
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA C-to-O atom-swapping reaction sequence enabled by Ni-catalyzed decarbonylation of lactones†
Quang H.
Luu
 and
Junqi
Li
*
and
Junqi
Li
*
Department of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: junqili@iastate.edu
First published on 6th January 2022
Abstract
Advances in site-selective functionalization reactions have enabled single atom changes on the periphery of a complex molecule, but reaction manifolds that enable such changes on the core framework of the molecule remain sparse. Here, we disclose a strategy for carbon-to-oxygen substitution in cyclic diarylmethanes and diarylketones to yield cyclic diarylethers. Oxygen atom insertion is accomplished by methylene and Baeyer–Villiger oxidations. To remove the carbon atom in this C-to-O “atom swap” process, we developed a nickel-catalyzed decarbonylation of lactones to yield the corresponding cyclic diaryl ethers. This reaction was enabled by mechanistic studies with stoichiometric nickel(II) complexes that led to the optimization of a ligand capable of promoting a challenging C(sp2)–O(aryl) reductive elimination. The nickel-catalyzed decarbonylation was applied to 6–8 membered lactones (16 examples, 32–99%). Finally, a C-to-O atom-swapping reaction sequence was accomplished on a natural product and a pharmaceutical precursor.
Introduction
The impact of single atom changes in small molecules on the intermolecular interactions with their biological targets is well-documented.1 With the development of site-selective functionalization reactions,2 the isosteric replacement of an atom on the periphery of a complex molecule has become increasingly practical. In contrast, the analogous process for an atom embedded in the molecular skeleton remains challenging.3 Although such an “atom swapping” process has been recognized as a highly desired transformation,4 a strategy demonstrating its feasibility has yet to be reported (Fig. 1). Herein, we design a sequence of atom insertion and atom deletion events to realize a carbon-to-oxygen atom swap (Fig. 1). We reason that oxidation of a methylene group to a ketone will enable oxygen atom insertion by Baeyer–Villiger rearrangement, which generates a lactone. The lactone must then undergo a decarbonylative C–O bond-forming reaction to remove the carbon atom. However, despite the growing number of reports on intermolecular decarbonylative coupling reactions,5 the proposed atom-swapping reaction sequence cannot be achieved due to the lack of a method for the decarbonylation of lactones.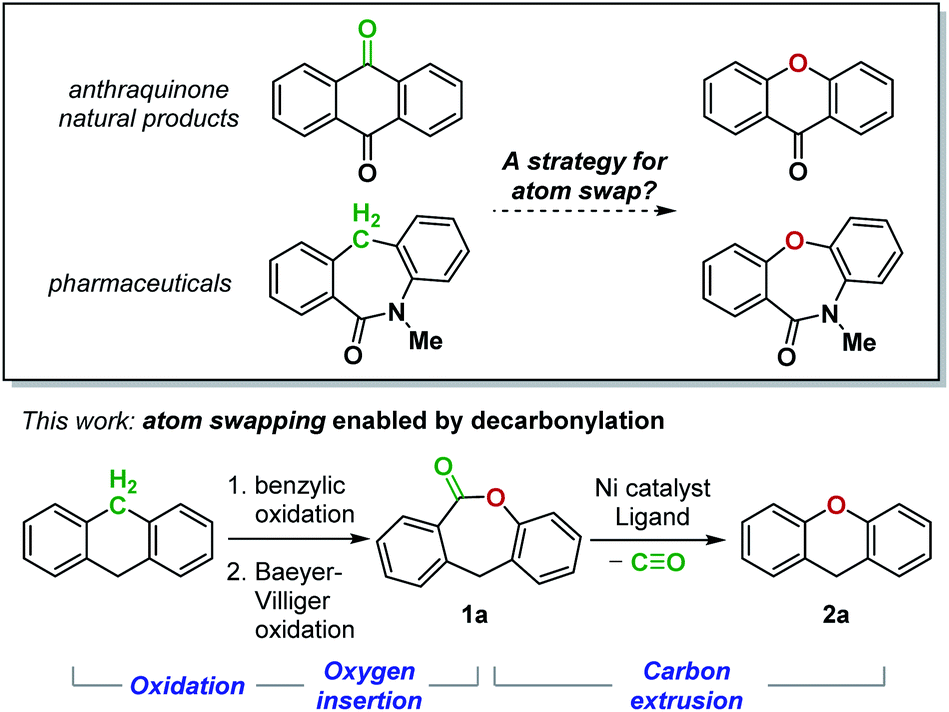 | ||
| Fig. 1 A two-stage strategy for a carbon-to-oxygen atom swap comprising oxidation and carbon extrusion steps. | ||
In 2017, Yamaguchi and coworkers pioneered an intramolecular decarbonylative C–O coupling of esters, but non-heteroaryl substrates such as 1a (Fig. 1) remain unviable to date.6 In this work, we developed a catalytic system that overcomes this limitation. Lactone 1a was chosen as the substrate for optimization as the dibenzolactone substructure can be derived from both diarylketone- and diarylmethane-containing natural products and pharmaceuticals. We first sought to understand the putative catalytic cycle for decarbonylation to identify the problematic step. The main steps involve oxidative addition, migratory-deinsertion, and a Csp2–O(aryl) reductive elimination (Fig. 2).
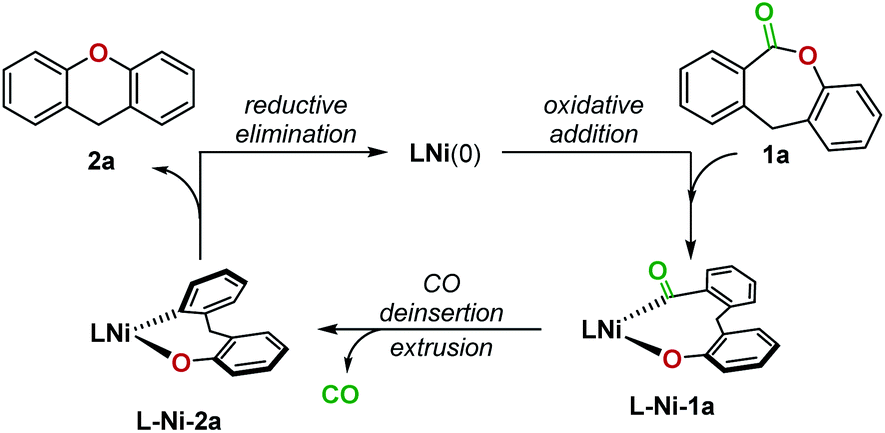 | ||
| Fig. 2 A proposed catalytic cycle for the Ni-catalyzed decarbonylation of lactone 1a to generate 2a. | ||
Results and discussion
The opposing ligand requirements7 in the different steps of the putative catalytic cycle make this transformation challenging. Oxidative addition into the relatively inert C(acyl)–O bond of an ester8,9 generally requires an electron-rich ligand on the metal complex.10 The ligand also has to promote a challenging Csp2–O reductive elimination from a Ni(II)(aryl)(aryloxo) complex,11–14 which is faster from a more electron-deficient metal center bearing bulky ligands.15 Dcype, a bisphosphine ligand used in Yamaguchi's decarbonylation reaction and a widely employed ligand for decarbonylative transformations,9c,16 did not give any yield of 2a from 1a. We thus proceeded to examine the individual steps of the proposed catalytic cycle to determine the problematic step. In a stoichiometric experiment, heating a mixture of Ni(cod)2, dcype, and lactone 1a at 80 °C in C6D6 generated dcype-Ni-2a and Ni(dcype)(CO)2, indicating that oxidative addition and decarbonylation occurred (Fig. 3A). The structure of dcype-Ni-2a was confirmed by independent synthesis, isolation, and characterization by X-ray crystallography (Fig. 3B). dcype-Ni-2a synthesized independently or formed in situ did not undergo reductive elimination to give 2a at temperatures up to 150 °C and resulted only in decomposition.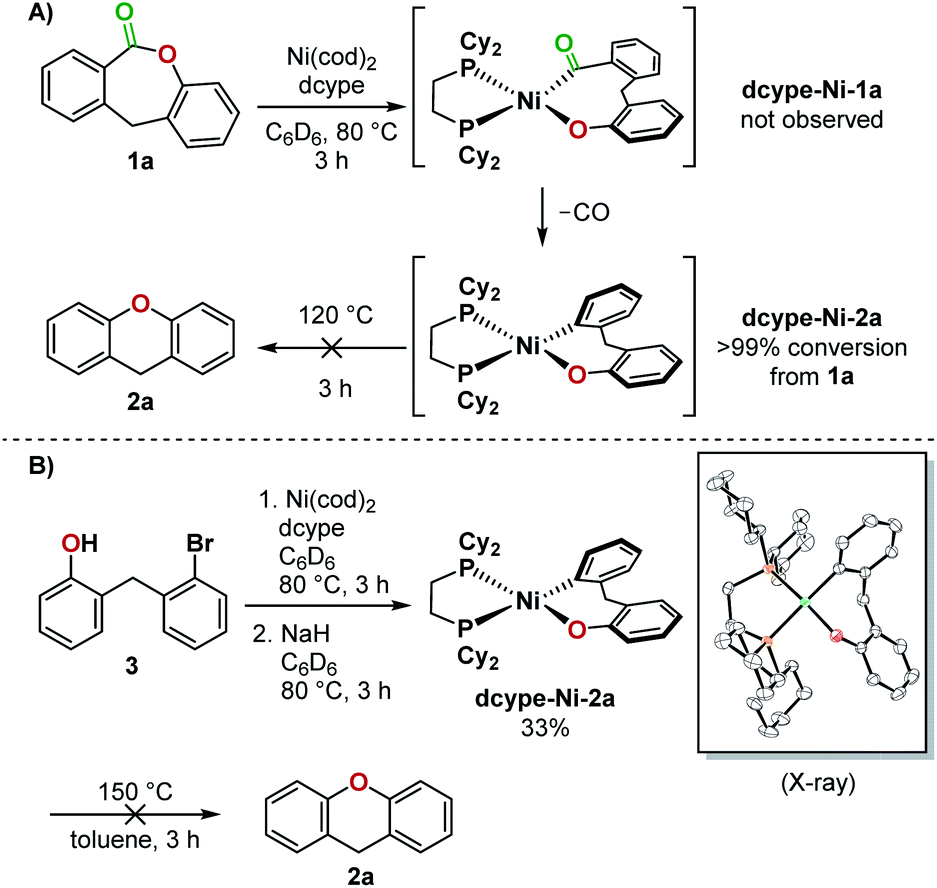 | ||
| Fig. 3 (A) Stoichiometric experiment using dcype and 1a, (B) independent synthesis of dcype-Ni-2a and a reductive elimination test. | ||
To solve this reductive elimination problem, we synthesized and tested phosphine ligand classes that were successful for carbon–heteroatom bond formations under Ni(0)/Ni(II) catalysis6,11a,b,17 (Fig. 4) and other ligands (see ESI for the full list, Section 3.1†). These ligands gave no or unsatisfactory yields of the desired product. However, bisphosphines bearing two bulky tetramethyltrioxaphosphaadamantyl moieties on an aryl backbone, first prepared by the Stradiotto group for C(aryl)–N cross couplings,18,19 were effective, with meso-L2 giving an 80% yield of the desired decarbonylation product (Fig. 4).
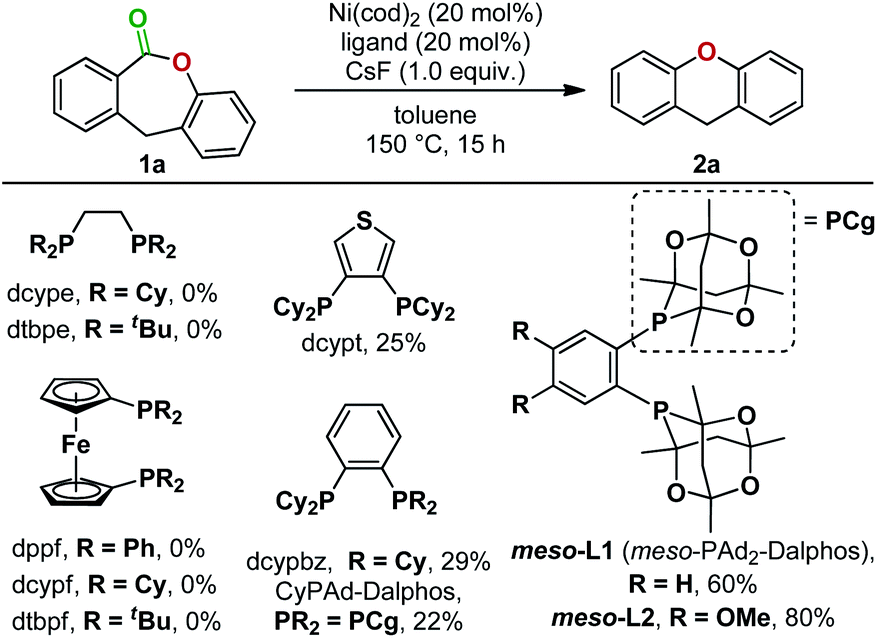 | ||
| Fig. 4 Ligand optimization. | ||
Stoichiometric experiments indicate that the successful formation of 2a with the bulkier, less electron-rich meso-L2 ligand compared to dcype can be attributed to its ability to promote reductive elimination from a Ni(II)(aryl)(aryloxo) complex. Heating a mixture of Ni(cod)2, meso-L2, and 1a at 110 °C in C6D6 resulted in no conversion, but increasing the reaction temperature to 120 °C then gave the product 2a and meso-L2-Ni(CO)2 with no other Ni(II) intermediates detected by 1H and 31P NMR (Fig. 5A). This was further confirmed by independent synthesis and isolation of meso-L2-Ni-2a from 3. At 110 °C, meso-L2-Ni-2a underwent reductive elimination cleanly to give 2a in 99% yield (Fig. 5B).
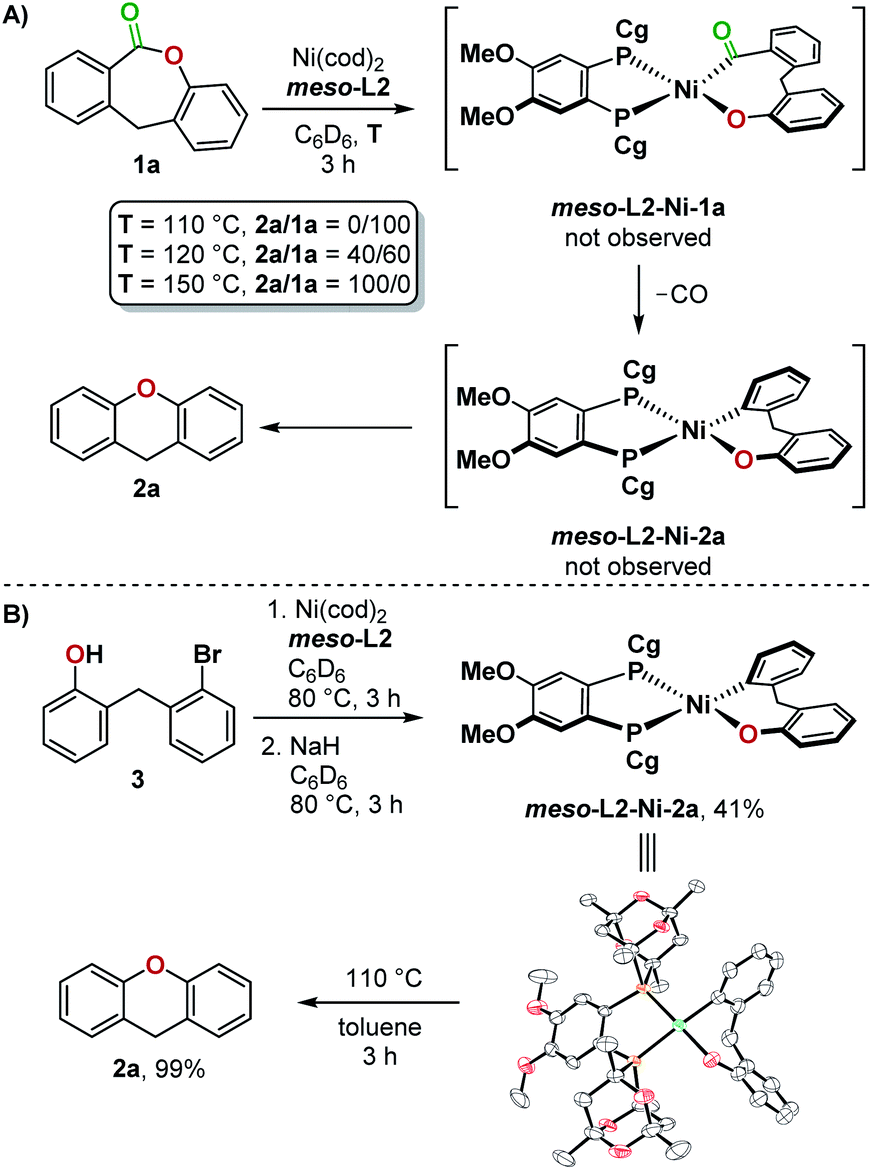 | ||
| Fig. 5 (A) Stoichiometric experiment using ligand meso-L2 and 1a, (B) independent synthesis of meso-L2-Ni-2a and a reductive elimination test. | ||
Further experiments uncovered more differences between dcype- and meso-L2-ligated nickel(II) complexes in reductive elimination. Heating dcype-Ni-2a with Cr(CO)6 at 80 °C resulted in the formation of lactone 1a in 90% yield. In contrast, meso-L2-Ni-2a underwent reductive elimination under the same conditions to give 2a in 98% yield (Fig. 6A). Since reductive elimination occurred at a lower temperature in the presence of CO than reductive elimination from meso-L2-Ni-2a in the absence of CO (Fig. 5B), a second catalytic cycle may be operating in the reaction employing Ni(cod)2 and meso-L2. In this second pathway, CO deinsertion from L-Ni-1a generates a five-coordinate complex20L-CO-Ni-2a, which undergoes reductive elimination to give 2a and complex L-Ni(CO)x. The inactive complex L-Ni(CO)x then re-enters the catalytic cycle after dissociation of carbon monoxide at high temperature (Fig. 6B).20c,d Reductive elimination from both a four-coordinate and five-coordinate dcype-ligated nickel complex are not feasible under the conditions examined.
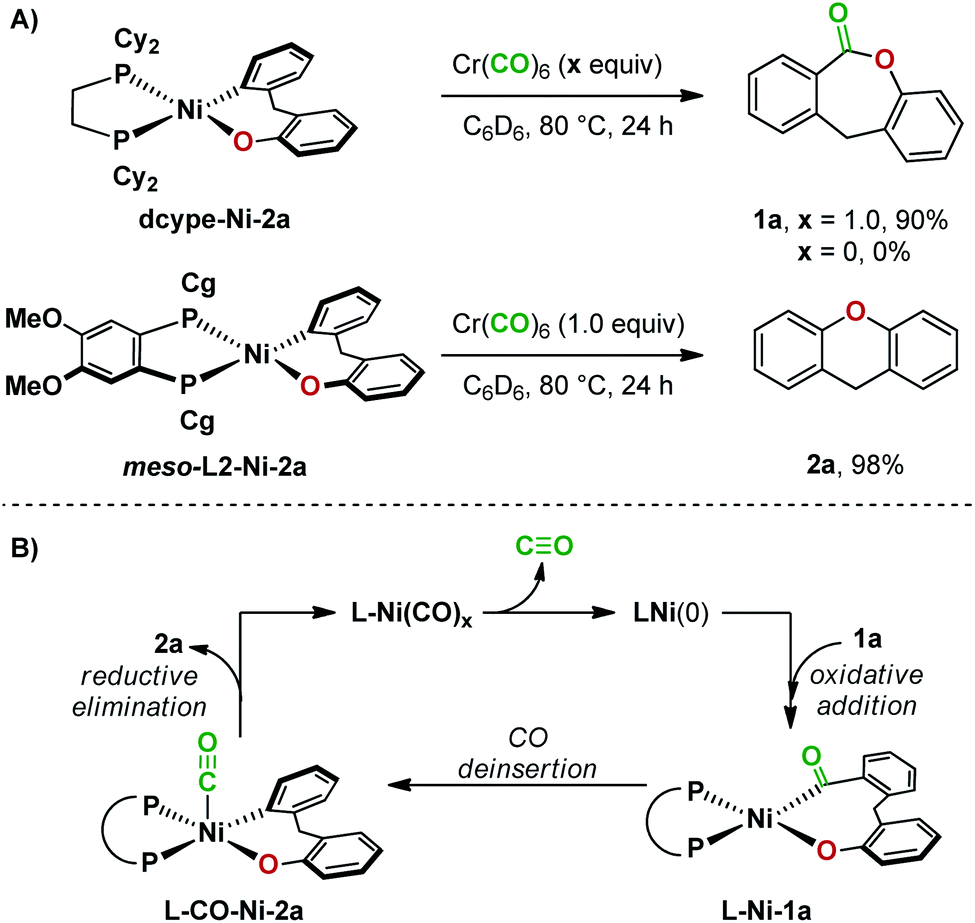 | ||
| Fig. 6 (A) Carbonylation reactions with nickel complexes. (B) An alternative catalytic cycle with Ni/meso-L2. | ||
Using Ni(cod)2 and meso-L2 as the catalyst, further optimization in solvent, temperature, concentration, and base led to 85% yield of 2a (see ESI, Section 3†). The optimized conditions were then applied to different lactone substrates (Fig. 7). Electron-donating and electron-withdrawing substituents on either aryl rings of 1 significantly affected the reaction yields.15 Two electron-withdrawing substituents at the para positions relative to the COO group gave a higher product yield than two electron-donating groups (2bvs.2c, 54% vs. 32%). Additionally, lactone 1d with R1 = CO2Me and R2 = OMe gave 67% yield of 2d.
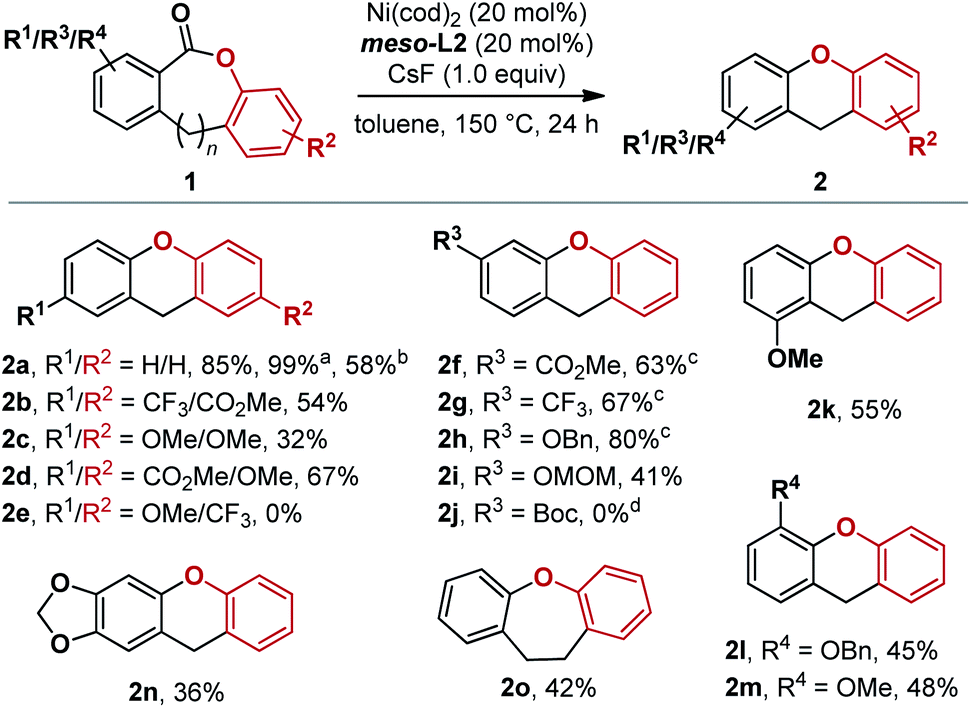 | ||
| Fig. 7 Substrate scope. Reactions were run on a 0.20 mmol scale unless otherwise noted. Work-up and purification were carried out as described in the ESI.†a 0.050 mmol scale. b 1.00 mmol scale. c 0.10 mmol scale. d 29% yield of the deprotected lactone was obtained. | ||
However, when the electronic character of the rings was reversed, the expected product 2e was not detected. Overall, substrates with electron-donating groups para to the COO moiety gave noticeably lower yields (2c, 2e, and 2n). A range of R3 substituents can be accommodated in the reaction, including both electron-donating and electron-withdrawing groups (2f–2i). Benzyl and MOM protecting groups are compatible (2h and 2i), but not the Boc protecting group (2j). A benzyloxy and methoxy group as the R4 substituent were tolerated with similar yields (45% and 48%, 2l and 2m). Dihydrodibenzooxepine 2o can also be synthesized under the same conditions, albeit in decreased yield (42%). meso-L2-Ni-2a was also a competent pre-catalyst in place of Ni(cod)2/meso-L2, giving essentially the same yield of 2d (see ESI, Section 5.7†) under otherwise identical conditions. Acyclic esters and alkyl lactones are currently outside the scope of this reaction (for details, see ESI Section 6.3†).
The successful development of the decarbonylation of lactones now opens the way for our proposed carbon-to-oxygen atom swap transformation. We first targeted the replacement of a ketone with an oxygen in an anthraquinone natural product derivative (Fig. 8A). A Baeyer–Villiger reaction21 on dimethylquinizarin 4 followed by reduction with triethylsilane in trifluoroacetic acid gave lactone 6. Subjecting 6 to the decarbonylation conditions we developed gave the desired diaryl ether in 52% yield. Oxidation with KMnO4 then afforded the desired xanthone 7. This sequence of reactions thus overall results in a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to O atom swap, transforming an anthraquinone to a xanthone. Our attempts to decarbonylate 5 without reduction to 6 led only to decomposition, suggesting that the decarbonylation is affected by the C–C–C bond angle at the carbon bridge in dibenzolactone 6.
O to O atom swap, transforming an anthraquinone to a xanthone. Our attempts to decarbonylate 5 without reduction to 6 led only to decomposition, suggesting that the decarbonylation is affected by the C–C–C bond angle at the carbon bridge in dibenzolactone 6.
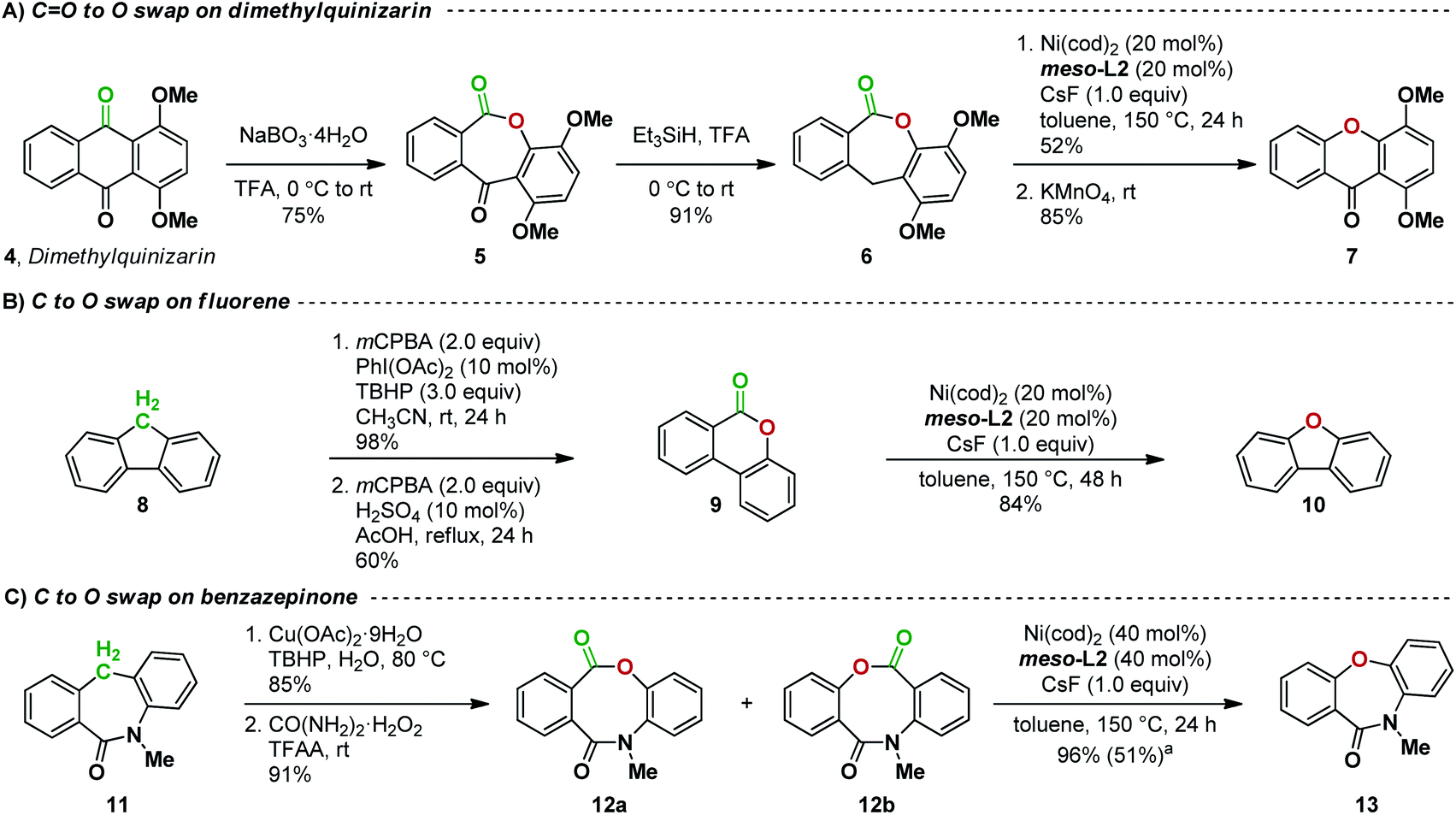 | ||
| Fig. 8 Atom-swapping reaction sequences in (A) a natural product derivative; (B) fluorene; and (C) a pharmaceutical precursor. a Yield in parentheses is from a reaction using 20 mol% of Ni(cod)2 and meso-L2. | ||
We next tested if CH2-to-O atom swap can be realized (Fig. 8B). Indeed, a sequence of oxidation, Baeyer–Villiger reaction, and decarbonylation smoothly converts fluorene 8 to dibenzofuran 10 in an overall yield of 49%. This example also demonstrates that our decarbonylation reaction can be applied to the ring contraction of 6-membered dibenzolactones.
We then expanded our atom-swapping method to the conversion of a dibenzazepinone to a dibenzoxazepinone. Dibenzazepinones such as 11 (Fig. 8C) are versatile intermediates in the synthesis of a range of pharmaceuticals containing a dibenzoazepine motif, including mianserin, etazepine, epinastine, and perlapine.22 Oxidation of dibenzazepinone 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22b followed by Baeyer–Villiger rearrangement gave a mixture of regioisomeric lactones 12a and 12b. The regioselectivity of the Baeyer–Villiger oxidation was inconsequential since subsequent decarbonylation of the mixture gave the desired C-to-O atom-swapping product 13 in 96% yield. No other by-product was observed, indicating a complete selectivity for the C(acyl)–O bond activation over the C(acyl)–N bond16j,23 during the decarbonylation step. In addition, the presence of two sp2 atoms in the bridge is not detrimental to decarbonylation, presumably due to the greater flexibility in the larger ring.
22b followed by Baeyer–Villiger rearrangement gave a mixture of regioisomeric lactones 12a and 12b. The regioselectivity of the Baeyer–Villiger oxidation was inconsequential since subsequent decarbonylation of the mixture gave the desired C-to-O atom-swapping product 13 in 96% yield. No other by-product was observed, indicating a complete selectivity for the C(acyl)–O bond activation over the C(acyl)–N bond16j,23 during the decarbonylation step. In addition, the presence of two sp2 atoms in the bridge is not detrimental to decarbonylation, presumably due to the greater flexibility in the larger ring.
Conclusion
In conclusion, we have achieved a carbon-to-oxygen atom swapping transformation comprising oxygen atom insertion and carbon atom deletion steps. To enable this overall process, we developed a Ni-catalyzed decarbonylation of lactones that overcomes the substrate limitations of previous ester decarbonylation reactions. The Ni/meso-L2 catalyst is capable of promoting a challenging Csp2–O(aryl) reductive elimination from either a four- or five-coordinate Ni(II) complex. This report establishes the framework for creating new atom-swapping processes in complex molecules using transition metal-catalyzed decarbonylation reactions.Data availability
Experimental data associated with this article are provided in the ESI.† Crystallographic data for meso-L2-Ni-2a and dcype-Ni-2a have been deposited at the CCDC under 2116181 and 2116182 and can be obtained from https://www.ccdc.cam.ac.uk/.Author contributions
J. Li conceptualized the project. Q. H. Luu developed the methodology and performed most of the experiments. Both authors analyzed the data and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge Dr Arkady Ellern for X-ray crystallographic analyses of compound dcype-Ni-2a and meso-L2-Ni-2a. We also thank Iowa State University for start-up funds and a postdoctoral seed grant award to Q. H. Luu.Notes and references
- (a) G. A. Patani and E. J. Lavoie, Chem. Rev., 1996, 96, 3147–3176 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (c) D. L. Boger, J. Org. Chem., 2017, 82, 11961–11980 CrossRef CAS PubMed.
- For reviews on late-stage modifications of complex molecules, see: (a) C. R. Shugrue and S. J. Miller, Chem. Rev., 2017, 117, 11894–11951 CrossRef CAS PubMed; (b) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS; (c) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Prim., 2021, 1, 43 CrossRef.
- (a) W. F. Bailey and J. J. Bischoff, J. Org. Chem., 1985, 50, 3009–3010 CrossRef CAS; (b) Z. J. Liu, X. Lu, G. Wang, L. Li, W. T. Jiang, Y. D. Wang, B. Xiao and Y. Fu, J. Am. Chem. Soc., 2016, 138, 9714–9719 CrossRef CAS PubMed; (c) G. Golime, H. Y. Kim and K. Oh, Org. Lett., 2018, 20, 942–945 CrossRef CAS PubMed.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (b) K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- For a general review on transition-metal catalyzed decarbonylation reactions, see H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed.
- R. Takise, R. Isshiki, K. Muto, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 3340–3343 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Oxidative addition of Nonpolar Reagents in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010, pp. 263–264 Search PubMed; (b) J. F. Hartwig, Reductive Elimination in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010, pp. 322–325 Search PubMed.
- For reviews of C(acyl)–O bond activation using Ni(0) or Pd(0), see: (a) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC; (b) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed.
- For recent reports on Ni-catalyzed activation of esters, see: (a) R. Isshiki, N. Inayama, K. Muto and J. Yamaguchi, ACS Catal., 2020, 10, 3490–3494 CrossRef CAS; (b) K. Matsushita, R. Takise, K. Muto and J. Yamaguchi, Sci. Adv., 2020, 6, eaba7614 CrossRef CAS PubMed; (c) C. A. Malapit, M. Borrell, M. W. Milbauer, C. E. Brigham and M. S. Sanford, J. Am. Chem. Soc., 2020, 142, 5918–5923 CrossRef CAS PubMed.
- T. Yamamoto, J. Ishizu, J. Kohara, S. Komiya and A. Yamamoto, J. Am. Chem. Soc., 1980, 102, 3758–3764 CrossRef CAS.
- For cross-coupling reactions involving C(aryl)–alkoxide reductive eliminations from Ni(II) complexes, see: (a) G. Mann and J. F. Hartwig, J. Org. Chem., 1997, 62, 5413–5418 CrossRef CAS; (b) P. M. MacQueen, J. P. Tassone, C. Diaz and M. Stradiotto, J. Am. Chem. Soc., 2018, 140, 5023–5027 CrossRef CAS PubMed; (c) T. Hashimoto, K. Shiota, K. Funatsu and Y. Yamaguchi, Adv. Synth. Catal., 2021, 363, 1625–1630 CrossRef CAS; (d) K. M. Morrison, R. T. McGuire, M. J. Ferguson and M. Stradiotto, ACS Catal., 2021, 11, 10878–10884 CrossRef CAS.
- For cross-coupling reactions involving C(vinyl)-alkoxide reductive eliminations from Ni(II) complexes, see: (a) S.-J. Han, R. Doi and B. M. Stoltz, Angew. Chem., Int. Ed., 2016, 55, 7437–7440 CrossRef CAS PubMed; (b) T. D. Lohrey, A. Q. Cusumano, W. A. Goddard and B. M. Stoltz, ACS Catal., 2021, 11, 10208–10222 CrossRef CAS.
- For an example of a cross-coupling reaction between phenols and aryl bromides in the presence of Ni(II) salts, see: F. Wu, K. Zhu, G. Wu, Y. Gao and H. Chen, Eur. J. Org. Chem., 2020, 519–522 CrossRef CAS.
- For alternative approaches to diarylether formation using nickel catalysis, see: (a) L. Liu and C. Nevado, Organometallics, 2021, 40, 2188–2193 CrossRef CAS; (b) D.-L. Zhu, S. Jiang, Q. Wu, H. Wang, H.-Y. Li and H.-X. Li, Org. Lett., 2021, 23, 8327–8332 CrossRef CAS PubMed.
- G. Mann, Q. Shelby, A. H. Roy and J. F. Hartwig, Organometallics, 2003, 22, 2775–2789 CrossRef CAS.
- (a) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573–13576 CrossRef CAS PubMed; (b) L. K. Meng, Y. Kamada, K. Muto, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2013, 52, 10048–10051 CrossRef CAS PubMed; (c) X. H. Pu, J. F. Hu, Y. Zhao and Z. Z. Shi, ACS Catal., 2016, 6, 6692–6698 CrossRef CAS; (d) H. F. Yue, L. Guo, S. C. Lee, X. Q. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972–3976 CrossRef CAS PubMed; (e) X. Q. Liu, J. Q. Jia and M. Rueping, ACS Catal., 2017, 7, 4491–4496 CrossRef CAS; (f) A. Chatupheeraphat, H. H. Liao, S. C. Lee and M. Rueping, Org. Lett., 2017, 19, 4255–4258 CrossRef CAS PubMed; (g) W. Srimontree, A. Chatupheeraphat, H. H. Liao and M. Rueping, Org. Lett., 2017, 19, 3091–3094 CrossRef CAS PubMed; (h) H. F. Yue, L. Guo, H. H. Liao, Y. F. Cai, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 4282–4285 CrossRef CAS PubMed; (i) A. Chatupheeraphat, H. H. Liao, W. Srimontree, L. Guo, Y. Minenkov, A. Poater, L. Cavallo and M. Rueping, J. Am. Chem. Soc., 2018, 140, 3724–3735 CrossRef CAS PubMed; (j) T. Morioka, S. Nakatani, Y. Sakamoto, T. Kodama, S. Ogoshi, N. Chatani and M. Tobisu, Chem. Sci., 2019, 10, 6666–6671 RSC.
- C. Brigham, C. Malapit, N. Lalloo and M. S. Sanford, ACS Catal., 2020, 10, 8315–8320 CrossRef CAS PubMed.
- J. S. K. Clark, M. J. Ferguson, R. McDonald and M. Stradiotto, Angew. Chem., Int. Ed., 2019, 58, 6391–6395 CrossRef CAS PubMed.
- L1 is commercially available as a mixture of the meso and racemic isomers under the name PAd2-Dalphos, https://www.sigmaaldrich.com/US/en/product/aldrich/919551. In this work, we found that the rac-L1 and rac-L2 gave significantly lower yields than meso-L1 and meso-L2 respectively. See ESI, Table S1.†.
- (a) T. Yamamoto, A. Yamamoto and S. Ikeda, J. Am. Chem. Soc., 1971, 93, 3350–3359 CrossRef CAS; (b) K. Tatsumi, A. Nakamura, S. Komiya, A. Yamamoto and T. Yamamoto, J. Am. Chem. Soc., 1984, 106, 8181–8188 CrossRef CAS; (c) K. Amaike, K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 13573–13576 CrossRef CAS PubMed; (d) C. A. Malapit, J. R. Bour, S. R. Laursen and M. S. Sanford, J. Am. Chem. Soc., 2019, 141, 17322–17330 CrossRef CAS PubMed; (e) J. G. Estrada, W. L. Williams, S. I. Ting and A. B. Doyle, J. Am. Chem. Soc., 2020, 142, 8928–8937 CrossRef CAS PubMed.
- H. L. Newson, D. A. Wild, S. Y. Yeung, B. W. Skelton, G. R. Flematti, J. E. Allan and M. J. Piggott, J. Org. Chem., 2016, 81, 3127–3135 CrossRef CAS PubMed.
- (a) A. K. Sinha and S. Nizamuddin, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1984, 23, 165 Search PubMed; (b) Y. Yu, L. Ma, J. Xia, L. Xin, L. Zhu and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 18261–18266 CrossRef CAS PubMed.
- (a) L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed; (b) C. Liu, G. Li, S. Shi, G. Meng, R. Lalancette, R. Szostak and M. Szostak, ACS Catal., 2018, 8, 9131–9139 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data, and NMR spectra of all new compounds. CCDC 2116181 and 2116182 contain the supplementary crystallographic data fordcype-Ni-2a and meso-L2-Ni-2a. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06968c |
| This journal is © The Royal Society of Chemistry 2022 |