
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn N-capping asparagine–lysine–proline (NKP) motif contributes to a hybrid flexible/stable multifunctional peptide scaffold†
Marlon H.
Cardoso
 *abcd,
Lai Y.
Chan
e,
Elizabete S.
Cândido
ab,
Danieli F.
Buccini
a,
Samilla B.
Rezende
a,
Marcelo D. T.
Torres
f,
Karen G. N.
Oshiro
ac,
Ítala C.
Silva
g,
Sónia
Gonçalves
g,
Timothy K.
Lu
h,
Nuno C.
Santos
g,
Cesar
de la Fuente-Nunez
f,
David J.
Craik
*e and
Octávio L.
Franco
*abc
*abcd,
Lai Y.
Chan
e,
Elizabete S.
Cândido
ab,
Danieli F.
Buccini
a,
Samilla B.
Rezende
a,
Marcelo D. T.
Torres
f,
Karen G. N.
Oshiro
ac,
Ítala C.
Silva
g,
Sónia
Gonçalves
g,
Timothy K.
Lu
h,
Nuno C.
Santos
g,
Cesar
de la Fuente-Nunez
f,
David J.
Craik
*e and
Octávio L.
Franco
*abc
aS-Inova Biotech, Programa de Pós-Graduação em Biotecnologia, Universidade Católica Dom Bosco Avenida Tamandaré 6000, Campo Grande – MS, 79117900, Brazil. E-mail: marlonhenrique6@gmail.com; d.craik@imb.uq.edu.au; ocfranco@gmail.com
bCentro de Análises Proteômicas e Bioquímicas, Programa de Pós-Graduação em Ciências Genômicas e Biotecnologia, Universidade Católica de Brasília, SGAN 916 Módulo B, Asa Norte, Brasília – DF, 70790160, Brazil
cPrograma de Pós-Graduação em Patologia Molecular, Faculdade de Medicina, Universidade de Brasília, Campus Darcy Ribeiro, Asa Norte, Brasília – DF, 70910900, Brazil
dInstituto de Biociências (INBIO), Universidade Federal de Mato Grosso do Sul, Cidade Universitária, 79070900, Campo Grande, Mato Grosso do Sul, Brazil
eInstitute for Molecular Bioscience, Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Queensland, Brisbane, QLD, 4072, Australia
fMachine Biology Group, Departments of Psychiatry and Microbiology, Institute for Biomedical Informatics, Institute for Translational Medicine and Therapeutics, Perelman School of Medicine, Departments of Bioengineering and Chemical and Biomolecular Engineering, School of Engineering and Applied Science, Penn Institute for Computational Science, University of Pennsylvania, Philadelphia, Pennsylvania, USA
gInstituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal
hSynthetic Biology Group, MIT Synthetic Biology Center, The Center for Microbiome Informatics and Therapeutics, Research Laboratory of Electronics, Department of Biological Engineering, Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge – MA, 02139, USA
First published on 4th August 2022
Abstract
Structural diversity drives multiple biological activities and mechanisms of action in linear peptides. Here we describe an unusual N-capping asparagine-lysine-proline (NKP) motif that confers a hybrid multifunctional scaffold to a computationally designed peptide (PaDBS1R7). PaDBS1R7 has a shorter α-helix segment than other computationally designed peptides of similar sequence but with key residue substitutions. Although this motif acts as an α-helix breaker in PaDBS1R7, the Asn5 presents exclusive N-capping effects, forming a belt to establish hydrogen bonds for an amphipathic α-helix stabilization. The combination of these different structural profiles was described as a coil/N-cap/α-helix scaffold, which was also observed in diverse computational peptide mutants. Biological studies revealed that all peptides displayed antibacterial activities. However, only PaDBS1R7 displayed anticancer properties, eradicated Pseudomonas aeruginosa biofilms, decreased bacterial counts by 100–1000-fold in vivo, reduced lipopolysaccharide-induced macrophages stress, and stimulated fibroblast migration for wound healing. This study extends our understanding of an N-capping NKP motif to engineering hybrid multifunctional peptide drug candidates with potent anti-infective and immunomodulatory properties.
Introduction
Short bioactive peptides have dynamic structures that include diverse conformational states such as random coils, α-helices, β-turns and β-sheets.1,2 The structures of peptides can be manipulated based on many parameters, ranging from amino acid composition to the biological environments into which they are inserted. For example, branched and bulky amino acids favor rigidity, whereas smaller amino acids elicit flexibility.3 Moreover, depending on the net charge, hydrophobicity, fluidity, salt concentration and pH of its environment, a single peptide may adopt different structural profiles to achieve the lowest free-energy state for a specific molecular complex (e.g., peptide-membrane, peptide–peptide or peptide–protein interactions).4Much effort has been devoted to designing peptide-based drugs having constrained scaffolds and well-defined secondary structures.5 Antimicrobial peptides (AMPs), for instance, are typically designed with the aim of having a well-defined α-helix conformation when in contact with a target microorganism to trigger membrane-associated mechanisms of action.6 Peptidomimetics have also been developed to stabilize a particular bioactive structure.7,8 Although these approaches have increased the number of peptides with promising therapeutic applications for infectious diseases, some studies have focused on a different direction. Specifically, it has been suggested that both structural flexibility and rigidity could co-exist in a single peptide scaffold, which can be triggered by specific amino acid motifs.9–12
Various amino acid motifs have been identified and incorporated in AMPs and correlated with specific biological functions.13 Higher flexibility has been achieved by adding glycine and flexible ω-amino and α,ω-diamino acid motifs into AMP sequences.11 Moreover, the incorporation of amino-terminal Cu(II) and Ni(II) (ATCUN) binding motifs has enabled the generation of optimized AMPs (catalytic metallodrugs).14,15 Immunomodulatory and antimicrobial properties have been enhanced in a wasp venom-derived peptide (mastoparan-L) by adding a hydrophobic amino-terminal pentapeptide motif (FLPII), which is conserved among diverse naturally occurring peptides.16
In addition to these examples, the use of capping amino acid motifs either at the N-terminus (N-cap) or C-terminus (C-cap) of linear peptides has been reported to assist α-helix stabilization. Capping effects in α-helical segments work via the saturation of NH donors and CO acceptors that can occur, for instance, through hydrogen bonding i, i + 2 and i, i + 4.17 Thus, the primary aspect for α-helix capping includes countering the helix macrodipole and providing additional hydrogen bonding interactions (at least one) for secondary structure stabilization.18 Among the amino acid residues commonly used for N-cap are polar residues with short side chains, including asparagine, serine, and threonine. Interestingly, proline, which is commonly described as an α-helix breaker, can also be a key residue for X-Pro capping motifs, where X represents amino acid residues with N- or C-cap effects.19 In this context, identifying novel amino acid motifs with possible capping effects is of great interest in the design of biologically active peptides, aiming to further explore the diversity of unusual structural scaffolds for peptide-based drugs development.
In the present study, three computationally designed peptides, PaDBS1R2, R3 and R7 (Fig. 1A), presenting flexibility-inducer residues (e.g., glycine and proline) at different positions and that could act as novel amino acid motifs, were selected for biophysical and biological studies. We recently reported the computer-aided design of these peptides aiming at antibacterial properties through an automated design methodology (Joker algorithm), thus generating nine analog sequences (designed PaDBS1R1 to R9)20 (Fig. 1A). Among the PaDBS1 peptides containing a glycine residue, PaDBS1R2 had the highest antibacterial potential against a bioluminescent Pseudomonas aeruginosa strain (minimal inhibitory concentration (MIC) = 1.4 μmol L−1).20 PaDBS1R3 was the only peptide containing two aliphatic leucine residues preceding an α-helix breaker proline residue (MIC = 5.8 μmol L−1); whereas PaDBS1R7 had the highest activity against P. aeruginosa among the peptides containing an α-helix breaker proline preceded by an asparagine (possible N-cap effects) at position 5 and a positively charged lysine at position 6 (MIC = 2.8 μmol L−1).
 | ||
| Fig. 1 Computer-aided design of PaDBS1 peptides and selection of PaDBS1R2, R3 and R7 for structural analysis. (A) For the computer-aided design of PaDBS1 peptides, 248 α-helical AMP sequences were retrieved from the Antimicrobial Peptide Database (APD),22 leading to the identification of an α-helical set (KK[ILV] × (3)[AILV]). This α-helical set was inserted into a non-AMP ribosomal protein fragment from P. aerophilum (GenBank ID AAL64633 – non-AMP blind sequence with predicted three turns of α-helix), generating nine analogs. From this set, PaDBS1R2, R3 and R7 presented flexibility-inducer residues (highlighted in red) that could act as novel amino acid motifs, also revealing similar physicochemical properties and antibacterial potential. CD spectra were obtained in ultrapure water (B), 30% TFE in water (v/v) (C) and 50 mmol L−1 SDS (D). αH secondary chemical shifts in 90% H2O (v/v) and 10% D2O (v/v) (E), 60% H2O (v/v) 30% TFE-d3 (v/v) and 10% D2O (v/v) (F), and amide proton (NH) temperature coefficients (G) of PaDBS1R2, R3 and R7 are also shown. Values more negative than −0.1 ppm (dashed lines in (E) and (F)) were interpreted as α-helix formation. Hydrogen bonds were considered for amide temperature coefficient values more positive than −4.6 ppb K−1 (dashed line in (G)). Conserved residues between all the peptides listed are in bold type. The superposition of the ten lowest energy NMR structures for PaDBS1R2 (PDB: 6CSK) (H), R3 (PDB: 6CSZ) (I) and R7 (PDB: 6CTI) (J) are also shown, along with their Adaptive Poisson–Boltzmann solver (APBS) electrostatic potentials, with potentials ranging from −5 kT/e (red) to +5 kT/e (blue) (H–J). | ||
Here, these peptides were investigated using a suite of structural and functional analyses to better understand how their differences in amino acid compositions would interfere with their structural scaffolds and how these parameters could be used as critical physicochemical and structural determinants for generating peptide drug candidates. We identified the role of an unusual N-capping asparagine-lysine-proline (NKP) motif in PaDBS1R7 that conferred this peptide with a hybrid structural scaffold (coil/N-cap/α-helix) and a broad spectrum of biological activities.
Results
An N-capping NKP motif contributes to a hybrid peptide scaffold
Three computationally designed peptides were selected for structural and functional analysis. A detailed scheme of the computer-aided design of the peptides is shown in Fig. 1A and is reported in our previous work.20 PaDBS1R2, R3 and R7 are cationic peptides with net positive charges +6 or +7, and with hydrophobic contents of 32% for PaDBS1R2 and R7, and 44% for R3. In terms of hydrophobic moment, similar values are observed for PaDBS1R2 and R3 (0.371 and 0.370, respectively), whereas the R7 peptide has a higher value (0.491) on the Eisenberg scale21 (Table S1†), indicating greater amphipathicity (Fig. S1†).After peptide synthesis by solid-phase (Fmoc) and molecular mass determination by liquid chromatography/mass spectrometry (LC/MS) (Fig. S2–4†), experimental biophysical studies were conducted. The secondary structures of the PaDBS1 peptides were investigated through circular dichroism (CD) spectroscopy. In hydrophilic conditions, the spectra indicated the presence of random coil structural profiles (Fig. 1B). However, as expected for linear cationic peptides, well-defined α-helix signatures were recorded in the presence of the secondary structure inducer 2,2,2-trifluoroethanol (TFE) (Fig. 1C) and in contact with anionic sodium dodecyl sulfate (SDS) micelles (Fig. 1D). Although similar CD spectra signatures were observed for all the peptides tested, different α-helical contents were obtained, with PaDBS1R7 being the least α-helical peptide (Fig. 1C and D).
To further investigate the structures, nuclear magnetic resonance (NMR) studies were carried out in aqueous solution (Fig. 1E and S5†) and 30% TFE in water (v/v) (Fig. 1F and S6†). 1H NMR spectra revealed that, in both conditions, PaDBS1R2 and R3 have similar and better defined spectra than PaDBS1R7, which shows broader peaks, mainly at the N-terminus (Fig. S5 and S6†). As observed in the CD analysis, the αH secondary shifts for all the peptides in hydrophilic conditions resulted in random coil arrangements (Fig. 1E). Chemical shifts for the peptides in 90% H2O (v/v) and 10% D2O (v/v) are summarized in Tables S2–4† for PaDBS1R2, R3 and R7, respectively.
We performed 1D and 2D NMR experiments in TFE/water mixtures to assess for possible random coil and α-helical segments for all the peptides tested. TFE is commonly used for studying the secondary structure of peptides as it displaces water molecules around the peptide backbone, favoring folding. The αH secondary chemical shifts for PaDBS1R2 and PaDBS1R3 revealed α-helical segments from residues 3 to 17, whereas PaDBS1R7 presented a shorter α-helical segment from residues 7 to 17, thus supporting the CD data (Fig. 1F). Amide proton temperature coefficient spectra were also recorded from 285 to 310 K, at 5 K intervals, and indicated higher structural flexibility for PaDBS1R7 at the N-terminus, as fewer residues are protected from the solvent and, consequently, are not involved in the intrapeptide hydrogen bonding (Fig. 1G). The complete chemical shift data for the peptides in 60% H2O (v/v), 30% TFE-d3 (v/v) and 10% D2O (v/v) are summarized in Tables S5 to S7† for PaDBS1R2, R3 and R7, respectively.
The αH secondary chemical shifts (obtained from TOCSY and NOESY experiments) and NMR temperature coefficient spectra, along with 13C-HSQC and 15N-HSQC NMR experiments, were used to determine the three-dimensional structures of the PaDBS1 peptides.
The structural statistics are summarized in Table S8.† Additional validations, including clashscore, Ramachandran outliers and side-chain outliers, are available in Protein Data Bank (PDB) under the PDB IDs: 6CSK, 6CSZ and 6CTI for PaDBS1R2, R3 and R7, respectively. The final structures for PaDBS1R2 and R3 display well-defined α-helix structures with α-helical segments from Met2 to Phe18 in R2 (Fig. 1H), and from Leu5 to Ala17 in R3 (Fig. 1I). For PaDBS1R7, the final structures indicate a highly flexible N-terminus region from Pro1 to Lys6, along with a highly stable and amphipathic α-helical segment from Pro7 to Ala17 (Fig. 1J, Table S1 and Fig. S1†). Considering that these peptides presented varying levels of structural flexibility in the NMR temperature coefficient analyses, unconstrained molecular dynamics (MD) simulations were carried out under similar NMR conditions to confirm that no virtual conformations were obtained. As shown in Fig. S7,† the MD simulations results revealed a high similarity between the experimental and theoretical structures for PaDBS1R2 (Fig. S7A†), R3 (Fig. S7B†) and R7 (Fig. S7C†), with root mean square deviations (RMSD) ranging from 0.56 to 0.98 Å for the α-helical segments, and from 0.93 to 1.12 Å for the complete structures. Interestingly, the presence of a flexible N-terminus tail in PaDBS1R7 does not seem to interfere with this peptide's α-helical segment stability, as the RMSD calculated for its backbone atoms in the α-helical segment was the lowest among the three peptides (Table S8†). These data encouraged us to investigate possible N-cap effects in PaDBS1R7 for α-helix stabilization.
Both PaDBS1R3 and R7 have a proline residue at position 7, commonly reported as an α-helix-breaker. A comparison of their N-terminus region shows that these peptides differ mainly by the presence of two aliphatic leucine residues at positions 5 and 6 in PaDBS1R3, and this seems to assist in the α-helical segment extension (no α-helix breakage at Pro7). By contrast, PaDBS1R7 has an asparagine and a lysine at those same positions, respectively (Table S1†). At positions 4 and 8, both peptides have positively charged residues. Considering the similarity between PaDBS1R3 and R7 sequences at the N-terminus and their substantial difference in terms of structural scaffold (Fig. 1I and J) we hypothesized that, in this case, not only Pro7 but also Asn5 and Lys6 seem to be involved in this unusual, hybrid scaffold observed for PaDBS1R7. Here, this combination of amino acid residues in PaDBS1R7 was defined as the 5NKP7 motif, from which we investigated possible N-cap effects.
We investigated the ten lowest energy NMR structures for PaDBS1R7, aiming at identifying potential Asn5 N-cap properties. Nine out of ten structures showed that Asn5 plays a crucial role for PaDBS1R7's α-helix stabilization through hydrogen bonds between Asn5 side-chain atoms and neighboring amino acid residues backbone atoms, thus characterizing an N-cap effect (Fig. 2A). As shown in Table S9,† this N-cap effect is mainly driven by hydrogen bonds between the Asn5 side-chain and Lys6, and Leu10 carbonyl backbone atoms (hydrogen bonding i, i + 1, and i, i + 5), with an average hydrogen bond distance of 3.24 Å.
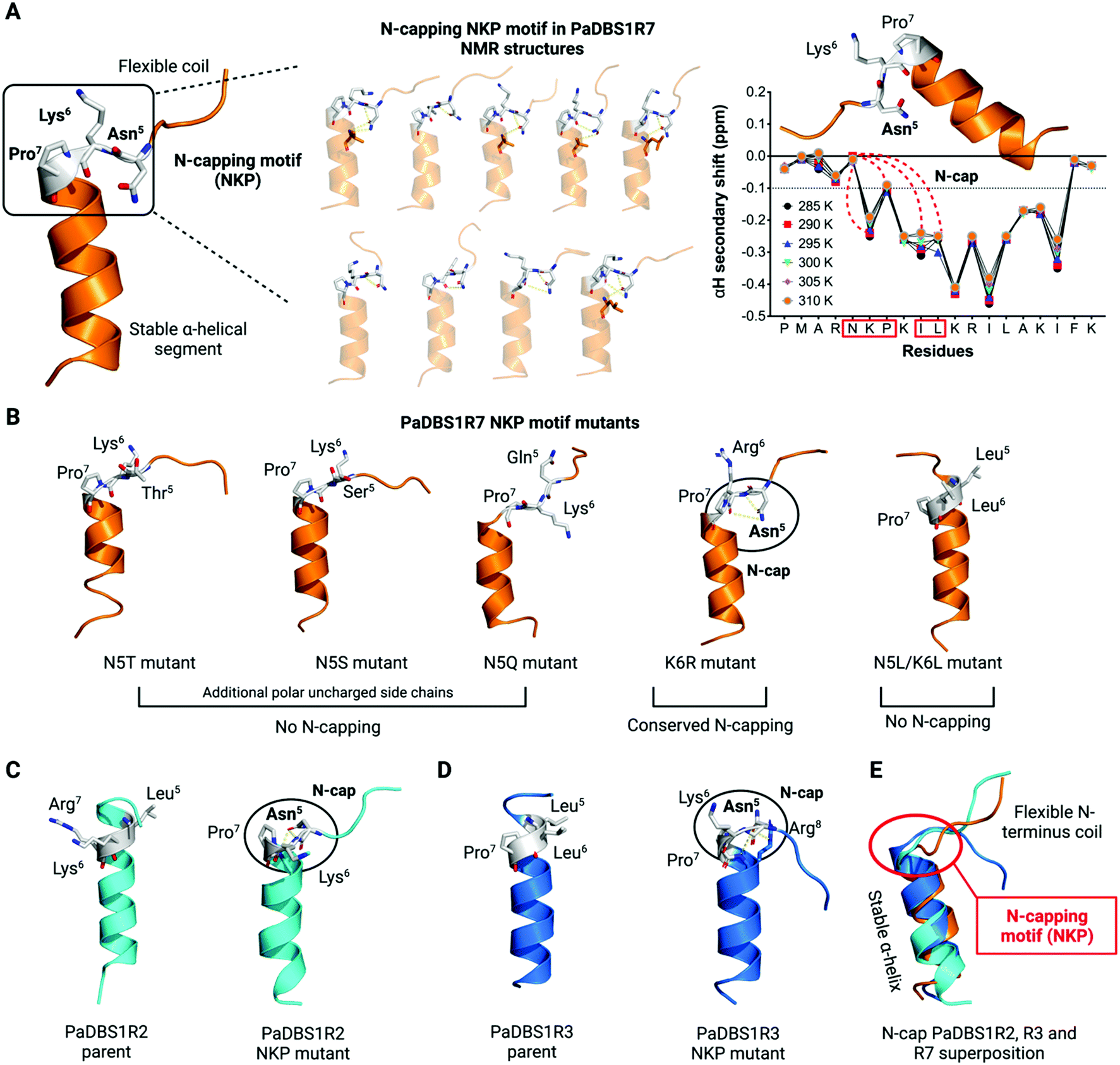 | ||
| Fig. 2 An NKP motif in PaDBS1R7 triggers the coil/N-cap/α-helix structural scaffold. (A) The lowest energy NMR structure for PaDBS1R7 presents an NKP motif that influences on the peptide's scaffold by interrupting the α-helical segment at Pro7, followed by hydrogen bonding between the Asn5 side-chain atoms and neighboring amino acid residues backbone atoms, thus characterizing an N-cap effect for α-helix stabilization. This coil/N-cap/α-helix structural scaffold was observed for all ten lowest energy PaDBS1R7 NMR structures, except for one structure that did not present the Asn5 N-cap effect. The αH secondary shifts at different temperatures are also shown, revealing that the coil/N-cap/α-helix scaffold is conserved from 285 to 310 K, with primary N-capping involving Asn5 side chain atoms and Lys6, Pro7, Ile9 and Leu10 backbone atoms (red dashes). (B) Computational PaDBS1R7 mutants at position 5 replacing asparagine by other polar uncharged residues were also generated, leading to N-cap effect loss. A K6R mutant was also simulated, resulting in a conserved Asn5 N-cap effect. A PaDBS1R7 mutant containing LLP at positions 5 to 7 resulted in α-helix extension, as for PaDBS1R3 NMR structures. The lowest energy NMR structures for PaDBS1R2 (C) and R3 (D) were also submitted to specific modifications, generating NKP mutants with α-helix breakage and Asn5 N-cap effects that led to a similar coil/N-cap/α-helix structural scaffold observed for the original PaDBS1R7 peptide (E). Yellow dashes represent hydrogen bonds. | ||
This analysis was extended for all 100 structures generated by NMR after structural refinement in water using CNS.23 Considering all the calculated structures, 89 structures supported the Asn5 N-cap effect for α-helix stabilization in PaDBS1R7, including a varying number of hydrogen bonds with Lys6, Pro7, Ile9 and Leu10 (Fig. 3). The hydrogen bonding summary, types of hydrogen bonds and “capping box” for PaDBS1R7 are also shown in Fig. 3, with a mean hydrogen bond distance of 3.21 Å (H-bond distance cut-off ≤ 3.5 Å). Bearing in mind that hydrogen bonds with acceptor–donor distances from 2.2 to 2.5 Å are considered “strong” and from 3.3 to 4.0 Å are considered “weak”, the average distance values obtained in the present study can be categorized as “moderate”.
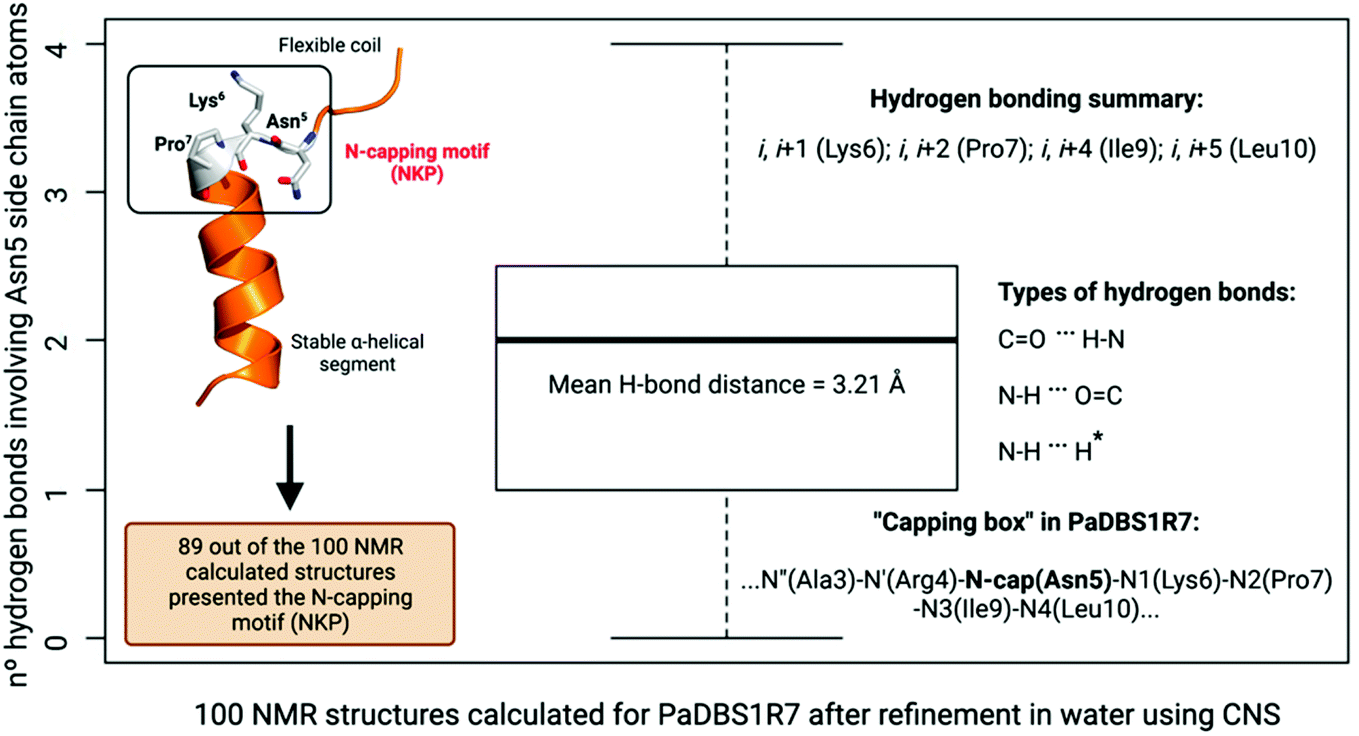 | ||
| Fig. 3 Box plot analysis for the 100 NMR structures calculated for PaDBS1R7. A total of 89 structures out of the 100 calculated structures presented N-cap effects triggered by Asn5 in contact (hydrogen bonds) with Lys6 or Pro7 or Ile9 or Leu10, with a mean hydrogen bond distance of 3.21 Å (H-bond distance cut-off ≤ 3.5 Å). The number of hydrogen bonds varied from 1 to 4 depending on the spatial orientation of the NMR structure analyzed, with hydrogen bonding i, i + 1 (Lys6); i, i + 2 (Pro7); i, i + 4 (Ile9); i, i + 5 (Leu10). The ND2 and OD1 atoms from Asn5 side-chain are donors and acceptors, respectively. The types of hydrogen bonds found are also shown. *Represents an unconventional N–H⋯N hydrogen bond involving the ND2 atom from Asn5 and the proline backbone nitrogen. The “capping box” for PaDBS1R7 based on all 100 NMR structures were also determined as follows: …N′′(Ala3)–N′(Arg4)–N-cap(Asn5)–N1(Lys6)–N2(Pro7)–N3(Ile9)–N4(Leu10)… | ||
Additionally, we plotted the NMR temperature coefficient data as a function of αH secondary shifts, revealing that the structural scaffold adopted by PaDBS1R7 remains highly stable from 285 to 310 K, also preserving the Asn5 N-cap effect through hydrogen bonding with Lys6, Pro7, Ile9 and Leu10 (Fig. 2A). By contrast, this structural behavior was not observed for PaDBS1R2, which had its α-helix compromised from 300 to 310 K (Fig. S8A†). PaDBS1R3 also presented higher αH chemical shifts variations from 295 to 310 K, thus indicating lower structural stability than PaDBS1R7 at varying temperatures (Fig. S8B†). Taken together, these experimental biophysics results suggest that the NKP motif present in PaDBS1R7 triggers an unusual coil/N-cap/α-helix (hybrid) structural scaffold.
Considering that not only asparagine but also threonine, serine and glutamine represent polar uncharged residues with varying levels of N-cap effects, we generated computational PaDBS1R7 mutants, including N5T, N5S and N5Q. All mutants were built from the lowest energy NMR structure obtained for PaDBS1R7 and submitted to MD simulations under similar NMR conditions. As seen in Fig. 2B, none of the mutants presented N-cap effects, supporting previous findings in which asparagine was described as the best N-cap residue.17,19,24 This was further confirmed by analyzing the hydrogen bond occupancies for all PaDBS1R7 mutants, revealing that only the parent peptide presents Asn5 side-chain atoms involved in hydrogen bonding with Lys6 (56.4% occupancy) and Leu10 (15.1% occupancy) backbone atoms (Fig. S9A–F†). A K6R PaDBS1R7 mutant was also simulated, showing that Lys6 substitution by a larger positively charged side chain (arginine) does not seem to compromise the Asn5 N-cap effects (Fig. 2B). Nevertheless, it is worth noting that no significant hydrogen bond occupancy was observed for Asn5 side-chain atoms throughout the MD simulations for this mutant (Fig. S9E†). Finally, another PaDBS1R7 mutant was generated to evaluate whether adding two leucine residues at positions 5 and 6 would result in the extension of the α-helical segment, as for PaDBS1R3 in our NMR data. As observed in Fig. 2B, the N5L/K6L mutant structurally behaved like PaDBS1R3, indicating that the presence of a single proline itself may not always lead to α-helix breakage or distortions, as usually reported. The MD simulation analyses are summarized in Table S10,† including α-helix content (%), RMSD and root mean square fluctuation (RMSF) for different peptide regions. Moreover, the hydrogen bond angles distribution are summarized in Fig. S10A.†
We performed additional MD simulations to test our hypothesis that inserting the NKP motif at positions 5 to 7 in other α-helical 19-amino acid residues would trigger the same hybrid coil/N-cap/α-helix structural scaffold characterized for PaDBS1R7. We first generated PaDBS1R2 and R3 NKP mutants (named PaDBS1R2-NKP and PaDBS1R3-NKP). The original peptides were used as a control in all the simulations. Compared to the parent PaDBS1R2 and R3 peptides (Fig. 2C and D), the PaDBS1R2-NKP and PaDBS1R3-NKP mutants had lower α-helical content (Fig. 2C and D) and higher overall RMSD and RMSF (Table S10†). In all cases, we observed that the NKP motif contributed to an N-terminus flexible coil followed by a stable α-helical segment, and that Asn5 displayed its N-cap effects, characterizing the coil/N-cap/α-helix scaffold (Fig. 2C and D; Table S11†). Moreover, the hydrogen bond occupancies for Asn5 from PaDBS1R2-NKP and PaDBS1R3-NKP were observed as 39.5% (Leu8–Asn5) and 76.9% (Arg8–Asn5), respectively (Fig. S11B and D†). A hydrogen bond occupancy of 13.3% was also observed for side-chain/side-chain atoms from Arg8 and Asn5, respectively, in PaDBS1R3-NKP (Fig S11D†). Main-chain/side-chain hydrogen bond occupancies were not observed for the parent peptides (Fig. S11A and C†).
For PaDBS1R2-NKP, we identified two hydrogen bonds between Asn5 and Lys6 or Leu8, whereas for PaDBS1R3-NKP, three hydrogen bonds were predicted and involved Asn5 and Lys6 or Arg8 (Table S11†). The N-capping for these mutants included hydrogen bonding i, i + 1, i, i + 2 and i, i + 3, with an average hydrogen bond distance of 3.5 Å. Two types of hydrogen bonds were identified, including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N and N–H⋯N. The latter represents a hydrogen bond established between the Asn5 ND2 (donor) atom and the Pro7 backbone nitrogen (acceptor). Although unusual, this N–H⋯N has been well characterized for peptides and proteins.25 It is also worth highlighting that this N–H⋯N was also found in eight of our PaDBS1R7 NMR structures. This finding reinforces the role of the Asn5 N-cap effects even through unusual hydrogen bond patterns. Finally, we performed a structural alignment between the lowest energy NMR structure for PaDBS1R7 and the NKP mutants simulated for PaDBS1R2 and R3. As a result, all the peptides containing the NKP motif adopted the coil/N-cap/α-helix structural scaffold here identified for PaDBS1R7, with a 1.54 Å RMSD for the aligned structures (Fig. 2E).
O⋯H–N and N–H⋯N. The latter represents a hydrogen bond established between the Asn5 ND2 (donor) atom and the Pro7 backbone nitrogen (acceptor). Although unusual, this N–H⋯N has been well characterized for peptides and proteins.25 It is also worth highlighting that this N–H⋯N was also found in eight of our PaDBS1R7 NMR structures. This finding reinforces the role of the Asn5 N-cap effects even through unusual hydrogen bond patterns. Finally, we performed a structural alignment between the lowest energy NMR structure for PaDBS1R7 and the NKP mutants simulated for PaDBS1R2 and R3. As a result, all the peptides containing the NKP motif adopted the coil/N-cap/α-helix structural scaffold here identified for PaDBS1R7, with a 1.54 Å RMSD for the aligned structures (Fig. 2E).
Apart from the NMR structures reported for PaDBS1R2, R3 and R7, previous studies from our group have also determined the three-dimensional structures for the PaDBS1 analogs R1 (PDB: 2N9R)26 and R6 (PDB: 6CFA).27 Therefore, we also generated computational NKP mutants for these additional PaDBS1 peptides (Fig. 4A). Moreover, aiming at extending our proof-of-concept regarding the NKP motif, we searched for other α-helical 19-amino acid residues AMPs with determined structures in APD to obtain NKP mutants outside the PaDBS1 peptide family. Four peptides matched the parameters we set, including HP (2–20)28,29 (PDB: 1P0G) and phylloseptins-1 to 3 (PDB: 2JQ0, 2JPY and 2JQ1, respectively).30 Because of high similarity in their sequence and structure, only phylloseptin-1 (PDB: 2JQ0) was selected for further analysis. We observed that the insertion of the NKP motif within these peptides' structures resulted in the coil/N-cap/α-helix scaffold in all cases (Fig. 4A and B). Three hydrogen bonds characterized the Asn5 N-cap effect in PaDBS1R1–NKP, PaDBS1R6–NKP and HP (2–20)–NKP mutants (all featuring at least one N–H⋯N hydrogen bond involving Pro7, at 100 ns of MD simulations), whereas two hydrogen bonds were predicted for phylloseptin-1-NKP (Table S11†). The hydrogen bonding followed the patterns i, i + 1, i, i + 2 and i, i + 3 for all NKP mutants simulated, with a hydrogen bond average distance of 3.4 Å. Additionally, all NKP mutants presented reduced α-helical contents, and increased overall RMSD and RMSF values compared to the parent peptides used as control (Table S10†). The hydrogen bond angles distribution are summarized in Fig. S10C and D.† Moreover, the overlap of these additional NKP peptide mutant structures reinforced that the NKP motif contributes to a hybrid (flexible/stable) coil/N-cap/α-helix scaffold (Fig. 4C). We also calculated the hydrogen bond occupancies for all parent peptides and their computational NKP mutants (Fig. 4D–G; S12A and C; S13A and C†). PaDBS1R1–NKP presented the lowest hydrogen bond occupancy for Asn5 (10.4%) (Fig. 4D), whereas PaDBS1R6–NKP and phylloseptin-1–NKP revealed moderate occupancies, ranging from 24.2 to 36% (Fig. 4E and G). Finally, the highest hydrogen bond occupancy was reported for the HP (2–20)–NKP peptide (Leu8–Asn5; main-chain/side-chain), with a 76.6% occupancy.
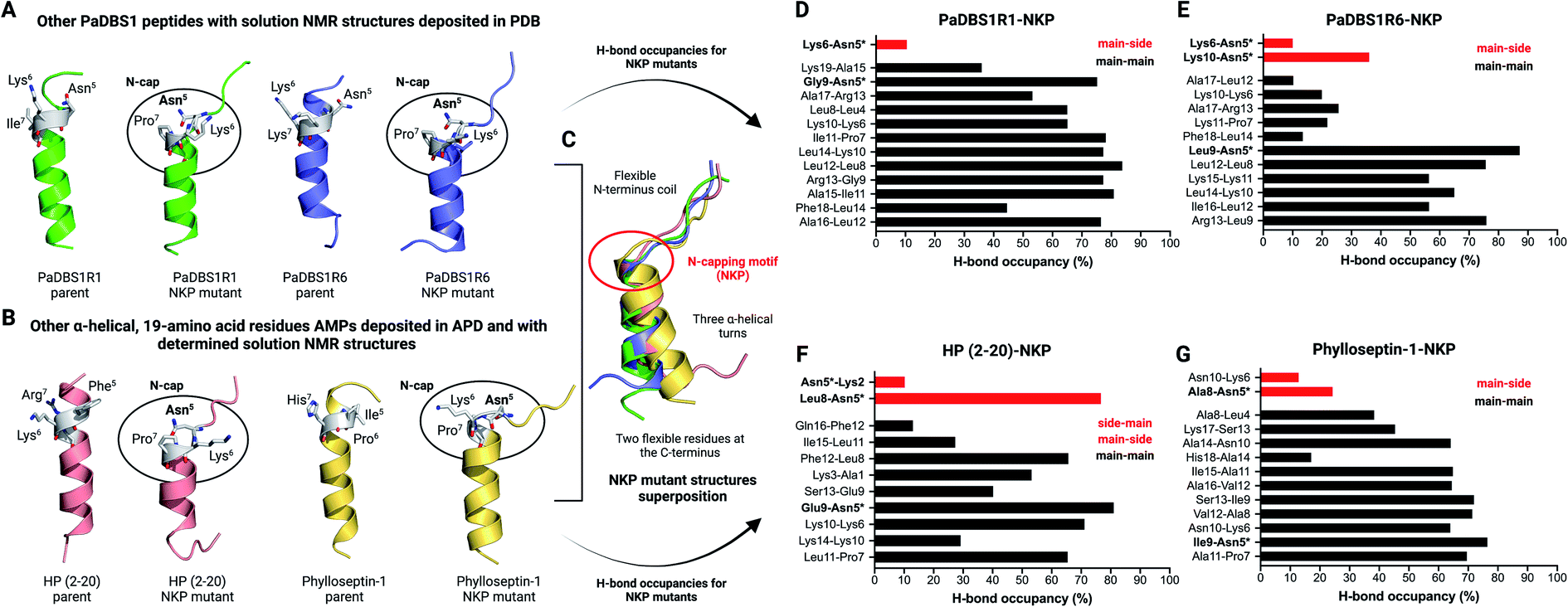 | ||
| Fig. 4 Additional MD simulations with four NKP peptide mutants. (A) Apart from the PaDBS1 peptides selected for the present work, our group has already characterized two other peptides from the same generation, named PaDBS1R1 (PDB: 2N9R)26 and PaDBS1R6 (PDB: 6CFA).27 These peptides have their solution NMR structures deposited in PDB and were used to generate computational peptide mutants containing the NKP motif at positions 5 to 7. (B) In addition to the PaDBS1 peptide family, we also selected two other α-helical, 19-amino acid residues peptides (HP (2–20)28,29 (PDB: 1P0G) and phylloseptin-1 (PDB: 2JQ0)30) with determined NMR structures deposited in PDB and built their NKP mutants. In all cases, the NKP motif here identified in the PaDBS1R7 sequence triggered the same coil/N-cap/α-helix structural scaffold in the peptide mutants. In (C), the structural alignment for all the NKP mutants is shown, revealing a conserved flexible N-terminus region, followed by three α-helical turns stabilized by the N-cap effect of Asn5. The hydrogen bond occupancies throughout the MD simulations for PaDBS1R1–NKP (D), PaDBS1R6–NKP (E), HP (2–20)–NKP (F) and phylloseptin-1–NKP (G) are also shown. Red bars and black bars represent main-chain/side-chain or side-chain/main-chain, and main-chain/main-chain hydrogen bonding, respectively. *Indicates Asn5 side-chain atoms involved in hydrogen bonding. | ||
Different structural scaffolds effect on interaction with mimetic vesicles
After investigating and characterizing the role of an NKP motif and its N-cap effects for α-helix stabilization, we performed additional CD experiments and leakage assays in the presence of large unilamellar vesicles (LUVs) of different lipid compositions and proportions. LUVs mimicking different biological conditions were used (e.g., mammalian cells, bacterial cells, and cancer cells), thus providing insights on the possible biological activities for the different structural profiles described above, with a focus on the coil/N-cap/α-helix scaffold. CD analyses revealed that PaDBS1R7 preserved its lower α-helical content in all conditions tested compared to PaDBS1R2 and R3 (Fig. 5A–C). CD θ signals at 222 nm (local minimum for α-helices) were not observed for the peptides in solution, in the presence of pure 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and in contact with POPC/cholesterol (Chol) vesicles (70:30). By contrast, in the presence of LUVs containing anionic lipids all the peptides presented α-helical signatures at 222 nm (Fig. 5A–C), especially for the POPC/1-palmitoyl-2-oleoyl-phosphatidylglycerol (POPG) (70:30) system, which is considered a general anionic membrane model. As reported previously, PaDBS1R7 was the least α-helical peptide (Fig. 5C).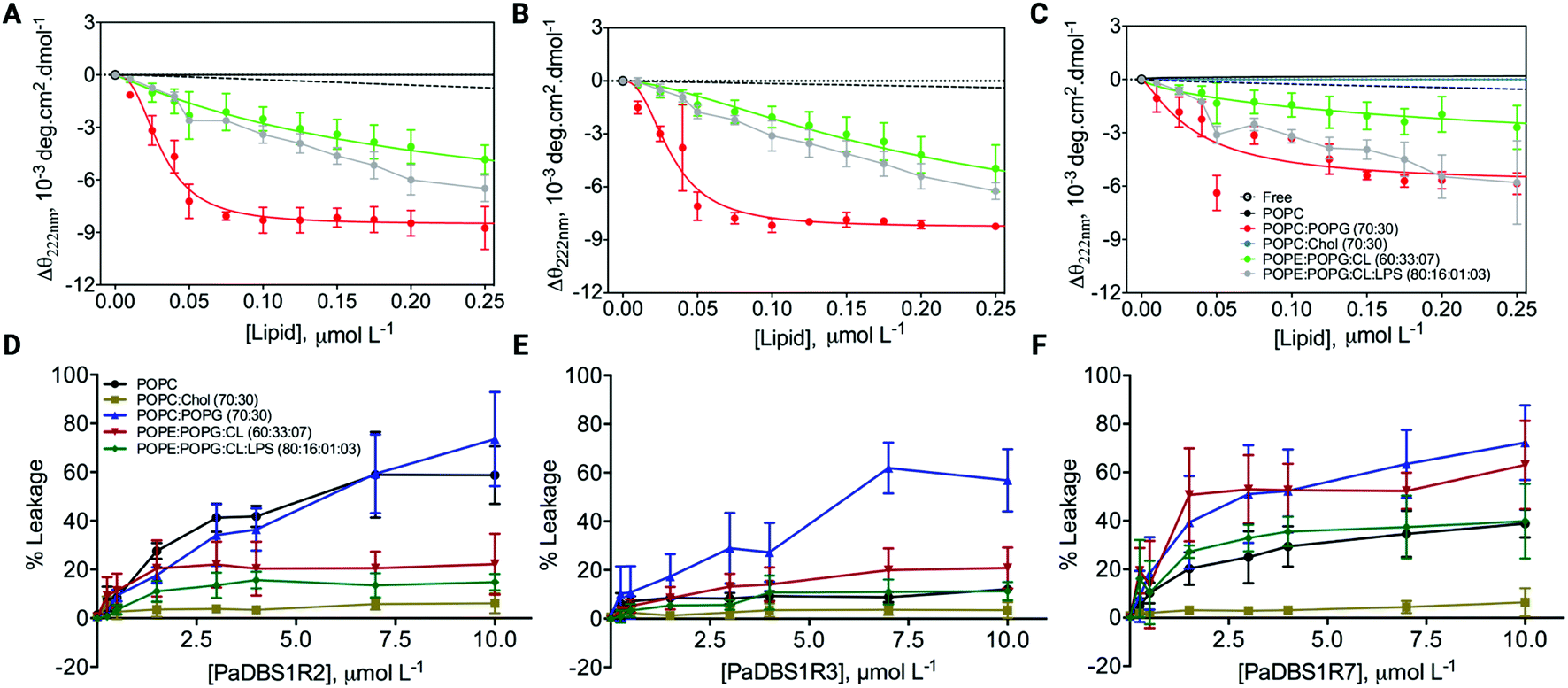 | ||
| Fig. 5 CD spectra and leakage assays of PaDBS1R2, R3 and R7 in the presence of LUVs of different lipid compositions. Comparison of the CD θ signal at 222 nm (local minimum for α-helices) for PaDBS1R2 (A), R3 (B) and R7 (C) at different lipid vesicle concentrations. Percentages of CF release were calculated at different lipid proportions in the presence of increasing concentrations (0 to 10 μmol L−1) of PaDBS1R2 (D), R3 (E) and R7 (F). Lipid vesicles used included pure POPC, POPC:Chol (70:30), POPC:POPG (70:30), POPE:POPG:CL (60:33:7), and POPE:POPG:CL:LPS (80:16:1:3). | ||
Interestingly, despite presenting a less structured arrangement, increasing PaDBS1R7 concentrations (Fig. 5F) caused higher amounts of carboxyfluorescein (CF) release in all negatively charged LUVs (from 39.8 to 72.2% leakage), followed by PaDBS1R2 (Fig. 5D) and R3 (Fig. 5E). Zwitterionic LUVs consisting of POPC enriched with cholesterol, mimicking mammalian cells, had the lowest CF release when treated with the tested peptides (Fig. 5D–F). Finally, when cardiolipin (CL) and lipopolysaccharide (LPS; Gram-negative bacteria endotoxin) were added to negatively charged LUVs, only PaDBS1R7 triggered high CF release (Fig. 5F). LUVs containing CL and LPS mixed with 1-palmitoyl-2-oleoyl-phosphatidylehtanolamine (POPE) and POPG mimic inner membrane (IM) and outer membrane (OM), respectively. Bearing this in mind and considering the data shown in Fig. 5F, the PaDBS1R7 peptide seems to bind with higher affinity and cause greater disruption of IM than OM in mimetic bacterial membrane models.
In summary, we observed that the reduced α-helical content in PaDBS1R7 compared with its other PaDBS1 peptide analogs did not affect its ability to disrupt negatively charged LUVs selectively. These data indicate possible biological activities toward bacterial and cancer cells (negatively charged surfaces), without compromising the viability of healthy mammalian cells (zwitterionic surfaces). To investigate this hypothesis further, all PaDBS1 peptides were tested in diverse biological assays to establish a correlation between their different structural profiles and their antibacterial, antibiofilm, hemolytic, cytotoxic (healthy and cancer cells), anti-infective (in vivo), immunomodulatory and healing properties.
Peptide effect on bacterial growth, cellular surface integrity and mammalian cells viability
PaDBS1R2, R3 and R7 were computationally designed20 aiming at antibacterial activities. Therefore, we first evaluated the activity of these peptides against Gram-positive and Gram-negative bacteria. As shown in Table 1, all three peptides were active against the bacterial strains tested, except for the Enterobacter cloacae clinical isolate and the carbapenemase-producing Klebsiella pneumoniae (Table 1). PaDBS1R3 showed the lowest antibacterial activity spectrum and inhibited only six bacterial strains out of the 11 tested. The most promising activities (lower MICs) were obtained for PaDBS1R2 and R7 from 2 to 8 μmol L−1 against susceptible Escherichia coli, K. pneumoniae and P. aeruginosa, as well as resistant Acinetobacter baumannii, E. coli and P. aeruginosa (Table 1). Only PaDBS1R2 and R7 inhibited methicillin-resistant Staphylococcus aureus (MRSA) growth at 16 μmol L−1 (Table 1).| Bacterial strains | PaDBS1R2 | PaDBS1R3 | PaDBS1R7 |
|---|---|---|---|
| a MIC: minimal inhibitory concentration. | |||
| A. baumannii (003326263) | 4 | 32 | 4 |
| E. cloacae (1383251) | >32 | >32 | >32 |
| E. coli (ATCC 25922) | 4 | 8 | 4 |
| E. coli (KpC+001812446) | 2 | 2 | 2 |
| K. pneumoniae (ATCC 13883) | 8 | 32 | 8 |
| K. pneumoniae (KpC+001825971) | >32 | >32 | >32 |
| MRSA (713623) | 16 | >32 | 16 |
| P. aeruginosa PA14 | 4 | 32 | 8 |
| P. aeruginosa PAO1 | 4 | 32 | 8 |
| S. aureus (ATCC 25923) | 16 | >32 | 16 |
| S. aureus (ATCC 12600) | 4 | >32 | 16 |
Among the bacterial strains tested in the MIC assay, E. coli and P. aeruginosa represent model microorganisms with clinical relevance for in-depth functional AMP studies. All peptides tested were mainly active against E. coli, whereas PaDBS1R2 and R7 displayed promising antibacterial effects against two P. aeruginosa strains (Table 1). To develop a structure–function-mechanism relationship, atomic force microscopy (AFM) studies were carried out to evaluate the effects of the peptides on E. coli and P. aeruginosa. The peptides were tested individually at their MIC and 10-fold higher values. We observed that E. coli treated with the peptides at their MIC did not show morphological damage (Fig. 6A), whereas PaDBS1R2 and R3 induced an increase in P. aeruginosa surface roughness (Fig. 6B). Upon increasing the peptide concentration 10-fold, E. coli cells were clearly disrupted by all the peptides, with a high loss of surface integrity (Fig. 6A). When these 10-fold higher concentrations were used to treat P. aeruginosa, both PaDBS1R2 and R3 preserved their evident surface disruption properties (Fig. 6B). By contrast, P. aeruginosa treated with PaDBS1R7 presented partial surface destabilization (Fig. 6B).
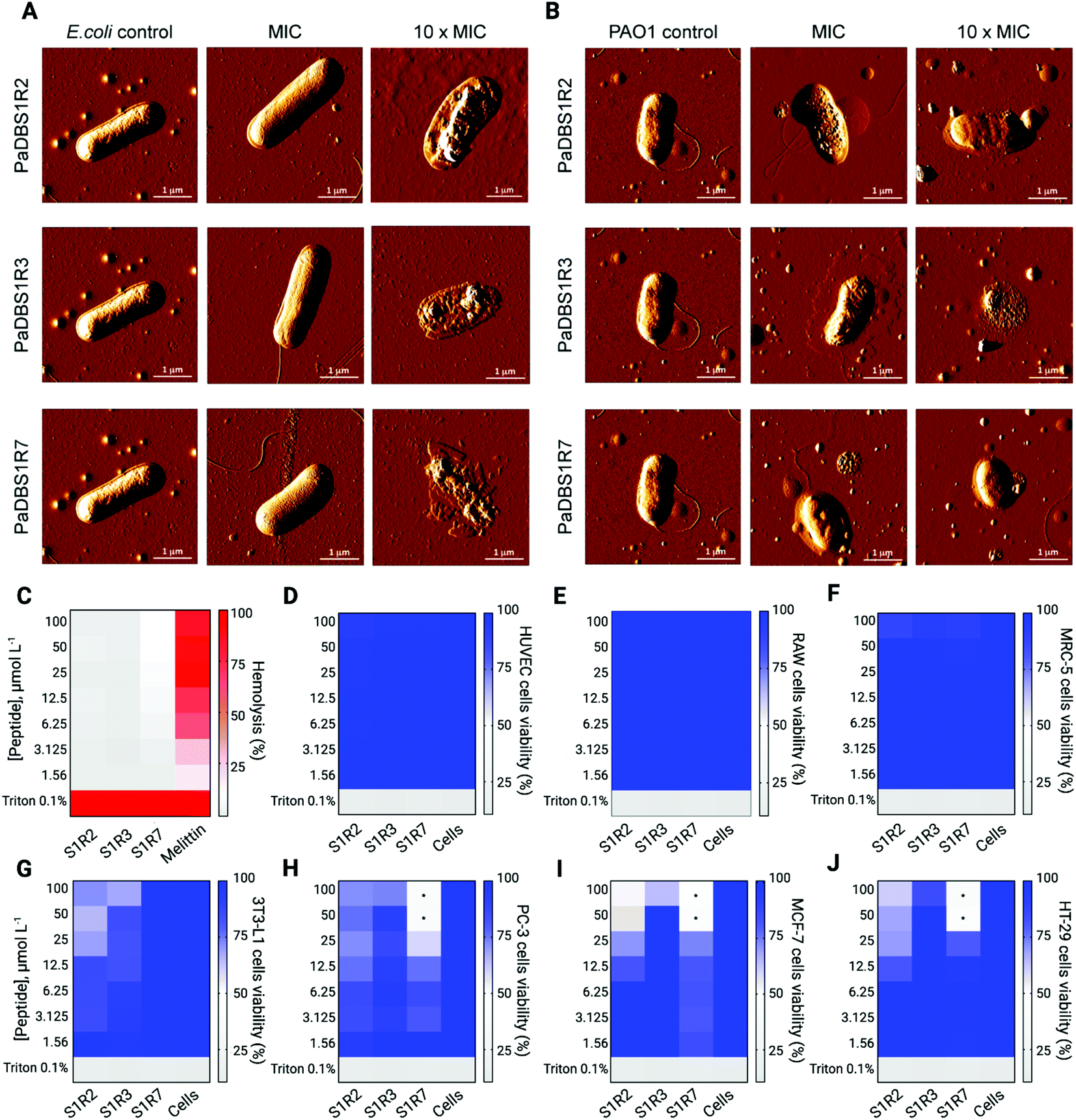 | ||
| Fig. 6 AFM images of E. coli and P. aeruginosa cells untreated and treated with PaDBS1R2, R3 and R7, and hemolytic and cell viability assays using human erythrocytes, healthy mammalian cell lines and cancer cell lines. E. coli cells (A) treated with PaDBS1R2, R3 and R7 at their MIC and 10-fold MIC. The same conditions were used for P. aeruginosa cells (B) treated with PaDBS1R2, R3 and R7. The same controls (untreated E. coli and P. aeruginosa) were used for all analyses. Total scanning area of 5 × 5 μm2; scale bar: 1 μm. The hemolytic activity of PaDBS1R2 (S1R2), R3 (S1R3) and R7 (S1R7) was evaluated using fresh human erythrocytes (C). Triton 1% and the cytolytic peptide melittin (from 0.15 to 20 μmol L−1) were used as positive controls. Cell viability assays were performed using healthy mammalian cells, including HUVEC (D), RAW 267.4 (E), MRC-5 (F) and 3T3-L1 cells (G); and cancer cells, including PC-3 (H), MCF-7 (I) and HT-29 cells (J). All cell lines were treated with PaDBS1R2, R3 and R7 from 1.56 to 100 μmol L−1. Triton 0.1% was used as the positive control. *Indicate peptide concentrations at which half-maximal inhibitory concentrations (IC50) were determined. | ||
Prior to in vivo analysis of the peptides' anti-infective potential, their toxicity towards healthy mammalian cells was evaluated. No hemolytic effects were observed (Fig. 6C), thus encouraging further studies in animal models. The peptides were nontoxic toward human umbilical vein endothelial cells (HUVEC) (Fig. 6D), murine macrophages (RAW 264.7) (Fig. 6E), human fibroblasts (MRC-5) (Fig. 6F) and mice adipocytes (3T3-L1) (Fig. 6G) cell lines at the maximum concentration tested (100 μmol L−1). Finally, considering the amphipathic, α-helical profile of the tested peptides, which is a known feature that favors interactions with negatively charged membranes as observed in our leakage studies, we also carried out cell viability assays with three cancer cell lines. As a result, only PaDBS1R7 compromised the viability of all the cancer cells (Fig. 6H–J) tested at 50 μmol L−1 (half maximal inhibitory concentration: IC50), thus attributing to this peptide an extra function, in addition to its above-described antibacterial properties.
The exclusive antibiofilm potential of PaDBS1R7, in vivo assay and cellular response modulation
Biologically active peptides with antibacterial properties have commonly been evaluated for their effects on bacterial biofilms, which represent more than 80% of all infections in humans. In particular, P. aeruginosa biofilms have been studied for their crucial role in cystic fibrosis, bacterial keratitis and cutaneous infections.31,32 The antibiofilm potential of PaDBS1R2, R3 and R7 was evaluated against two-day-old P. aeruginosa biofilms in flow chambers (Fig. 7A). All peptides were tested at 16 μmol L−1, as this was the common concentration at which they inhibited P. aeruginosa PAO1 growth (Table 1). Compared to the untreated control (Fig. 7B), we observed that PaDBS1R2 was not active against biofilms, PaDBS1R3 caused a slight dispersion of biofilm-constituting cells, whereas PaDBS1R7 completely eradicated P. aeruginosa biofilms (Fig. 7B).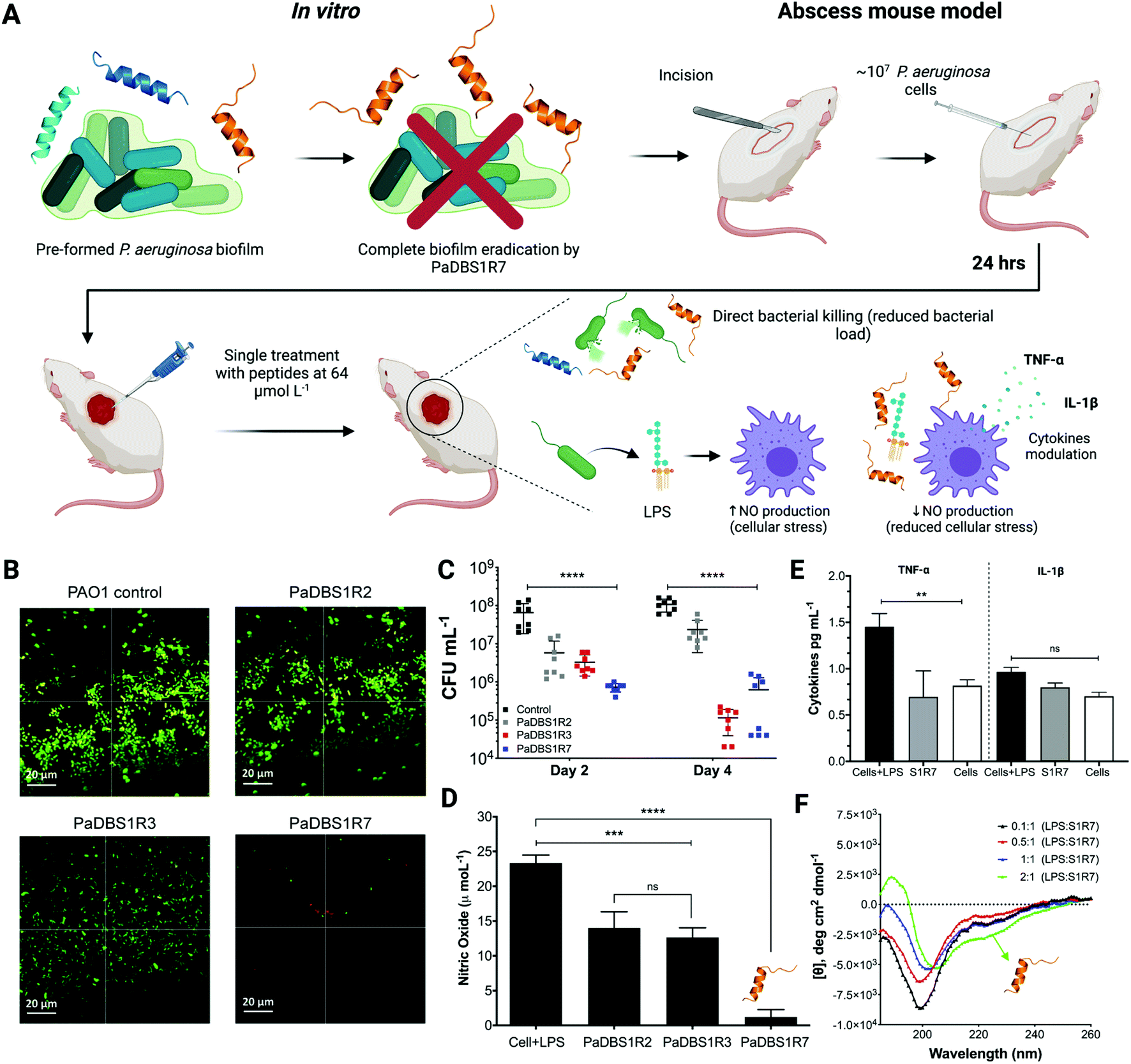 | ||
| Fig. 7 Flow cell analysis of two-day-old P. aeruginosa biofilms, skin abscess mouse model with P. aeruginosa, nitric oxide (NO) and cytokines production by macrophages in the presence of LPS, and LPS titration using CD experiments. P. aeruginosa preformed biofilm (A) prior to treatment (control) and after treatment with PaDBS1R2, R3, and R7, at 16 μmoL−1 (B). Scale bar: 20 μm. The anti-infective potential of PaDBS1R2, R3 and R7 (64 μmol L−1; single dose) on 6 week-old female CD-1 mice, two and four days post-infection (C). LPS-induced macrophages NO production in the absence and presence of PaDBS1R2, R3 and R7 at 64 μmol L−1 (D). TNF-α and IL-1β modulation in the presence of PaDBS1R7 at 64 μmol L−1 (E). LPS titrations were performed recording the CD spectra of PaDBS1R7 in the presence of increasing concentrations of LPS, up to a molar ratio LPS/PaDBS1R7 of 2:1 (F). Statistical significance was assessed using one-way analysis of variance (ANOVA), followed by Dunnett's multiple comparison tests. **p < 0.05; ***p < 0.001 ****p < 0.0001. The error bars represent standard deviation values. ns: not significant. | ||
Considering the antibacterial potential of PaDBS1R2, R3 and R7 and their lack of toxicity towards healthy mammalian cells (Fig. 6C–G), in vivo experiments were carried out using a skin abscess mouse model with P. aeruginosa (PA14), followed by the measurement of recruited macrophage response triggered by bacterial LPS. All tested peptides, at 64 μmol L−1, reduced the P. aeruginosa count from 10 to 100-times on day two post-infection, with a slight prevalence for PaDBS1R7 (Fig. 7C). At day four post-infection, PaDBS1R2 showed a decrease in its anti-infective potential, whereas both PaDBS1R3 and R7 reduced P. aeruginosa counts by 100 to >1000 times, highlighting the higher efficiency of these two peptides under the conditions tested.
Gram-negative bacteria, including P. aeruginosa, produce and secrete LPS, a known endotoxin that induces cellular stress in the host during an infective process.33 Considering the role of LPS in reactive oxygen species formation (e.g., nitric oxide (NO)) and cellular stress, we also evaluated the potential of PaDBS1R2, R3 and R7 in modulating macrophage responses to LPS according to NO levels produced by these cells. As shown in Fig. 7D, macrophages in the presence of LPS present high levels of NO production. By contrast, macrophages in the presence of LPS treated with PaDBS1R2 or R3 at 64 μmol L−1 revealed a decrease in NO production (Fig. 7D). However, our most promising result was observed by incubating macrophages in the presence of LPS with PaDBS1R7 (64 μmol L−1), which led to a drastic reduction of NO production (Fig. 7D) and, consequently, macrophage stress. These data are initial evidence that our peptide containing the N-cap NKP motif and with a hybrid structural scaffold is not only effective at directly compromising bacterial cells viability in skin infections, but also contributes to lower cellular stress in the host and, therefore, favors recovery (Fig. 7A).
NO production is directly correlated with the up-/down-regulation of proinflammatory cytokines that cause cell injury, thus slowing down the healing process in the site of infection. Here, apart from measuring NO production, we also evaluated the expression levels of the proinflammatory cytokines TNF-α and IL-1β. As shown in Fig. 7E, macrophages stimulated with LPS in the presence of PaDBS1R7 present basal levels of TNF-α, thus preventing an LPS-induced inflammatory response. Nevertheless, significant IL-1β modulation by PaDBS1R7 was not reported at the maximal concentration tested, which may suggest that this peptide acts on a specific proinflammatory signaling pathway in the cell (Fig. 7E). It is also worth highlighting that the PaDBS1R7 peptide analogs (PaDBS1R2 and R3), which do not adopt the coil/N-cap/α-helix structural scaffold, could not modulate the expression levels of any of the cytokines tested (Fig. S14A and B†).
Considering the effectiveness of PaDBS1R7 at modulating macrophage response in the presence of LPS, we aimed at characterizing the secondary structure of this peptide in the presence of LPS micelles. LPS titrations were performed recording the CD spectra of PaDBS1R7 in the presence of increasing LPS concentrations, up to a molar ratio LPS/PaDBS1R7 of 2:1. Random coil signatures were recorded from 0.1:1 to 1:1 LPS-to-peptide molar ratio (Fig. 7F). By contrast, an α-helical signature was obtained at 2:1 LPS-to-peptide molar ratio (Fig. 7F). To investigate this structural data further, computational simulations were carried out. The simulations included E. coli and P. aeruginosa lipid A modeling, followed by molecular docking and MD simulations for PaDBS1R7/lipids A systems. A total of 100 molecular docking simulations were performed and the molecular complexes ranked according to their binding affinities in kcal mol−1. The affinities for the complex PaDBS1R7/E. coli lipid A ranged from −7.3 to −8.1 kcal mol−1, whereas lower affinities from −7.3 to −7.6 kcal mol−1 were calculated for the complex PaDBS1R7/P. aeruginosa lipid A (Fig. S15A†). Moreover, the binding affinities with higher frequency throughout the simulations were −7.8 and −7.5 kcal mol−1 for PaDBS1R7 in contact with E. coli and P. aeruginosa lipids A, respectively (Fig. S15A†). Based on this, a representative molecular complex was selected for each of those binding affinities and submitted to 500 ns of MD simulations in aqueous solution (0.15 M NaCl ionic strength). Both systems presented similar RMSD and RMSF profiles (Fig. S15B and C†). Moreover, 13 and 11 atomic interactions were predicted for the molecular complexes PaDBS1R7/E. coli lipid A and PaDBS1R7/P. aeruginosa lipid A, respectively, including hydrogen bonds (O–H⋯OC; CO⋯H–O; N–H⋯O–C; C–O−…H–O), one electrostatic interaction (N+–H = |||||:O), and hydrophobic interactions, with a mean bond distance of 3.5 Å for all interactions (Table S12†). In terms of three-dimensional arrangement, our simulations support the CD data (low CD θ signals at 222 nm), revealing a small, central α-helical segment (two turns) for PaDBS1R7 in both systems (Fig. S15D and E†).
PaDBS1R7 stimulates fibroblast migration
Up to this point, the PaDBS1R7 peptide was characterized for its unusual coil/N-cap/α-helix structural scaffold, which represents a significant determinant that differentiates this peptide from its two analogs, PaDBS1R2 and R3. Moreover, in terms of functionality, PaDBS1R7 displayed antibacterial, antibiofilm and anticancer properties, without having hemolytic activity or toxicity toward healthy mammalian cells. This peptide is also effective at directly killing bacterial cells in cutaneous infections and modulates macrophages' stress response to LPS by reducing NO and TNF-α expression levels. Therefore, considering its multifunctionality compared to PaDBS1R2 and R3, and its effectiveness in the in vivo and immunomodulatory assays (characterizing wound healing potential), PaDBS1R7 was examined in fibroblast migration experiments.The wound scratch assay consists of an initial evaluation of a compound's potential in assisting the healing processes by measuring fibroblast (MRC-5 cells) migration in microplates (Fig. 8A).34 Three experimental groups were established, including a negative control consisting of MRC-5 cells in Dulbecco's Modified Eagle's Medium (DMEM) media not supplemented with fetal bovine serum (FBS), a positive control consisting of MRC-5 cell in DMEM media supplemented with FBS and pyruvate, and a group composed of MRC-5 cells treated with PaDBS1R7 at 64 μmol L−1 (same concentration used in the in vivo and immunomodulatory assays) (Fig. 8A). As shown in Fig. 8B, PaDBS1R7 stimulated 68.18 ± 2.01% MRC-5 cell migration after 24 h incubation, whereas the negative and positive control induced 26.7 ± 5.14% and 28.33 ± 4.96% MRC-5 cell migration at the same incubation time, respectively. After 48 h of incubation, complete (100%) cell migration was observed for MRC-5 cells treated with PaDBS1R7. The negative control presented similar cell migration rates after 24 and 48 h of incubation (25.93 ± 3.87%), whereas the positive control presented a 2-fold cell migration rate increase after 48 h of incubation (59.26 ± 14.58%) (Fig. 8B).
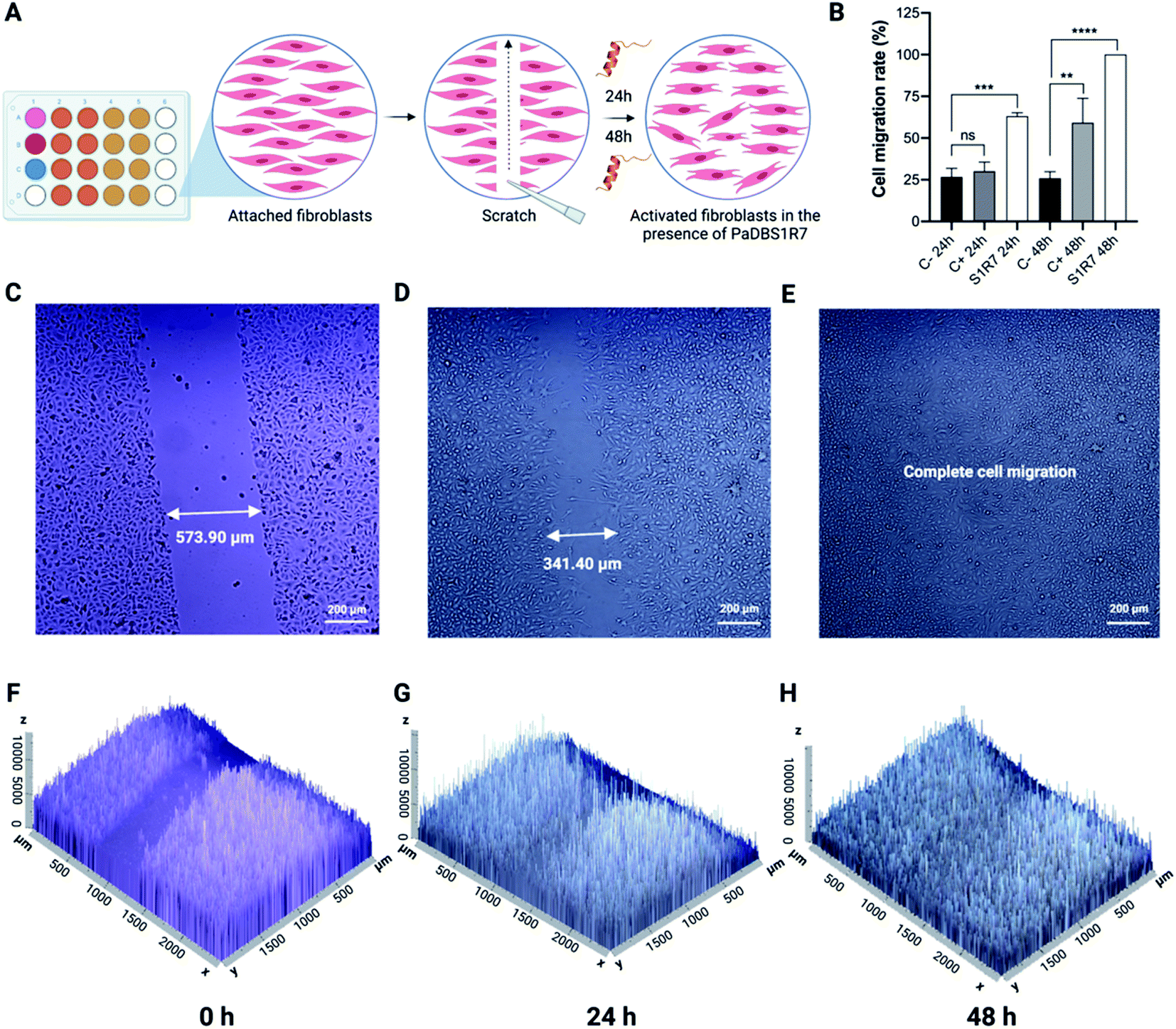 | ||
| Fig. 8 Wound scratch assay using MCR-5 cells (human fibroblasts) incubated with PaDBS1R7 (64 μmol L−1) for 0, 24 and 48 h. The assay was performed in 24-well microplates in which the fibroblasts were cultured until 90% confluence was reached (A). A comparison in cell migration rates between the control groups and PaDBS1R7 treated group is shown (B). Inverted microscopy images were captured to measure the gap distances in the scratches at 0 (C), 24 (D) and 48 h (E). 2.5D images were also obtained to visualize the gap distances and cell density at the incubation times 0 (F), 24 (G) and 48 h (H). Statistical significance was assessed using one-way analysis of variance (ANOVA), followed by Dunnett's multiple comparison tests. **p < 0.05; ***p < 0.001; ****p < 0.0001. The error bars represent standard deviation values. ns: not significant. | ||
Inverted microscopy images were captured to calculate the gap distance at times 0, 24 and 48 h for all the conditions tested. As observed in Fig. 8C–E, MRC-5 cells treated with PaDBS1R7 reduced the gap distance at the scratch region from 573.90 μm to 341.40 μm within 24 h of incubation. After 48 h of incubation, no gap was detected, indicating complete MRC-5 cells migration. These results are also expressed as 2.5D images in Fig. 8F–H, demonstrating how MRC-5 cells migration and density increase in the presence of PaDBS1R7. Inverted microscopy images for the negative and positive controls are shown in Fig. S16A–F and S17A–F,† respectively.
Discussion
Various amino acid motifs have been inserted into target peptide sequences to optimize a given function or to extend the spectrum of bioactivities. RWRW motifs have been used to generate β-sheet peptides capable of interacting with bacterial membranes through cation-π interactions.35 AMPs containing tryptophan–proline motifs have been characterized as non-lytic peptides capable of translocating through bacterial membranes to act on protein synthesis, also triggering protein misfolding.36 Another well-described motif that confers potent and selective antibacterial activities to AMPs is KLAK,37 which has also been studied in its D-enantiomer form for improved proteolytic resistance and extended anticancer properties.38 Previous works from our group have used both amino-terminal Cu(II) and Ni(II) (ATCUN) binding motifs (GGH and VIH),14 and a hydrophobic pentapeptide immunomodulatory motif (FLPII) to generate catalytic metallodrugs and fine-tune the pharmacological potential of wasp venom-derived peptides,16 respectively. Hybrid peptide scaffolds have also been obtained using a hairpin/β-turn motif to accommodate different α- and β-amino acid residues and, therefore, modulate peptide folding and stability.2Among the amino acid motifs found in peptides and proteins are those with capping effects at the N-terminus (N-cap) or C-terminus (C-cap) for α-helix and β-sheet stabilization. The nomenclature for N-cap and C-cap effects in peptides and proteins was first described by Richardson & Richardson in the 1980s39 and includes …N′′–N′–N-cap–N1–N2–N3–N4…, where (…) represents amino acid residues outside the “capping box” that goes from N′′ to N4, the N′′–N′–N-cap region represents a non-helical segment, and the N1–N2–N3–N4 region represents the α-helical segment stabilized by the N-cap. The same has been proposed for C-cap, but in the reverse order compared to N-cap (C-cap: …C4–C3–C2–C1–C-cap-C′–C′′…).17 Furthermore, hydrogen bonding i, i + 1, i, i + 2, i, i + 3 and i, i + 4 within the capping box is the most described in the literature.17
Based on the above considerations, in the present study we identified and characterized a capping box featuring an unusual 5NKP7 motif. The Pro7 triggers the extension of the Lys6 dihedral angles, thus terminating the N-terminus α-helical segment in PaDBS1R7 and resulting in a highly flexible and dynamic coil from residues 1 to 6. In turn, this extension and amino-terminal unfold favors the Asn5 N-cap effects, which forms a belt to establish hydrogen bonds with Lys6, Pro7, Ile9 and Leu10. Thus, the capping box for PaDBS1R7 can be described as: …N′′(Ala3)–N′(Arg4)–N-cap(Asn5)–N1(Lys6)–N2(Pro7)–N3(Ile9)–N4(Leu10)…
N-cap motifs containing proline residues can stabilize α-helical segments in peptides and proteins. Proline cannot donate a proton to make a main-chain/main-chain hydrogen bond. Therefore, residues preceding proline usually present extended ϕ and ψ dihedral angles, as observed in our study. In a C-cap capping box, the proline residue is often found at position C′. This has been described for Pro C-capping motifs.19 Moreover, a bridged-tricyclic diproline (Pro–Pro) N-cap motif has been shown to stabilize linear α-helical peptides efficiently.40 Interestingly, different from what is observed in C-cap capping boxes, our proline residue occupies the N2 position in our N-cap capping box. This allows the Asn5 to exert its N-cap effects on Pro7 in PaDBS1R7 through an unconventional N–H⋯N hydrogen bond,25 assisting α-helix stabilization. This unusual N-cap NKP motif, in addition to this peptide's physicochemical properties, resulted in the biophysical characterization of a novel, hybrid coil/N-cap/α-helix scaffold that, in our study, presented multiple biological activities.
Although the study of N-cap amino acid motifs has led to biochemical and biophysical advances, little is known about the range of biological activities that these N-capped peptides may display. Here, we showed that PaDBS1R7 exerts antibacterial, antibiofilm and anticancer activities, reduces bacterial counts in the site of skin infections and modulates LPS-induced macrophages stress response, as well as fibroblasts migration for wound healing. The interest in all these activities is further strengthened by the absence of toxicity towards healthy mammalian cells, suggesting PaDBS1R7 selectivity for anionic cell membranes (e.g., bacteria and cancer cells). This multifunctionality was exclusive for PaDBS1R7, thus providing strong evidence that the N-cap NKP motif is the main parameter differentiating this peptide from its analogs (PaDBS1R2 and R3) in terms of structural profile and of biological activities spectrum.
Another critical point that must be considered is the influence of the different structural scaffolds here described in peptide amphipathicity and, therefore, in the distribution of hydrophilic and hydrophobic residues within the peptide's structure. Although PaDBS1R7 is the least α-helical, its hydrophobic moment in the α-helical section is significantly higher (0.749) than the other two peptides studied (0.431 and 0.454), which is attributed to the N-cap effects elicited by Asn5 and that stabilizes the α-helical segment. Amphipathicity has been commonly described as a crucial determinant of peptides' multifunctionality.41,42 For instance, optimal modulation of amphipathicity and hydrophobicity allows improved antibacterial activities in short AMPs, also enhancing selectivity and specificity for Gram-negative pathogens.43 Moreover, studies have reported a linear correlation between amphipathicity and antibacterial activity, thus identifying intersection points for selecting promising scaffolds for AMP rational design.42
The higher amphipathicity of PaDBS1R7 associated with its coil/N-cap/α-helix scaffold not only favored antibacterial activity, but also extended the peptide's biological activities. PaDBS1R7 was the only peptide tested capable of completely eradicating two-day-old P. aeruginosa biofilms. Bacteria differ significantly in metabolism and resistance profile between their planktonic and biofilm modes of growth.44,45 Consequently, this difference also affects how antibacterial and antibiofilm agents, including AMPs, will exert their functions on these two different biological scenarios.46 Therefore, although the antibiofilm mechanism of PaDBS1R7 is still unknown, we hypothesize that its higher amphipathicity and unusual structural scaffold are intrinsically involved in its ability to disperse and eradicate biofilms.
The antibacterial effectiveness of the tested peptides was also evaluated in a skin abscess mouse model, demonstrating that both PaDBS1R3 and R7 have higher and more prolonged activity than PaDBS1R2. Although PaDBS1R3 and R7 led to similar bacterial count reduction in this animal infection model, in vitro analysis with macrophages recruited from mice revealed an additional function to PaDBS1R7. Macrophages in the presence of LPS produce NO and proinflammatory cytokines (e.g., TNF-α and IL-1β).47 Some AMPs can regulate the cellular response to this bacterial endotoxin (LPS).47 Here, at the same concentration administrated as in the in vivo assay, PaDBS1R7 also reduced macrophage NO production and TNF-α expression levels, thus suggesting that it cannot only act directly on bacterial growth, but also modulates cellular stress and, therefore, favors the host organism in fighting the infection. Interestingly, all the peptides have similar CD signatures in contact with POPE/POPG/CL/LPS vesicles. Nevertheless, only PaDBS1R7 induced ∼40% leakage in this condition, suggesting this peptide's higher interaction with LPS.
Considering the biological conditions tested, PaDBS1R2 and R3 were characterized as AMPs selective for planktonic bacteria. By contrast, PaDBS1R7 was selective for anionic membranes (enriched or not with LPS or CL), as this peptide acted both on bacterial cells (in vitro and in vivo) and cancer cells (in vitro). It is not unusual for AMPs to have anticancer properties.33,48 Cancer cells lose the membrane asymmetry typical of a healthy mammalian cell plasma membrane (with anionic lipids concentrated on its inner leaflet), exposing part of the anionic phospholipids to its outer monolayer. The surface exposure of these lipids favors the electrostatic binding of cationic peptides, including AMPs, further inactivating or disrupting the cell. However, in the present study, PaDBS1R7 was the only peptide to have anticancer properties against three cancer cell lines. Therefore, we have strong evidence that this additional activity is also correlated with the two main distinct determinants associated with this peptide: the coil/N-cap/α-helix structural scaffold and a significantly higher amphipathicity compared to its analogs.
Conclusions
This study extends our understanding of the rules governing AMPs with N-cap motifs. We highlight the importance of considering a coil/N-cap/α-helix structural scaffold as a critical parameter for improving a particular function (antibacterial) and expanding the spectrum of biological activities for a given AMP. In the peptides studied here, this unusual structural scaffold is triggered by an N-cap NKP motif, which resulted in a multifunctional AMP selective for anionic surfaces and capable of exerting antibacterial, antibiofilm, anticancer and anti-infective (skin infection) effects, also reducing LPS-induced macrophage stress and recruiting fibroblasts to assist wound healing. The biological multifunctionality associated with the N-cap NKP motif makes PaDBS1R7 a highly attractive coil/N-cap/α-helix scaffold for peptide-based drug design.Data availability
All experimental data is available in the ESI.†Author contributions
Conceptualization, M. H. C., L. Y. C., D. J. C. and O. L. F.; methodology, M. H. C., L. Y. C., N. C. S., C. F-N., D. J. C. and O. L. F.; formal analysis, M. H. C., L. Y. C., D. F. B, S. B. R., M. D. T. T., I. C. S., S. G and C. F-N.; resources, M. H. C., T. K. L., N. C. S., C. F-N., D. J. C. and O. L. F.; writing – original draft, M. H. C.; writing – review and editing, M. H. C. with input from all authors. Visualization, M. H. C.; supervision, M. H. C., L. Y. C., S. G., T. K. L., N. C. S., C. F-N., D. J. C. and O. L. F.; project administration, M. H. C., L. Y. C., S. G., N. C. S., C. F-N., D. J. C. and O. L. F. Funding acquisition, M. H. C., T. K. L., N. C. S., C. F-N., D. J. C. and O. L. F.Conflicts of interest
Timothy K. Lu is a co-founder of Senti Biosciences, Synlogic, Engine Biosciences, Tango Therapeutics, Corvium, BiomX, Eligo Biosciences, Bota.Bio, and NE47Bio. T.K.L. also holds financial interests in nest.bio, Armata, IndieBio, MedicusTek, Quark Biosciences, Personal Genomics, Thryve, Lexent Bio, MitoLab, Vulcan, Serotiny, Pulmobiotics, Provectus Algae, Invaio, and NSG Biolabs. Cesar de la Fuente-Nunez provides consulting services to Invaio Sciences and is a member of the Scientific Advisory Boards of Nowture S.L. and Phare Bio. The de la Fuente Lab has received research funding or in-kind donations from Peptilogics, United Therapeutics, and Procter & Gamble.Acknowledgements
This work was supported by grants from Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento e Tecnológico (CNPq) and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT), Brazil, DTRA (DTRA HDTRA1-15-1-0050) (to T.K.L.), and Fundação para a Ciência e a Tecnologia – Ministério da Ciência, Tecnologia e Ensino Superior (FCT-MCTES, Portugal). D. J. C. is an Australian Research Council Australian Laureate Fellow (FL150100146, CE200100012). L. Y. C. was supported by the Advance Queensland Women’s Academic Fund (WAF-6884942288). I. C. S. acknowledges FCT-MCTES fellowship PD/BD/136880/2018. We also thank Robin Kramer for her assistance with the skin abscess mouse model. Cesar de la Fuente-Nunez holds a Presidential Professorship at the University of Pennsylvania and acknowledges funding from the Procter & Gamble Company, United Therapeutics, a BBRF Young Investigator Grant, the Nemirovsky Prize, Penn Health-Tech Accelerator Award, and the Dean’s Innovation Fund from the Perelman School of Medicine at the University of Pennsylvania. Research reported in this publication was supported by the Langer Prize (AIChE Foundation), the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138201 and the Defense Threat Reduction Agency (DTRA; HDTRA11810041 and HDTRA1-21-1-0014). All figures were prepared in BioRender.com.Notes and references
- C. D. Fjell, J. A. Hiss, R. E. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2011, 11, 37–51 CrossRef PubMed.
- Q. Tang, Y. Zhong, D. P. Miller, R. Liu, E. Zurek, Z. L. Lu and B. Gong, Org. Lett., 2020, 22, 1003–1007 CrossRef CAS PubMed.
- T. Roncevic, D. Vukicevic, N. Ilic, L. Krce, G. Gajski, M. Tonkic, I. Goic-Barisic, L. Zoranic, Y. Sonavane, M. Benincasa, D. Juretic, A. Maravic and A. Tossi, J. Med. Chem., 2018, 61, 2924–2936 CrossRef CAS PubMed.
- D. J. Paterson, M. Tassieri, J. Reboud, R. Wilson and J. M. Cooper, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8324–E8332 CAS.
- Y. Zheng, X. Meng, Y. Wu, Y. Zhao and C. Wu, Chem. Sci., 2018, 9, 569–575 RSC.
- C. H. Chen, M. C. Melo, N. Berglund, A. Khan, C. de la Fuente-Nunez, J. P. Ulmschneider and M. B. Ulmschneider, Curr. Opin. Struct. Biol., 2020, 61, 160–166 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37, 1186–1197 CrossRef CAS PubMed.
- S. B. Rezende, K. G. N. Oshiro, N. G. O. Junior, O. L. Franco and M. H. Cardoso, Chem. Commun., 2021, 57, 11578 RSC.
- S. T. Amos, L. S. Vermeer, P. M. Ferguson, J. Kozlowska, M. Davy, T. T. Bui, A. F. Drake, C. D. Lorenz and A. J. Mason, Sci. Rep., 2016, 6, 37639 CrossRef CAS PubMed.
- K. G. N. Oshiro, E. S. Candido, L. Y. Chan, M. D. T. Torres, B. E. D. Monges, S. G. Rodrigues, W. F. Porto, S. M. Ribeiro, S. T. Henriques, T. K. Lu, C. de la Fuente-Nunez, D. J. Craik, O. L. Franco and M. H. Cardoso, J. Med. Chem., 2019, 62, 8140–8151 CrossRef CAS PubMed.
- A. Oddo, T. T. Thomsen, H. M. Britt, A. Lobner-Olesen, P. W. Thulstrup, J. M. Sanderson and P. R. Hansen, ACS Med. Chem. Lett., 2016, 7, 741–745 CrossRef CAS PubMed.
- M. H. Cardoso, E. S. Candido, L. Y. Chan, M. Der Torossian Torres, K. G. N. Oshiro, S. B. Rezende, W. F. Porto, T. K. Lu, C. de la Fuente-Nunez, D. J. Craik and O. L. Franco, ACS Infect. Dis., 2018, 4, 1727–1736 CrossRef CAS PubMed.
- N. Y. Yount and M. R. Yeaman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7363–7368 CrossRef CAS PubMed.
- C. M. Agbale, J. K. Sarfo, I. K. Galyuon, S. A. Juliano, G. G. O. Silva, D. F. Buccini, M. H. Cardoso, M. D. T. Torres, A. M. Angeles-Boza, C. de la Fuente-Nunez and O. L. Franco, Biochemistry, 2019, 58, 3802–3812 CrossRef CAS PubMed.
- J. Portelinha, S. S. Duay, S. I. Yu, K. Heilemann, M. D. J. Libardo, S. A. Juliano, J. L. Klassen and A. M. Angeles-Boza, Chem. Rev., 2021, 121, 2648–2712 CrossRef CAS PubMed.
- O. N. Silva, M. D. T. Torres, J. Cao, E. S. F. Alves, L. V. Rodrigues, J. M. Resende, L. M. Liao, W. F. Porto, I. C. M. Fensterseifer, T. K. Lu, O. L. Franco and C. de la Fuente-Nunez, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26936–26945 CrossRef CAS PubMed.
- V. Spicer, Y. W. Lao, D. Shamshurin, P. Ezzati, J. A. Wilkins and O. V. Krokhin, Anal. Chem., 2014, 86, 11498–11502 CrossRef CAS PubMed.
- B. L. Kier, I. Shu, L. A. Eidenschink and N. H. Andersen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10466–10471 CrossRef CAS PubMed.
- J. Prieto and L. Serrano, J. Mol. Biol., 1997, 274, 276–288 CrossRef CAS PubMed.
- W. F. Porto, I. C. M. Fensterseifer, S. M. Ribeiro and O. L. Franco, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2043–2052 CrossRef CAS PubMed.
- D. Eisenberg, R. M. Weiss and T. C. Terwilliger, Nature, 1982, 299, 371–374 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson and G. L. Warren, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 905–921 CrossRef CAS PubMed.
- A. Chakrabartty, A. J. Doig and R. L. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11332–11336 CrossRef CAS PubMed.
- R. N. Deepak and R. Sankararamakrishnan, Biophys. J., 2016, 110, 1967–1979 CrossRef CAS PubMed.
- L. N. Irazazabal, W. F. Porto, I. C. M. Fensterseifer, E. S. F. Alves, C. O. Matos, A. C. S. Menezes, M. R. Felicio, S. Goncalves, N. C. Santos, S. M. Ribeiro, V. Humblot, L. M. Liao, A. Ladram and O. L. Franco, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 178–190 CrossRef CAS PubMed.
- I. C. M. Fensterseifer, M. R. Felicio, E. S. F. Alves, M. H. Cardoso, M. D. T. Torres, C. O. Matos, O. N. Silva, T. K. Lu, M. V. Freire, N. C. Neves, S. Goncalves, L. M. Liao, N. C. Santos, W. F. Porto, C. de la Fuente-Nunez and O. L. Franco, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1375–1387 CrossRef CAS PubMed.
- D. G. Lee, H. N. Kim, Y. Park, H. K. Kim, B. H. Choi, C. H. Choi and K. S. Hahm, Biochim. Biophys. Acta, Proteins Proteomics, 2002, 1598, 185–194 CrossRef CAS PubMed.
- S. C. Park, M. H. Kim, M. A. Hossain, S. Y. Shin, Y. Kim, L. Stella, J. D. Wade, Y. Park and K. S. Hahm, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 229–241 CrossRef CAS PubMed.
- J. M. Resende, C. M. Moraes, M. V. Prates, A. Cesar, F. C. Almeida, N. C. Mundim, A. P. Valente, M. P. Bemquerer, D. Pilo-Veloso and B. Bechinger, Peptides, 2008, 29, 1633–1644 CrossRef CAS PubMed.
- A. Thanabalasuriar, B. N. V. Scott, M. Peiseler, M. E. Willson, Z. Zeng, P. Warrener, A. E. Keller, B. G. J. Surewaard, E. A. Dozier, J. T. Korhonen, L. I. Cheng, M. Gadjeva, C. K. Stover, A. DiGiandomenico and P. Kubes, Cell Host Microbe, 2019, 25, 526–536 CrossRef CAS PubMed.
- O. Oluyombo, C. N. Penfold and S. P. Diggle, mBio, 2019, 10, e01828–e01918 CrossRef CAS PubMed.
- A. L. Tornesello, A. Borrelli, L. Buonaguro, F. M. Buonaguro and M. L. Tornesello, Molecules, 2020, 25, 2850 CrossRef CAS PubMed.
- R. Pan, Y. Zhang, M. Zheng, B. Zang and M. Jin, Front. Pharmacol., 2017, 8, 264 CrossRef PubMed.
- D. I. Chan, E. J. Prenner and H. J. Vogel, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 1184–1202 CrossRef CAS PubMed.
- A. K. Mishra, J. Choi, E. Moon and K. H. Baek, Molecules, 2018, 23, 815 CrossRef PubMed.
- M. M. Javadpour, M. M. Juban, W. C. Lo, S. M. Bishop, J. B. Alberty, S. M. Cowell, C. L. Becker and M. L. McLaughlin, J. Med. Chem., 1996, 39, 3107–3113 CrossRef CAS PubMed.
- H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, Nat. Med., 1999, 5, 1032–1038 CrossRef CAS PubMed.
- J. S. Richardson and D. C. Richardson, Science, 1988, 240, 1648–1652 CrossRef CAS PubMed.
- V. Hack, C. Reuter, R. Opitz, P. Schmieder, M. Beyermann, J. M. Neudorfl, R. Kuhne and H. G. Schmalz, Angew. Chem., Int. Ed. Engl., 2013, 52, 9539–9543 CrossRef CAS PubMed.
- M. D. T. Torres, C. N. Pedron, Y. Higashikuni, R. M. Kramer, M. H. Cardoso, K. G. N. Oshiro, O. L. Franco, P. I. Silva Junior, F. D. Silva, V. X. Oliveira Junior, T. K. Lu and C. de la Fuente-Nunez, Commun. Biol., 2018, 1, 221 CrossRef PubMed.
- I. A. Edwards, A. G. Elliott, A. M. Kavanagh, J. Zuegg, M. A. Blaskovich and M. A. Cooper, ACS Infect. Dis., 2016, 2, 442–450 CrossRef CAS PubMed.
- Z. Jiang, C. T. Mant, M. Vasil and R. S. Hodges, Chem. Biol. Drug Des., 2018, 91, 75–92 CrossRef CAS PubMed.
- I. Poquet, L. Saujet, A. Canette, M. Monot, J. Mihajlovic, J. M. Ghigo, O. Soutourina, R. Briandet, I. Martin-Verstraete and B. Dupuy, Front. Microbiol., 2018, 9, 2084 CrossRef PubMed.
- C. Wang, Q. Zhang, Y. Wang, X. Tang, Y. An, S. Li, H. Xu, Y. Li, W. Luan, X. Wang, M. Liu and L. Yu, Electrophoresis, 2019, 40, 2736–2746 CrossRef CAS PubMed.
- C. de la Fuente-Nunez, M. H. Cardoso, E. S. Candido, O. L. Franco and R. E. Hancock, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1061–1069 CrossRef CAS PubMed.
- N. G. Oliveira-Junior, M. S. Freire, J. A. Almeida, T. M. B. Rezende and O. L. Franco, Cytokine, 2018, 111, 309–316 CrossRef CAS PubMed.
- M. R. Felicio, O. N. Silva, S. Goncalves, N. C. Santos and O. L. Franco, Front. Chem., 2017, 5, 5 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S12, Fig. S1–S17,† and supplementary references. See https://doi.org/10.1039/d1sc06998e |
| This journal is © The Royal Society of Chemistry 2022 |