramagnetic Resonance, and Magnetic Studies of Binuclear Bis(η5-cyclopentadienyl)titanium(III) Compounds with Bridging Pyrazolate, Biimidazolate, and Bibenzimidazolate Anions, Inorg. Chem., 1978, 17, 2078–2084 CrossRef.
- D. E. Bergbreiter and J. M. Killough, Reactions of Potassium-Graphite, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef CAS.
- COSMO V1.61, Software for the CCD Detector Systems for Determining Data Collection Parameters, Bruker Analytical X-ray Systems, Madison, WI, 2009 Search PubMed.
- APEX2 V2010.11-3, Software for the CCD Detector System, Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed.
- R. H. Blessing, SAINT V 7.68a, Software for the Integration of CCD Detector System, Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed.
- R. H. Blessing, SADABS V2.008/2, Program for absorption corrections using Bruker-AXS CCD, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1995, 51, 33–38 CrossRef PubMed.
- CrysAlisPro Software System, Rigaku Corporation, Oxford, 2020.
- SCALE3 ABSPACK Empirical Absorption Correction, CrysAlis Pro - Software Package, Rigaku Corporation, Oxford, 2020 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXT – Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian program suite (revision B01), 2016 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephen, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density formula, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. Tao and J. P. Perdew, Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design an assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y–Pd, J. Chem. Phys., 2007, 126, 124101–124112 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. G. A. Smith, L. A. Burns, K. Patkowski and C. D. Sherrill, Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory, J. Phys. Chem. Lett., 2016, 7, 2197–2203 CrossRef CAS PubMed.
- A. E. Reed and F. Weinhold, Natural localized molecular orbitals, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- S. E. Lorenz, B. M. Schmiege, D. S. Lee, J. W. Ziller and W. J. Evans, Synthesis and reactivity of bis(tetramethylcyclopentadienyl) yttrium metallocenes including the reduction of Me3SiN3 to [(Me3Si)2N] with [(C5Me4H)2Y(THF)]2(μ-η2:η-N2), Inorg. Chem., 2010, 49, 6655–6663 CrossRef CAS PubMed.
- J. Fabian, TDDFT-calculations of vis/NIR absorbing compounds, Dyes Pigm., 2010, 84, 36–53 CrossRef CAS.
- D. V. Konarev, L. V. Zorina, M. S. Batov, S. S. Khasanov, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Optical and magnetic properties of: Trans-indigo⋅− radical anions. Magnetic coupling between trans-indigo⋅− (S = 1/2) mediated by intermolecular hydrogen N–H⋯OC bonds, New J. Chem., 2019, 43, 7350–7354 RSC.
- M. Chatterjee, P. Mondal, K. Beyer, A. Paretzki, W. Kaim and G. K. Lahiri, A structurally characterised redox pair involving an indigo radical: indigo based redox activity in complexes with one or two [Ru(bpy)2] fragments, Dalton Trans., 2017, 46, 5091–5102 RSC.
- M. A. Lemes, N. Mavragani, P. Richardson, Y. Zhang, B. Gabidullin, J. L. Brusso, J. Moilanen and M. Murugesu, Unprecedented intramolecular pancake bonding in a {Dy2} single-molecule magnet, Inorg. Chem. Front., 2020, 7, 2592–2601 RSC.
- S. M. Greer, R. T. Oakley, J. van Tol, M. Shatruk and S. Hill, Investigating the thermally- and light-induced interconversion of bisdithiazolyl radicals and dimers with high-field EPR, Polyhedron, 2018, 153, 99–103 CrossRef CAS.
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, Intermolecular π-to-π bonding between stacked aromatic dyads. Experimental and theoretical binding energies and near-IR optical transitions for phenalenyl radical/radical versus radical/cation dimerizations, J. Am. Chem. Soc., 2004, 126, 13850–13858 CrossRef CAS PubMed.
- Q. Xiang, J. Guo, J. Xu, S. Ding, Z. Li, G. Li, H. Phan, Y. Gu, Y. Dang, Z. Xu, Z. Gong, W. Hu, Z. Zeng, J. Wu and Z. Sun, Stable Olympicenyl Radicals and Their π-Dimers, J. Am. Chem. Soc., 2020, 142, 11022–11031 CrossRef CAS PubMed.
- K. E. Preuss, Pancake bonds: π-stacked dimers of organic and light-atom radicals, Polyhedron, 2014, 79, 1–15 CrossRef CAS.
- W. Kaim and S. Kohlmann, Four Bridging Bis Chelate Ligands with Very Low Lying π Orbitals. MO Perturbation Calculations, Electrochemistry, and Spectroscopy of Mononuclear and Binuclear Group 6 Metal Tetracarbonyl Complexes, Inorg. Chem., 1987, 26, 68–77 CrossRef CAS.
- W. Kaim, The transition metal coordination chemistry of anion radicals, Coord. Chem. Rev., 1987, 76, 187–235 CrossRef CAS.
- A. Singh, S. Panda, S. Dey and G. K. Lahiri, Metal-to-Ligand Charge Transfer Induced Valence Tautomeric Forms of Non-Innocent 2,2′-Azobis(benzothiazole) in Ruthenium Frameworks, Angew. Chem., Int. Ed., 2021, 60, 11206–11210 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- W. J. Evans, M. Fang, G. L. Zucchi, F. Furche, J. W. Ziller, R. M. Hoekstra and J. I. Zink, Isolation of dysprosium and yttrium complexes of a three-electron reduction product in the activation of dinitrogen, the (N2)3− radical, J. Am. Chem. Soc., 2009, 131, 11195–11202 CrossRef CAS PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 2, 123–141 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2123273 (1), 2123274 (2). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc07245e |
| This journal is © The Royal Society of Chemistry 2022 |