
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton interactions in helical crystals of a hydrogen-bonded eumelanin monomer†
Devika
Sasikumar
 ,
Kavya
Vinod
,
Jeswin
Sunny
and
Mahesh
Hariharan
*
,
Kavya
Vinod
,
Jeswin
Sunny
and
Mahesh
Hariharan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala P.O., Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: mahesh@iisertvm.ac.in
First published on 27th January 2022
Abstract
Eumelanin, a naturally occurring group of heterogeneous polymers/aggregates providing photoprotection to living organisms, consist of 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole-2-carboxylic acid (DHICA) building blocks. Despite their prevalence in the animal world, the structure and therefore the mechanism behind the photoprotective broadband absorption and non-radiative decay of eumelanin remain largely unknown. As a small step towards solving the incessant mystery, DHI is crystallized in a non-protic solvent environment to obtain DHI crystals having a helical packing motif. The present approach reflects the solitary directional effect of hydrogen bonds between the DHI chromophores for generating the crystalline assembly and filters out any involvement of the surrounding solvent environment. The DHI single crystals having an atypical chiral packing motif (P212121 Sohncke space group) incorporate enantiomeric zig-zag helical stacks arranged in a herringbone fashion with respect to each other. Each of the zig-zag helical stacks originates from a bifurcated hydrogen bonding interaction between the hydroxyl substituents in adjacent DHI chromophores which act as the backbone structure for the helical assembly. Fragment-based excited state analysis performed on the DHI crystalline assembly demonstrates exciton delocalization along the DHI units that connect each enantiomeric helical stack while, within each stack, the excitons remain localized. Fascinatingly, over the time evolution for generation of single-crystals of the DHI-monomer, mesoscopic double-helical crystals are formed, possibly attributed to the presence of covalently connected DHI trimers in chloroform solution. The oligomeric DHI (in line with the chemical disorder model) along with the characteristic crystalline packing observed for DHI provides insights into the broadband absorption feature exhibited by the chromophore.
Eumelanin, which represents a broad class of natural pigments found in the animal kingdom, acts as a biological shield for protecting the skin cells against harsh UV radiation.1 Eumelanin, a black coloured pigment, obtained from the oxidative polymerization of 5,6-dihydroxyindoles (DHIs) and 5,6-dihydroxyindole carboxylic acid (DHICA) is one of the extensively explored archetypes of the melanin family (Fig. 1a).2 Eumelanin has synergistic merits of possessing broadband UV absorption and proficient dissipation of the excessive electronic energy via non-radiative deactivation of the excited states, thereby resulting in the photoprotective nature of the pigment.3 Apart from the photoprotective behaviour, melanin possesses exceptional antioxidant activity via its free radical scavenging traits.4–7 However, much less has been understood about the fundamental photophysics and structural features of eumelanin due to the enormous heterogeneity in the molecular framework8,9 coupled with poor solubility in common solvents.10 Recent years have witnessed a growing interest towards unravelling the excited state processes occurring in the eumelanin pigment upon interaction with light.11,12 A better correlation between the structure–property relationship and photoexcited state processes in eumelanin can guide the development of inspired functional materials for potential application in biomedical and dermo-cosmetic fields.1,13–15
 | ||
| Fig. 1 (a) Chemical diagrams of 5,6-dihydroxyindole (DHI) and 5,6-dihydroxyindole carboxylic acid (DHICA). (b) Various unconventional hydrogen bonding interactions identified in DHI crystals. | ||
In the natural world, the chromophoric architecture of eumelanin can symbolize an organized and efficient organic system for photoprotection that nature developed through evolution. The outcome of the research done so far indicates the presence of continuous π-stacks of oligomers in eumelanin which induce different levels of aggregation to construct the eumelanin framework.16 Furthermore, eumelanin has been reported to exhibit weak fluorescence which indicates the presence of competing non-radiative channels that provide efficient de-excitation pathways for repopulating the ground states.17–20 The broadband absorption of eumelanin has theoretically and experimentally been evaluated, in part, to be a result of π-stacking interactions between the oligomers of DHI/DHICA in multiple oxidation states.21–24 While dealing with biomacromolecules, non-covalent interactions such as hydrogen bonding and π–π stacking take the centre stage in controlling the supramolecular architecture, especially in the 3-dimensional structures of proteins and DNA. Modulating the balance between each of these noncovalent interactions over another will produce significant changes not only in the structure but also in the functional properties.
Hydrogen bonding is simultaneously both ubiquitous and diverse and therefore its significance in biochemical systems comes as no surprise particularly due to the surrounding aqueous environment. Apart from the classical hydrogen bonding interactions, an array of hydrogen bond-like weak interactions which include a delocalized π-system acting as the acceptor group to the X–H hydrogen donor (X = O, N, C) is identified to provide additional contributions for stabilizing the biomolecular structure and controlling intrinsic functions (Fig. 1b).25,26 Investigations aimed at identification (using X-ray crystallography) followed by energetic quantification of the major stabilizing interactions such as those with the aromatic π-rings in biological complexes are of paramount importance for developments in diverse areas including drug design. There has been extensive research conducted on eumelanin building blocks showcasing their ability to form hydrogen bonds through the –OH and –NH functional groups.27,28 Most reports almost exclusively focus on the hydrogen bonding with the solvent environment surrounding the eumelanin monomer units.29 Findings from these theoretical studies have demonstrated the role of several deactivation pathways in the presence of a protic solvent, namely –OH and –NH bond elongation and 5-/6-membered ring puckerings.18
Chemical and spectral evidence from the eumelanin polymeric structures identified so far points to five main levels of chemical disorder leading to the supramolecular structure, which includes (i) disorder from the simultaneous presence of different building blocks; (ii) molecular size disorder; (iii) disorder from the position of coupling; (iv) electronic/redox disorder of the constituent units and (v) supramolecular disorder.30 Given the complex structure of melanin, a bottom-up approach using the building blocks or basic constituent molecules of eumelanin is a pertinent strategy for the mechanistic study of the photoproperties of eumelanin. This can be followed by understanding the more intricate structures of melanin formed from the constituents with less complex approaches. Due to the abundant presence of water in the natural media, the corresponding solute–solvent interactions can have profound significance in driving the fast polymerization and the consequent heterogeneity of natural melanin. The tedious task of extracting melanin from natural sources and the lack of solubility of the polymeric melanin material in organic/aqueous solvents have called for basic model systems to understand the complex eumelanin architecture. In this regard, we have adopted a facile approach to decode the perpetual puzzle by single crystal X-ray crystallographic and spectroscopic analyses of DHI crystalline aggregates derived from a non-aqueous environment (chloroform). Due to the highly autooxidative31 nature of eumelanin precursors even in the slight presence of protic solvents, the simple model implemented here precludes the contribution of solute–solvent hydrogen bonding interaction towards the formation and resultant structure of DHI crystalline aggregates. In chloroform, each DHI molecule experiences weak interactions solely from the neighbouring DHI chromophores thereby leading to helical aggregation.
Our efforts towards recognizing and monitoring the photogenerated excitons and charge-transfer dynamics in crystalline and contorted polyaromatic assemblies32–36 prompted us to explore the structure–optical property relationship in the eumelanin precursor molecule DHI. Unlike the commonly understood π-stacking in eumelanin derivatives, the single crystals of DHI arrange in a helical zig-zag fashion with a completely edge-to-face aggregate structure driven by both conventional and unconventional hydrogen bonds (Fig. 1b). The structural heterogeneity imposed by the different hydrogen bonds has led to varied levels of exciton delocalization between the neighbouring chromophores in the crystalline DHI aggregates. Along with the diffracting single crystals of monomeric DHI, covalently connected trimeric units of DHI are also identified in chloroform solution, which form double-helical crystals in the mesoscopic scale. Such double-helical architectures are omnipresent in nature as exemplified by the DNA structure.
DHI was synthesized by following a previously reported procedure having L-dopa as the starting material (Scheme S1, Appendix C1–C4 and C7, ESI†).37 Slow evaporation from dry chloroform solution of DHI produced colourless diamond shaped single crystals of DHI (Fig. S1a†). Interestingly, the DHI molecule with no chiral centre atypically crystallized in the Sohncke space group, P212121 (Table S1†). Single-crystal X-ray structure analysis revealed the presence of conventional and unconventional hydrogen bonds directing the crystalline self-assembly of DHI chromophores about a zig-zag helical backbone. Fig. 2 presents the four different types of non-covalent dimers (D1–4) distinguished within the DHI crystal. The zig-zag helical stacks proceed along the crystallographic a-axis (Fig. S2†) and are fabricated by bifurcated O–H⋯O hydrogen bonds (dO1⋯H = 2.25 Å, dO2⋯H = 2.34 Å, <O1–H–O2 = 67.89°) between the two hydroxyl substituents in the DHI chromophore (D3 in Fig. 2). Such bifurcated hydrogen bonded assemblies are prevalent in the secondary and tertiary structures of proteins.38 Interestingly, in the DHI crystal, enantiomeric helical stacks (Fig. S2†) that are arranged with respect to different screw axes are observed, wherein each stack aligns in a herringbone fashion to the other zig-zag helix (as represented by the dimers D1, D2 and D4). The stacks are interconnected majorly through the unconventional hydrogen bonds such as C–H⋯π (dC–H⋯π = 2.66–2.99 Å), O–H⋯π (dO–H⋯π = 2.59 Å), C–H⋯N (dC–H⋯N = 2.72 Å), C–H⋯O (dC–H⋯O = 2.66 Å) and the classical N–H⋯O (dN–H⋯O = 2.72 Å) hydrogen bonds. The absence of π–π stacking interaction is validated by the Hirshfeld surface analysis wherein the formation of the DHI crystalline assembly is majorly stabilized by the C⋯H (40.5%), H⋯H (29.7%), N⋯H (4.3%) and O⋯H (25.4%) noncovalent interactions (Fig. S3 and Table S2†).
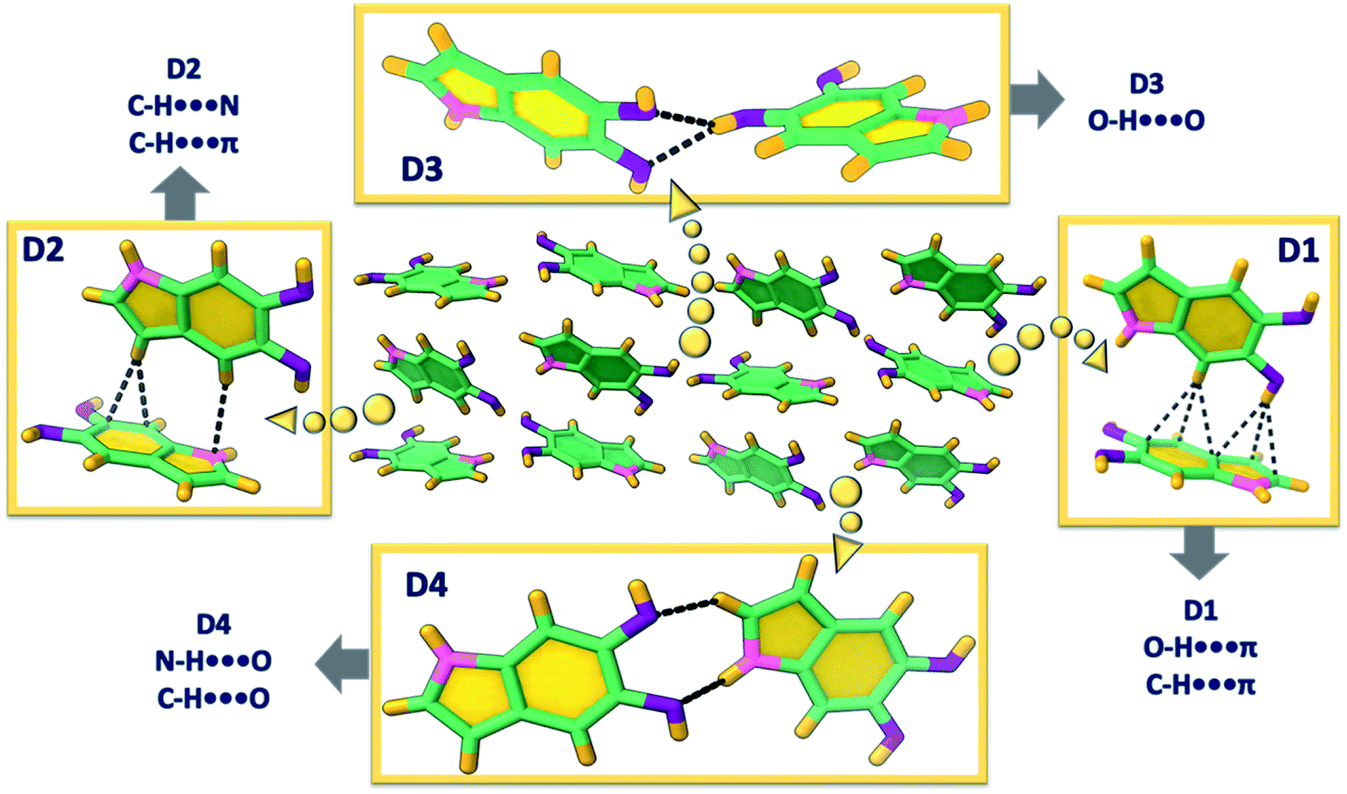 | ||
| Fig. 2 Different orientations of DHI (D1–4) and the directing hydrogen bonds observed in the single crystal. | ||
Detailed examination of the interchromophoric interactions supporting the zig-zag helical stacks in DHI crystals using Bader's quantum theory of atoms in molecules (QTAIM) analysis revealed the presence of supramolecular synthons in the crystal system (Fig. S4†). This is evidenced by the (3,+1) ring critical points in each of the representative dimers. For a molecular self-assembly to occur efficiently, recognition between the intermolecular functionalities is important, which often culminates in the formation of smaller repetitive units or supramolecular synthons.39 The recognition information which is then carried by these units forms the kernel of self-aggregation or crystallization processes. In the case of DHI crystals, all the dimer assemblies D1–D4 display synthon formation with the D1 and D3 synthons showing greater energetic stabilities. The dimer unit representing the helical backbone, D3, forms two supramolecular synthons orchestrated by the bifurcated O–H⋯O hydrogen bonds and a weak C–H⋯O interaction (Fig. S4†). The five- and six-membered rings so formed fabricate the helical zig-zag backbone of the DHI crystal. Similarly, the dimer D1 also forms two synthons through a classical N–H⋯O interaction along with the weak C–H⋯π and O–H⋯π interactions. D2 and D4 dimers are stabilized by one synthon each, materialized by C–H⋯π and C–H⋯N interactions in D2 and N–H⋯O and C–H⋯O interactions in D4.
The synthon formation and the concomitant electron delocalization involving the π-rings in dimers D1 and D2 have resulted in the aromatic stabilization of the π-rings upon comparison with the monomer DHI. The nucleus independent chemical shift (NICS(1)) values evaluated for D1–4 and monomer DHI in the ground state indicate the aromatic stabilization of the π-surface leading to the favourable alternate stacking of the enantiomeric zig-zag helices facilitated by the unconventional hydrogen bonds. The negative NICS(1) values for the six- and five-membered (6C, 5C) rings of molecule A (Fig. S5 and Table S3†) increased to −27.22 ppm (6C) and −29.13 ppm (5C) when compared to the monomer DHI (6C: −26.14 ppm, 5C: −28.03 ppm). Similarly, in D2, the π-surface of molecule B that is involved in the weak interaction undergoes aromatic stabilization (6C: −29.43 ppm, 5C: −31.48 ppm). Truncated symmetry adapted perturbation theory (SAPT(0)) analysis40 performed on the DHI dimers shows higher stabilization for D1 (ESAPTint = −9.70 kcal mol−1) and D3 (ESAPTint = −6.57 kcal mol−1) dimers, which could be attributed to the two supporting supramolecular synthons in both the dimers (Table S4†). The total stabilization of D1 and D3 orientations is facilitated by the higher contributions of electrostatic energy (E(1)elc = −6.17 to −6.14 kcal mol−1) and induction energy (E(1)elc = −1.60 to −1.38 kcal mol−1) towards the total SAPT energy. The dominant role of the stronger classical hydrogen bonds in the fabrication of D1 and D3 synthons when compared to the other DHI orientations (having equal contribution from the weak unconventional interactions) explains the observed energy distribution in the SAPT(0) analysis.
The crystalline architecture of the DHI precursor molecule identified herein could provide a sound model for understanding the inherent nature of the excited energy states leading to the characteristic photo-function of the eumelanin pigment. Several experimental and theoretical investigations on eumelanin aggregates revealed the occurrence of excitation energy transfer within the aggregates.41,42 The extent of energy delocalization within the four dimer orientations in the DHI crystal structure is determined using the fragment-based excited state analysis developed by Plasser, executed in the TheoDORE package.43–45 The expanse of excitation delocalization amongst the fragment units is described by the value of participation ratio (PR). The contribution towards the exciton delocalization from the fragments is defined by the mean position or the POS value, wherein the indicated number shows the involvement of one or more units. The charge-transfer or Frenkel character of the excited states is defined by the CT number which assumes values closer to one for pure CT states and closer to zero for pure Frenkel states. In dimers D1 and D2, the first singlet excited state of highest oscillator strength (S2) shows Frenkel exciton character with effective delocalization of the excitons between the individual constituting units during the excitation process (Tables S5 and S6†). The low CT values of the S2 states in D1 (PR = 1.79, POS = 1.33, CT = 0.08) and D2 (PR = 1.98, POS = 1.45, CT = 0.02) along with a PR close to a value of two indicated the delocalization of the Frenkel excitons on the two monomers. Hence, there exists a possible excitation energy delocalization along the adjacent enantiomeric stacks in the DHI crystal. However, for D3 with the bifurcated hydrogen bonding interaction between the monomeric units, both S1 and S2 states have significant oscillator strength. The Frenkel excitons in S1 and S2 states remain localized on only one fragment of D3 (Table S7†), while in D4, there exists a partial delocalization of the Frenkel excitons in the S1 state (Table S8†). Thus, it is understood that within each enantiomeric stack the initial Frenkel excitons remain localized on one fragment. The natural transition orbitals (NTOs) of dimers D1–4 give an idea about the nature of the dominant orbital transitions for the allowed electronic excitations. The allowed electronic excitation in all the dimers could only be well represented by taking two distinct orbital transitions with significant coefficients into consideration. The absence of a single dominant orbital transition in the dimers proposed the need for fragment-based hole–electron analysis for the better understanding of the electronic energy delocalization. The hole–electron isosurface analysis provides a pictorial representation of the corresponding delocalized and localized nature of the initial Frenkel excitons in the four DHI orientations (Fig. 3). The delocalization of the hole and electron density on both the fragments puts forward the possibility of effective energy transfer in dimers D1, D2 and D4.46 In dimer D3, a very weak delocalization of the electron–hole densities was observed suggesting a localized exciton formation.
 | ||
| Fig. 3 Hole–electron isosurface plots of the DHI orientations in the crystal. (a) D1, (b) D2, (c) D3 and (d) D4. | ||
Several factors including slight exposure to air,47 humidity and/or light have been observed to cause the autooxidative polymerization of DHI, wherein lowered temperatures decrease the kinetics of this solid-state polymerization. Although, a major fraction of the solid oligomer mixture remains as the DHI monomer (≥80%), oligomeric units up to DHI-hexamers have been identified with varying solubilities in different solvents. MALDI-MS spectra (Appendix C5, ESI†) sequentially collected for the oligomer mixture in CHCl3 and DMSO indicated the major presence of DHI trimers (in CHCl3; m/z = 442.138) and DHI-hexamers (in DMSO; m/z = 882.382) along with the smaller counterparts. Interestingly, over the time course for nucleation and the subsequent growth mechanism of the single crystals of monomeric DHI in chloroform, oligomerization of DHI is found to occur concurrently to form the covalently connected DHI trimer (DHI-T). Along with the diffracting single crystals of DHI, small right-handed double-helical crystals (non-diffracting, Fig. S1b–f and S6†) are observed for the first time, which could be attributed to the self-assembled morphology of DHI-T (Fig. 4a). Observed only in chloroform, we speculate that the prolonged exposure of the chlorinated solvent plays a significant role in the chemical transformation of the DHI monomer to the covalent trimer, possibly through a radical initiated reaction.48 The 1H-NMR spectrum obtained for the bulk crystalline sample dissolved in CDCl3 along with the observed MALDI-MS data of the DHI trimer evidenced the formation of DHI-T, although in very low yields when compared to the monomer (Appendix C5 and C6†).
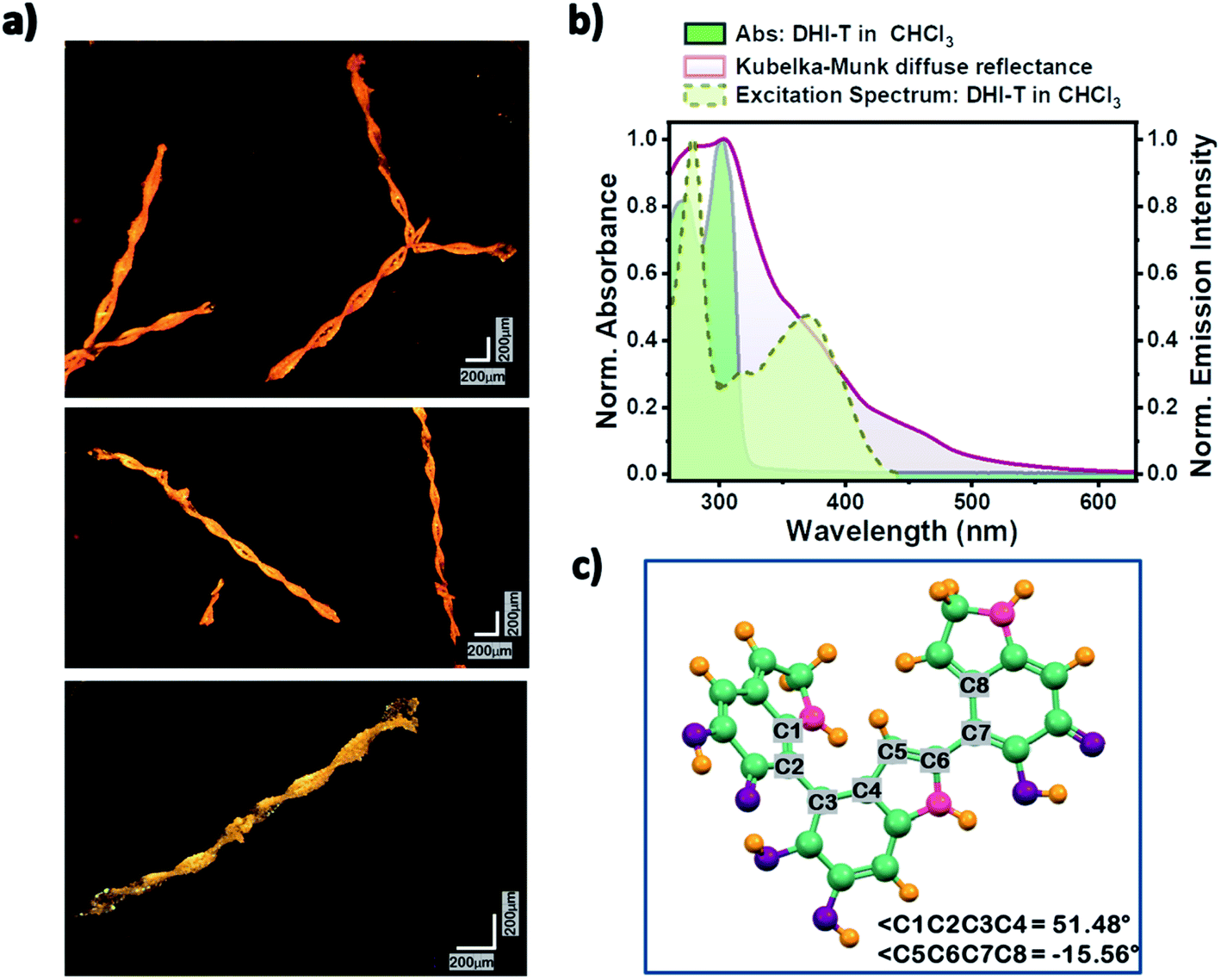 | ||
| Fig. 4 (a) Optical microscopy images of the right-handed double-helical crystals of the DHI trimer. (b) Normalized absorbance and excitation spectra showing DHI-T formation in chloroform solution and the Kubelka–Munk transformed diffuse reflectance spectrum of the DHI bulk crystal. (c) Optimized structure of DHI-T at the CAM-B3LYP/6-311g+(d,p) level of theory. | ||
With the understanding that DHI readily aggregates/crystallizes in chloroform, the directionality of the hydrogen bonding interactions (to the π-ring) within the definite spatial arrangements of D1 and D2 orientations hints towards the mode/position of coupling for the associated generation of DHI-T (Appendix C9†). The molecular structure of the DHI trimer that best fits the observed characterization data (Fig. S7a and Appendix C8, ESI†) is in line with the chemical disorder model having semiquinone and catechol units connected covalently as an extension of the D1 and D2 noncovalent interactions. Geometry optimization of the predicted structure of the DHI-T performed using the CAM-B3LYP/6-311g+(d,p) level of theory in Gaussian 16 suite shows a twisted conformation having the possibility of forming intramolecular hydrogen bonds from the –OH and –NH functionalities (Fig. 4 and S7b†). Separation of DHI-T from DHI is a real challenge since exposure of DHI to the adsorbent in column chromatography accelerates the oxidative polymerization of DHI, resulting in a black, insoluble material difficult to characterize. Also, the presence of higher oligomer units of DHI (hexamers etc.) was not identified in the multiple data sets collected for the DHI sample dissolved in CHCl3.
Solvent-dependent steady-state UV-vis absorption and fluorescence emission measurements of DHI were performed and the line shapes of the absorption spectra of monomeric DHI in different solvents match consistently (Fig. S8†). Two major absorption bands at λa1 ∼ 270 nm and λa2 ∼ 300 nm form the characteristic absorption spectrum of the DHI monomer. The fluorescence emission of monomeric DHI exhibits a single broad spectral feature peaking at λem ∼ 330 nm in a majority of the solvents. The relative fluorescence quantum yields are found to be exceptionally low in chloroform, dichloromethane, THF and water (Table S9†) indicating the presence of non-radiative decay channels for dissipating the excitation energy. In chloroform, the emergence of a red-shifted tail in the absorption spectrum of the DHI-monomer is observed over time possibly signifying the onset of DHI-T formation. The fluorescence emission in CHCl3 also shows a new band peaking at λem ∼ 460 nm along with the emission band at λem ∼ 335 nm (Fig. S9†). A broad red-shifted band arising at 370 nm in the excitation spectrum of DHI solution collected in chloroform at λem ∼ 460 nm (Fig. 4b) evidenced the presence of the DHI trimer. A similar decrease in fluorescence quantum yield in chloroform and the concomitant emergence of new bands in the fluorescence emission profiles have been noted previously in tryptophan and other indole species synthesized for eumelanin investigation.49,50 In such cases, the photoionization of excited indole leads to the ejection of a solvated electron which attacks the chloroform molecule, releasing a chloride ion, and further undergoes reactions to yield photoproducts.
Spectroscopic investigation of the crystalline DHI (containing both monomer single crystals and covalent trimer crystals) sample showed broad, red-shifted absorption bands spanning from 210 to 560 nm (Fig. 4b and S10†). The solid-state absorption spectrum shows two prominent bands at λa1 ∼ 280 nm and λa2 ∼ 305 nm which could be attributed to the red-shifted absorption bands of the crystalline DHI monomer (compared to the monomer absorption bands in the solution state). The observed red-shift in the absorption band of the DHI crystal arises from the nonplanar packing motif and the ensuing intermolecular interactions in the solid state. The presence of a broad shoulder band centred at λa3 ∼ 375 nm in the absorption spectrum could be assigned to the double-helical crystal of the covalent DHI trimer. The crystalline state fluorescence emission spectrum of DHI spans from 390 to 490 nm (Fig. S10†). The excitation energies and the allowed vertical transitions of the monomer and DHI-T have been computed at the CAM-B3LYP/6-311g+(d,p) level of theory. Unlike the precursor DHI monomer which undergoes higher energy electronic transitions (at 270 nm and 300 nm), the favorable transition in the covalent trimer is red-shifted with the S0 → S1 electronic excitation occurring at ∼434 nm. Hence, the spectroscopic and theoretical investigation of bulk crystalline DHI indicates that the broad absorption profile of the eumelanin precursor could be ascribed to the combined effects of the non-planar chromophore stacking and the presence of covalent DHI trimers that exist as double-helical aggregates. The photoprotective nature of eumelanin arises from the signature broadband absorption of eumelanin which spans throughout the UV and visible region tailing around 800 nm as explained by the chemical disorder model. In line with this understanding, the spectroscopic data of the DHI crystal also exhibit broadband absorption which expands up to 600 nm unlike the DHI monomer. Moreover, the low relative fluorescence quantum yields of DHI suggest the presence of non-radiative decay pathways within the DHI units. The oligomeric trimer which in itself shows structural heterogeneity aggregates as double helical structures and shows a red-shifted absorption band which is comparable to the computed TDDFT vertical excitation energies. Thus, our report on the characterization of DHI and the oligomeric trimer could possibly be beneficial in advancing melanin structure characterization and elucidating the photoprotective function of eumelanin.
The solid-state CD spectrum of crystalline DHI (in KBr, Fig. S11a†) showed the signatures for the presence of helical packing51–55 (possibly from the zig-zag helical motif along with the double helical arrangement). However, the basis of the CD couplet of significant intensity spanning from ∼250 to 600 nm (including ranges outside of the absorption maxima) could not be exclusively assigned to chiral absorption from the chromophoric packing.56 In the case of macromolecular systems having long-range organization, differential scattering of incident left and right circularly polarized light can provide significant contributions to the observed circular dichroism.57–59 The occurrence of broad CD bands outside the absorption bands of the macromolecule can signify the possible role of differential scattering in the circular dichroism.60 Although, for the DHI sample, the characteristic CD spectrum has been found to be concurrent for the different data sets collected using freshly prepared crystalline samples on different days (Fig. S11a†), the ratios of the intensities of the positive and negative bands have been observed to vary. Such a heterogeneity in the ellipticity values of the positive and negative bands could be attributed to the possible presence of different enantiomeric assemblies that exhibit varying abilities to undergo chiral absorption and differential scattering.57 The possibility of having linear dichroism (LD) artifacts in the CD data was evaluated for the DHI sample (Fig. S11b†).61 The LD artifact fell within the error bar of the order of 10−3–10−4 mdeg and hence, the contribution of LD to the strong CD signal of DHI could be ignored.62,63 Also, the idea of having a chiral nucleation centre, probably from any conformationally chiral DHI oligomer units, leading to the double-helical aggregation and the consequent mesoscopic chirality could not be ignored while assessing the origin of the observed CD spectrum. The existing uncertainties in solving the source of the double-helical aggregation of the DHI chromophores and identifying the intermolecular forces acting behind the same remain a challenge that requires detailed examination in future studies.
Conclusions
In conclusion, we introduce the helical crystalline packing arrangement of an achiral eumelanin precursor, 5,6-dihydroxyindole (DHI), derived from a non-protic environment, chloroform. The present account well describes the solitary effect of interchromophoric interactions on the crystalline self-aggregation of DHI molecules by filtering out the external hydrogen bonding association with the solvent environment. This approach resulted in a helical assembly of achiral DHI chromophores, unlike any of the previously understood structural motifs of eumelanin precursors. The diffraction quality single crystals of DHI obtained from chloroform solution encompass enantiomeric zig-zag helical stacks aligned in a herringbone fashion with respect to each stack. The origin of each of the zig-zag stacks occurs from a backbone structure constructed by bifurcated hydrogen bonding interactions between the hydroxyl substituents in adjacent DHI chromophores. The excitation energy delocalization pathways evaluated for the crystalline arrangement of DHI show effective exciton delocalization along the chromophores connecting each of the enantiomeric stacks. However, minimal exciton delocalization was observed within each enantiomeric stack.Along with the single crystals of the DHI monomer, formation of covalently connected DHI trimer aggregates (DHI-T) is also observed in the chloroform solution. DHI-T forms small double-helical crystals on the mesoscopic scale. The best-fit molecular structure of the DHI covalent trimer as evident from the spectroscopic characterization points to the presence of catechol and semiquinone moieties, in line with the renowned chemical disorder model proposed for eumelanin. The covalent DHI trimer shows a red-shifted absorption band (∼370 nm) when compared to the DHI monomer (∼270–300 nm) as demonstrated by the spectroscopic and theoretical investigations. Thus, with the unceasing ambiguity still existing for the eumelanin molecular framework and the consequent photophysics, the crystalline evidence presented here of a eumelanin precursor molecule could serve as a potential candidate for decoding the melanin mystery.
Data availability
All experimental/computational data and procedures are available in the ESI† under the sections A and B.Author contributions
D. S. and M. H. conceived the project; D. S., K. V., J. S. and M. H. analysed the results and wrote the paper; M. H. supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. H. acknowledges the Nanomission project (DST/NM/TUE/EE01/2019) of the Department of Science and Technology (DST), Government of India, for financial support. We thank Mr Alex P. Andrews for the X-ray diffraction analysis. We greatly acknowledge the support for high-performance computing time at the Padmanabha cluster at IISER TVM, India. We thank Dr Reji Varghese, Dr Babu Varghese, Dr Vignesh A. and Ms Prajakta Bodkhe for their constructive suggestions. D. S. and K. V. acknowledge IISER-TVM and CSIR for financial assistance.References
- M. d'Ischia, A. Napolitano, V. Ball, C. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- W. Cao, X. Zhou, N. C. McCallum, Z. Hu, Q. Z. Ni, U. Kapoor, C. M. Heil, K. S. Cay, T. Zand, A. J. Mantanona, A. Jayaraman, A. Dhinojwala, D. D. Deheyn, M. D. Shawkey, M. D. Burkart, J. D. Rinehart and N. C. Gianneschi, J. Am. Chem. Soc., 2021, 143, 2622–2637 CrossRef CAS PubMed.
- A. Pezzella, A. Iadonisi, S. Valerio, L. Panzella, A. Napolitano, M. Adinolfi and M. D'Ischia, J. Am. Chem. Soc., 2009, 131, 15270–15275 CrossRef CAS PubMed.
- B. F. E. Curchod and T. J. Martínez, Chem. Rev., 2018, 118, 3305–3336 CrossRef CAS PubMed.
- L. Panzella, G. Gentile, G. D'Errico, N. F. Della Vecchia, M. E. Errico, A. Napolitano, C. Carfagna and M. D'Ischia, Angew. Chem., Int. Ed., 2013, 52, 12684–12687 CrossRef CAS PubMed.
- Q. Z. Ni, B. N. Sierra, J. J. La Clair and M. D. Burkart, Chem. Sci., 2020, 11, 7836–7841 RSC.
- C.-T. Chen, F. J. Martin-Martinez, G. S. Jung and M. J. Buehler, Chem. Sci., 2017, 8, 1631–1641 RSC.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- N. F. Della Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. D'Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS.
- P. Meredith and T. Sarna, Pigm. Cell Res., 2006, 19, 572–594 CrossRef CAS PubMed.
- F. R. Kohl, C. Grieco and B. Kohler, Chem. Sci., 2020, 11, 1248–1259 RSC.
- C. Grieco, F. R. Kohl, A. T. Hanes and B. Kohler, Nat. Commun., 2020, 11, 4569 CrossRef CAS PubMed.
- L. A. Baker, B. Marchetti, T. N. V. Karsili, V. G. Stavros and M. N. R. Ashfold, Chem. Soc. Rev., 2017, 46, 3770–3791 RSC.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Chem. Sci., 2013, 4, 3796 RSC.
- K.-Y. Ju, M. C. Fischer and W. S. Warren, ACS Nano, 2018, 12, 12050–12061 CrossRef CAS PubMed.
- A. Corani, A. Huijser, T. Gustavsson, D. Markovitsi, P.-Å. Malmqvist, A. Pezzella, M. D'Ischia and V. Sundström, J. Am. Chem. Soc., 2014, 136, 11626–11635 CrossRef CAS PubMed.
- P. Ghosh and D. Ghosh, Phys. Chem. Chem. Phys., 2019, 21, 26123–26132 RSC.
- A. Datar and A. Hazra, J. Phys. Chem. A, 2017, 121, 2790–2797 CrossRef CAS PubMed.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698 RSC.
- R. Micillo, L. Panzella, M. Iacomino, G. Prampolini, I. Cacelli, A. Ferretti, O. Crescenzi, K. Koike, A. Napolitano and M. Ischia, Sci. Rep., 2017, 7, 41532 CrossRef CAS PubMed.
- C.-T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 3859 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- A. A. R. Watt, J. P. Bothma and P. Meredith, Soft Matter, 2009, 5, 3754 RSC.
- M. J. Plevin, D. L. Bryce and J. Boisbouvier, Nat. Chem., 2010, 2, 466–471 CrossRef CAS PubMed.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2000, 122, 11450–11458 CrossRef CAS.
- A. A. Voityuk, Photochem. Photobiol. Sci., 2013, 12, 1303–1309 CrossRef CAS PubMed.
- A. Choudhury and D. Ghosh, Chem. Commun., 2020, 56, 10481–10484 RSC.
- M. Gauden, A. Pezzella, L. Panzella, M. T. Neves-Petersen, E. Skovsen, S. B. Petersen, K. M. Mullen, A. Napolitano, M. D’Ischia and V. Sundström, J. Am. Chem. Soc., 2008, 130, 17038–17043 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and M. Buehler, Angew. Chem., Int. Ed., 2020, 59, 11196–11205 CrossRef PubMed.
- M. Sugumaran, J. Evans, S. Ito and K. Wakamatsu, Int. J. Mol. Sci., 2020, 21, 7321 CrossRef CAS PubMed.
- A. Mohan, D. Sasikumar, V. Bhat and M. Hariharan, Angew. Chem., Int. Ed., 2020, 59, 3201–3208 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- A. R. Mallia, P. S. Salini and M. Hariharan, J. Am. Chem. Soc., 2015, 137, 15604–15607 CrossRef CAS PubMed.
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS PubMed.
- A. Benny, R. Ramakrishnan and M. Hariharan, Chem. Sci., 2021, 12, 5064–5072 RSC.
- K. Wakamatsu and S. Ito, Anal. Biochem., 1988, 170, 335–340 CrossRef CAS PubMed.
- E. S. Feldblum and I. T. Arkin, Proc. Natl. Acad. Sci., 2014, 111, 4085–4090 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- C.-T. Chen, C. Chuang, J. Cao, V. Ball, D. Ruch and M. J. Buehler, Nat. Commun., 2014, 5, 3859 CrossRef CAS PubMed.
- J. Wang and L. Blancafort, Angew. Chem., Int. Ed., 2021, 60, 18800–18809 CrossRef CAS PubMed.
- S. Wirsing, M. Hänsel, V. Belova, F. Schreiber, K. Broch, B. Engels and P. Tegeder, J. Phys. Chem. C, 2019, 123, 27561–27572 CrossRef CAS.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- F. Plasser and H. Lischka, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef.
- B.-L. L. Seagle, K. A. Rezai, E. M. Gasyna, Y. Kobori, K. A. Rezaei and J. R. Norris, J. Am. Chem. Soc., 2005, 127, 11220–11221 CrossRef CAS PubMed.
- A. Pezzella, O. Crescenzi, L. Panzella, A. Napolitano, E. J. Land, V. Barone and M. D'Ischia, J. Am. Chem. Soc., 2013, 135, 12142–12149 CrossRef CAS PubMed.
- R. A. Edwards, G. Jickling and R. J. Turner, Photochem. Photobiol., 2002, 75, 362 CrossRef CAS PubMed.
- S. P. Nighswander-Rempel, I. B. Mahadevan, P. V. Bernhardt, J. Butcher and P. Meredith, Photochem. Photobiol., 2008, 84, 620–626 CrossRef CAS PubMed.
- N. S. S. Nizar, M. Sujith, K. Swathi, C. Sissa, A. Painelli and K. G. Thomas, Chem. Soc. Rev., 2021, 50, 11208–11226 RSC.
- S. J. George, R. De Bruijn, Ž. Tomović, B. Van Averbeke, D. Beljonne, R. Lazzaroni, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 17789–17796 CrossRef CAS PubMed.
- C. Kulkarni, K. K. Bejagam, S. P. Senanayak, K. S. Narayan, S. Balasubramanian and S. J. George, J. Am. Chem. Soc., 2015, 137, 3924–3932 CrossRef CAS PubMed.
- A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456–460 CrossRef CAS PubMed.
- K. V. Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133–1139 CrossRef PubMed.
- J. Crassous, A. Amon and J. Crassous, Phys. Rev. A, 2012, 85, 023806 CrossRef.
- R. Rubires, J.-A. Farrera and J. M. Ribó, Chem.–Eur. J., 2001, 7, 436–446 CrossRef CAS.
- B. Auguié, J. L. Alonso-Gómez, A. Guerrero-Martínez and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2011, 2, 846–851 CrossRef PubMed.
- L. Y. Wang, K. W. Smith, S. Dominguez-Medina, N. Moody, J. M. Olson, H. Zhang, W. S. Chang, N. Kotov and S. Link, ACS Photonics, 2015, 2, 1602–1610 CrossRef CAS.
- C. Bustamante, I. Tinoco and M. F. Maestre, Proc. Natl. Acad. Sci., 1983, 80, 3568–3572 CrossRef CAS PubMed.
- Y. Sang, D. Yang, P. Duan and M. Liu, Chem. Sci., 2019, 10, 2718–2724 RSC.
- S. I. S. Hendrikse, L. Su, T. P. Hogervorst, R. P. M. Lafleur, X. Lou, G. A. Van Der Marel, J. D. C. Codee and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 13877–13886 CrossRef CAS PubMed.
- T. Narushima and H. Okamoto, Sci. Rep., 2016, 6, 35731 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2120651. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06755a |
| This journal is © The Royal Society of Chemistry 2022 |