
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective electrochemical capture and release of uranyl from aqueous alkali, lanthanide, and actinide mixtures using redox-switchable carboranes†
Megan
Keener‡
 a,
Maxwell
Mattejat‡
a,
Shao-Liang
Zheng
b,
Guang
Wu
a,
Trevor W.
Hayton
a and
Gabriel
Ménard
*a
a,
Maxwell
Mattejat‡
a,
Shao-Liang
Zheng
b,
Guang
Wu
a,
Trevor W.
Hayton
a and
Gabriel
Ménard
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106, USA. E-mail: menard@chem.ucsb.edu
bDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA
First published on 25th February 2022
Abstract
We report the selective electrochemical biphasic capture of the uranyl cation (UO22+) from mixed-metal alkali (Cs+), lanthanide (Nd3+, Sm3+), and actinide (Th4+, UO22+) aqueous solutions to an organic, 1,2-dichloroethane (DCE), phase using the ortho-substituted nido-carborane anion, [1,2-(Ph2PO)2-1,2-C2B10H10]2− (POCb2−). The reduced POCb2− is generated by electrochemical reduction of the closo-carborane, POCb, prior to mixing with the aqueous mixed-metal solution. Subsequent UO22+ release from the captured product, [UO2(POCb)2]2−, was performed by galvanostatic bulk electrolysis of the DCE phase and back-extraction of UO22+ to a fresh aqueous phase. The selective capture and release of UO22+ was confirmed by combined ICP-OES and NMR spectral analyses of the aqueous and organic phases, respectively, against the newly synthesized nido-carborane complexes, [[CoCp*2][Cs(POCb)]]2, [CoCp*2]3[Nd(POCb)3], [CoCp*2]3[Sm(POCb)3], and [CoCp*2]2[Th(POCb)3].
Introduction
With over 440 operational reactors worldwide, nuclear energy currently provides 11% of all electricity. Several countries have proposed to increase nuclear energy production to meet their Paris Agreement targets for decarbonizing their economies, with the most ambitious being India and China that propose eight- and five-fold increases in domestic nuclear capacity, respectively.1 While nuclear energy is often considered a low-carbon energy alternative to fossil fuels,2,3 the disposal of spent nuclear fuel (SNF), as well as the inadvertent release of radioactive material to the environment (e.g., release of 137Cs at Chernobyl and Fukushima), make this technology imperfect.Uranium, in its dioxide form (UO2), is both the main component in nuclear fuel, as well as SNF, where the concentration drops to approximately 95%. New fission products generated include: Pu (0.9%); the minor actinides (0.1% (Np, Am, Cm)); lanthanides, Tc, Mo, I, Cs and others (together ca. 4%).4 As of 2020, approximately 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of SNF have been cumulatively generated worldwide, of which only ∼25% have been reprocessed using the decades-old Plutonium Uranium Redox EXtraction (PUREX) process.5 While this commercial, liquid–liquid process is extremely efficient at extracting and recycling UO22+ using stoichiometric extractants, in turn reducing SNF loads, PUREX involves the selective extraction of a pure Pu stream which raises significant proliferation concerns from major stakeholders, such as the U.S.6 While other reprocessing schemes addressing these concerns have been developed (e.g., UREX), none are commercial. To this day, proliferation concerns have superseded reprocessing efforts in places like the U.S., forcing countries to instead increase their SNF storage capacity, thus deferring action on the nuclear waste issue.4,6,7 New strategies for the selective separation and recovery of UO22+ from SNF, without the parallel extraction of a Pu stream, could therefore significantly aid in reducing net SNF generated from reactors, minimizing demands on long-term geological repositories, and in turn closing the fuel cycle.
000 tons of SNF have been cumulatively generated worldwide, of which only ∼25% have been reprocessed using the decades-old Plutonium Uranium Redox EXtraction (PUREX) process.5 While this commercial, liquid–liquid process is extremely efficient at extracting and recycling UO22+ using stoichiometric extractants, in turn reducing SNF loads, PUREX involves the selective extraction of a pure Pu stream which raises significant proliferation concerns from major stakeholders, such as the U.S.6 While other reprocessing schemes addressing these concerns have been developed (e.g., UREX), none are commercial. To this day, proliferation concerns have superseded reprocessing efforts in places like the U.S., forcing countries to instead increase their SNF storage capacity, thus deferring action on the nuclear waste issue.4,6,7 New strategies for the selective separation and recovery of UO22+ from SNF, without the parallel extraction of a Pu stream, could therefore significantly aid in reducing net SNF generated from reactors, minimizing demands on long-term geological repositories, and in turn closing the fuel cycle.
We recently reported a new, biphasic, electro/chemical method for capturing UO22+ using the ortho-substituted nido-carborane anion, [1,2-(Ph2PO)2-1,2-C2B10H10]2− (POCb2−) generating the captured species, [UO2Xn(POCb)(2−n/2)]2− (n = 0, 2; X = Cl, OAc; Fig. 1a). Electrochemical oxidation of this species was initiated to generate the oxidized closo-carborane (POCb), initiating the release of UO22+ to the aqueous layer. Repeated capture and release of UO22+ in monophasic organic solution further demonstrated the potential applicability and recyclability of this extractant.8 In this study, we wanted to explore the selective biphasic capture and release of UO22+ using the POCb2−/POCb system from aqueous solutions of alkali, lanthanide, and actinide metals more closely mimicking SNF streams. The choice of metals, and the reasons for using each, are as follows: (1) natural abundance 133Cs+ (100%) was used to mimic the highly radiotoxic 137Cs isotope which is responsible for much of the human health, environmental, and hot SNF disposal issues;7,9,10 (2) Nd3+ and Sm3+ were chosen due to their abundance in SNF;7 (3) Th4+ was used due to its abundance in SNF,7 and also because it functions as a Pu4+ surrogate in light of our inability to handle this highly controlled element in house.11–13 Herein, we describe both the coordination chemistry of POCb2− to these individual metals, as well as the highly selective electrochemical capture of UO22+ with POCb2− from the mixed-metal aqueous solution to an organic phase. The electrochemical release of UO22+ to a fresh aqueous phase is also described (Fig. 1b).
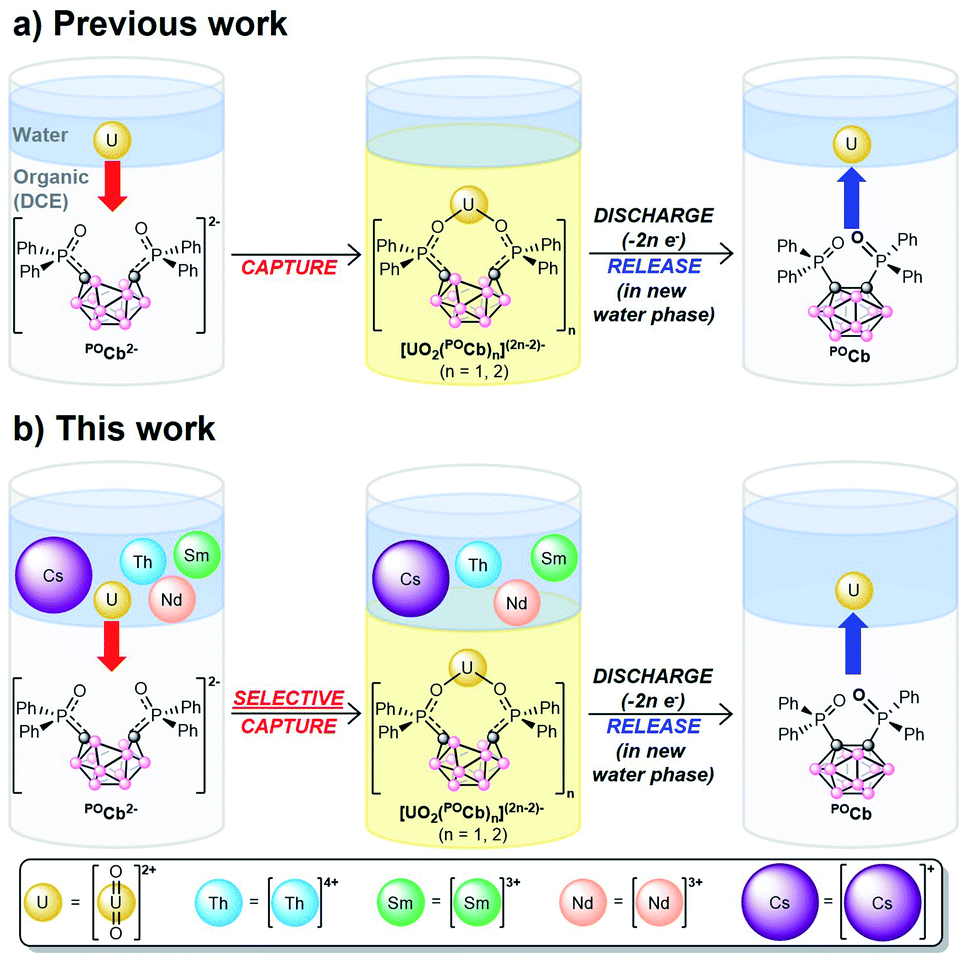 | ||
| Fig. 1 (a) Our previous work demonstrating the biphasic, electrochemical capture and release of UO22+ using the POCb2−/POCb system. (b) This work highlighting the selective capture and release of UO22+ from aqueous solutions containing alkali, lanthanide, and actinide metals. | ||
Results and discussion
The coordination chemistry of the POCb2− ligand was investigated using the previously reported [CoCp*2]+ salt, [CoCp*2]2[POCb] (Cp* = η5-C5Me5),8 in tandem with the nitrate salts of Cs+, Nd3+, Sm3+, and Th4+. All complexes were synthesized following an analogous synthetic procedure in MeCN. The Cs complex was generated by addition of an equimolar solution of [CoCp*2]2[POCb] to a solution of CsNO3 in MeCN at r.t. Following the selective recrystallization and separation of the [CoCp*2][NO3] byproduct, the desired product was isolated and unambiguously identified by single crystal X-ray diffraction (XRD) studies as the dimeric salt, [[CoCp*2][Cs(POCb)]]2 (Fig. 2a). The symmetric dimer features a central diamond-shaped core structure with two Cs atoms at the apical positions held in place by oxide donors from each ligand (Cs1–O(1, 1′) = 2.9893(18), 3.0844(19) Å), as well as Cs–H–B bonds14,15 (Cs1–B(3, 3′) = 3.681(3), 3.631(3) Å; Cs1–H(3, 3′) = 2.924, 3.08(3) Å). Other interactions outside the diamond core are provided by the additional oxide donor (Cs1–O2 = 2.9356(18) Å), as well as an additional B contact (Cs1–B4′ = 3.726(3) Å). We note that a Cs–H (3.199 Å) contact arising from a phenyl meta-C–H bond of an adjacent dimer is also observed, generating a polymeric structure (see Fig. S7† (not shown in Fig. 2a)). The nidoPOCb2− ligand charged state is maintained as indicated by the long C1–C2 distance (2.862 Å),8 which is well outside the range of a C–C bond. Together, we tentatively assign a coordination number (CN) of 9 to the large Cs cation. Due to the imposed crystal symmetry, identical bond metrics are found for Cs1′. The bonding types and lengths, the polymeric structure, and the assigned CN are similar to previously reported data for Cs.14–17 Lastly, while the solid-state structure displays inequivalent P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O donor groups in the POCb2− ligands, we note that the diamagnetic complex displays a single resonance in the 31P NMR spectrum at 31.7 ppm in MeCN-d3 indicating higher symmetry in solution, perhaps due to the breakup of the polymeric structure initiated by the coordinating solvent (Fig. S10†).
O donor groups in the POCb2− ligands, we note that the diamagnetic complex displays a single resonance in the 31P NMR spectrum at 31.7 ppm in MeCN-d3 indicating higher symmetry in solution, perhaps due to the breakup of the polymeric structure initiated by the coordinating solvent (Fig. S10†).
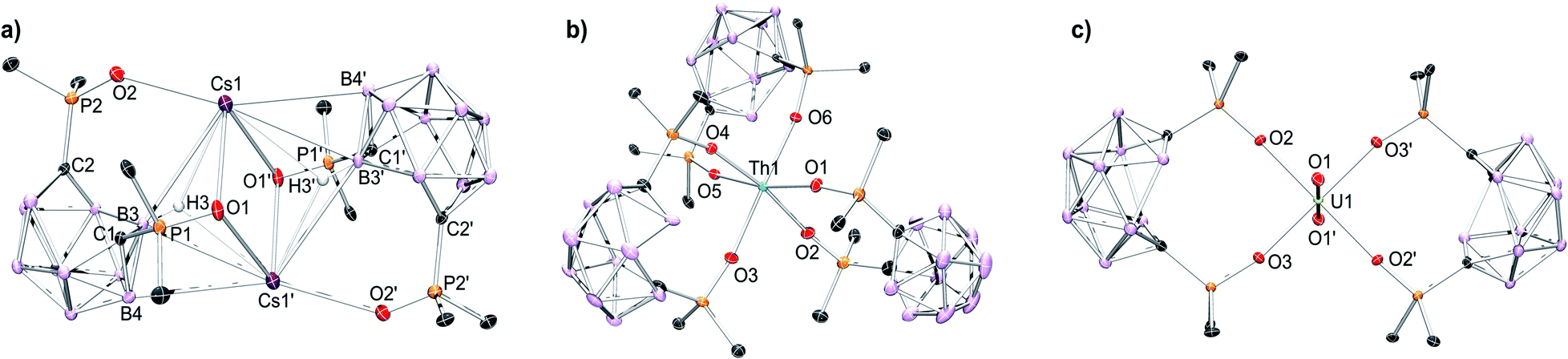 | ||
| Fig. 2 Solid-state molecular structures obtained by XRD studies of: (a) [[CoCp*2][Cs(POCb)]]2; (b) [CoCp*2]2[Th(POCb)3], and; (c) [CoCp*2]2[UO2(POCb)2].8 [CoCp*2]+ counter cations, phenyl C–H linkages, co-crystallized solvent molecules, and all H atoms, except those shown in (a), are omitted for clarity. Pertinent bond lengths and angles are discussed in the manuscript. The polymeric structure of [[CoCp*2][Cs(POCb)]]2 (a), filling an additional coordination site at Cs, is shown in Fig. S7.† | ||
The lanthanide (Nd3+, Sm3+) and actinide (Th4+) complexes were next synthesized using an identical procedure. Three equivalents of [CoCp*2]2[POCb] were added to one equivalent of M(NO3)n (M = Nd, Sm (n = 3); Th (n = 4)) in MeCN at r.t. The [CoCp*2][NO3] byproduct was again selectively crystallized and separated prior to isolation of the final products, which were all unambiguously identified by single crystal XRD studies as: [CoCp*2]3[Nd(POCb)3] (Fig. S8†); [CoCp*2]3[Sm(POCb)3] (Fig. S9†), and; [CoCp*2]2[Th(POCb)3] (Fig. 2b). All complexes have a central 6-coordinate metal center in pseudo-octahedral geometries. Average M–O bond distances of 2.327 Å (Nd), 2.315 Å (Sm), and 2.297 Å (Th) are similar to reported values18–24 and follow an expected periodic trend based on decreasing ionic radii and increasing ionic charge (for Th). Long carborane C–C distances (average 2.86 Å) in each case are again indicative of a nidoPOCb2− configuration.8 The solid-state structure of the previously reported uranyl complex, [CoCp*2]2[UO2(POCb)2], is also shown in Fig. 2c as a comparison to the new complexes reported here.8 Spectroscopically, [CoCp*2]3[Nd(POCb)3] and [CoCp*2]3[Sm(POCb)3] display 31P NMR resonances at 140.6 and 27.9 ppm, respectively. These values are notably different from each other, likely due to their varying paramagnetism. These values are also much different from the diamagnetic [CoCp*2]2[Th(POCb)3] (51.3 ppm) and [CoCp*2]2[UO2(POCb)2] (52.0 ppm) complexes.8
Building on our previous work (Fig. 1a),8 we investigated the selective electrochemical capture of UO22+ from mixed aqueous alkali (Cs+), lanthanide (Nd3+, Sm3+), and actinide (Th4+, UO22+) solutions mimicking in part SNF. Mixed-metal aqueous stock solutions were first prepared by dissolving equimolar quantities of the common starting materials, CsNO3, Nd(NO3)3(THF)3, Sm(NO3)3(THF)3, Th(NO3)4(H2O)x, and UO2(NO3)2(THF)2 in Milli-Q deionized water, either with a NaOAc buffer (0.5 M, pH = 5.2) or without (pH = 2.6). The buffer in the former was used for two reasons: (1) to mimic our previous results which required the use of a buffer to control for the pH-dependent extinction coefficient (ε) of UO22+ which was monitored by UV-vis spectroscopy,8,25,26 and; (2) to compare the extraction efficacy of our system at varying pH values. In contrast to our previous work, we used inductively-coupled plasma optical emission spectrometry (ICP-OES) to directly, and more accurately, measure trace metal concentrations in the aqueous phases pre-extraction (pre-X), post-extraction (post-X), and following back-extraction (back-X, vide infra).
Three separate 1,2-dichloroethane (DCE) solutions were next loaded with POCb (1 equiv.), [PPN][PF6] (0.5 equiv. [PPN]+ = [Ph3PNPPh3]+) as internal standard for NMR spectroscopy (vide infra), and [Bu4N][PF6] (0.1 M) as supporting electrolyte, and were loaded into one of two compartments of divided H-cells. Each counter compartment was loaded with a heterogeneous carbon additive (Ketjenblack) which served as a capacitive buffer8,27 and which was mixed in DCE with 0.1 M [Bu4N][PF6]. All H-cells were configured with physical glass-frit separators and contained reticulated vitreous carbon electrodes on each side (see ESI† for full experimental detail and H-cell setup). We note that each of these experiments were run in triplicate. The POCb solutions were electrochemically reduced by galvanostatic bulk electrolysis (GBE) to a theoretical state-of-charge (SOC) of ca. 77% assuming a 100% coulombic efficiency (Fig. S5†). Subsequent analyses of the carborane solutions by unlocked 31P{1H} NMR spectroscopy revealed the clean conversion of POCb to the reduced nido-carborane, POCb2−, each in approximate 76% yield and in line with the SOC. We note a loss of ca. 10% of combined carborane resonances (POCb and POCb2−) following charging and relative to the starting solutions and internal standard, perhaps due to ill-defined electrochemical side reactions. Each charged solution was then removed from its respective H-cell and mixed with either: (1) a non-buffered (pH = 2.6) aqueous mixed-metal solution with ca. 1.25 equiv. of each metal relative to POCb2− (Fig. 3a); (2) a NaOAc-buffered (pH = 5.2) aqueous mixed-metal solution with ca. 1.25 equiv. of each metal relative to POCb2− (Fig. 3b), or; (3) a NaOAc-buffered (pH = 5.2) aqueous mixed-metal solution with ca. 0.60 equiv. of each metal relative to POCb2− (Fig. 3c). Significant yellowing of the organic phases was observed after 1.5 h of rapid biphasic mixing (Fig. 1b).
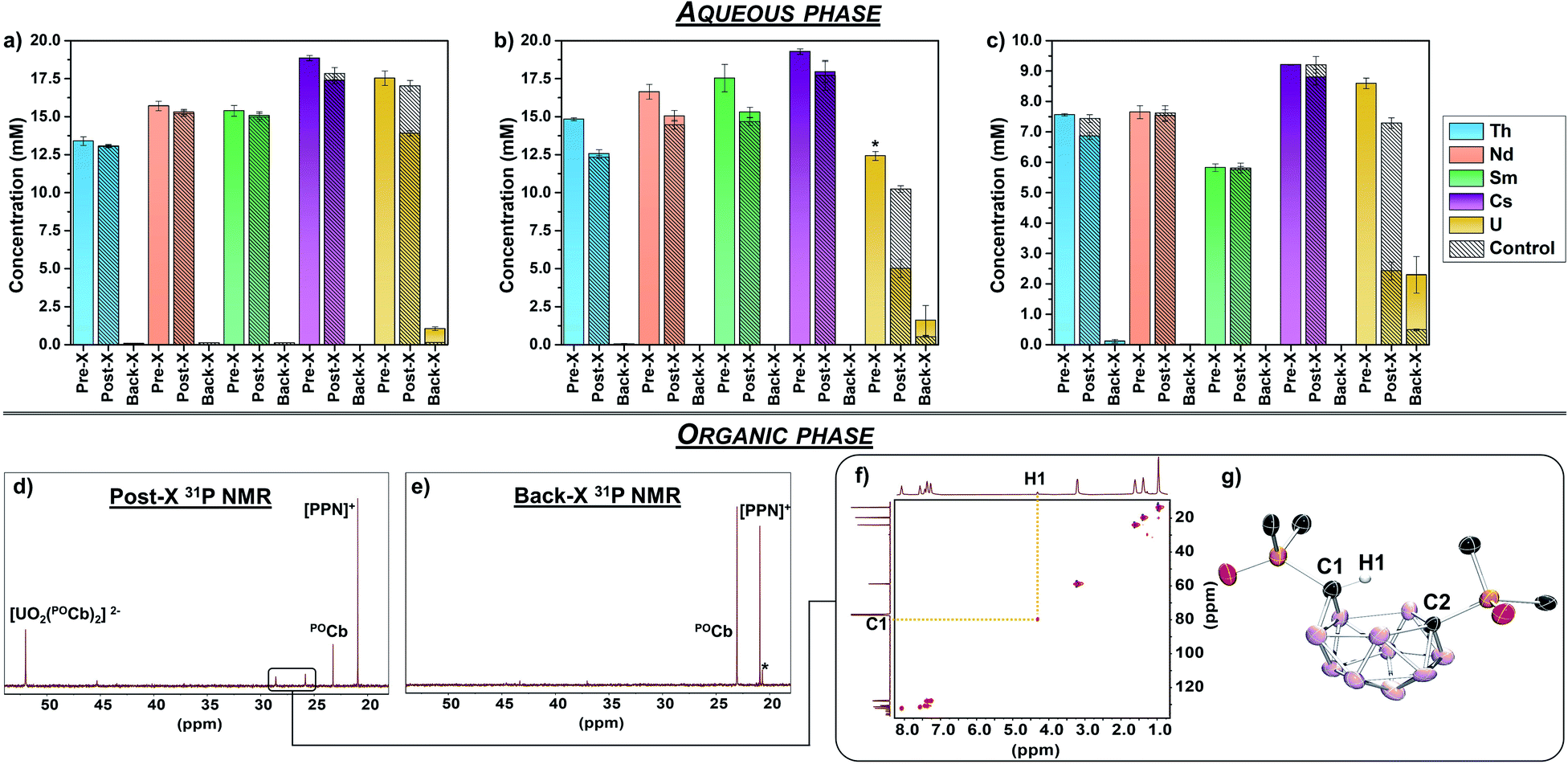 | ||
| Fig. 3 ICP-OES, spectroscopic, and crystallographic data for the selective electrochemical capture and release of UO22+ from mixed-metal (Cs+, Nd3+, Sm3+, Th4+, UO22+) aqueous solutions using the POCb/POCb2− system in DCE. (a–c) Average concentrations (from triplicate runs) of each metal species initially (pre-X) and following post-X and back-X using the following conditions and assuming 1.0 equiv. of POCb2−: (a) a non-buffered (pH = 2.6) aqueous mixed-metal solution with ca. 1.25 equiv. of each metal; (b) a NaOAc-buffered (pH = 5.2) aqueous mixed-metal solution with ca. 1.25 equiv. of each metal (*slightly lower for UO22+ due to saturation concentration); (c) a NaOAc-buffered (pH = 5.2) aqueous mixed-metal solution with ca. 0.6 equiv. of each metal. (d) Representative 31P{1H} NMR spectrum of the DCE layer following post-X of UO22+ from the aqueous, buffered mixed-metal solution (0.6 equiv.). No resonances attributable to Cs+, Nd3+, Sm3+, or Th4+ extraction are present. (e) Representative 31P{1H} NMR spectrum of the DCE layer following GBE and UO22+ back-X to a fresh, buffered aqueous phase (*unknown by-product). (f) 1H–13C HSQC NMR spectrum of [Bu4N][POCbH] in CDCl3 revealing C1–H1 correlation. (g) Solid-state molecular structure obtained by XRD studies of [Bu4N][POCbH] ([Bu4N]+ counter cation, phenyl C–H linkages, and all H atoms, except H1, are omitted for clarity). | ||
The aqueous mixed-metal phases were analyzed by ICP-OES prior to mixing with the organic phases (pre-X), following biphasic mixing (post-X), as well as following back-X (vide infra). Analysis of the non-buffered solution (Fig. 3a) post-X revealed an average decrease in UO22+ concentration of 20.7%, with minimal observed changes to the concentrations of Th4+, Nd3+, Sm3+, and Cs+ relative to pre-X. The minor decreases in the concentrations of these latter metals were within error to the observed changes in the controls, which were performed in parallel using identical aqueous and organic solutions (but without added POCb or POCb2−), as observed by the hashed bars in Fig. 3a–c. In contrast to this non-buffered solution, analysis of the buffered solution containing ca. 1.25 equiv. of each metal (Fig. 3b) post-X revealed an improved extraction of UO22+ with an average decrease in concentration of 59.7% relative to pre-X (note that the pre-X [UO22+] reached saturation here at a slightly lower concentration than the other metals). While extraction of Th4+, Nd3+, Sm3+, and Cs+ also increased here relative to the non-buffered solution, the observed changes were again within error and consistent with the control experiments, thus suggesting that the observed extraction of these ions was not driven by coordination to POCb2−. We next probed the effect of modifying the POCb2−:metal ratios. We note that the observed ratios of POCb2−:UO22+ are either 1:1 (ref. 8) (Cs+ also, Fig. 2a) or 2:1 (Fig. 2c), whereas all other complexes reported here (Th4+, Sm3+, Nd3+) are 3:1. Reducing the mixed-metal aqueous buffered solution concentration to ca. 0.6 equiv. of each metal to POCb2− revealed an increased post-X extraction of UO22+ – 71.6% relative to pre-X (Fig. 3c) – compared to the 1.25 equiv. extraction (Fig. 3b). While no significant changes in Nd3+, Sm3+, and Cs+ concentrations were observed here relative to the controls, we did observe a slight decrease in Th4+ concentration (9.2% vs. pre-X) which was greater than the control (1.6%) and beyond the detection error limit. While these data suggest that POCb2− may drive the extraction of some Th4+ under these higher ratios, the selectivity for UO22+ under these conditions still dominates, as evidenced by the calculated separation factor (SF), derived from the distribution ratios of metals: SFU/Th = 25.28
In each experiment, the organic phases were analyzed by 31P{1H} NMR spectroscopy prior to GBE, following GBE, following extraction (post-X stage), following GBE discharge (vide infra), and following back-X (see Fig. S2–S4† for representative spectra). A representative post-X spectrum is shown in Fig. 3d and revealed the formation of (integrated ratios relative to initially formed POCb2− are in parentheses): a main product at 52.0 ppm (54%), residual POCb (18%), and minor new byproduct peaks (28%) (vide infra, Fig. 3d). The main new resonance at 52.0 ppm in DCE matches the chemical shift of the bis-carborane complex, [CoCp*2]2[UO2(POCb)2], in MeCN-d3 and referenced to [PPN]+ (this salt is insoluble in DCE). Given that the calculated ICP-OES-determined quantity of captured UO22+ is 0.50 equiv. and 0.49 equiv. relative to the electrochemically generated POCb2− (1.0 equiv.) for the buffered 1.25 equiv. (Fig. 3b) and 0.60 equiv. (Fig. 3c) reactions, respectively, we propose that the resonance at 52.0 ppm most likely represents the bis-ligated anion, [UO2(POCb)2]2−. Together, these results indicate that electrochemically generated POCb2−selectively captures UO22+ from a mixed alkali, lanthanide, and actinide aqueous phase. While the nature of this selectivity remains under investigation, we suspect that optimal covalent bonding interactions between the PO units and the U center – very recently investigated in the PUREX context29,30 – are likely at play.
In addition to this selective capture, these biphasic experiments revealed the formation of minor byproduct resonances in the 31P{1H} NMR spectrum of the DCE phase at 28.6 and 25.8 ppm (Fig. 3d). These resonances consistently had a 1:1 ratio suggesting that this may be a single product with inequivalent P centers. We also observed that these resonances became dominant when mixing a DCE solution of POCb2− with an aqueous buffered phase in the absence of additional metal. Thus, treatment of a DCE solution containing [Bu4N]2[POCb] to the NaOAc-buffered aqueous solution without additional metals cleanly generated the byproduct, along with residual POCb. The unknown byproduct was isolated by separation of the DCE phase, removal of the solvent, and selective crystallization by vapor diffusion of pentane into a saturated THF solution of the crude mixture. Analysis of the colorless single crystals by XRD studies revealed the formation of the protonated, monoanionic carborane species, [Bu4N][POCbH] (Fig. 3g), featuring protonation at one of the nido-carborane C centers. The H1 atom at C1 was located in the difference map and was further observed by 1H and 1H–13C HSQC NMR spectroscopy (Fig. 3f). The distinctly different geometries at C1 versus C2 leads to the observed asymmetry in the product and is responsible for the distinct 31P NMR resonances observed. To the best of our knowledge, this is the first example of protonation of the ortho-substituted nido-carborane unit, [1,2-L2-1,2-C2B10H10]2−, at one of its C centers.
The release of extracted UO22+ was next probed electrochemically (Fig. 1b). The DCE phase containing the extracted UO22+ was separated from the aqueous phase and returned to the H-cell where it was galvanostatically discharged to achieve a theoretical final SOC of ca. 0% (Fig. S6†). The DCE layer was next removed from the H-cell and a fresh, buffered (0.1 M NaOAc) or non-buffered aqueous solution was mixed with it rapidly for 15 h. Analysis of the aqueous layer by ICP-OES revealed the back-X of 22–38% of UO22+ relative to post-X values, with similar values observed regardless of the use of buffered or non-buffered aqueous solutions (Fig. 3a–c). The highest UO22+ back-X observed were in the 0.6 equiv. separations (ca. 38%), wherein concurrent back-X of Th4+ was also observed, albeit in smaller quantities (ca. 16%) relative to post-X (Fig. 3c). With the exception of this case, the back-X of all metals except UO22+ was negligible compared to the controls. Further analysis of the DCE layer by 31P{1H} NMR spectroscopy revealed the conversion back to the starting closo-carborane, POCb (Fig. 3e), as well as a minor unknown byproduct at 20.2 ppm (ca. 10% of total carborane peaks). These results demonstrate the electrochemical back-X of selectively captured UO22+ to an aqueous phase.
Conclusion
In summary, we have demonstrated the selective biphasic electrochemical capture and release of UO22+ from mixed-metal aqueous media using the redox-switchable POCb/POCb2− system. This system may offer a unique, electrochemical, non-stoichiometric extraction platform – distinguished from current PUREX technology – for UO22+ separation. Further studies are underway to further optimize this proof-of-principle system and to probe the origin of this selectivity, as well as to expand the mixed-metal system and better mimic SNF mixtures. New metal capture and release chemistry of energy importance is also being investigated.Data availability
All of the experimental data have been included in the ESI.† Crystallographic data can be obtained from the CCDC (2071514–2071518).Author contributions
M. K. synthesized and fully characterized all new complexes. M. K. and M. M. performed the biphasic electrochemical extraction experiments. M. M. performed all ICP-OES analyses. S.-L. Z. and G. W. performed all crystallographic refinements. T. W. H. provided some starting materials and assisted with data analysis. M. K., M. M., and G. M. wrote the manuscript with input from all authors. G. M. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1900651), the US-Israel Binational Science Foundation (2018221), and the US Department of Energy, Office of Basic Energy Sciences (DE-SC0001861, DE-SC0021649) for funding. The MRL Shared Experimental Facilities are supported by the MRSEC Program of the NSF under Award No. DMR 1720256; a member of the NSF-funded Materials Research Facilities Network (http://www.mrfn.org).References
- IAEA, Nuclear Power and the Paris Agreement, https://www.iaea.org/sites/default/files/16/11/np-parisagreement.pdf, accessed 2/5/2021, 2021 Search PubMed.
- A. Adamantiades and I. Kessides, Energy Policy, 2009, 37, 5149–5166 CrossRef.
- D. J. Rose, Science, 1974, 184, 351–359 CrossRef CAS PubMed.
- I. Kumari, B. V. R. Kumar and A. Khanna, Nucl. Eng. Des., 2020, 358, 110410 CrossRef CAS.
- W. B. Lanham and T. C. Runion, Purex Process for Plutonium and Uranium Recovery (No. ORNL-479 (Del.)), Oak Ridge National Lab, USA, 1949 Search PubMed.
- J.-P. Glatz, in Comprehensive Nuclear Materials, ed. R. J. M. Konings and R. E. Stoller, Elsevier, Oxford, 2nd edn, 2020, pp. 305–326 Search PubMed.
- P. Carbol, D. H. Wegen, T. Wiss, P. Fors, C. Jegou and K. Spahiu, in Comprehensive Nuclear Materials, ed. R. J. M. Konings and R. E. Stoller, Elsevier, Oxford, 2nd edn, 2020, pp. 347–386 Search PubMed.
- M. Keener, C. Hunt, T. G. Carroll, V. Kampel, R. Dobrovetsky, T. W. Hayton and G. Ménard, Nature, 2020, 577, 652–655 CrossRef CAS PubMed.
- Y. Wu, X. Zhang, S.-Y. Kim and Y. Wei, J. Nucl. Sci. Technol., 2016, 53, 1968–1977 CrossRef CAS.
- M. A. Denecke, N. Bryan, S. Kalmykov, K. Morris and F. Quinto, in Experimental and Theoretical Approaches to Actinide Chemistry, ed. J. K. Gibson and W. A. Jong, Wiley, 2018, pp. 378–444 Search PubMed.
- F. Lahrouch, O. Sofronov, G. Creff, A. Rossberg, C. Hennig, C. Den Auwer and C. Di Giorgio, Dalton Trans., 2017, 46, 13869–13877 RSC.
- P. A. Bingham, R. J. Hand, M. C. Stennett, N. C. Hyatt and M. T. Harrison, MRS Online Proc. Libr., 2011, 1107, 421 CrossRef.
- A. E. V. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- N. S. Hosmane, T. Demissie, H. Zhang, J. A. Maguire, W. N. Lipscomb, F. Baumann and W. Kaim, Organometallics, 1998, 17, 293–295 CrossRef CAS.
- A. R. Oki, O. Sokolova, B. Gilbes, A. Aduroja, G. Abdelaziz and T. J. Emge, Inorg. Chem. Commun., 2002, 5, 694–697 CrossRef CAS.
- R. Neufeld, R. Michel, R. Herbst-Irmer, R. Schöne and D. Stalke, Chem. - Eur. J., 2016, 22, 12340–12346 CrossRef CAS PubMed.
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O'Hara, Inorg. Chem., 2016, 55, 5719–5728 CrossRef CAS PubMed.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Polyhedron, 2003, 22, 3475–3482 CrossRef CAS.
- Z. Spichal, M. Necas, J. Pinkas and Z. Zdrahal, Polyhedron, 2006, 25, 809–814 CrossRef CAS.
- K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem. - Eur. J., 2011, 17, 521–528 CrossRef CAS PubMed.
- Y.-Z. Pan, Q.-Y. Hua, L.-S. Lin, Y.-B. Qiu, J.-L. Liu, A.-J. Zhou, W.-Q. Lin and J.-D. Leng, Inorg. Chem. Front., 2020, 7, 2335–2342 RSC.
- I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248–6252 CrossRef CAS.
- A.-G. D. Nelson, T. H. Bray, F. A. Stanley and T. E. Albrecht-Schmitt, Inorg. Chem., 2009, 48, 4530–4535 CrossRef CAS PubMed.
- J. Diwu, J. J. Good, V. H. DiStefano and T. E. Albrecht-Schmitt, Eur. J. Inorg. Chem., 2011, 2011, 1374–1377 CrossRef.
- F. Quilès, C. Nguyen-Trung, C. Carteret and B. Humbert, Inorg. Chem., 2011, 50, 2811–2823 CrossRef.
- D. D. Pant and D. P. Khandelwal, Proc. - Indian Acad. Sci., Sect. A, 1959, 50, 323–335 CrossRef.
- C. Hunt, M. Mattejat, C. Anderson, L. Sepunaru and G. Ménard, ACS Appl. Energy Mater., 2019, 2, 5391–5396 CrossRef CAS.
- N. A. Thiele, D. J. Fiszbein, J. J. Woods and J. J. Wilson, Inorg. Chem., 2020, 59, 16522–16530 CrossRef CAS PubMed.
- D. Raychaudhuri, G. Gopakumar, S. Nagarajan and C. V. S. Brahmmananda Rao, J. Phys. Chem. A, 2020, 124, 7805–7815 CrossRef CAS PubMed.
- Y. Zhang, W. Duan, Y. Yang, T. Jian, Y. Qiao, G. Ren, N. Zhang, L. Zheng, W. Yan, J. Wang, J. Chen, S. G. Minasian and T. Sun, Inorg. Chem., 2022, 61, 92–104 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, electrochemical details, NMR, UV-vis, XRD data. CCDC 2071514–2071518. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc07070c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |