
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetalation-induced denitrogenative reductive coupling of isocyanides on a silylene-bridged nickel cluster†
Kento
Shimamoto
a and
Yusuke
Sunada
 *abc
*abc
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan
bInstitute of Industrial Science, The University of Tokyo, 4-6-1, Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: sunada@iis.u-tokyo.ac.jp
cJST PRESTO, Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 15th March 2022
Abstract
The denitrogenative reductive coupling of two molecules of CNtBu to afford a disilylketenimine with an aza-disilacyclobutane skeleton was achieved on a multinuclear silylene-bridged Ni cluster framework in the absence of any strong reducing reagents. During this reaction, sequential cleavage of a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and formation of a CC bond involving two molecules of CNtBu were achieved on a nickel cluster surrounded by four silylene moieties. First, the cleavage of the CN bond of one molecule of CNtBu provided a silylene-supported carbide and an NtBu moiety on the dinuclear nickel skeleton. Further metalation induced coupling between the carbide moiety and an additional molecule of CNtBu on the pentanuclear nickel-cluster framework to form a
N bond and formation of a CC bond involving two molecules of CNtBu were achieved on a nickel cluster surrounded by four silylene moieties. First, the cleavage of the CN bond of one molecule of CNtBu provided a silylene-supported carbide and an NtBu moiety on the dinuclear nickel skeleton. Further metalation induced coupling between the carbide moiety and an additional molecule of CNtBu on the pentanuclear nickel-cluster framework to form a 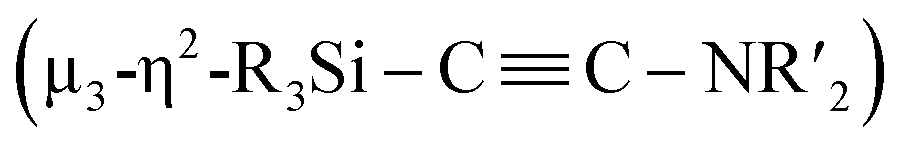 moiety via formation of a CC bond. Thermolysis of this pentanuclear cluster produced a disilylketenimine with an aza-disilacyclobutane skeleton in 58% yield.
moiety via formation of a CC bond. Thermolysis of this pentanuclear cluster produced a disilylketenimine with an aza-disilacyclobutane skeleton in 58% yield.
Introduction
The activation of strong chemical bonds such as CO, CN, and NN constitutes a field of chemistry with a strong focus on the transformation of relatively simple molecules into value-added chemicals.1 For instance, the Fischer–Tropsch reaction, in which CO molecules are directly converted into hydrocarbons by activation of the CO bond via contact with H2 is an industrially important process.2 During this reaction, H2 acts as the combined source of electrons and protons. As in the Fischer–Tropsch process, deoxygenative reductive coupling of CO via CO bond cleavage and subsequent C–C bond formation has generally been achieved on heterogeneous metal catalysts, such as active metal surfaces and metal nanoparticles. In contrast, the development of deoxygenative reductive coupling reactions of CO on well-defined molecular compounds such as transition-metal complexes remains challenging, despite the fact that the development of homogeneous systems may facilitate the detailed investigation of the underlying reaction mechanism.3–10 However, scientists have recently achieved coupling reactions of CO using several defined transition-metal and lanthanide complexes, which has led to a better understanding of the mechanisms of CO activation in each case. For instance, Agapie et al. developed a deoxygenative reductive coupling of two molecules of CO at the single molybdenum center of a Mo(II)–dicarbonyl complex with the aid of KC8 and R3SiCl to form alkyne iPr3SiO–CC–SiiPr3 or bis(silyl)ketene (SiMe3)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO as the deoxygenative coupling products (Scheme 1).4 The authors succeeded in isolating some intermediary species, and the activation of CO was found to be achieved as follows: first, the activation of the CO bond of one of the two CO ligands on the Mo(II)–dicarbonyl complex occurs in the presence of KC8 and R3SiCl to produce a Mo(IV)–carbyne complex that contains a CO ligand; then, further reduction of the Mo(IV)–carbyne complex by KC8 and subsequent addition of R3SiCl triggers the coupling of a carbyne and CO ligands on the Mo center to produce the deoxygenative reductive coupling product. This is quite a unique process, but there remains the issue that stoichiometric amounts of strong reductants such as KC8 are required.
CO as the deoxygenative coupling products (Scheme 1).4 The authors succeeded in isolating some intermediary species, and the activation of CO was found to be achieved as follows: first, the activation of the CO bond of one of the two CO ligands on the Mo(II)–dicarbonyl complex occurs in the presence of KC8 and R3SiCl to produce a Mo(IV)–carbyne complex that contains a CO ligand; then, further reduction of the Mo(IV)–carbyne complex by KC8 and subsequent addition of R3SiCl triggers the coupling of a carbyne and CO ligands on the Mo center to produce the deoxygenative reductive coupling product. This is quite a unique process, but there remains the issue that stoichiometric amounts of strong reductants such as KC8 are required.
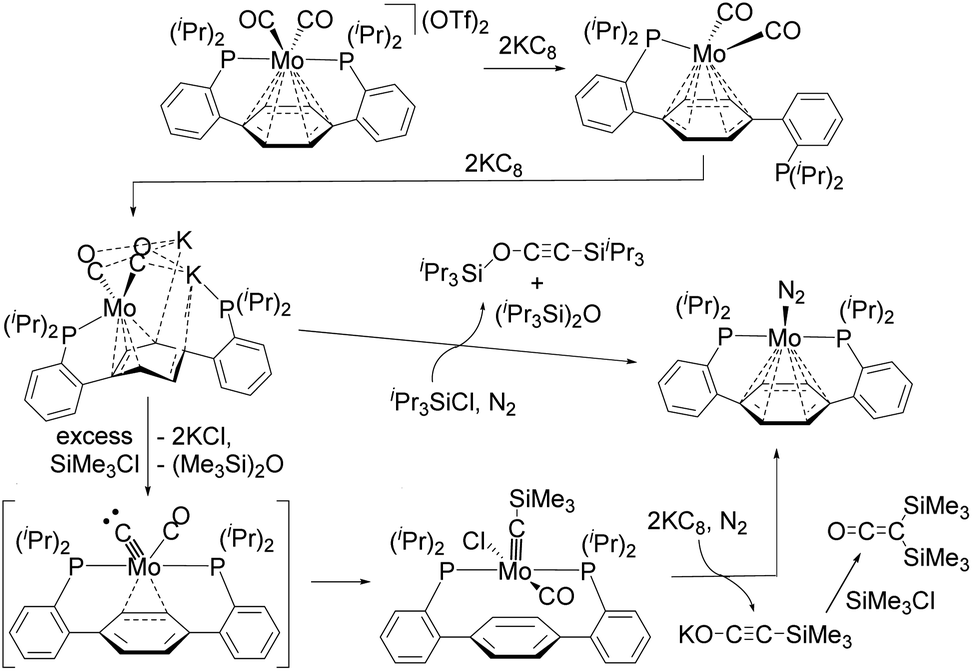 | ||
| Scheme 1 Deoxygenative reductive coupling of two molecules of CO at a single molybdenum center reported by Agapie et al.4 | ||
Another example has recently been reported by Kays et al., in which the scission of a CO bond and the formation of a C–C bond were achieved on a coordinatively unsaturated dinuclear iron(II) aryl complex.5 In addition, some early transition-metal6,7 or lanthanide complexes8 consisting of Y, Ti, Zr, Hf, Ta, Ln, Sm, and Lu have also been reported to achieve the deoxygenative reductive coupling of CO. However, the use of strong reducing reagents such as KC8 and Na/Hg is often required to synthesize reactive electron-rich precursors, and the release of the coupling product is generally problematic due to the high oxophilicity of the early-transition-metal centers. Furthermore, substantial effort has recently been devoted to developing methods for the activation of the CN bond of isocyanides, given that isocyanides (CNR) are isoelectronic with CO.10,11
Considering the fact that, as shown in the Fischer–Tropsch reaction, deoxygenative coupling of CO is effectively realized on the active surface of nano-sized metal compounds such as metal nanoparticles, we hypothesized that the cooperative function of the multiple metal centers in molecular metal clusters could be an approach to effectively activate strong chemical bonds without using strong reducing reagents. Recently, we have developed an efficient synthesis of metal clusters via the reaction of low-valent metal species with organosilicon compounds bearing multiple Si–Si bonds.12 For instance, a palladium cluster consisting of eleven palladium atoms was synthesized selectively via the reaction of [Pd(CNtBu)2]3 with a bicyclic ladder polysilane.12a In addition, we have synthesized a silylene-bridged planar tetranuclear palladium cluster, Pd4(SiR2)3(CNtBu)4 (R = iPr, cyclopentyl), via the reaction of [Pd(CNtBu)2]3 with a cyclotetrasilane, Si4R8. We discovered that this tetranuclear palladium cluster exhibits good catalytic performance for the hydrogenation of various alkenes. In contrast, the corresponding mononuclear palladium disilyl complex as well as the trinuclear palladium complex supported by isocyanide ligands show significantly decreased catalytic performance.12d These results indicate that silylene-bridged clusters can be expected to show higher reactivity than conventional metal compounds. Therefore, we planned to construct defined metal clusters based on this method, which might then enable the activation of strong chemical bonds on the cluster framework. Given that isocyanides (CNR) are isoelectronic with CO, the former were chosen as model substrates for CO. In this paper, we wish to report the synthesis and reactions of nickel complexes and clusters obtained from the reaction of the zero-valent nickel species Ni(cod)2 with cyclotetrasilane 1. A mononuclear nickel complex was obtained when Ni(cod)2 was treated with 1 in the presence of N-heterocyclic carbene Me2IMEt as an auxiliary ligand. In contrast, denitrogenative reductive coupling of two molecules of CNtBu took place on the silylene-bridged multinuclear nickel skeleton when CNtBu was used instead of Me2IMEt to afford a disilylketenimine with an aza-disilacyclobutane skeleton. Isolation of possible intermediary species revealed that activation of the CN bond of CNR to form carbide and imido moieties was realized on the dinuclear nickel framework. Further metalation-induced denitrogenative coupling of CNR to form a coordinated alkyne moiety 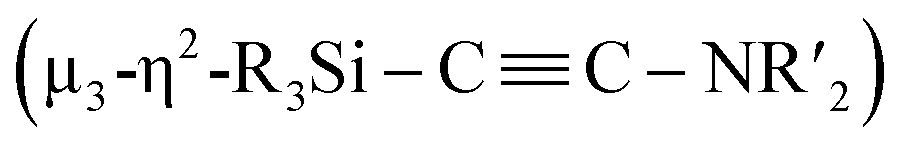 afforded a pentanuclear nickel cluster.
afforded a pentanuclear nickel cluster.
Results and discussion
Synthesis of mononuclear nickel complex 2
We have recently reported that the reaction of a cyclotetrasilane with a Pd(0) precursor can lead to the selective formation of a planar tetranuclear palladium cluster.11d We also found that judicious choice of the substituents on the silicon center is crucial to develop an efficient synthesis method for the clusters; namely, the reaction of iPr-substituted cyclotetrasilane Si4(iPr)8 (1a) with the Pd(0) precursor [Pd(CNtBu)2]3 required a temperature of 65 °C to reach completion, whereas the reaction of [Pd(CNtBu)2]3 with Si4(cyclopentyl)8 (1b) proceeded smoothly even at room temperature. Although it is known that nickel species often show in comparison less reactivity toward insertion into Si–Si bonds, we recently found that the insertion of nickel species into the Si–Si bond of a hypervalent disilane took place when a combination of Ni(cod)2 and Me2IMEt was used as the nickel(0) precursor.11c Thus, we first performed the reactions using Ni(cod)2/Me2IMEt with a series of cyclotetrasilanes. The reaction did not proceed when the reactions were conducted with 1a and 1b; in contrast, the reaction proceeded when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of Ni(cod)2 and Me2IMEt was stirred with Si4Ph8 (1c) at 80 °C for 48 h. In the 1H NMR spectrum of the crude product, the formation of a single product was observed, and mononuclear nickel complex 2 was isolated from the mixture in 48% yield after recrystallization (Scheme 2). It should be mentioned here that no further insertion of the nickel species to form multinuclear nickel clusters was observed when the reaction of 1c was conducted with 4 equiv. of Ni(cod)2 and 8 equiv. of Me2IMEt.
:2 mixture of Ni(cod)2 and Me2IMEt was stirred with Si4Ph8 (1c) at 80 °C for 48 h. In the 1H NMR spectrum of the crude product, the formation of a single product was observed, and mononuclear nickel complex 2 was isolated from the mixture in 48% yield after recrystallization (Scheme 2). It should be mentioned here that no further insertion of the nickel species to form multinuclear nickel clusters was observed when the reaction of 1c was conducted with 4 equiv. of Ni(cod)2 and 8 equiv. of Me2IMEt.
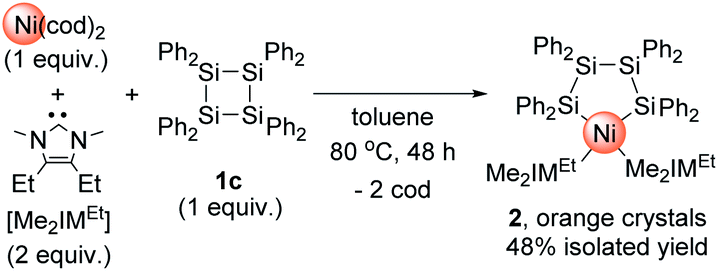 | ||
| Scheme 2 Reaction of Ni(cod)2/Me2IMEt with 1c to form mononuclear nickel complex 2. | ||
The molecular structure of 2 in the solid state was unequivocally determined via single-crystal X-ray diffraction analysis, and the thermal ellipsoid plot is shown in Fig. S7 in the ESI.† The formation of tetrasilanickelacycle was confirmed, and Ni–Si bond distances of 2.2952(7) and 2.2917(7) Å were observed. In the 29Si NMR spectrum, two singlets appeared at −2.00 and −26.12 ppm; the former can be ascribed to the silicon atom coordinated to the nickel center, whereas the latter was attributed to the silicon atom located on the ligand backbone.13
C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N bond cleavage on the dinuclear nickel framework to form 3
N bond cleavage on the dinuclear nickel framework to form 3
In the reaction of 1c with Ni(cod)2/Me2IMEt, the insertion of only one nickel species into the Si–Si bonds occurred. In contrast, we observed the insertion of multiple nickel species when CNtBu was used instead of Me2IMEt. Namely, treatment of a 2:5 mixture of Ni(cod)2 and CNtBu with 1c in toluene at 65 °C for 48 h gave rise to the formation of dinuclear nickel complex 3 (Scheme 3). In the 1H NMR spectrum of the final reaction mixture, the formation of dinuclear nickel cluster 3 and disilylketenimine 4 (vide infra) as the major products was suggested, and the ratio of 3 to 4 was found to be ca. 1:1. Isolation of 3 followed by heating a C6D6 solution of 3 to 60 °C or 80 °C indicated that 4 was formed via thermolysis of 3 (vide infra).
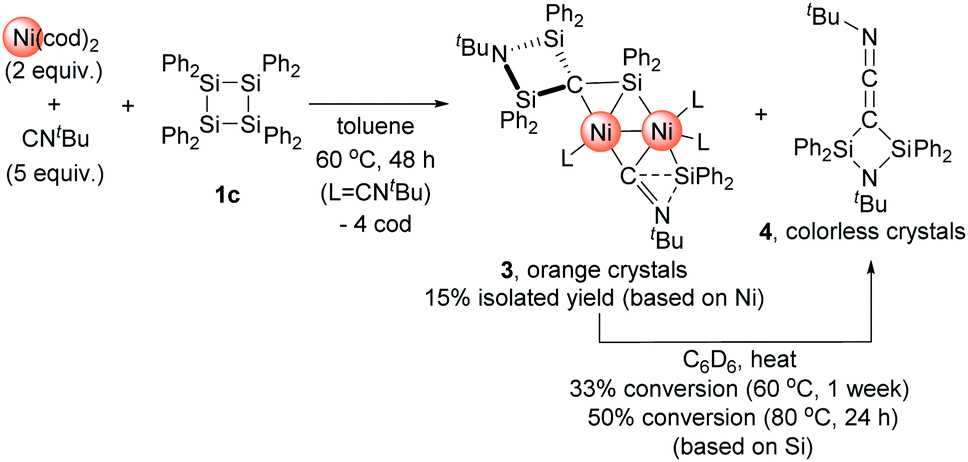 | ||
| Scheme 3 Reaction of 1c with 2 equiv. of Ni(cod)2 and 5 equiv. of CNtBu to form dinuclear Ni cluster 3 and thermolysis of 3 to afford 4. | ||
The reaction mixture obtained from the reaction of 1c with a 2:5 mixture of Ni(cod)2 and CNtBu was dissolved in pentane, and subjected to column chromatography on silica gel in order to remove the small amount of unassignable by-product, which is not very soluble in pentane. Cluster 3 was isolated in 15% yield in the form of orange crystals by cooling the obtained pentane solution to −20 °C. In contrast, no reaction took place when 1a or 1b was used instead of 1c, presumably due to the large steric hindrance of 1a and 1b around the silicon centers. The molecular structure of 3 in the solid state was determined via single-crystal X-ray diffraction analysis (Fig. 1).
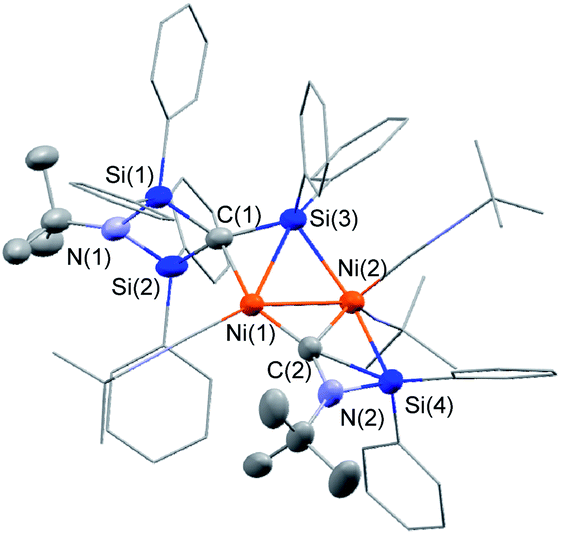 | ||
| Fig. 1 Molecular structure of 3 with thermal ellipsoids at 50% probability. All carbon and nitrogen atoms are shown in wireframe style except for the C(1), C(2), N(1) and N(2) atoms and the two tBu groups; all hydrogen atoms are omitted for clarity. | ||
The most notable structural feature of 3 is that the cleavage of the CN bond apparently occurred on the dinuclear nickel framework surrounded by four SiPh2 moieties, together with the formation of a carbide moiety supported by three silicon atoms, as well as an imido (NtBu) moiety connected to two silicon atoms. The Si–C bond distances around the carbide atom (Si(1)–C(1) = 1.817(3) Å; Si(2)–C(1) = 1.824(3) Å; and Si(3)–C(1) = 1.803(3) Å) fall within the expected range for silicon–carbon single bonds. The Si(3)–C(1) unit exhibits a bonding interaction with the Ni(1) center (Ni(1)–Si(3) = 2.3888(17) Å; Ni(1)–C(1) = 2.029(3) Å). The calculated Okuniewski parameter14 for C(1) (τ ≈ 0.63) indicates distorted tetrahedral coordination geometry around the C(1) atoms. Additionally, the C(2)–N(2) moiety, which bridges two nickel centers, exhibits some bonding interaction with the Si(4) atom (Si(4)–C(2) = 2.091(3) Å; Si(4)–N(2) = 1.743(3) Å). Thus, the C(2)–N(2) bond was significantly elongated (1.311(4) Å) compared to those of the other η1-coordinated CNtBu ligands in 3 (1.133(4)–1.144(4) Å). This bonding interaction may imply the importance of the silylene (SiPh2) moiety for the activation of the CN bond of isocyanides. The sufficiently short Ni–Ni bond distance of 2.4229(18) Å is indicative of the presence of a metal–metal bonding interaction.15
To gain more insight into the bonding interactions in 3, DFT calculations were carried out using the M06 functional. The calculated Wiberg bond index (WBI) for the Ni–Ni bonds (0.25) indicates the presence of bonding interactions. The WBI analysis revealed that no bonding interaction was present between C(1) and N(1), indicating that complete C–N bond cleavage had occurred. The WBIs of Si(3)–C(1) (0.79), Ni(1)–C(1) (0.33), and Ni(1)–Si(3) (0.26) indicate some bonding interaction between C(1) and Ni(1) as well as Si(3) and Ni(1). The WBIs of the C(2)–Si(4) (0.31) and N(2)–Si(4) (0.46) bonds indicate the presence of a bonding interaction between the C(2)–N(2) moiety and the Si(4) atom, which led to a decrease of the WBI for C(2)–N(2) (1.49).
The molecular structure of 3 in the solid state, determined by single-crystal X-ray diffraction analysis, shows three inequivalent CNtBu ligands as well as one NtBu group. In the 1H NMR spectrum of 3, signals for the tBu moieties appear at 0.39, 0.88, 1.34, and 1.59 ppm with an integral ratio of 18:9:9:9. In the 13C NMR spectrum of 3, four singlets for the methyl groups of the tBu moieties appear at 29.38, 29.66, 29.96, and 36.22 ppm, together with four peaks derived from C(CH3)3 at 52.80, 54.77, 55.69, and 60.88 ppm. In the 29Si NMR spectrum of 3, three singlets were observed at −57.02, −6.80, and −1.48 ppm. These spectral features clearly indicate that the dinuclear structure of 3 is maintained in solution. The IR spectrum showed two strong absorption bands at 2210.5 cm−1 and 2075 cm−1.
As mentioned above, the 1H NMR spectrum of the crude product obtained from the reaction of 1c with 2 equiv. of Ni(cod)2 in the presence of 5 equiv. of CNtBu showed the formation of disilylketenimine 4. We found that thermolysis of the isolated dinuclear nickel cluster 3 afforded 4. For instance, 4 was formed in 33% yield upon keeping a C6D6 solution of pure 3 at 60 °C for 1 week. When the reaction temperature was raised to 80 °C, the thermolysis of 3 was completed within 24 h to afford 4 in 50% yield. It is noteworthy that in the presence of 5 equiv. of CNtBu, the formation of 4 was suppressed during the thermolysis of 3 in C6D6 at 80 °C. This result might indicate that the dissociation of the CNtBu ligand from the nickel center might be involved in the reaction mechanism to afford 4 upon thermolysis of 3. Furthermore, compound 4 was isolated in 38% yield (based on Si) as colorless crystals via thermolysis of the crude product obtained by the reaction of 1c with 2 equiv. of Ni(cod)2 in the presence of 5 equiv. of CNtBu at 80 °C for 48 h. Thus, dinuclear nickel cluster 3 might be an intermediary species to form 4 under thermal conditions. Although the detailed reaction mechanism is not clear at present, one can imagine that Ni-bound carbide (C(1)) and Ni-bound CNtBu from one Ni site on 3 might couple to give 4. In other words, denitrogenative homocoupling of two molecules of CNtBu occurred on the Ni cluster to form disilylketenimine 4.
The molecular structure of 4 in the solid state, determined via single-crystal X-ray diffraction analysis, is shown in Fig. S9 in the ESI.† In the 1H NMR spectrum of 4, two singlets for the methyl protons of the tBu groups appear at 0.88 and 1.14 ppm with an integral ratio of 9:9, whereas signals for the phenyl moieties emerge as multiplets at 7.22–7.29 ppm and 8.12–8.15 ppm with an integral ratio of 12:8. In the 13C NMR spectrum of 4, two signals for the CCNtBu moieties were observed at 54.77 and 161.15 ppm, and two magnetically inequivalent tBu groups were observed with signals at 30.91 and 34.14 ppm for the C(CH3) units, and at 52.30 and 54.77 ppm for the C(CH3) units. In the 29Si NMR spectrum of 4, only one singlet was observed at −13.12 ppm, which suggests two magnetically equivalent silicon atoms. In the IR spectrum of 4, one strong absorption band appeared at 2024 cm−1; this value is comparable to that observed for the disilylketenimine reported by Driess et al. (vide infra).9
Metalation-induced formation of a CC bond to afford 5
Then, we turned our attention to the reaction of 1c with an increased amount of Ni species to examine the possibility of constructing nickel clusters with higher nuclearity. Namely, we treated a mixture of 5 equiv. of Ni(cod)2 and 7 equiv. of CNtBu with 1c, which furnished pentanuclear nickel cluster 5 in 22% isolated yield as dark red crystals (Scheme 4).
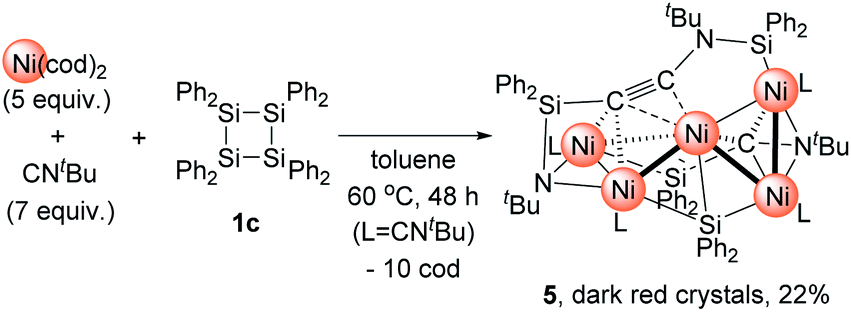 | ||
| Scheme 4 Construction of Ni5 cluster 5 and subsequent thermolysis to produce 4. | ||
The molecular structure of 5 in the solid state was determined via single-crystal X-ray diffraction analysis (Fig. 2). Cluster 5 contains five nickel atoms and the NtBu group generated via cleavage of the CN bond of CNtBu bridges Ni(1) and Ni(2) while being coordinated to the Si(1) center of the “SiPh2” moiety. Strikingly, the formation of a new CC bond occurred between C(1) and C(2). The C(1)–C(2) bond distance (1.304(5) Å) is slightly lengthened compared to those in C–C triple bonds with amino- and/or silyl-substituents.16 The Si(1)–C(1)–C(2) and C(1)–C(2)–N(2) bond angles were measured to be 146.7(2) and 152.2(2)°, respectively. In addition, the bond distance between C(2) and N(2) (1.340(4) Å) is comparable to those found in amino- and silyl substituted alkynes (1.321(3)–1.362(2) Å).16 These structural parameters indicate that the C(1)–C(2) bond can be considered to have carbon–carbon triple bond character, and it is coordinated to the Ni(1), Ni(2), and Ni(3) centers in an η2-fashion as a  unit in the molecular structure of 5. This bonding situation was also supported by theoretical calculations (vide infra). In addition, the elongated C(3)–N(3) bond (1.341(4) Å) suggests that the isocyanide consisting of the C(3)–N(3) bond engages in some bonding interaction with the three nickel atoms Ni(3), Ni(4), and Ni(5). C(3) also exhibits a bonding interaction with Si(2), which is reflected in a bond distance of 1.899(4) Å. The Ni–Ni bond distances (2.4564(8)–2.6564(6) Å) are short enough to invoke bonding interactions.15
unit in the molecular structure of 5. This bonding situation was also supported by theoretical calculations (vide infra). In addition, the elongated C(3)–N(3) bond (1.341(4) Å) suggests that the isocyanide consisting of the C(3)–N(3) bond engages in some bonding interaction with the three nickel atoms Ni(3), Ni(4), and Ni(5). C(3) also exhibits a bonding interaction with Si(2), which is reflected in a bond distance of 1.899(4) Å. The Ni–Ni bond distances (2.4564(8)–2.6564(6) Å) are short enough to invoke bonding interactions.15
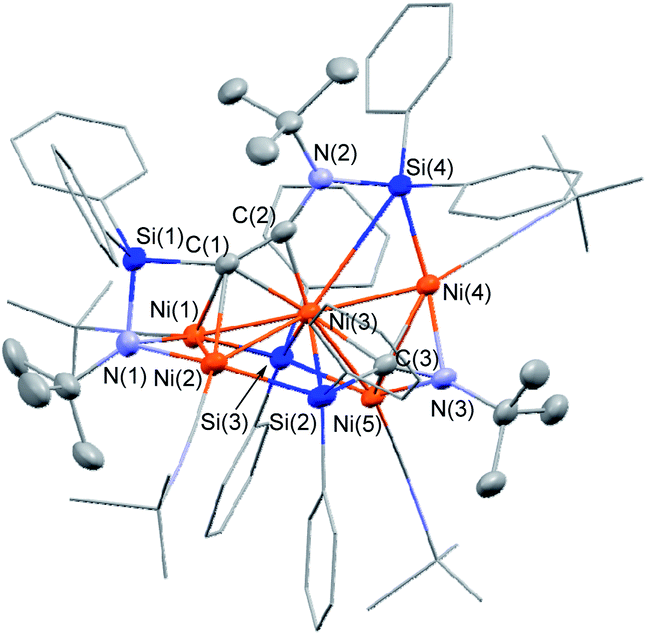 | ||
| Fig. 2 Molecular structure of 5 with thermal ellipsoids at 50% probability. All carbon and nitrogen atoms are shown in wireframe style except for the C(1), C(2), N(1), and N(2) atoms; all hydrogen atoms are omitted for clarity. | ||
To elucidate the bonding interactions in 5, DFT calculations were carried out using the PBE0, B3PW91, M06, and B3LYP functionals. The optimized structural parameters obtained using the M06 functional were in good agreement with the data obtained from the XRD analysis (for details, see the ESI†). The calculated WBIs for the Ni–Ni bonds (0.13–0.16) support the presence of bonding interactions. It should also be noted that the WBI of the C(1)–C(2) bonds was 1.73, while those of the Ni(3)–C(1) and Ni(3)–C(2) bonds were 0.34 and 0.46, respectively. In addition, the WBIs for the Ni(1)–C(1) and Ni(2)–C(1) were estimated to be 0.14 and 0.27, respectively. These parameters suggest triple-bond character for the C(1)–C(2) bond, which is coordinated to the nickel centers in a μ3-η2-coordination mode. In addition, the WBI of the C(3)–N(3) bond (1.39) indicates that the C(3)N(3) bond was effectively activated by the three surrounding nickel atoms, i.e., Ni(3), Ni(4), and Ni(5). Indeed, the WBIs between these three nickel centers and C(3) or N(3) support the presence of bonding interactions (for details, see the ESI†).
The molecular structure of 5, determined by single-crystal X-ray diffraction analysis, indicates that there are seven inequivalent tBu moieties in 5. In the 1H NMR spectrum of 5, six singlets for the methyl protons of the tBu group appeared at 0.72, 0.77, 0.81, 0.87, 1.30, and 1.78 ppm with an integral ratio of 9:9:9:18:9:9, suggesting that all tBu groups in 5 are magnetically inequivalent, even in solution. In the 13C NMR spectrum of 5, seven signals appeared for the methyl group of the tBu units at 29.49, 29.56, 29.64, 29.74, 30.08, 32.04, and 36.34 ppm. Four singlets appeared in the 29Si NMR spectrum of 5 at −59.26, −36.68, 0.43, and 137.93 ppm, which is consistent with the solid-state structure. In the IR spectrum of 5, two strong absorption bands appeared at 2071 cm−1 and 2102 cm−1, which were assigned to the CNtBu groups.
With Ni5 cluster 5 in hand, we investigated whether its thermolysis would enable the formation of disilylketenimine 4. Indeed, 4 was formed in 42% yield, as evident from the integral value of the tBu signal relative to that of an internal standard anisole, after keeping a C6D6 solution of 5 at 60 °C for 1 week. At 80 °C, the thermolysis of 5 was completed within 24 h to afford 4 in 58% yield. In both cases, the nickel species decomposed during the course of this reaction (Scheme 5). This result clearly indicates that the  alkyne moiety in 5 is converted into disilylketenimine 4 to produce the denitrogenative coupling product involving two CNtBu molecules.
alkyne moiety in 5 is converted into disilylketenimine 4 to produce the denitrogenative coupling product involving two CNtBu molecules.
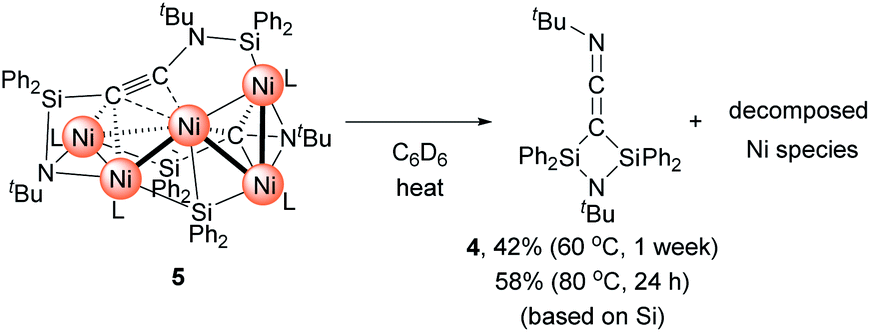 | ||
| Scheme 5 Thermolysis of Ni5 cluster 5 to produce 4. | ||
When 1c was treated with 5 equiv. of Ni(cod)2 and 7 equiv. of CNtBu, pentanuclear nickel cluster 5, which includes a 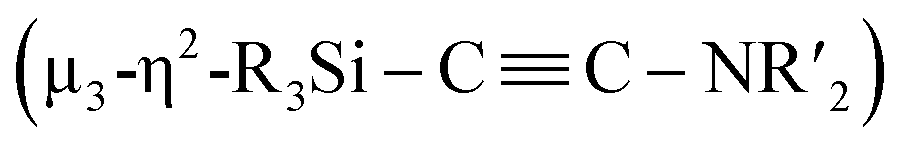 unit, was formed. The generation of a
unit, was formed. The generation of a 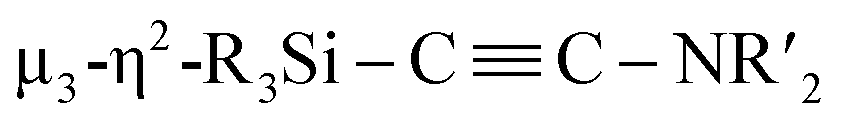 moiety could be interpreted in terms of the denitrogenative reductive coupling of two molecules of CNtBu, i.e., in terms of the coupling of one molecule of CNtBu with a carbide moiety that could be generated via bond cleavage of the CN bond in CNtBu. It should be noted here that electrophile-induced coupling between a carbyne (CR) moiety and η1-coordinated isocyanides on the molybdenum center of a mononuclear complex has already been reported.17 This transformation might be relevant to our reaction, i.e., the coupling of the Si-surrounded carbide moiety and an additional molecule of CNtBu to form the metal-coordinated alkyne moiety. However, the carbyne moiety in the aforementioned report does not directly originate from the isocyanide. These results imply that denitrogenative reductive coupling of two molecules of CNtBu via activation of the inactive CN bond effectively occurred on the nickel cluster framework supported by four silylene (SiPh2) units derived from 1c. Subsequently, the thermolysis of 5 led to the formation of disilylketenimine 4. The formation of disilylketenimine 4 might be interpreted in terms of liberation of the
moiety could be interpreted in terms of the denitrogenative reductive coupling of two molecules of CNtBu, i.e., in terms of the coupling of one molecule of CNtBu with a carbide moiety that could be generated via bond cleavage of the CN bond in CNtBu. It should be noted here that electrophile-induced coupling between a carbyne (CR) moiety and η1-coordinated isocyanides on the molybdenum center of a mononuclear complex has already been reported.17 This transformation might be relevant to our reaction, i.e., the coupling of the Si-surrounded carbide moiety and an additional molecule of CNtBu to form the metal-coordinated alkyne moiety. However, the carbyne moiety in the aforementioned report does not directly originate from the isocyanide. These results imply that denitrogenative reductive coupling of two molecules of CNtBu via activation of the inactive CN bond effectively occurred on the nickel cluster framework supported by four silylene (SiPh2) units derived from 1c. Subsequently, the thermolysis of 5 led to the formation of disilylketenimine 4. The formation of disilylketenimine 4 might be interpreted in terms of liberation of the  unit followed by tautomerization.
unit followed by tautomerization.
It should be mentioned here that Driess and co-workers reported that divalent silicon compounds, i.e., silylenes, located adjacently through a xanthene or ferrocenyl linker promote deoxygenative homocoupling involving two molecules of CO or heterocoupling involving one molecule of CO and one molecule of CNXyl (Xyl = 2,6-dimethylphenyl) to form CCO or CCNXyl units via cleavage of the CO bond.9 It is thus probable that the silylene “SiPh2” moieties generated upon the insertion of the nickel species into the Si–Si bonds in 1c might play a crucial role to activate the inactive CN bond in CNtBu. Ito et al. reported another example of the scission of the CN bond of isocyanides assisted by organosilicon moieties, i.e., a Pd-catalyzed CN bond cleavage in the reaction of aryl isocyanides with linear tetra- or hexasilanes, while alkyl isocyanides remained intact.18 These studies suggest that the reaction mechanism of our dinuclear nickel system should be different.
Conclusions
In conclusion, sequential cleavage of CN bonds and formation of CC bonds involving two molecules of CNtBu were achieved on a multinuclear nickel cluster framework surrounded by four silylene moieties, followed by the formation of a disilylketenimine via thermolysis. First, the formation of Ni2 cluster 3 was confirmed by the reaction of 1c with a 2:5 mixture of Ni(cod)2 and CNtBu in toluene at 65 °C. One outstanding structural feature of Ni2 cluster 3 is that the carbide and imido (NtBu) moieties were generated via cleavage of the CN bond in CNtBu. Then, 1c was treated with a 5:7 mixture of Ni(cod)2 and CNtBu in toluene at 65 °C, which resulted in the formation of Ni5 cluster 5, which contains a  unit. We found that thermolysis of 3 or 5 leads to the formation of disilylketenimine 4. Although the transformation from 3 to 5 by adding 3 equiv. of Ni(cod)2 and 2 equiv. of CNtBu was unsuccessful,‡ the structural features of 3 and 5 could imply the following reaction sequence of CN bond cleavage and CC bond formation. First, cleavage of the CN bond occurs on the Ni2 center to afford a silylene-supported carbide and the NtBu moiety. On the other hand, incorporation of five nickel moieties may activate the CN bond and simultaneously form the
unit. We found that thermolysis of 3 or 5 leads to the formation of disilylketenimine 4. Although the transformation from 3 to 5 by adding 3 equiv. of Ni(cod)2 and 2 equiv. of CNtBu was unsuccessful,‡ the structural features of 3 and 5 could imply the following reaction sequence of CN bond cleavage and CC bond formation. First, cleavage of the CN bond occurs on the Ni2 center to afford a silylene-supported carbide and the NtBu moiety. On the other hand, incorporation of five nickel moieties may activate the CN bond and simultaneously form the  unit. A subsequent thermolysis of the formed Ni clusters could then lead to disilylketenimine 4 as the final product. In total, denitrogenative reductive coupling of two molecules of CNtBu was realized on the nickel cluster framework surrounded by four SiPh2 moieties without the use of any strong reducing reagents such as KC8. As isocyanides are isoelectronic to CO, the results shown here could provide some insight into the transformation of CO with the aid of multiple metal atoms on active metal surfaces, such as in the Fischer–Tropsch process.
unit. A subsequent thermolysis of the formed Ni clusters could then lead to disilylketenimine 4 as the final product. In total, denitrogenative reductive coupling of two molecules of CNtBu was realized on the nickel cluster framework surrounded by four SiPh2 moieties without the use of any strong reducing reagents such as KC8. As isocyanides are isoelectronic to CO, the results shown here could provide some insight into the transformation of CO with the aid of multiple metal atoms on active metal surfaces, such as in the Fischer–Tropsch process.
Data availability
All experimental and theoretical data are provided in the ESI.†Author contributions
K. Shimamoto conducted all the experiments. All the authors analysed the data. Y. Sunada supervised this study and wrote the manuscript. All the authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST, PRESTO Grant Number JPMJPR20A9, Japan.Notes and references
- (a) Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, ed. W. B. Tolman, Wiley-VCH, 2006 Search PubMed; (b) J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. Morris Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed; (c) G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- (a) A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed; (b) E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC; (c) M. E. Dry, Catal. Today, 1990, 6, 183–206 CrossRef CAS; (d) R. A. van Santen, A. J. Markvoort, I. A. W. Filot, M. M. Ghouriab and E. J. M. Hensen, Phys. Chem. Chem. Phys., 2013, 15, 17038–17063 RSC.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC.
- (a) J. A. Buss and T. Agapie, Nature, 2016, 529, 72–75 CrossRef CAS PubMed; (b) J. A. Buss, G. A. Bailey, J. Oppenheim, D. G. VanderVelde, W. A. Goddard III and T. Agapie, J. Am. Chem. Soc., 2019, 141, 15664–15674 CrossRef CAS PubMed; (c) J. A. Buss and T. Agapie, J. Am. Chem. Soc., 2016, 138, 6466–16477 Search PubMed; (d) G. A. Bailey and T. Agapie, Organometallics, 2021, 40, 2881–2887 CrossRef CAS; (e) G. A. Bailey, J. A. Buss, P. H. Oyala and T. Agapie, J. Am. Chem. Soc., 2021, 143, 13091–13102 CrossRef CAS PubMed.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 8, 1 Search PubMed.
- D. R. Neithamer, R. E. Lapointe, R. A. Wheeler, D. S. Richeson, G. D. Van Duyne and P. T. Wolczanski, J. Am. Chem. Soc., 1989, 111, 9056–9072 CrossRef CAS.
- (a) S. Hu, T. Shima and Z. Hou, J. Am. Chem. Soc., 2020, 142, 19889–19894 CrossRef CAS PubMed; (b) T. Shima and Z. Hou, J. Am. Chem. Soc., 2006, 128, 8124–8125 CrossRef CAS PubMed; (c) T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2005, 127, 17198–17199 CrossRef CAS PubMed; (d) F. Calderazzo, U. Englert, A. Guarini, F. Marchetti, G. Pampaloni and A. Segre, Angew. Chem., Int. Ed. Engl., 1994, 33, 1188–1189 CrossRef; (e) F. Calderazzo, U. Englert, A. Guarini, F. Marchetti, G. Pampaloni, A. Segre and G. Tripepi, Chem.–Eur. J., 1996, 2, 412–419 CrossRef CAS.
- (a) W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS; (b) W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed.
- (a) Y. Wang, A. Kostenko, T. J. Hadlington, M. P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2021, 141, 626–634 CrossRef PubMed; (b) M. P. Luecke, A. Kostenko, Y. Wang, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 12940–12944 CrossRef CAS PubMed.
- H. Asakawa, K. H. Lee, Z. Lin and M. Yamashita, Nat. Commun., 2014, 5, 1 Search PubMed.
- (a) R. D. Adams, P. Mathur and B. E. Segmüller, Organometallics, 1983, 2, 1259–1261 CrossRef ; selected examples for reductive coupling of isocyanides mediated by the transition metal complexes, see: ; (b) S. L. Staun, G. T. Kent, A. Gomez-Torres, G. Wu, S. Fortier and T. W. Hayton, Organometallics, 2021, 40, 2934–2938 CrossRef CAS; (c) W. Chen, Y. Zhao, W. Xu, J. H. Su, L. Shen, L. Liu, B. Wu and X.-J. Yang, Chem. Commun., 2019, 55, 9452–9455 RSC; (d) S. Hasegawa, Y. Ishida and H. Kawaguchi, Chem. Commun., 2021, 57, 8296–8299 RSC; (e) Y. Yamamoto, H. Yamazaki and T. Sakurai, J. Am. Chem. Soc., 1982, 104, 2329–2330 CrossRef CAS; (f) J. Shen, G. P. A. Yap and K. H. Theopold, J. Am. Chem. Soc., 2014, 136, 3382–3384 CrossRef CAS PubMed.
- (a) Y. Sunada, R. Haige, K. Otsuka, S. Kyushin and H. Nagashima, Nat. Commun., 2013, 4, 3014 CrossRef PubMed; (b) K. Shimamoto and Y. Sunada, Chem.–Eur. J., 2019, 25, 3761–3765 CrossRef CAS PubMed; (c) R. Usui and Y. Sunada, Chem. Commun., 2020, 56, 8464–8467 RSC; (d) C. Yanagisawa, S. Yamazoe and Y. Sunada, ChemCatChem, 2021, 13, 1152–1156 CrossRef; (e) K. Shimamoto and Y. Sunada, Chem. Commun., 2021, 57, 7649–7652 RSC; (f) R. Usui and Y. Sunada, Inorg. Chem., 2021, 60, 15101–15106 CrossRef CAS PubMed.
- H. Ogino and H. Tobita, Adv. Organomet. Chem., 1998, 42, 223–290 CrossRef CAS.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- (a) R. Beck and S. A. Johnson, Organometallics, 2012, 31, 3599–3609 CrossRef CAS; (b) I. Bach, R. Goddard, C. Kopiske, K. Seevogel and K. R. Pörschke, Organometallics, 1999, 18, 10–20 CrossRef CAS.
- (a) B. Waldecker, F. Kraft, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2018, 57, 12538–12542 CrossRef CAS PubMed; (b) C. Wang, W. Mao, L. Xiang, Y. Yang, J. Fang, L. Maron, X. Leng and Y. Chen, Chem.–Eur. J., 2018, 24, 13903–13917 CrossRef CAS PubMed.
- (a) C. M. Bastos, N. Daubenspeck and A. Mayr, Angew. Chem., Int. Ed., 1993, 32, 743–745 CrossRef; (b) A. C. Filippou, W. Grünleitner, C. Völkl and P. Kiprof, Angew. Chem., Int. Ed., 1991, 30, 1167–1169 CrossRef; (c) A. C. Filippou, B. Lungwitz and G. Kociok-Köhn, Eur. J. Inorg. Chem., 1999, 1905–1910 CrossRef CAS; (d) A. C. Filippou, C. Völkl, W. Grünleitner and P. Kiprof, J. Organomet. Chem., 1992, 434, 201–223 CrossRef CAS.
- (a) M. Suginome and Y. Ito, Chem. Rev., 2000, 100, 3221–3256 CrossRef CAS PubMed; (b) Y. Ito, M. Suginome, T. Matsuura and M. Murakami, J. Am. Chem. Soc., 1991, 113, 8899–8908 CrossRef CAS; (c) Y. Ito, M. Suginome, M. Murakami and M. Shiro, J. Chem. Soc., Chem. Commun., 1989, 1494–1495 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2120736–2120739. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06935g |
| ‡ Reaction of Ni2 cluster 3 with 3 equiv. of Ni(cod)2 and 2 equiv. of CNtBu was performed at 60 °C in C6D6 and monitored via1H NMR spectroscopy. However, only a complex mixture was formed in this reaction, and the formation of Ni5 cluster 5 could not be confirmed. In this reaction, the thermolysis of 3 to produce disilylketenimine 4 proceeded, which may prevent the transformation of 3 to 5 in the presence of additional Ni(cod)2 and CNtBu. |
| This journal is © The Royal Society of Chemistry 2022 |