
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNonspherical anion sequestration by C–H hydrogen bonding†
Saber
Mirzaei
 ,
Victor M.
Espinoza Castro
and
Raúl
Hernández Sánchez
*
,
Victor M.
Espinoza Castro
and
Raúl
Hernández Sánchez
*
Department of Chemistry, University of Pittsburgh, 219 Parkman Ave., Pittsburgh, Pennsylvania 15260, USA. E-mail: raulhs@pitt.edu
First published on 26th January 2022
Abstract
Macrocyclic arenes laid the foundations of supramolecular chemistry and their study established the fundamentals of noncovalent interactions. Advancing their frontier, here we designed rigidified resorcin[4]arenes that serve as hosts for large nonspherical anions. In one synthetic step, we vary the host's anion affinity properties by more than seven orders of magnitude. This is possible by engineering electropositive aromatic C–H bond donors in an idealized square planar geometry embedded within the host's inner cavity. The hydrogen atom's electropositivity is tuned by introducing fluorine atoms as electron withdrawing groups. These novel macrocycles, termed fluorocages, are engineered to sequester large anions. Indeed, experimental data shows an increase in the anion association constant (Ka) as the number of F atoms increase. The observed trend is rationalized by DFT calculations of Hirshfeld Charges (HCs). Most importantly, fluorocages in solution showed weak-to-medium binding affinity for large anions like [PF6]− (102< Ka <104 M−1), and high affinity for [MeSO3]− (Ka >106).
Introduction
Macrocyclic arenes are quintessential compounds in the development of host–guest chemistry.1–5 Guests are usually hosted within the inner cavity of the macrocycle through non-covalent interactions, e.g., hydrogen bonding, π⋯π stacking, and C–H⋯π interactions.6 Among these non-covalent interactions, hydrogen bonding is the strongest one,7 especially when considering oxygen and nitrogen, where the difference in Pauling electronegativity with hydrogen (Δχ) is 1.24 and 0.84,8 respectively. Lehn et al. recognized in 1976 that incorporating hydrogen bonding capabilities (N–H bonds) within cryptands resulted in weak-to-medium affinity for spherical anions (e.g., halides),9,10 this realization opened the door to “anion coordination chemistry”.11 Later reports demonstrated fluorinated macrocycles hosting fluoride ion purely through C–H⋯F− hydrogen bonding,12 which is remarkable noting that Δχ for C and H is just 0.35. However, due to the relatively weak binding affinities, molecular designs for anion binding based solely on C–H hydrogen bonding were marginalized in supramolecular host–guest chemistry.13,14 This notion has been challenged, and in the past few decades a range of hosts with remarkable affinities towards spherical anions (Ka >106 M−1) have been reported by J. L. Sessler,15–17 P. A. Gale,18 A. P. Davis,18–20 B. D. Smith,20 A. H. Flood,21–25 V. Sindelar,26,27 J. Yoon,28 J. You,29 M. Pittelkow,30 H. Jiang,31 M. Stępień,32 and others.33Molecular recognition of nonspherical anions have transformed the design of rotaxane assemblies,34 advanced novel catalytic processes by stabilization of in situ generated anions,35 laid the ground work supporting anion–anion stabilization theories,36 facilitated ion pair dissociation in battery electrolytes,37 and provided systems of remarkable selectivity towards the recognition of bifluoride,38 organophosphates,39 dicarboxylates,40 the biologically relevant GTP anion,41 nitrate,42,43 and sulfate.44 Despite these noteworthy advances, hosts capable of binding nonspherical anions with high affinity are rare and usually lack synthetic tunability requiring a complete host redesign to tune binding affinities, therefore efforts to develop hosts for nonspherical anions remains a challenge. Herein, we report a strategy to create novel supramolecular anion cages through a straightforward and versatile synthetic procedure in which we discovered binding preferences for nonspherical anions.
Results and discussion
Host–guest design
Fluorocages, as we refer to these supramolecular hosts, were conceived by trying to maximize the host–guest properties of resorcin[n]arenes45 towards anionic species. Note that while their host–guest capabilities for neutral guests are well-established,6 their anion hosting abilities are not nearly as developed.46,47 Towards this goal, we designed and synthesized a modular family of resorcin[4]arene-based cages, 1–8 (Scheme 1), all having the same binding cage geometry capable of accommodating large guests, and also able to tune the overall framework to systematically and monotonically increase the anion binding affinity. | ||
| Scheme 1 Synthesis of fluorocages 1–8. | ||
Fluorocages structure
Fluorocages 1–8 were synthesized from S1 (ref. 48) or S2 (ref. 49) through Suzuki–Miyaura cross-coupling reactions with the corresponding aromatic flanking unit, Fn as described in Scheme 1, in yields ranging from 38 to 53% (see ESI†). All hosts define a cavity comprised of eight C–H donors, four aromatic colored in blue (CAr–H) and four aliphatic colored in dark purple (CCH2–H). Our hypothesis is that installing electron withdrawing groups (EWGs) on the aromatic flanking units would produce sufficiently high electropositive hydrogen atoms in CAr–H capable of binding anionic species with high affinity. Moreover, fluorocages 1–8 are designed as rigid scaffolds to minimize entropic penalties that may arise from conformational flexibility and host rearrangement upon guest binding.50,51
1H, 13C, and 19F NMR spectra in CDCl3 of 1–4, 6, and 8 reveals their expected ideal C4v molecular symmetry in solution (see ESI†). Fluorocages 5 and 7 display a more complex behavior (Fig. S26–S28 and S32–S34†), however in-depth analysis of their 1H NMR spectra corroborates their assignment. High quality crystals for single-crystal X-ray diffraction for 1–8 provided further confirmation of their molecular structure (Fig. 1). Crystals of 1–8 were all obtained by slow evaporation of a MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 solution at room temperature. Note that all fluorocage structures display one molecule of MeCN bound within their inner cavity, except 7 which displayed heavy disorder that prevented correct modeling of the MeCN molecule. The molecular structures of 3, 5, and 7 displayed rotational disorder around their aromatic flanking units. For example, fluorocage 3 displays in the major occupancy structure three F atoms pointing into the inner cavity while one F atom is pointing away. Nonetheless, this behavior is not manifest in solution, as the NMR data indicates a fully C4-symmetric structure. In contrast, 5 and 7 display solid state major occupancy structures with idealized Cs and C2v point group symmetries, respectively, which is reconciled with their complex solution behavior indicating that rotation around the biaryl moiety is hampered and that all possible rotational isomers coexist in solution, even at 100 °C (Fig. S69 and S71†). Up to six different rotamers are possible for 5 and 7. DFT calculations at the M06-2X/6-311++G(3df,2p)+CPCM(solvent)//M06-2X/6-31+G(d,p) level of theory, where solvent = CHCl3 or DMSO, showed that these rotamers are relatively close in energy, thus supporting the experimental observations in solution (Tables S2 and S3†).
:CH2Cl2 solution at room temperature. Note that all fluorocage structures display one molecule of MeCN bound within their inner cavity, except 7 which displayed heavy disorder that prevented correct modeling of the MeCN molecule. The molecular structures of 3, 5, and 7 displayed rotational disorder around their aromatic flanking units. For example, fluorocage 3 displays in the major occupancy structure three F atoms pointing into the inner cavity while one F atom is pointing away. Nonetheless, this behavior is not manifest in solution, as the NMR data indicates a fully C4-symmetric structure. In contrast, 5 and 7 display solid state major occupancy structures with idealized Cs and C2v point group symmetries, respectively, which is reconciled with their complex solution behavior indicating that rotation around the biaryl moiety is hampered and that all possible rotational isomers coexist in solution, even at 100 °C (Fig. S69 and S71†). Up to six different rotamers are possible for 5 and 7. DFT calculations at the M06-2X/6-311++G(3df,2p)+CPCM(solvent)//M06-2X/6-31+G(d,p) level of theory, where solvent = CHCl3 or DMSO, showed that these rotamers are relatively close in energy, thus supporting the experimental observations in solution (Tables S2 and S3†).
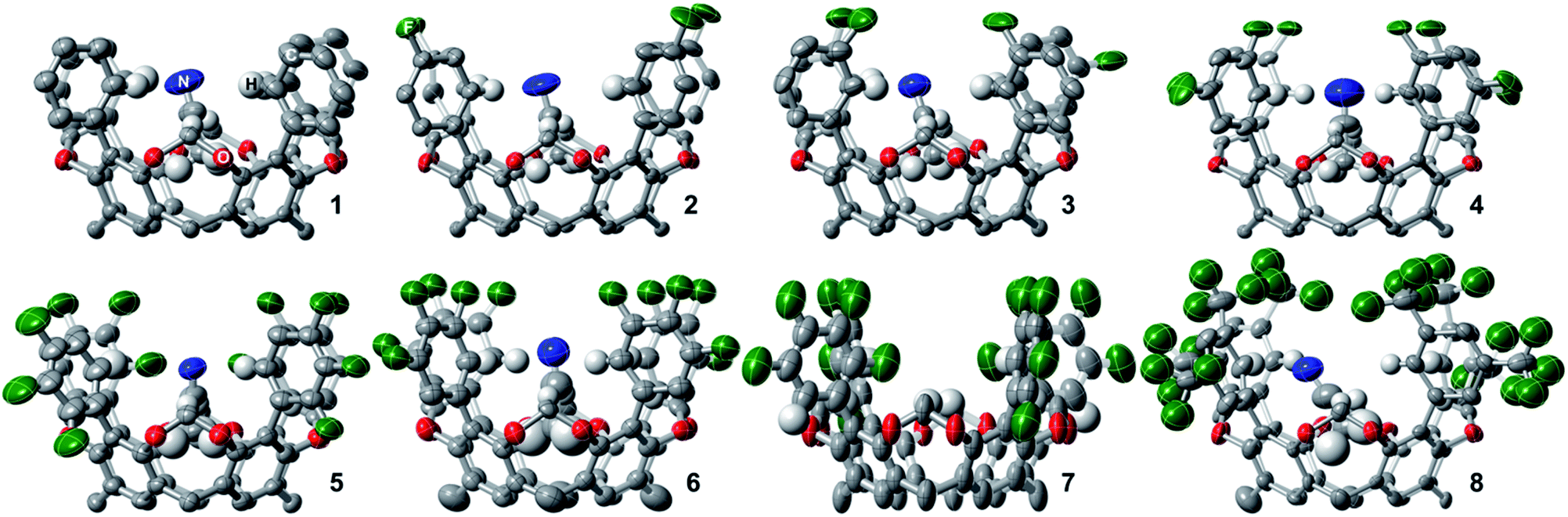 | ||
| Fig. 1 Molecular crystal structures of fluorocages 1–8. Thermal ellipsoids are set at 50% probability level (except for 6 which are at 30%). The R groups (n-pentyl) and H atoms are omitted for clarity, except those H atoms within the fluorocage's inner cavity. The C, N, O, F, and H atoms are colored grey, blue, red, green, and white, respectively. | ||
| Salt | Solvent | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| a ND = Not determined. b Strong binding that prevents direct titration via1H NMR, even at 100 °C the anion remains bound. 106 M−1 is taken as the maximum reliable limit for NMR titration experiments.52 c Not soluble in DMSO-d6 even at 100 °C. | |||||||||
| [n-Bu4N] [PF6] | CDCl3 | 0 | 0 | 0 | 84(12) | 0 | 1.510(1) × 104 | 1.64(11) × 103 | 280(15) |
| [n-Bu4N] [MeSO3] | DMSO-d6 | NDa | NDa | NDa | >106b | >106b | >106b | >106b | NDc |
Anion affinity towards square planar electropositive cavity
The design and engineering of the electropositive cavity within 1–8 is best exemplified by the hosted MeCN molecule, which has its electronegative N atom residing in the same plane as the cavity's electropositive H atoms (colored in blue in Scheme 1). The average C-to-N distances (CAr–H and NMeCN) for all fluorocages is 4.0 ± 0.2 Å [4.0(2) Å (number in parenthesis indicates the estimated standard deviation in the final digit)], while the average C-to-π system centroid (CMeCN and centroid of each aromatic ring comprising the resorcin[4]arene base) is 3.55(4) Å. Anion sequestration was first tested by 1H NMR titration experiments adding [n-Bu4N][PF6] to 2–8 in CDCl3 (solvent dielectric constant, εr = 4.7) at 20 °C (Table 1 and Fig. S38–S44†). There is negligible affinity for [PF6]− by 1–3, however 4, 6, and 8 display weak-to-medium binding peaking at 6 with Ka of 1.510(1) × 104 M−1, while 4 and 8 display Ka of 84(12) and 280(15) M−1, respectively. To our surprise, 5 displays no binding of [PF6]−, while 7 only reaches Ka = 1.64(11) × 103 M−1, likely as a result of the energetic penalty involved in rearranging their initial conformational distribution. Note that only a few synthetic hosts are known to bind large anions such as [PF6]−.22,34,46,53 Overall, the adduct [PF6⊂6]− demonstrates that the square planar arrangement of electropositive H atoms serves to sequester large anionic species.Encouraged by the binding results of [PF6]−, we tested the ability of fluorocage 6 to bind other noncoordinating nonspherical anions. For reference, the large iodide anion, with a thermochemical radii (r)54 of 2.11(19) Å displays a Ka = 161(19) M−1 (Fig. S45†); and as the spherical anion shrinks, e.g. Br− (r = 1.90(19) Å), the observed Ka goes to zero (Fig. S46†). Linear [N3]− and [SCN]−, bent [NO2]−, and trigonal planar [NO3]− have negligible-to-weak binding affinities (Table 2, Fig. S47–S50†), likely resulting from their size mismatch with the host's cavity. However, strong binding is observed as the anion's size reaches a radius of ∼2.2 Å judging from the series [BF4]− (r = 2.05(19) Å, Fig. S51†), [HSO4]− (r = 2.21(19) Å, Fig. S52†), [ClO4]− (r = 2.25(19) Å, Fig. S53†), [ReO4]− (r = 2.27(19) Å, Fig. S54†), [IO4]− (r = 2.31(19) Å, Fig. S55†), and [SbF6]− (r = 2.52(19) Å, Fig. S56†). Note that anion binding affinity decreases by about two orders of magnitude when the anion's size surpass [PF6]−, as observed in [SbF6]− which is only ∼4% larger than [PF6]−. Thus, we establish that the optimum anion's size fitting in the host's cavity must be 2.2< r <2.4 Å. Other nonspherical anions, such as acetate, [MeCO2]−, also binds to 6 (Ka = 4.8(3) × 103 M−1, Fig. S57†), however the closely related [MeSO3]− binds so strongly to 6 in CDCl3 that its affinity falls beyond the reliable measurable limit via1H NMR (106 M−1), see Fig. 2a.52 Note that a marked downfield complexation-induced chemical shift occurs for protons “a” (Δδ ≈ 0.57 ppm) and “bin” (Δδ ≈ 0.69 ppm) as established for H atoms involved in direct hydrogen bonding with the bound anion (Fig. 2a). In contrast, proton “bout” shifts upfield as a result of increased electron density on the C atom (methylene) due to anion binding.56 We conclude that sulfonate's structure is better poised to interact with the four electropositive H atoms as opposed to the flat structure of carboxylate. Note that p-toluenesulfonate, [pTsO]−, with its much larger organic group, also binds to 6 displaying a Ka of 1.88(4) × 103 M−1, where its p-toluene fragment points away from the fluorocage's cavity (Fig. S59 and S60, Table S4†).
| Host | Solvent | I−/Br− | [N3]−/[SCN]− | [NO2]−/[NO3]− | [BF4]− (2.05) | [HSO4]− (2.21) | [ClO4]− (2.25)/[ReO4]− (2.27) | [IO4]− (2.31) | [PF6]− (2.42)/[SbF6]− (2.52) | [MeCO2]−/[MeSO3]− | [pTsO]−a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a pTs = p-toluenesulfonyl. | |||||||||||
| 6 | CDCl3 | 161(19)/0 | 8(6)/290(8) | 40(40)/76(6) | 990(23) | 7(3) × 104 | 7.91(9) × 103/1.08(2) × 103 | 1.00(3) × 103 | 1.510(1) × 104/230(20) | 4.8(3) × 103/>106 | 1.88(4) × 103 |
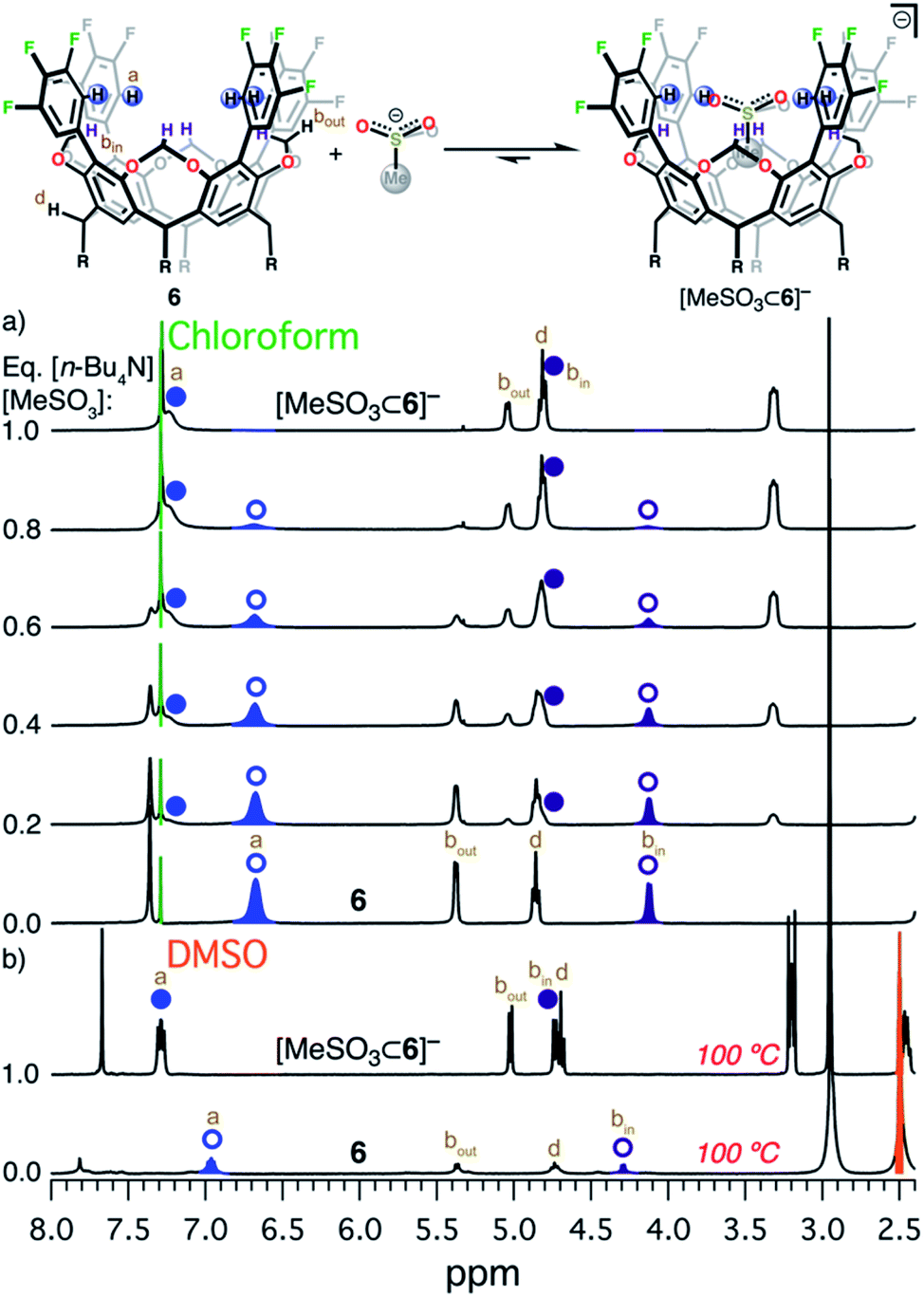 | ||
| Fig. 2 Equilibrium between 6 and [MeSO3]−. (a) 1H NMR titration of [n-Bu4N][MeSO3] into 6 in CDCl3 at 20 °C. (b) 1H NMR of 6 (bottom)55 and [n-Bu4N][MeSO3⊂6] (top) in DMSO-d6 at 100 °C. Circular symbols correspond to the blue and dark purple hydrogen atoms in the scheme above. Open symbols indicate free host 6, while solid symbols correspond to the adduct [n-Bu4N][MeSO3⊂6]. Relevant H atoms are labeled a, bin, and bout. | ||
Sulfonate anion appears to have the optimum size to fit in the cavity described by the four electropositive H atoms in CAr–H. Intrigued by this observation, we decided to expand our efforts and investigate the binding properties of [MeSO3]− towards 1–5 and 7–8 in CDCl3 at 20 °C. Host 1 failed to bind [MeSO3]− (Fig. S61†). Fluorocages 2 and 3 displayed binding of [MeSO3]− with Ka of 22(4) and 667(38) M−1 (Fig. S62 and S63†), respectively, while host 8 exhibited slow exchange57,58 in the NMR time scale with Ka ≈ 2.5(2) × 103 M−1 (Fig. S67 and Table S4†). In contrast, fluorocages 4 and 7, similar to 6, revealed binding affinities well-above 106 M−1 (Fig. S64 and S66,† respectively). Note that 5 displayed no binding of [MeSO3]− in CDCl3 (Fig. S65†). To our surprise, NMR experiments in the much more polar solvent DMSO (εr = 46.8) display 4–7 strongly binding [MeSO3]−, even when the solution is heated to 100 °C (in situ1H NMR, Fig. 2b and Fig. S68–S71†). This finding led us to conclude that binding of [MeSO3]− to hosts 4–7 in DMSO-d6 exceeds Ka of 106 M−1. Assuming the general anion binding in solution model put forward by Flood et al.59 holds true for [MeSO3]−, we expect Ka for [MeSO3]− binding to hosts 4–7 in CDCl3 to surpass 106 M−1 by several orders of magnitude. We note that binding is mostly driven by interaction with the four electropositive H atoms in CAr–H and not by C–H⋯π interactions found between the Me group in MeSO3− and the π system centroids within the resorcin[4]arene backbone since the average C-to-π distance for [MeSO3⊂host]−, where host = 4, and 6–8, is 3.62(3) Å; which is larger than that found in the neutral adduct [MeCN⊂host], for host = 1–6, and 8 (vide supra). Altogether, the qualitative picture portrayed by this NMR data reveals that the equilibrium between host + [MeSO3]− and [MeSO3⊂host]−, for hosts 4–7, is strongly displaced towards the host–guest adduct.
The strong interaction with methanesulfonate allowed us to isolate and crystallize the adducts [n-Bu4N][MeSO3⊂host], for host = 4, and 6–8 (Fig. S9–S12†). Shown in Fig. 3a is the molecular structure and relevant distances for [MeSO3⊂6]−. Note how the three O atoms from the sulfonate group reside in the square plane described by the four electropositive H1X atoms, X = A–D (colored in blue in Scheme 1). This sulfonate anion accommodation maximizes the CAr–H⋯OMeSO3− hydrogen bonding, with remarkably short C-to-O average distances of 3.31(4), 3.24(3), 3.18(7), and 3.34(6) Å for 4, 6, 7, and 8 (Fig. 3b), respectively.60 Note that the sum of the van der Waals radius for C and O is 3.22 Å.61 Fluorocages adopt a cone-shaped structure; to determine the expansion of this cone, we measured the distances between rigid carbon atoms C6X (Fig. 3a), X = A–D, which define an almost ideal square (C–Csquare). Plotting these data together, we observe a V-shaped trend in the C-to-O distance, with 7 at the minimum, and no discernible correlation in C–Csquare distances (Fig. 3b), meaning that strengthening of the hydrogen bonds within the host's cavity only requires rotational movement of the aromatic flanking units as opposed to a breathing in or out distortion.
 | ||
| Fig. 3 (a) Side and top view of the molecular crystal structure of [MeSO3⊂6]− obtained at 220 K. Thermal ellipsoids are set at 50% probability level. The [n-Bu4N]+, R groups (n-pentyl), and H atoms are omitted for clarity, except those H atoms within the inner cavity. (b) Comparison of structural molecular metrics. Average C–Csquare (red and maroon) and C⋯O (green) distances. Open symbols correspond to 1–8, and filled symbols to [MeSO3⊂host]−, for host = 4, and 6–8. | ||
DFT-supported structure–function relationship
To gain insight into the trends observed in this family of fluorocages, we performed DFT calculations (M06-2X/6-31+G(d,p) level of theory) in 1–8 to obtain their Hirshfeld Charges (HCs) in an effort to access quantitative data about the electropositivity of the H atoms involved in hydrogen bonding (Fig. 4). Note that HCs are recommended as they yield chemically meaningful partial charges.62 Following the series from 1 to 7, we observe a monotonic increase in HC at the H atoms in CAr–H (blue). Unexpectedly, the calculated HCs for fluorocage 8 reside in between those of 3 and 4. Notably, HCs for 8 explain its weaker binding of [MeSO3]− compared to 4–7. The HC values for the hydrogen atoms in CCH2–H (dark purple) are provided in Fig. 4 as control since these should not be affected by the nature of the EWG in the aromatic flanking unit. Overall, this trend correlates well with the binding affinity studies of 1–8, and with the metrics observed in the host-guest adducts [n-Bu4N][MeSO3⊂host], for host = 4, and 6–8 (Fig. 3b). Note that the HCs on 5 and 7 does not correlate well with their binding properties, which we attribute to the conformational distribution observed in the as-synthesized hosts and the large aromatic flanking unit rotational barrier (biaryl bond) of ∼22 kcal mol−1, compared to ∼12 kcal mol−1 for all other hosts (Table S5†). | ||
| Fig. 4 DFT-calculated Hirshfeld charges for 1–10 (M06-2X/6-31+G(d,p) level of theory). Blue CAr–H, dark purple CCH2–H, pink CCC–H, and red Ctriazole–H hydrogen atoms correspond to those shown in Scheme 1 or in the chemdraw drawings in this figure. | ||
In an effort to compare hosting capabilities of fluorocages with other rigid anion-binding hosts operating solely by C–H hydrogen bonding, we analyzed two recent macrocyclic hosts reported in the literature (Fig. S72†): (1) pentagonal cyanostar 9, capable of forming a strong 2:1 host:guest adduct with Cl− (40% MeOH/CDCl3, β2 ≈ 108 M−2);22 and (2) cryptand-like triazole cage 10,63 which binds Cl− remarkably strong (CDCl3, Ka ≈ 1017 M−1).25 HCs of the H atoms involved in C–H hydrogen bonding increase from 9 to 10, as shown in Fig. 4, supporting the relative trend observed in their anion binding properties. Furthermore, in the case of 10, the most electropositive H atoms reside in Ctriazole–H and are on par to those found in fluorocage 4. However, while 4 has four of these electropositive H atoms, 10 has six of them making its internal cavity more electropositive. Most importantly, 5–7 display higher HCs relative to 10 suggesting that further exploration of these fluorocages have the potential to uncover strong affinities for anions of appropriate size to fit in the square planar geometry defined by the blue electropositive H atoms.
Conclusions
Herein we describe a family of anion hosts – termed fluorocages – sharing the same overall structure, however with anion affinities tuned by several orders of magnitude through a straightforward single four-fold Suzuki–Miyaura cross-coupling synthetic step. These fluorocages are able to bind nonspherical anions, such as sulfonate groups, which display remarkable affinity and size complementarity to their cavity. Further development of this general strategy, and based on the structure–function relationship reported herein, will guide the development of novel, more potent, and selective hosts for anion sequestration from polluted ecosystems, e.g., sulfonate-based PFAS,64–66 currently an unmet pressing challenge.Data availability
All the data have been included in the ESI.†Author contributions
S. M. and V. M. E. C. performed the synthesis, titrations, and measurements; S. M. performed the DFT analysis; S. M. and R. H. S. carried out the crystallographic studies; S. M. and R. H. S. conceived the research hypothesis; R. H. S. prepared the manuscript; all authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support from the Center for Research Computing and startup funds from the University of Pittsburgh. S. M. acknowledges the support from the Dietrich School of Arts & Sciences Graduate Fellowship and the Andrew Mellon Pre-doctoral Fellowship.Notes and references
- J. J. Rebek, Chem. Commun., 2000, 637–643 RSC.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- G.-W. Zhang, P.-F. Li, Z. Meng, H.-X. Wang, Y. Han and C.-F. Chen, Angew. Chem., Int. Ed., 2016, 55, 5304–5308 CrossRef CAS PubMed.
- C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- C. Gropp, B. L. Quigley and F. Diederich, J. Am. Chem. Soc., 2018, 140, 2705–2717 CrossRef CAS PubMed.
- J. Řezáč and P. Hobza, Chem. Rev., 2016, 116, 5038–5071 CrossRef PubMed.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- E. Graf and J. M. Lehn, J. Am. Chem. Soc., 1976, 98, 6403–6405 CrossRef CAS.
- B. Dietrich, B. Dilworth, J.-M. Lehn, J.-P. Souchez, M. Cesario, J. Guilhem and C. Pascard, Helv. Chim. Acta, 1996, 79, 569–587 CrossRef CAS.
- K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- W. B. Farnham, D. C. Roe, D. A. Dixon, J. C. Calabrese and R. L. Harlow, J. Am. Chem. Soc., 1990, 112, 7707–7718 CrossRef CAS.
- O. B. Berryman, A. C. Sather, B. P. Hay, J. S. Meisner and D. W. Johnson, J. Am. Chem. Soc., 2008, 130, 10895–10897 CrossRef CAS PubMed.
- J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
- P. A. Gale, J. L. Sessler, V. Kral and V. Lynch, J. Am. Chem. Soc., 1996, 118, 5140–5141 CrossRef CAS.
- C.-H. Lee, H.-K. Na, D.-W. Yoon, D.-H. Won, W.-S. Cho, V. M. Lynch, S. V. Shevchuk and J. L. Sessler, J. Am. Chem. Soc., 2003, 125, 7301–7306 CrossRef CAS PubMed.
- D.-W. Yoon, D. E. Gross, V. M. Lynch, J. L. Sessler, B. P. Hay and C.-H. Lee, Angew. Chem., Int. Ed., 2008, 47, 5038–5042 CrossRef CAS PubMed.
- S. J. Edwards, H. Valkenier, N. Busschaert, P. A. Gale and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 4592–4596 CrossRef CAS PubMed.
- A. J. Ayling, M. N. Pérez-Payán and A. P. Davis, J. Am. Chem. Soc., 2001, 123, 12716–12717 CrossRef CAS PubMed.
- J. P. Clare, A. J. Ayling, J.-B. Joos, A. L. Sisson, G. Magro, M. N. Pérez-Payán, T. N. Lambert, R. Shukla, B. D. Smith and A. P. Davis, J. Am. Chem. Soc., 2005, 127, 10739–10746 CrossRef CAS PubMed.
- Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649–2652 CrossRef CAS PubMed.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef CAS PubMed.
- S. Lee, B. E. Hirsch, Y. Liu, J. R. Dobscha, D. W. Burke, S. L. Tait and A. H. Flood, Chem. - Eur. J., 2016, 22, 560–569 CrossRef CAS PubMed.
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159 CAS.
- J. Svec, M. Necas and V. Sindelar, Angew. Chem., Int. Ed., 2010, 49, 2378–2381 CrossRef CAS PubMed.
- T. Lizal and V. Sindelar, Isr. J. Chem., 2018, 58, 326–333 CrossRef CAS.
- N. R. Song, J. H. Moon, J. Choi, E. J. Jun, Y. Kim, S.-J. Kim, J. Y. Lee and J. Yoon, Chem. Sci., 2013, 4, 1765–1771 RSC.
- H. Zhou, Y. Zhao, G. Gao, S. Li, J. Lan and J. You, J. Am. Chem. Soc., 2013, 135, 14908–14911 CrossRef CAS PubMed.
- M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed.
- J. Shang, W. Zhao, X. Li, Y. Wang and H. Jiang, Chem. Commun., 2016, 52, 4505–4508 RSC.
- H. Gregolińska, M. Majewski, P. J. Chmielewski, J. Gregoliński, A. Chien, J. Zhou, Y.-L. Wu, Y. J. Bae, M. R. Wasielewski, P. M. Zimmerman and M. Stępień, J. Am. Chem. Soc., 2018, 140, 14474–14480 CrossRef PubMed.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195–5206 RSC.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- N. Lopez, D. J. Graham, R. McGuire, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450 CrossRef CAS PubMed.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- B. Qiao, G. M. Leverick, W. Zhao, A. H. Flood, J. A. Johnson and Y. Shao-Horn, J. Am. Chem. Soc., 2018, 140, 10932–10936 CrossRef CAS PubMed.
- R. O. Ramabhadran, Y. Liu, Y. Hua, M. Ciardi, A. H. Flood and K. Raghavachari, J. Am. Chem. Soc., 2014, 136, 5078–5089 CrossRef CAS PubMed.
- W. Zhao, B. Qiao, C.-H. Chen and A. H. Flood, Angew. Chem., Int. Ed., 2017, 56, 13083–13087 CrossRef CAS PubMed.
- S. K. Kim, B.-G. Kang, H. S. Koh, Y. J. Yoon, S. J. Jung, B. Jeong, K.-D. Lee and J. Yoon, Org. Lett., 2004, 6, 4655–4658 CrossRef CAS PubMed.
- J. Y. Kwon, N. J. Singh, H. N. Kim, S. K. Kim, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2004, 126, 8892–8893 CrossRef CAS PubMed.
- M. Arunachalam and P. Ghosh, Inorg. Chem., 2010, 49, 943–951 CrossRef CAS PubMed.
- S.-Y. Zhuang, Y. Cheng, Q. Zhang, S. Tong and M.-X. Wang, Angew. Chem., Int. Ed., 2020, 59, 23716–23723 CrossRef PubMed.
- H. Xie, T. J. Finnegan, V. W. Liyana Gunawardana, R. Z. Pavlović, C. E. Moore and J. D. Badjić, J. Am. Chem. Soc., 2021, 143, 3874–3880 CrossRef CAS PubMed.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- O. Hayashida, A. Shivanyuk and J. J. Rebek, Angew. Chem., Int. Ed., 2002, 41, 3423–3426 CrossRef CAS PubMed.
- S. S. Zhu, H. Staats, K. Brandhorst, J. Grunenberg, F. Gruppi, E. Dalcanale, A. Lützen, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2008, 47, 788–792 CrossRef CAS PubMed.
- J. A. Bryant, M. T. Blanda, M. Vincenti and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2167–2172 CrossRef CAS.
- C. B. Aakeröy, P. D. Chopade, N. Schultheiss and J. Desper, Eur. J. Org. Chem., 2011, 2011, 6789–6793 CrossRef.
- D. H. Williams and M. S. Westwell, Chem. Soc. Rev., 1998, 27, 57–64 RSC.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- B. Qiao, J. R. Anderson, M. Pink and A. H. Flood, Chem. Commun., 2016, 52, 8683–8686 RSC.
- H. K. Roobottom, H. D. B. Jenkins, J. Passmore and L. Glasser, J. Chem. Educ., 1999, 76, 1570 CrossRef CAS.
- Host 6 has extremely low solubility in DMSO, even at 100 °C Search PubMed.
- K. Choi and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 10241–10249 CrossRef CAS PubMed.
- M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559–3563 CrossRef CAS PubMed.
- The exchange barrier for anion binding likely results from steric impediment of the CF3 groups in 8, as shown in Fig. S12.†.
- Y. Liu, A. Sengupta, K. Raghavachari and A. H. Flood, Chem, 2017, 3, 411–427 CAS.
- The H-to-O distance comprising formally the hydrogen bonds seen here are remarkably short. However, since the H atoms are not refined (they are simply fixed to the structure through AFIX commands), we decided not to relay on those figures and instead use the more reliable C-to-O distances Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- Host 10 has been reported with R = –CONCy2, however to reduce computational costs, Cy was truncated to Me Search PubMed.
- C. F. Kwiatkowski, D. Q. Andrews, L. S. Birnbaum, T. A. Bruton, J. C. DeWitt, D. R. U. Knappe, M. V. Maffini, M. F. Miller, K. E. Pelch, A. Reade, A. Soehl, X. Trier, M. Venier, C. C. Wagner, Z. Wang and A. Blum, Environ. Sci. Technol. Lett., 2020, 7, 532–543 CrossRef CAS PubMed.
- D. Q. Andrews and O. V. Naidenko, Environ. Sci. Technol. Lett., 2020, 7, 931–936 CrossRef CAS.
- A. Bălan Simona, C. Mathrani Vivek, F. Guo Dennis and M. Algazi André, Environ. Health Perspect., 2021, 129, 025001 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, 1H, 13C, 19F, COSY, and titration NMR spectra, crystallographic details, DFT calculations. CCDC 2104069–2104080. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc07041j |
| This journal is © The Royal Society of Chemistry 2022 |