
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntiviral cyclic peptides targeting the main protease of SARS-CoV-2†
Jason
Johansen-Leete
 ab,
Sven
Ullrich
c,
Sarah E.
Fry
ab,
Rebecca
Frkic
cd,
Max J.
Bedding
ab,
Anupriya
Aggarwal
e,
Anneliese S.
Ashhurst
abf,
Kasuni B.
Ekanayake
cd,
Mithun C.
Mahawaththa
cd,
Vishnu M.
Sasi
cd,
Stephanie
Luedtke
g,
Daniel J.
Ford
ab,
Anthony J.
O'Donoghue
g,
Toby
Passioura
abhi,
Mark
Larance
ij,
Gottfried
Otting
cd,
Stuart
Turville
e,
Colin J.
Jackson
cd,
Christoph
Nitsche
*c and
Richard J.
Payne
*ab
ab,
Sven
Ullrich
c,
Sarah E.
Fry
ab,
Rebecca
Frkic
cd,
Max J.
Bedding
ab,
Anupriya
Aggarwal
e,
Anneliese S.
Ashhurst
abf,
Kasuni B.
Ekanayake
cd,
Mithun C.
Mahawaththa
cd,
Vishnu M.
Sasi
cd,
Stephanie
Luedtke
g,
Daniel J.
Ford
ab,
Anthony J.
O'Donoghue
g,
Toby
Passioura
abhi,
Mark
Larance
ij,
Gottfried
Otting
cd,
Stuart
Turville
e,
Colin J.
Jackson
cd,
Christoph
Nitsche
*c and
Richard J.
Payne
*ab
aSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
bAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, Sydney, NSW 2006, Australia
cResearch School of Chemistry, Australian National University, Canberra, ACT 2601, Australia. E-mail: christoph.nitsche@anu.edu.au
dAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, Australian National University, Canberra, ACT 2601, Australia
eKirby Institute, Sydney, NSW 2052, Australia
fSchool of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia
gSkaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA
hSchool of Life and Environmental Sciences, The University of Sydney, Sydney, NSW 2006, Australia
iSydney Analytical, The University of Sydney, Sydney, NSW 2006, Australia
jCharles Perkins Centre, The University of Sydney, Sydney, NSW 2006, Australia
First published on 28th February 2022
Abstract
Antivirals that specifically target SARS-CoV-2 are needed to control the COVID-19 pandemic. The main protease (Mpro) is essential for SARS-CoV-2 replication and is an attractive target for antiviral development. Here we report the use of the Random nonstandard Peptide Integrated Discovery (RaPID) mRNA display on a chemically cross-linked SARS-CoV-2 Mpro dimer, which yielded several high-affinity thioether-linked cyclic peptide inhibitors of the protease. Structural analysis of Mpro complexed with a selenoether analogue of the highest-affinity peptide revealed key binding interactions, including glutamine and leucine residues in sites S1 and S2, respectively, and a binding epitope straddling both protein chains in the physiological dimer. Several of these Mpro peptide inhibitors possessed antiviral activity against SARS-CoV-2 in vitro with EC50 values in the low micromolar range. These cyclic peptides serve as a foundation for the development of much needed antivirals that specifically target SARS-CoV-2.
Introduction
The COVID-19 pandemic, caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused widespread morbidity and mortality as well as devastation to the global economy since the disease was first reported in late 2019 in Wuhan, China.1 At the time of writing there has been more than 400 million confirmed cases and over 6 million deaths worldwide as a result of COVID-19.2 There has been significant effort from the global research community to develop effective vaccines for COVID-19; this has been enormously successful, with adenoviral vectored vaccines, protein vaccines and mRNA vaccines now in widespread use across the world. Whilst vaccines will enable protective immunity in most, there will be populations where vaccine-based immunity may fail, and these individuals will be vulnerable to SARS-CoV-2 infection in the future. It is furthermore unclear what changes will appear in the virus in contemporary SARS-CoV-2 viral variants (highlighted by the recent emergence of the delta (B.1.617.2)3 and omicron (B.1.1.529) variants of concern), and how those variants will navigate both convalescent and vaccine immune responses. Given that this is the third coronavirus that has crossed via zoonoses in the 21st century, antiviral development against SARS-CoV-2, and future coronaviruses with pandemic potential, are desperately needed in addition to prophylactic vaccines.While there have been significant efforts toward the discovery of effective antivirals for SARS-CoV-2, the vast majority of molecules that have completed (or are currently being assessed in) clinical trials were originally developed for other infectious and inflammatory disease indications and are being repurposed for COVID-19. For example, at the time of writing, remdesivir, originally trialled for Ebola, is currently the only antiviral drug to be approved by the U.S. Food and Drug Administration (FDA) for the treatment of COVID-19. While the molecule has been shown to possess some activity during early infection, it has shown limited to no efficacy in a number of trials,4,5 as well as in infections in patients hospitalized with COVID-19.6 Other repurposed antiviral drugs that have entered trials include the HIV combination therapy lopinavir–ritonavir,7,8 type I interferon treatments,9,10 and the antimalarial hydroxychloroquine;11–13 however, these have not demonstrated improvement in disease progression over standard care. In fact, it has recently been suggested that many repurposing efforts may be compromised by experimental artefacts reflecting the physicochemical properties of certain drugs rather than specific target-based activities.14 To date, the most effective therapeutic intervention for improving COVID-19 patient outcomes in a hospital setting is the use of the corticosteroid dexamethasone, which reduces inflammation-mediated lung injury associated with SARS-CoV-2 infection in patients with elevated levels of C-reactive protein.15,16 Based on the above, there is an urgent need for the discovery of effective antivirals for COVID-19, ideally with mechanisms of action that specifically target proteins critical in the SARS-CoV-2 lifecycle.
Infection of human cells by SARS-CoV-2 is initiated by interaction between the receptor binding domain of the trimeric viral spike protein (S) with the host cell-surface receptor angiotensin converting enzyme 2 (ACE2) (Fig. 1). Following receptor binding of the virus, the spike protein is activated by cleavage between the S1 and S2 domains leading to host cell entry via two distinct pathways: (1) an endocytic pathway through endosomal–lysosomal compartments with spike cleavage facilitated by lysosomal cathepsins, or (2) a cell surface pathway following activation by a serine protease such as transmembrane protease serine 2 (TMPRSS2).17–19 Following proteolysis, the N-terminus of the cleaved S2 domain is embedded into the cell membrane and leads to fusion of the membranes of the virus and the host cell, followed by transfer of viral RNA into the cytoplasm.20
 | ||
| Fig. 1 Mechanism of SARS-CoV-2 entry into host cells and replication (cell-surface entry pathway mediated by TMPRSS2 shown). The proteolytic activity of SARS-CoV-2 Mpro on the two polyproteins pp1a and pp1ab is shown in the box, including the structure of the covalent peptidomimetic Mpro inhibitor PF-07321332 (nirmatrelvir) developed by Pfizer. | ||
Viral gene expression within the host cell results in the translation of two overlapping polyproteins, pp1a and pp1ab. Embedded within these polyproteins are sixteen non-structural proteins critical for viral replication, the majority of which form the viral replication and transcription complex (RTC) including the viral RNA-dependent RNA polymerase (RdRp, nsp12) and helicase (nsp13).20 These proteins become functional only after proteolytic release by two viral proteases. The first of these is a domain of nsp3 called the papain-like protease (PLpro) which cleaves the pp1a and pp1ab at three sites, releasing nsp1, nsp2 and nsp3.20,21 The second is the SARS-CoV-2 main protease (Mpro), also called nsp5 or the chymotrypsin-like protease (3CLpro), which cleaves pp1a and pp1ab at a minimum of 11 distinct cleavage sites to release nsp4-16 (Fig. 1).20 Interestingly, Mpro has also been found to aid in immune evasion by inhibiting type I IFN production, contributing to the impaired type I IFN response that has become a hallmark of severe SARS-CoV-2 infection, with persistent viral load and poor patient outcomes.22–25 Mpro forms a catalytically active homodimer which cleaves with high specificity at Leu–Gln↓Xaa (where ↓ represents the cleavage site and Xaa can be Ser, Ala or Asn).26–28 Such sequence specificity has not been observed for any human proteases and therefore peptide or peptidomimetic based inhibitors are predicted to inhibit SARS-CoV-2 Mpro with high selectivity and with minimal off-target effects in humans.20,29
The key role of Mpro for the replication and viability of SARS-CoV-2 has naturally led to the search for novel inhibitors of the protease. Perhaps the most promising of these are the peptidomimetic compounds developed by Pfizer, inspired by PF-00835231 (IC50 = 4–8 nM against SARS-CoV-2 Mpro) that was originally developed against SARS-CoV-1 Mpro, which is homologous to SARS-CoV-2 Mpro (96% amino acid identity).30–32 Specifically, a phosphate prodrug of this inhibitor (PF-07304814, lufotrelvir) has recently completed two phase 1 studies (clinical trials identifiers: NCT04535167, NCT04627532). A second-generation orally available peptidomimetic Mpro inhibitor developed by Pfizer (PF-07321332, nirmatrelvir, Fig. 1)33 has very recently received emergency use authorization following positive results in phase 3 clinical studies for treatment of COVID-19 (clinical trials identifiers: NCT04960202 NCT05011513, NCT05047601).34 Both molecules possess a γ-lactam as a mimic of the glutamine (Gln) residue found at the P1 position in physiological cleavage sites and also inhibit SARS-CoV-2 Mprovia a covalent mechanism through electrophilic warheads embedded within the inhibitors.30 Specifically, PF-00835231 possesses a hydroxymethyl ketone moiety, while PF-07321332 contains a nitrile warhead, both of which react with the catalytic cysteine (Cys145) to inactivate the protease. In addition to these clinical candidates, a number of other peptidomimetic inhibitors of SARS-CoV-2 Mpro are currently under preclinical investigation,27,35–38 including repurposed drugs such as boceprevir, a serine protease inhibitor approved in 2011 for the treatment of hepatitis C,36,39,40 and the feline anticoronaviral drug GC376.36,39
Macrocyclic peptides are attractive chemotypes for medicinal chemistry efforts due to their ability to bind targets with high affinity and selectivity, whilst exhibiting greater proteolytic stability and membrane permeability than their linear counterparts.41–43 In this work we describe several potent cyclic peptide inhibitors of the SARS-CoV-2 Mpro, identified through the use of the  nonstandard
nonstandard ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) eptide
eptide ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ntegrated
ntegrated ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) iscovery (RaPID) technology, which couples mRNA display with flexizyme-mediated genetic code reprogramming.43–45 Importantly, we also report a crystal structure of the SARS-CoV-2 Mpro dimer bound to our most potent cyclic peptide inhibitor that highlights the residues important for binding both at the catalytic site and across the dimer interface. Finally, we demonstrate that three of the cyclic peptides identified exhibited antiviral activity against SARS-CoV-2 in vitro, with an additional peptide gaining antiviral activity upon conjugation to a cell penetrating peptide.
iscovery (RaPID) technology, which couples mRNA display with flexizyme-mediated genetic code reprogramming.43–45 Importantly, we also report a crystal structure of the SARS-CoV-2 Mpro dimer bound to our most potent cyclic peptide inhibitor that highlights the residues important for binding both at the catalytic site and across the dimer interface. Finally, we demonstrate that three of the cyclic peptides identified exhibited antiviral activity against SARS-CoV-2 in vitro, with an additional peptide gaining antiviral activity upon conjugation to a cell penetrating peptide.
Results and discussion
Selection against a chemically cross-linked SARS-CoV-2 Mpro homodimer
In order to identify cyclic peptide inhibitors of SARS-CoV-2 Mpro, we sought to utilize RaPID, which allows the screening of >1012 cyclic peptides for affinity against a protein target of interest immobilized on magnetic beads. However, functional SARS-CoV-2 Mpro is a homodimer with relatively weak affinity between the monomers (KD = 2.5 μM), and we hypothesized that the protein may exist in a predominantly monomeric (i.e. inactive) form when immobilized on magnetic beads.27 Consistent with this, we found that the catalytic activity (measured by fluorescent substrate cleavage) of C-terminally His6-tagged Mpro was significantly diminished (ca. 30%) when immobilized on His-tag pull-down Dynabeads™ compared to that of the wild-type protein in solution (see ESI, Fig. S1a†); this indicated that a significant proportion of the immobilized Mpro was unable to form an active homodimer. We therefore investigated the use of a chemical cross-linking strategy to covalently lock SARS-CoV-2 Mpro in the homodimeric state to ensure that RaPID resulted in selection of ligands to the catalytically active form of the protease. The presence of proximal lysine residues near the dimer interface in the crystal structure of SARS-CoV-2 Mpro (PDB: 6Y2E) prompted us to investigate a number of amine-reactive bifunctional cross-linkers of differing lengths for this purpose.27 Efficient cross-linking was obtained by disuccinimidyl glutarate (DSG) (Fig. 2a and b), with subsequent mapping by LC-MS/MS revealing intermolecular cross-links between lysine residues on the different monomer chains (K97–K97*, K97–K90*). We also observed cross-links between K12 and K97, which are positioned in close proximity on different monomers of the dimer, as well as within the same monomer unit; however, in this case we could not differentiate between intermolecular or intramolecular cross-links by mass spectrometry (see ESI, Fig. S1b†). Importantly, cross-linked Mpro exhibited catalytic activity comparable to that of wild-type Mpro (in solution), following immobilization on magnetic beads (see ESI, Fig. S1a†). Based on these data, we moved on to RaPID selections against the cross-linked SARS-CoV-2 Mpro with a view to discovering novel cyclic peptide inhibitors.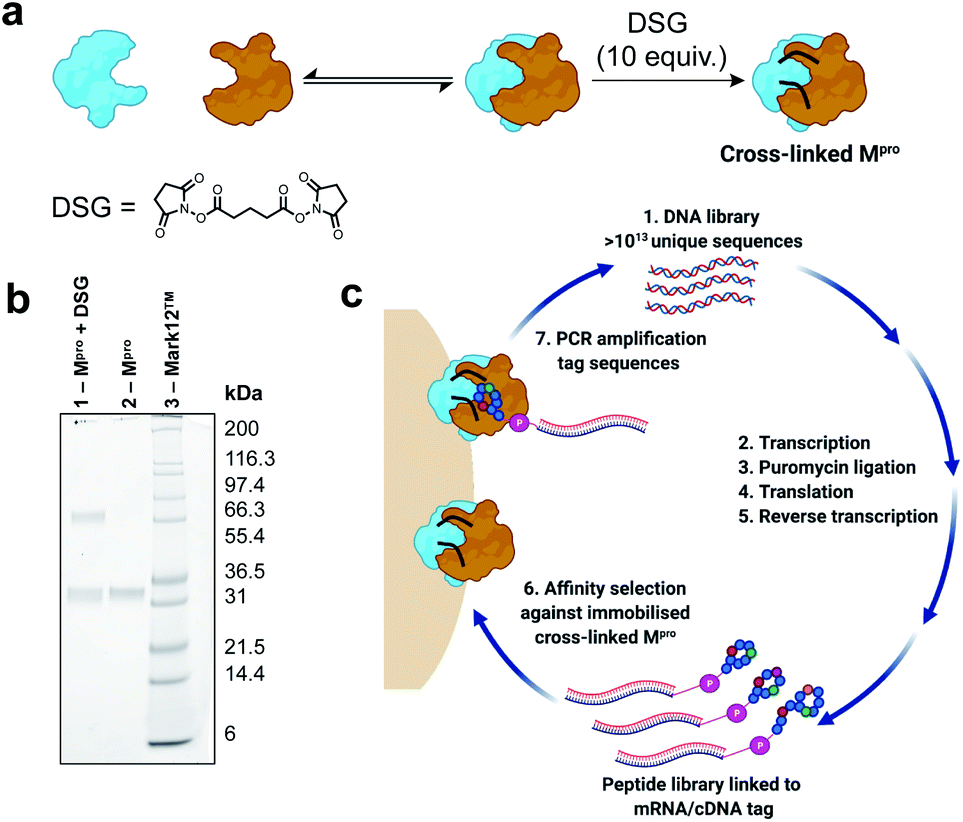 | ||
| Fig. 2 (a) Scheme for cross-linking of SARS-CoV-2 Mpro with disuccinimidyl glutarate (DSG). (b) SDS-PAGE gel of cross-linking reaction of Mpro. Lane 1: reaction of SARS-CoV-2 Mpro (25 μM) with DSG (10 equivalents relative to Mpro monomer) for 1 h at 37 °C in aqueous buffer (20 mM HEPES, 100 mM NaCl, pH 7.6). Lane 2: SARS-CoV-2 Mpro. Lane 3: Mark12 ™ ladder. (c) Workflow for the discovery of macrocyclic peptide ligands of cross-linked SARS-CoV-2 Mpro using RaPID mRNA display. | ||
To select for peptide binders of SARS-CoV-2 Mpro we performed parallel display using the RaPID mRNA display technology, employing peptide libraries initiated with either N-chloroacetyl-L-tyrosine or N-chloroacetyl-D-tyrosine to induce thioether macrocyclization through reaction with the side chain of a downstream cysteine residue. Tyr was chosen as the initiating amino acid due to its high translation efficiency in place of N-formyl-Met,46,47 while the use of D-Tyr in addition to L-Tyr expanded the accessible chemical space of the library. For each selection, a semi-randomized DNA library of >1013 unique sequences were transcribed into mRNA followed by ligation to a puromycin linker (Fig. 2c). Translation of the mRNA-puromycin hybrids in vitro, followed by reverse transcription, yielded a library of peptide-mRNA:cDNA conjugates that were counter-selected against His Pull-down Dynabeads™ (to remove bead-binding peptides) before an affinity selection step, in which the libraries were panned against bead-immobilized cross-linked SARS-CoV-2 Mpro. PCR of the bead-bound fraction yielded an enriched DNA library that was used as the starting point for the subsequent rounds of selection (Fig. 2c). After seven rounds of selection, next generation sequencing of each of the final DNA libraries was performed, however, enrichments of the recovered sequences remained low (see Fig. S2, ESI†). Therefore, an additional two rounds of selection were performed after which a number of peptide sequences were enriched that were predicted to be high affinity ligands of the SARS-CoV-2 Mpro based on high relative abundance. It should be noted that the requirement for additional rounds of selection, as well as the absence of a strong consensus sequence in the final libraries, is highly target dependent. In this case this may have arisen from some structural/functional heterogeneity in the protein following cross-linking (i.e. remaining inactive monomeric Mpro).
Synthesis and in vitro evaluation of cyclic peptide inhibitors of SARS-CoV-2 Mpro
We selected eight peptides from the enriched libraries for synthesis (five L-Tyr initiated and three D-Tyr initiated) based on their abundance in the final DNA library and diversity in sequence (Fig. 3a and b). Peptides 1–8 were synthesized via Fmoc-strategy solid-phase peptide synthesis on Rink amide resin with the N-terminal L- or D-Tyr residue derivatized with chloroacetic acid to facilitate thioether cyclization with the thiol of a downstream cysteine residue. Deprotection and cleavage from resin followed by cyclization provided the target thioether-linked cyclic peptides 1–8 following purification by reverse-phase HPLC. The synthetic cyclic peptides were next evaluated for inhibitory activity against SARS-CoV-2 Mpro using a previously reported fluorescence resonance energy transfer (FRET)-based assay.27,48 Gratifyingly, we observed potent inhibition of proteolytic activity for six of the eight peptides in the series, with IC50 values for peptides 1–6 ranging from 0.070–12.7 μM (Fig. 3c). Interestingly, despite being enriched in the selection, lariat peptide 7 and head to tail cyclic peptide 8 did not show appreciable inhibition of the protease (IC50 > 50 μM). We next assessed the selectivity of the two most potent inhibitors, 1 and 6, against a panel of coronaviral and human proteases (Tables 1 and 2, Fig. S3 and S4†). Both peptides exhibited high selectivity for SARS-CoV-2 Mpro. Whilst 1 exhibited inhibition of SARS-CoV-1 Mpro (owing to the high sequence similarity to SARS-CoV-2 Mpro), both 1 and 6 were inactive at a concentration of 10 μM against MERS-CoV Mpro, SARS-CoV-2 PLpro, TMPRSS2, furin and cathepsins B and E, whilst exhibiting only modest inhibition of cathepsin L (IC50 = 10.9 and 5.7 μM for 1 and 6, respectively). Peptides 1 and 6 were also assessed for stability in human plasma; both exhibited high stability with 85% of 1 and 80% of 6 remaining after 24 hours (see ESI, Fig. S5†).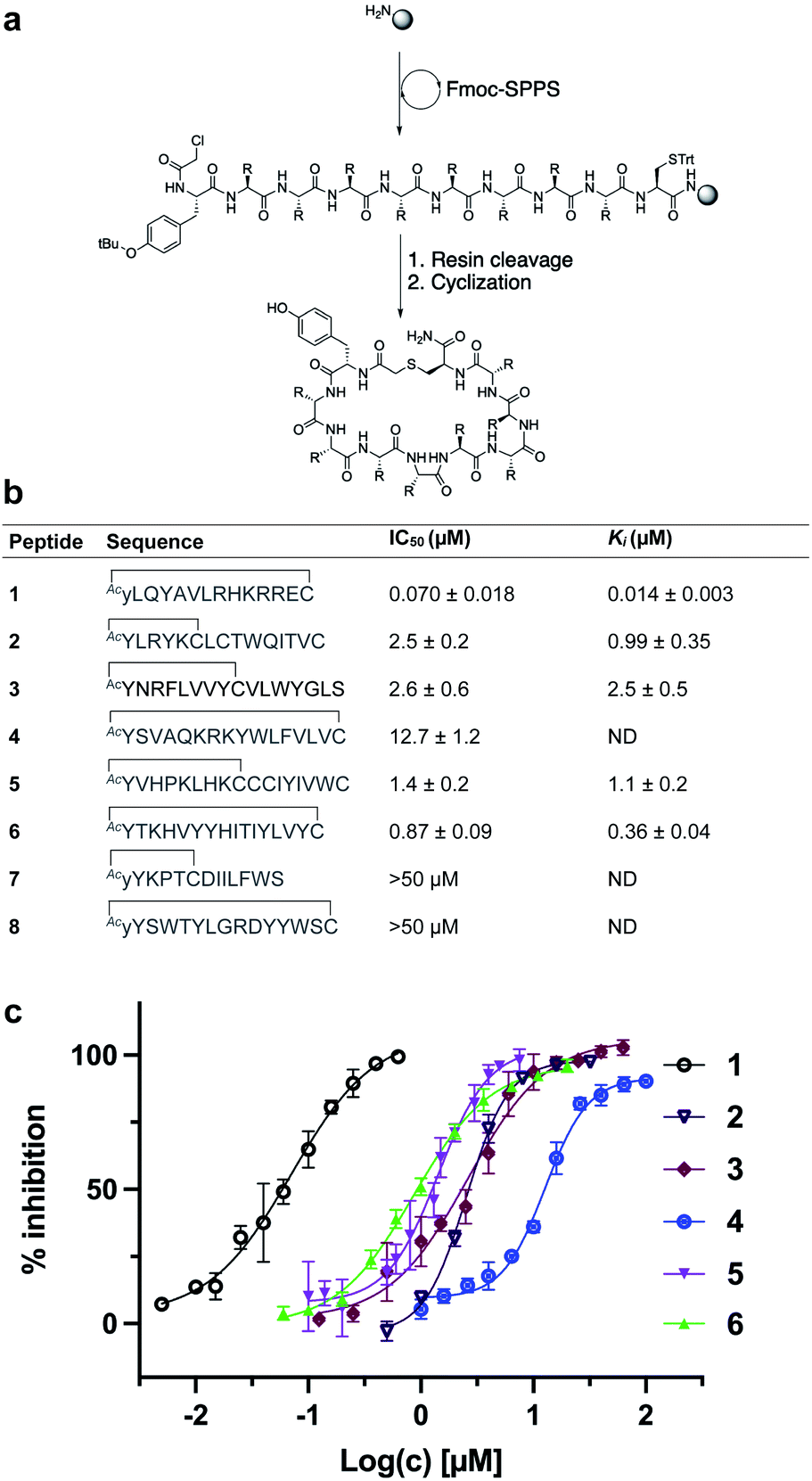 | ||
| Fig. 3 (a) Synthesis of cyclic peptides 1–8via Fmoc-SPPS (see ESI† for full synthetic details). (b) Sequences of peptides 1–8 in one letter amino acid code with associated IC50 and Ki values ± SEM. Thioether cyclization is represented as a black line. (c) In vitro inhibition data of SARS-CoV-2 Mpro for peptides 1–6. NB: cyclic peptides 7 and 8 showed no inhibition of SARS-CoV-2 Mpro at 50 μM, ND = not determined. | ||
| Peptide | SARS-CoV-1 Mpro IC50/μM | MERS-CoV Mpro IC50/μM | SARS-CoV-2 PLpro IC50/μM |
|---|---|---|---|
| 1 | 0.078 ± 0.002 | >10 | >10 |
| 6 | >10 | >10 | >10 |
| Peptide | TMPRSS2 IC50/μM | Furin IC50/μM | Cathepsin B IC50/μM | Cathepsin L IC50/μM | Cathepsin E IC50/μM |
|---|---|---|---|---|---|
| 1 | >10 | >10 | >10 | 10.9 ± 1.12 | >10 |
| 6 | >10 | >10 | >10 | 5.7 ± 0.45 | >10 |
The most potent SARS-CoV-2 Mpro inhibitor in the series was cyclic peptide 1, which exhibited an IC50 of 70 ± 18 nM and a Ki of 14 ± 3 nM against the protease. Given the superior inhibitory potency of this molecule over the other cyclic peptides that emerged from the RaPID screen (>15-fold), this molecule was selected for further structure–activity investigations.
Probing the binding interaction of lead cyclic peptide 1
We next performed experiments to probe the key interactions of lead cyclic peptide inhibitor 1 with SARS-CoV-2 Mpro. Given the critical role dimerization plays in Mpro catalytic activity, we first investigated the effect of 1 on the monomer–dimer equilibrium of Mpro by multi-angle laser light scattering with size exclusion chromatography (SEC-MALLS). Interestingly, the addition of two molar equivalents of peptide 1 to a solution of wild-type Mpro (present in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of monomer:dimer) resulted in a shift in the dimer–monomer equilibrium to afford a solution of exclusively homodimeric protease (Fig. 4a). This data indicates that cyclic peptide 1 stabilizes the homodimeric form of SARS-CoV-2 Mpro and suggests that peptide 1 may bind to the active site present in the dimeric, but not monomeric forms of the protein. To corroborate this data, we used 3D NMR spectroscopy (specifically TROSY-HNCO spectra) to analyze SARS-CoV-2 Mpro after titration with peptide 1 (Fig. 4b). This revealed shifts of backbone NMR resonances of residues near the active site, consistent with binding of peptide 1 at this location. However, shifts were also observed for residues located far from the active site (see ESI, Fig. S6†), suggesting that the protein responds to the binding of 1 with global allosteric changes. In contrast, titration of the obligate monomeric mutant R298A of SARS-CoV-2 Mpro49 with peptide 1 led to no shifts in NMR resonances (see ESI, Fig. S6, Tables S1, S2 and Fig. S7† for full assignment of monomeric Mpro mutant). Notably, R298, which is mutated in the obligate monomer, is buried within the dimer interface in the structure of the wild-type protease and therefore the lack of binding of 1 to the monomeric protein is unlikely to be owing to a loss of interactions with this amino acid residue. Taken together, these SEC-MALLS and NMR data therefore suggest that peptide 1 selectively binds to and inhibits the active homodimer of SARS-CoV-2 Mpro but does not bind to the inactive monomer.
:1 ratio of monomer:dimer) resulted in a shift in the dimer–monomer equilibrium to afford a solution of exclusively homodimeric protease (Fig. 4a). This data indicates that cyclic peptide 1 stabilizes the homodimeric form of SARS-CoV-2 Mpro and suggests that peptide 1 may bind to the active site present in the dimeric, but not monomeric forms of the protein. To corroborate this data, we used 3D NMR spectroscopy (specifically TROSY-HNCO spectra) to analyze SARS-CoV-2 Mpro after titration with peptide 1 (Fig. 4b). This revealed shifts of backbone NMR resonances of residues near the active site, consistent with binding of peptide 1 at this location. However, shifts were also observed for residues located far from the active site (see ESI, Fig. S6†), suggesting that the protein responds to the binding of 1 with global allosteric changes. In contrast, titration of the obligate monomeric mutant R298A of SARS-CoV-2 Mpro49 with peptide 1 led to no shifts in NMR resonances (see ESI, Fig. S6, Tables S1, S2 and Fig. S7† for full assignment of monomeric Mpro mutant). Notably, R298, which is mutated in the obligate monomer, is buried within the dimer interface in the structure of the wild-type protease and therefore the lack of binding of 1 to the monomeric protein is unlikely to be owing to a loss of interactions with this amino acid residue. Taken together, these SEC-MALLS and NMR data therefore suggest that peptide 1 selectively binds to and inhibits the active homodimer of SARS-CoV-2 Mpro but does not bind to the inactive monomer.
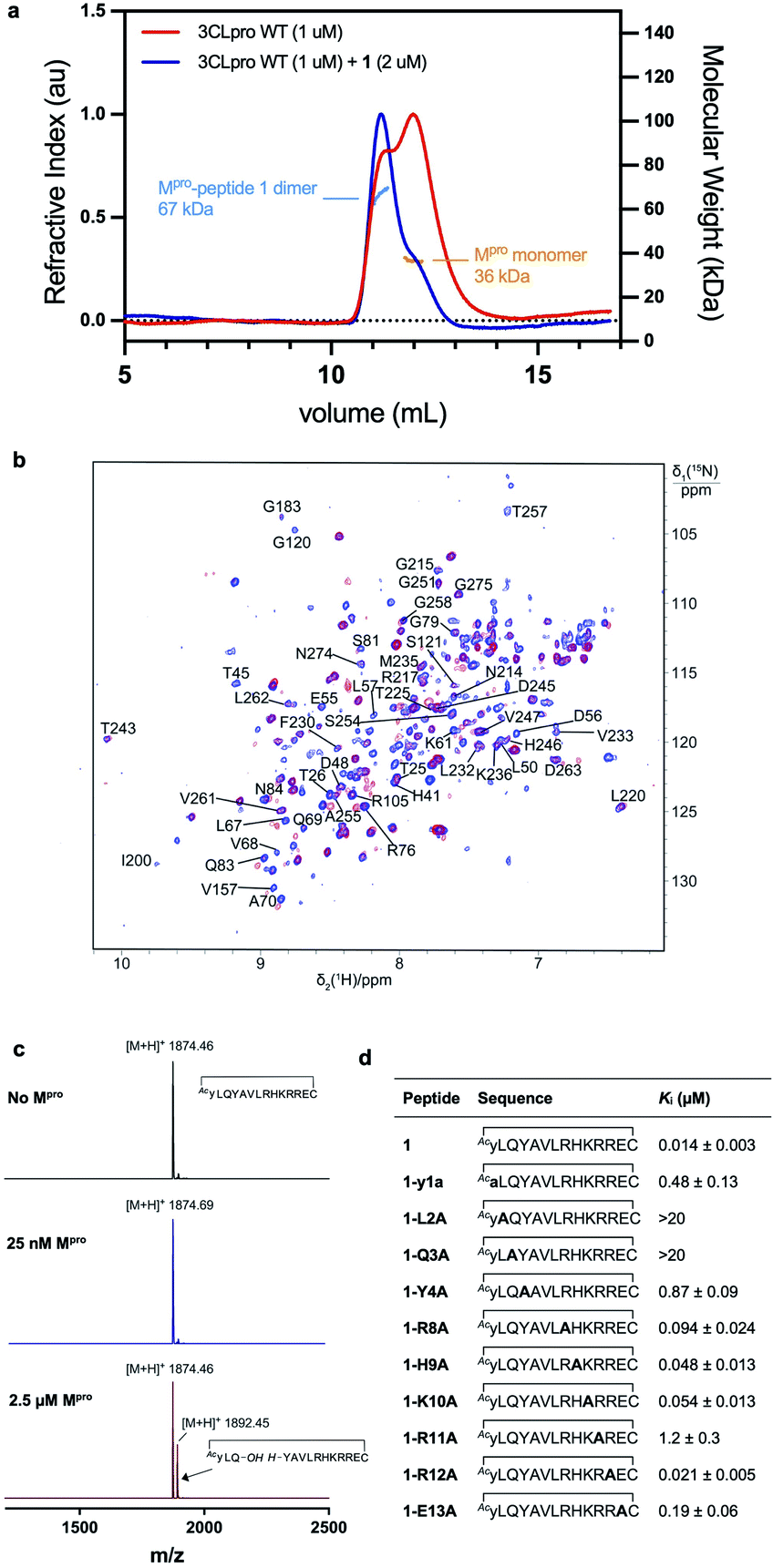 | ||
| Fig. 4 (a) SEC-MALLS of SARS-CoV-2 Mpro with and without peptide 1. SARS-CoV-2 Mpro (red line) exists in equilibrium between monomeric and homodimeric forms giving rise to two peaks (in ca. 1:1.25 ratio of monomer to dimer) in the size-exclusion chromatogram at a concentration of 1 μM in aqueous buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl). MALLS analysis indicates an approximate molecular weight of 36 kDa for the monomer (calculated molecular weight = 33.8 kDa). After addition of two molar equivalents of 1, Mpro converges predominantly to a homodimer (blue line) with a MALLS reading of 66 kDa (calculated MW of SARS-CoV-2 Mpro dimer = 67.6 kDa, MW of peptide 1 = 1874 Da) indicative of formation of the SARS-CoV-2 Mpro homodimer upon binding 1. (b) Cyclic peptide inhibitor 1 binds to the dimeric form of SARS-CoV-2 Mpro. Overlay of projections onto the 15N–1H plane of 3D TROSY-HNCO spectra of 0.3 mM solutions of 15N/13C/2H-labelled wild-type Mpro. Blue and red contour lines show the spectra recorded in the absence and presence of equimolar inhibitor 1, respectively. Assignments are shown for peaks that shift or disappear in response to the inhibitor. (c) Monitoring of the cleavage of cyclic peptide inhibitor 1 with SARS-CoV-2 Mpro. MALDI-TOF mass spectrum of cyclic peptide 1 (top spectrum). Negligible cleavage of 1 was observed following incubation with SARS-CoV-2 Mpro under standard assay conditions (25 nM SARS-CoV-2 Mpro, 5 μM 1, 20 mM Tris–HCl pH 7.6, 100 mM NaCl, 1 mM DTT, 1 mM EDTA) for 1 h at 37 °C (middle spectrum). Slow cleavage of 1 was observed (ca. ∼30% after 1 h) in the presence of a high concentration of SARS-CoV-2 Mpro to 2.5 μM (bottom spectrum). (d) Inhibitory activity of alanine mutants of lead cyclic peptide 1. Sequences and associated inhibitory constants for peptide 1 analogues, whereby all polar residues within the randomized region of 1 were each systematically mutated to alanine and their inhibitory activity against SARS-CoV-2 Mpro assessed (Cys14 was not mutated as this residue is required for cyclization, Ala substitutions are shown in bold). | ||
Cyclic peptide 1 includes the cleavage recognition motif of Mpro found in the natural viral protein substrates, namely a Gln in the P1 position and a Leu in the P2 position.27 It was therefore hypothesized that this Leu–Gln motif embedded within 1 mimics the natural substrate and binds to the catalytic site of the protease. Importantly, this proposal is supported by the purely competitive inhibition mode observed for 1 (see ESI, Fig. S8†). However, this also raises the possibility that the protease may be able to cleave 1 next to the Gln–Leu recognition sequence. To test this, we incubated 1 with wild type SARS-CoV-2 Mpro and assessed cleavage by MALDI-TOF-MS (Fig. 4c and S9, ESI†). Peptide 1 exhibited notable resistance to proteolysis by Mpro with negligible cleavage of peptide 1 observed under standard assay conditions (25 nM Mpro) after 5 h. However, incubation of 1 with a high concentration of Mpro (2.5 μM) resulted in slow cleavage of peptide 1, with 30% peptide cleavage observed after 1 h. Analysis of the cleavage reaction by LC-MS/MS confirmed that proteolysis had indeed occurred between Gln3 and Tyr4 (see ESI Fig. S10†). We also synthesized an authentic standard of the resulting cleavage product, which was verified to be identical to Mpro-cleaved 1 by LC-MS/MS (see ESI Fig. S10b†). Finally, we assessed whether the linear peptide product resulting from Mpro cleavage of 1 possessed inhibitory activity against SARS-CoV-2 Mpro. Interestingly, the peptide exhibited an IC50 of 23.2 ± 5 μM, ∼330-fold higher than the IC50 of 70 nM for 1 (see ESI, Fig. S10c†). This two-orders of magnitude loss in activity upon linearization suggests that the conformation of cyclic peptide 1 is pre-organised for optimal interaction with the protease.
In order to assess the importance of each residue in 1 for Mpro inhibitory activity, we systematically replaced all residues in the peptide with alanine (except alkyl side chain-containing amino acids Ala5, Val6 and Leu7) and determined the inhibitory activity of the resulting mutants against Mpro (Fig. 4d and S11, ESI†). Consistent with the known recognition sequence for SARS-CoV-2 Mpro and supported by the mass spectrometry results described above, mutation of either Leu2 or Gln3 to Ala (that would be predicted to bind in the S2 and S1 recognition sites, respectively) led to more than two orders of magnitude reduction in inhibitory activity. Remarkably, mutation of Tyr4 (which would be predicted at  ) also led to a significant loss in inhibitor activity (IC50 = 1.9 μM); while this is consistent with the established importance of aromaticity in
) also led to a significant loss in inhibitor activity (IC50 = 1.9 μM); while this is consistent with the established importance of aromaticity in 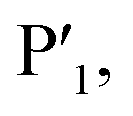 small residues such as alanine are often found in substrates at this position, and the dramatic reduction in inhibitory potency was therefore unexpected.27,28 Interestingly, mutation of Arg11, which is distal from the most prominent recognition residues of the cyclic peptide, also led to a significant reduction in inhibitory activity (IC50 = 3.4 μM) suggesting that this residue makes important interactions with the protease and/or serves a crucial role in the adoption of the active conformation of the cyclic peptide. In contrast, mutation of Arg8, His9, Lys10, Arg12 or Glu13 resulted in equipotent activity or only a modest reduction in inhibitory activity (IC50 values = 90–390 nM).
small residues such as alanine are often found in substrates at this position, and the dramatic reduction in inhibitory potency was therefore unexpected.27,28 Interestingly, mutation of Arg11, which is distal from the most prominent recognition residues of the cyclic peptide, also led to a significant reduction in inhibitory activity (IC50 = 3.4 μM) suggesting that this residue makes important interactions with the protease and/or serves a crucial role in the adoption of the active conformation of the cyclic peptide. In contrast, mutation of Arg8, His9, Lys10, Arg12 or Glu13 resulted in equipotent activity or only a modest reduction in inhibitory activity (IC50 values = 90–390 nM).
Co-crystallization of peptide 1 and a selenoether analogue with SARS-CoV-2 Mpro
In order to further interrogate the binding mode of inhibitor 1, we used X-ray diffraction to solve the co-crystal structure of the SARS-CoV-2 Mpro-1 complex to 3.4 Å. Unfortunately, the limited resolution of the 3.4 Å structure hindered the interpretation of the electron density within the active site. We therefore also solved the structure of a synthetic selenoether analogue of 1 (Se-1) in complex with SARS-CoV-2 Mpro, at 2.35 Å resolution, in which the cysteine residue involved in thioether peptide macrocyclization was replaced with selenocysteine.50 We rationalized that the modest S to Se substitution would maintain the protease inhibitory profile observed for cyclic peptide 1, but that the greater electron density of Se would aid its placement in the active site. It is noteworthy that while there have been several high-resolution structures of SARS-CoV-2 Mpro in complex with small molecules or short peptides with covalent warheads reported, this represents the first high-resolution structure of the protease complexed with a large non-covalently bound peptide. Importantly, Se-1 displayed identical inhibitory activity against SARS-CoV-2 Mpro as 1 (Ki of Se-1 = 17 nM vs. 14 nM for 1) as well as very similar activity against the panel of other proteases tested (Tables 1 and 2, ESI, Fig. S3, S4 and S12†) suggesting its interactions with Mpro are also very similar to parent inhibitor 1. The SARS-CoV-2 Mpro-Se-1 complex diffracted to 2.35 Å resolution in the same space group and with similar cell dimensions as the SARS-CoV-2 Mpro-1 complex, thus providing a detailed view of the interactions between the inhibitor and the protease (see Table S3, ESI†). It was observed that the crystal was pseudo-merohedrally twinned (−h,−k,h + l; twin fraction 0.493) and indexed in the P1211 space group. The crystal dimensions and packing are unique and not observed in any of the hundreds of deposited SARS-CoV-2 Mpro structures, with a notably long b axis. This complex most likely crystallized in a novel crystal form because the peptide sterically blocks many of the crystal contacts present in previously described SARS-CoV-2 Mpro crystal structures. There were four protein subunits within the asymmetric unit comprising two physiological dimers (Fig. 5a).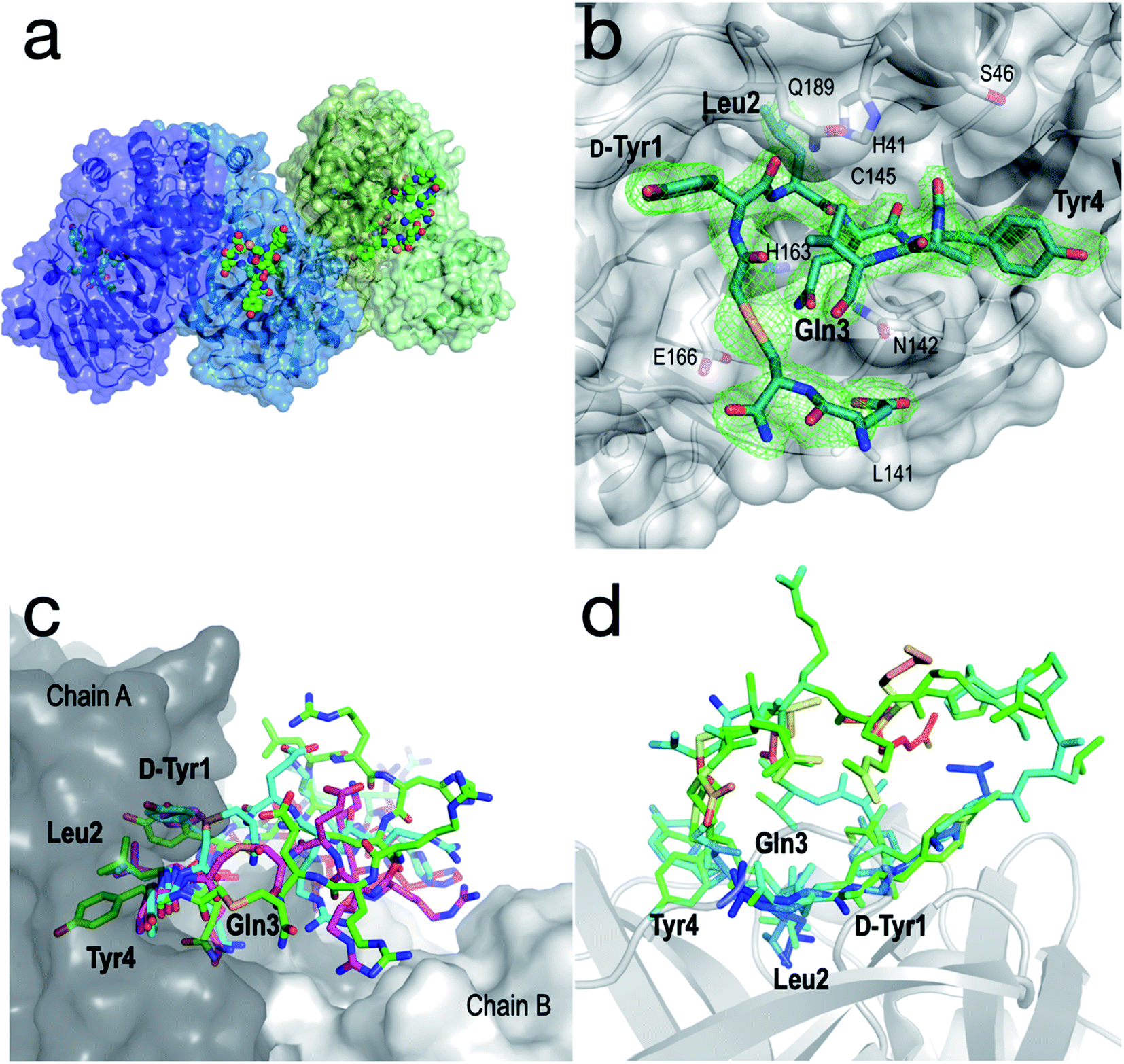 | ||
| Fig. 5 Structural analysis of the SARS-CoV-2 Mpro-Se-1 complex (PDB ID: 7RNW). (a) The asymmetric unit of the SARS-CoV-2 Mpro-Se-1 crystal structure, containing two physiological dimers shown as blue and green. The Se-1 peptide is shown in a sphere representation. (b) Zoom of the active site with electron density (omit map generated using twin refinement in phenix, contoured to 3.0 σ) showing bound peptide (residues 7–12 disordered). Key active site residues of SARS-CoV-2 Mpro are shown as sticks, and key positions of the Se-1 peptide (residues 1–4) are labelled in bold text. (c) Representative conformations (300 ns) from each of the triplicate simulations suggest that Tyr1, Leu2, Gln3 and Tyr4 are stably bound in the active site of SARS-CoV-2 Mpro, while the remainder of the peptide is more mobile and makes transient interactions across the dimer interface. (d) Se-1 peptide bound to chains A, B and D colored by B-factor, showing the residues 1–4 are stable and bound within the S3, S2, S1 and S−1 subsites, respectively, whereas the polar half of the peptide (Arg8–Glu13) is either too disordered to accurately model, or is modelled with high B-factors. Chain C was omitted because crystal packing interactions distorted the B-factors of the peptide. | ||
Although the peptide is not fully resolved in the structure, 9–13 residues were observable in various chains with good electron density; the selenoether linkage and the first 5 residues (D-Tyr1, Leu2, Gln3, Tyr4, Ala5) were very stable, with the remainder of the peptide (7–12) appearing somewhat disordered (Fig. 5b). The peptide displayed a consensus binding pose across three subunits of the crystal structure. The fourth subunit displayed an alternative binding pose in which Leu2 and Gln3 were present in identical positions, but the flanking D-Tyr1 and Tyr4 residues adopted alternative conformations (see ESI, Fig. S13a and b†). Closer inspection revealed a non-Pro cis-peptide bond between Tyr4 and Ala3 in chains A, C, D, which results in a tight kink in the helix, while in chain B this bond remains in the trans-configuration (see ESI, Fig. S13c and d†). The Gln3 of the peptide occupies the canonical S1 subsite of the protease (following the numbering by Lee et al.)51 in every peptide:protein complex, facilitated by interactions with His163, Glu166, and Asn142 (Fig. 5b). Likewise, Leu2 is always bound in the S2 subsite comprised of His41, His164 and Gln189. D-Tyr1 is in the solvent-exposed S3 site bordered by Gln189, Ala191 and Pro16, while the peptide twists at the selenoether/D-Tyr1 linkage, turning away from the canonical S4 site to be positioned adjacent to Glu166. Tyr4 is therefore positioned in the 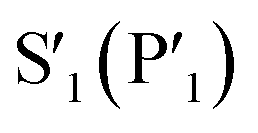 position in ¾ chains, fully consistent with the slow proteolysis between Gln3 and Tyr4 observed by mass spectrometry (Fig. 4c). In chain B, the peptide backbone and Tyr4 have swapped positions (see ESI, Fig. S13a and b†).
position in ¾ chains, fully consistent with the slow proteolysis between Gln3 and Tyr4 observed by mass spectrometry (Fig. 4c). In chain B, the peptide backbone and Tyr4 have swapped positions (see ESI, Fig. S13a and b†).
To obtain a better understanding of the interaction of the full peptide with the protein in the absence of crystal contacts, we modelled the missing residues as accurately as possible with the available density and used this structure as the starting point for triplicate 333 ns molecular dynamics (MD) simulations52 (∼1 ms total simulation time; Fig. 5c and S14, ESI†). The results were consistent with the crystal structure, i.e. residues 1–4 were all relatively stable within their respective binding pockets within the substrate cleft of SARS-CoV-2 Mpro, with the mobility of the peptide increasing on either side of these residues (Fig. 5d). Notably, the simulations showed regular transient interactions between the peptide and the second chain of the physiological dimer.
The structure and MD simulations also provide a molecular explanation for the inhibitory activity of the alanine mutants of 1. Specifically, the structure shows that the central interactions are formed by Leu2 and Gln3, consistent with the large reductions in activity when these positions are mutated to Ala, while D-Tyr1 and Tyr4 also form significant interactions with the protease on either side of these residues. Other positions (8, 9, 10, 12 and 13) that were observed to have little influence on inhibitory activity are either disordered or solvent-exposed. MD simulations suggest that Arg11, the mutation of which to Ala had a significant effect on inhibition (Fig. 4d), makes contacts across the dimer interface (see ESI Fig. S13e†), interacting with the side chain and main chain carbonyl of Q256. This is consistent with electron density showing Arg11 within hydrogen bonding distance of the neighbouring chain B in one dimer (see ESI Fig. S13f†). These observations are consistent with the SEC-MALLS and NMR data that shows the peptide binds the dimeric form of SARS-CoV-2 Mpro exclusively for its mode of inhibition (Fig. 4a and b).
Antiviral activity of cyclic peptide SARS-CoV-2 Mpro inhibitors
Given the promising inhibitory activity of cyclic peptides 1–6 against SARS-CoV-2 Mpro, we next assessed the antiviral activity of the four most potent peptides against SARS-CoV-2 in vitro. Specifically, we used ACE2 and TMPRSS2 overexpressing HEK293T cells infected with SARS-CoV-2 to assess each peptide. This cell line is hyper-permissive to SARS-CoV-2 infection with extensive viral syncytia forming after 18 hours post viral infection. Dose-dependent sigmoidal inhibition curves can be generated through enumeration of remaining single cell nuclei using a live nuclear dye and a standard high content fluorescent microscope. In this setting, we observed a dose-dependent protection for three of the four peptides, with EC50 values ranging from 11.8–33.1 μM. However, Peptide 1, the most active compound in the enzyme inhibition assay, was inactive up to a concentration of 50 μM, most likely due to low cell permeability owing to the large number of polar and charged residues in this molecule. Cell-penetrating peptides have been developed that can transport molecular cargo that may be too large or polar to enter cells passively (as is the case of 1) by entering cells via alternate mechanisms such as endocytosis.53 To facilitate cell entry of 1, we therefore conjugated its C-terminus to penetratin, a 16 amino acid cell-penetrating peptide (CPP) derived from the Drosophila Antennapedia homeodomain54 (to afford pen-1). It should be noted that during the selection process the C-termini of the peptides are conjugated to a large mRNA:cDNA tag, and therefore addition of a CPP to the C-terminus of the peptide (to afford pen-1) was deemed unlikely to affect binding and inhibition of Mpro. Pleasingly, pen-1 had equivalent in vitro inhibitory activity against SARS-CoV-2 Mpro to peptide 1 (Ki = 9 nM for pen-1vs. 14 nM for 1, Fig. S15A, ESI†) indicating that addition of a CPP to the C-terminus of the peptide did not affect binding and inhibition of the protease. We also prepared a penetratin conjugate of cyclic peptide 6, the second most active peptide in the enzyme inhibition assay (Ki = 360 nM). While parent cyclic peptide 6 showed antiviral activity in the cell-based assay (EC50 of 33.1 μM), we anticipated that the conjugation to penetratin might further improve potency. We also assessed the cytotoxicity of 1, pen-1, 6 and pen-6 on HEK293-ACE2-TMPRSS2 cells, with none of the compounds affecting cell viability at a concentration of 50 μM (see ESI, Fig. S16†). Pleasingly, we found that pen-1 and pen-6 both exhibited significantly improved antiviral activity, with EC50 values of 15.2 μM and 6.6 μM, respectively (Fig. 6). As expected, the marked improvement in antiviral activity of peptide 1 by conjugation to the penetratin CPP correlated with enhanced cellular uptake, whereby LC-MS/MS analysis showed a 5.5-fold increase in levels of pen-1 compared to 1 in cell lysates (see ESI, Fig. S17a–c†). Interestingly, peptides 2 and 5 possessed significant antiviral activity (11.8 μM and 13.0 μM, respectively) despite displaying only moderate inhibitory activity against Mpro. In order to rationalise this observation, we also assessed the cell permeability of 2 and 5 by LCMS/MS. Both 2 and 5 were detected in lysates at concentrations that exceeded that of the more potent Mpro inhibitors 1 and 6 (see ESI, Fig. S17d–f†). This suggests that peptides 2 and 5 may also serve as interesting leads for future development of cell-permeable cyclic peptide SARS-CoV-2 Mpro inhibitors.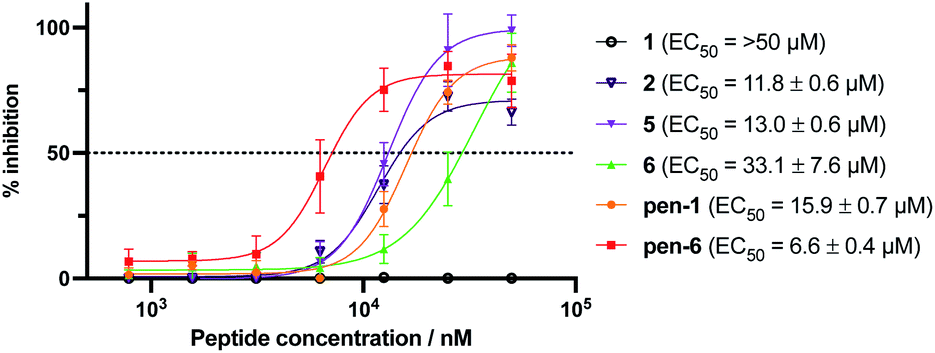 | ||
| Fig. 6 Antiviral activity of peptides 1, 2, 5, 6 and penetratin conjugates of 1 and 6 (pen-1 and pen-6). HEK293-ACE2-TMPRSS2 cells were incubated with varying concentrations of cyclic peptide Mpro inhibitors and infected with SARS-CoV-2. Inhibition curves and 50% effective concentrations (EC50) were determined by non-linear regression analysis using GraphPad Prism. Data are the means ± SD of experiments performed in quadruplicate. Negative control Ac-PEG2-Penetratin EC50: >50 μM (ESI, Fig. S15B†). | ||
Conclusions
In summary, we discovered macrocyclic peptide inhibitors of SARS-CoV-2 Mpro using RaPID mRNA display technology. A novel protein cross-linking strategy was employed to generate a covalently locked catalytically active dimer, facilitating display selection against solid-supported SARS-CoV-2 Mpro. This enabled the discovery of potent inhibitors of Mpro, including the 14-residue cyclic peptide 1 with a Ki of 14 nM that represents one of the most potent inhibitors reported against the protease to date. SEC-MALLS and NMR spectroscopic studies revealed that this molecule inhibits SARS-CoV-2 Mpro by exclusively binding to the catalytic protease dimer, highlighting the importance of controlling the nature of the immobilized protein construct for display selections; in this case by covalently cross-linking the Mpro homodimer. Co-crystal structures of Mpro and 1 and the selenocysteine derivative Se-1 solved by X-ray crystallography revealed the canonical interaction between a Gln residue and subsite S1, flanked by hydrophobic residues. This half of the peptide was relatively stable, while the hydrophilic face (8–12) was relatively mobile, interacting with solvent and across the dimer interface to facilitate an exclusive dimer binding mode for the inhibitor. The strain imposed by cyclization prevents the peptide from adopting a fully relaxed conformation and likely explains the very slow turnover, despite the inhibitor possessing canonical residues that bind to the key recognition subsites of the protease. Thus, cyclization of the peptide has essentially converted a peptide substrate into a highly potent SARS-CoV-2 Mpro inhibitor, highlighting a key benefit of the cyclic peptides that emerge from RaPID screens in medicinal chemistry studies. Several of the cyclic peptides also exhibited antiviral activity against SARS-CoV-2 in vitro and enhancing cellular uptake of cyclic peptides by conjugation to the CPP penetratin led to dramatic improvements in antiviral activity. This was especially pronounced for lead molecule 1, which was ineffective without the covalent CPP tag but exhibited an EC50 of 15.9 μM following fusion to penetratin. These results demonstrate that peptides 1 and 6 have the potential to be effective antivirals when conjugated to CPPs. It should be noted however, that while penetratin is a well-established CPP that has been successfully utilised for in vivo delivery,55,56 it is known to have a relatively short plasma half-life57,58 and, as such, pen-1 and pen-6 should only be regarded as tool molecules. Therefore, ongoing work in our laboratories seeks to investigate a range of other CPPs with a view to optimising cell-permeability and stability for future in vivo experiments. Nonetheless, the cyclic peptides discovered in this study serve as bona fide starting points for the rational design of peptide- or peptidomimetic-based antivirals for COVID-19 that target SARS-CoV-2 Mpro. Moreover, the crosslinked protease selection strategy developed as part of this work is expected to inform medicinal chemistry campaigns targeting other dimeric coronaviral main proteases. Future work in our laboratories will involve lead optimization of the discovered peptides for improved activity and cell permeability both by rational design and high-throughput affinity maturation59,60 with a view to developing molecules with potent antiviral activity in vivo.Data availability
Sequences of peptides emerging from RaPID selections, characterisation data for cyclic peptides and SARS-CoV-2 Mpro, inhibition and selectivity data, plasma stability data and molecular dynamics data are provided in the ESI.† Crystallographic coordinates and structure factors for the structure of the SARS-CoV-2 Mpro-Se-1 complex have been deposited in the PDB (ID: 7RNW).Author contributions
JJ-L., GO, ST, CJJ, CN and RJP conceived the project. JJ-L performed protein cross-linking and SEC-MALLS. JJ-L and TP performed RaPID selection experiments. JJ-L, SEF and MJB performed peptide synthesis. SU performed all Mpro inhibition assays. KBE and MCM produced wild-type and mutant variants of SARS-CoV-2 Mpro and performed NMR assignments. RF performed Mpro crystallographic studies. VMS performed molecular dynamics simulations. AA performed all SARS-CoV-2 inhibition assays. ASA assessed compounds for cytotoxicity and conducted cellular uptake experiments. ML performed proteomics experiments. JJ-L, SU, GO, ST, CJJ, CN and RJP wrote the paper with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the ARC Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012 to GO, CJJ and RJP) for funding and the John A. Lamberton Research Scholarship (to JJ-L) and Research Training Program from the Australian government (to JJ-L, SEF and MJB) for PhD scholarships. CN acknowledges ARC funding (DE190100015, DP200100348). This research was undertaken in part using cyclic peptide display screening facilities in Sydney Analytical, and the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. Next generation sequencing was conducted through the Ramaciotti Centre for Genomics. ML is a Cancer Institute New South Wales Future Research Leader Fellow. We thank SydneyMS for providing the instrumentation used in this study. We also acknowledge Karishma Patel for her assistance in the use of SEC-MALLS.References
- P. Zhou, X. L. Yang, X. G. Wang, B. Hu, L. Zhang, W. Zhang, H. R. Si, Y. Zhu, B. Li, C. L. Huang, H. D. Chen, J. Chen, Y. Luo, H. Guo, R. D. Jiang, M. Q. Liu, Y. Chen, X. R. Shen, X. Wang, X. S. Zheng, K. Zhao, Q. J. Chen, F. Deng, L. L. Liu, B. Yan, F. X. Zhan, Y. Y. Wang, G. F. Xiao and Z. L. Shi, Nature, 2020, 579, 270–273 CrossRef CAS PubMed.
- E. Dong, H. Du and L. Gardner, Lancet Infect. Dis., 2020, 20, 533–534 CrossRef CAS PubMed.
- E. C. Wall, M. Wu, R. Harvey, G. Kelly, S. Warchal, C. Sawyer, R. Daniels, P. Hobson, E. Hatipoglu and Y. Ngai, Lancet, 2021, 397, 2331–2333 CrossRef CAS.
- Y. Wang, D. Zhang, G. Du, R. Du, J. Zhao, Y. Jin, S. Fu, L. Gao, Z. Cheng and Q. Lu, Lancet, 2020, 395, 1569–1578 CrossRef CAS.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer, S. Kline, D. Lopez de Castilla, R. W. Finberg, K. Dierberg, V. Tapson, L. Hsieh, T. F. Patterson, R. Paredes, D. A. Sweeney, W. R. Short, G. Touloumi, D. C. Lye, N. Ohmagari, M. D. Oh, G. M. Ruiz-Palacios, T. Benfield, G. Fätkenheuer, M. G. Kortepeter, R. L. Atmar, C. B. Creech, J. Lundgren, A. G. Babiker, S. Pett, J. D. Neaton, T. H. Burgess, T. Bonnett, M. Green, M. Makowski, A. Osinusi, S. Nayak and H. C. Lane, N. Engl. J. Med., 2020, 383, 1813–1826 CrossRef CAS PubMed.
- O. Dyer, BMJ, 2020, 371, m4057 CrossRef PubMed.
- B. Cao, Y. Wang, D. Wen, W. Liu, J. Wang, G. Fan, L. Ruan, B. Song, Y. Cai and M. Wei, N. Engl. J. Med., 2020, 382, 1787–1799 CrossRef PubMed.
- P. W. Horby, M. Mafham, J. L. Bell, L. Linsell, N. Staplin, J. Emberson, A. Palfreeman, J. Raw, E. Elmahi and B. Prudon, Lancet, 2020, 396, 1345–1352 CrossRef CAS.
- G. Schreiber, Front. Immunol., 2020, 11, 595739 CrossRef CAS PubMed.
- P. D. Monk, R. J. Marsden, V. J. Tear, J. Brookes, T. N. Batten, M. Mankowski, F. J. Gabbay, D. E. Davies, S. T. Holgate, L. P. Ho, T. Clark, R. Djukanovic, T. M. A. Wilkinson, M. G. Crooks, D. P. S. Dosanjh, S. Siddiqui, N. M. Rahman, J. A. Smith, A. Horsley, T. W. Harrison, D. Saralaya, L. McGarvey, A. Watson, E. Foster, A. Fleet, D. Singh, S. Hemmings, S. Aitken, S. Dudley, R. Beegan, A. Thompson and P. M. B. Rodrigues, Lancet Respir. Med., 2021, 9, 196–206 CrossRef CAS PubMed.
- D. R. Boulware, M. F. Pullen, A. S. Bangdiwala, K. A. Pastick, S. M. Lofgren, E. C. Okafor, C. P. Skipper, A. A. Nascene, M. R. Nicol and M. Abassi, N. Engl. J. Med., 2020, 383, 517–525 CrossRef CAS PubMed.
- C. P. Skipper, K. A. Pastick, N. W. Engen, A. S. Bangdiwala, M. Abassi, S. M. Lofgren, D. A. Williams, E. C. Okafor, M. F. Pullen and M. R. Nicol, Ann. Intern. Med., 2020, 173, 623–631 CrossRef PubMed.
- A. B. Cavalcanti, F. G. Zampieri, R. G. Rosa, L. C. Azevedo, V. C. Veiga, A. Avezum, L. P. Damiani, A. Marcadenti, L. Kawano-Dourado and T. Lisboa, N. Engl. J. Med., 2020, 383, 2041–2052 CrossRef CAS PubMed.
- T. A. Tummino, V. V. Rezelj, B. Fischer, A. Fischer, M. J. O'Meara, B. Monel, T. Vallet, K. M. White, Z. Zhang, A. Alon, H. Schadt, H. R. O'Donnell, J. Lyu, R. Rosales, B. L. McGovern, R. Rathnasinghe, S. Jangra, M. Schotsaert, J. R. Galarneau, N. J. Krogan, L. Urban, K. M. Shokat, A. C. Kruse, A. García-Sastre, O. Schwartz, F. Moretti, M. Vignuzzi, F. Pognan and B. K. Shoichet, Science, 2021, 373, 541–547 CrossRef CAS PubMed.
- R. C. Group, N. Engl. J. Med., 2020, 384, 693–704 Search PubMed.
- M. J. Keller, E. A. Kitsis, S. Arora, J. T. Chen, S. Agarwal, M. J. Ross, Y. Tomer and W. Southern, J. Hosp. Med., 2020, 15, 489–493 Search PubMed.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N. H. Wu, A. Nitsche, M. A. Müller, C. Drosten and S. Pöhlmann, Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- A. Bayati, R. Kumar, V. Francis and P. S. McPherson, J. Biol. Chem., 2021, 296, 100306 CrossRef CAS PubMed.
- J. Shang, Y. Wan, C. Luo, G. Ye, Q. Geng, A. Auerbach and F. Li, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 11727–11734 CrossRef CAS PubMed.
- P. V’kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef PubMed.
- D. Shin, R. Mukherjee, D. Grewe, D. Bojkova, K. Baek, A. Bhattacharya, L. Schulz, M. Widera, A. R. Mehdipour, G. Tascher, P. P. Geurink, A. Wilhelm, G. J. van der Heden van Noort, H. Ovaa, S. Müller, K. P. Knobeloch, K. Rajalingam, B. A. Schulman, J. Cinatl, G. Hummer, S. Ciesek and I. Dikic, Nature, 2020, 587, 657–662 CrossRef CAS PubMed.
- Y. Wu, L. Ma, Z. Zhuang, S. Cai, Z. Zhao, L. Zhou, J. Zhang, P. H. Wang, J. Zhao and J. Cui, Signal Transduct. Target. Ther., 2020, 5, 221 CrossRef CAS PubMed.
- S. Y. Fung, K. L. Siu, H. Lin, M. L. Yeung and D. Y. Jin, Int. J. Biol. Sci., 2021, 17, 1547–1554 CrossRef CAS PubMed.
- D. Blanco-Melo, B. E. Nilsson-Payant, W. C. Liu, S. Uhl, D. Hoagland, R. Møller, T. X. Jordan, K. Oishi, M. Panis, D. Sachs, T. T. Wang, R. E. Schwartz, J. K. Lim, R. A. Albrecht and B. R. tenOever, Cell, 2020, 181, 1036–1045 CrossRef CAS PubMed.
- J. Hadjadj, N. Yatim, L. Barnabei, A. Corneau, J. Boussier, N. Smith, H. Péré, B. Charbit, V. Bondet, C. Chenevier-Gobeaux, P. Breillat, N. Carlier, R. Gauzit, C. Morbieu, F. Pène, N. Marin, N. Roche, T. A. Szwebel, S. H. Merkling, J. M. Treluyer, D. Veyer, L. Mouthon, C. Blanc, P. L. Tharaux, F. Rozenberg, A. Fischer, D. Duffy, F. Rieux-Laucat, S. Kernéis and B. Terrier, Science, 2020, 369, 718–724 CrossRef CAS PubMed.
- K. Anand, G. J. Palm, J. R. Mesters, S. G. Siddell, J. Ziebuhr and R. Hilgenfeld, EMBO J., 2002, 21, 3213–3224 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- S. Ullrich and C. Nitsche, Bioorg. Med. Chem. Lett., 2020, 30, 127377 CrossRef CAS PubMed.
- K. Anand, J. Ziebuhr, P. Wadhwani, J. R. Mesters and R. Hilgenfeld, Science, 2003, 300, 1763–1767 CrossRef CAS PubMed.
- B. Boras, R. M. Jones, B. J. Anson, D. Arenson, L. Aschenbrenner, M. A. Bakowski, N. Beutler, J. Binder, E. Chen, H. Eng, H. Hammond, J. Hammond, R. E. Haupt, R. Hoffman, E. P. Kadar, R. Kania, E. Kimoto, M. G. Kirkpatrick, L. Lanyon, E. K. Lendy, J. R. Lillis, J. Logue, S. A. Luthra, C. Ma, S. W. Mason, M. E. McGrath, S. Noell, R. S. Obach, M. N. O'Brien, R. O'Connor, K. Ogilvie, D. Owen, M. Pettersson, M. R. Reese, T. F. Rogers, R. Rosales, M. I. Rossulek, J. G. Sathish, N. Shirai, C. Steppan, M. Ticehurst, L. W. Updyke, S. Weston, Y. Zhu, K. M. White, A. García-Sastre, J. Wang, A. K. Chatterjee, A. D. Mesecar, M. B. Frieman, A. S. Anderson and C. Allerton, Nat. Commun., 2021, 12, 6055 CrossRef CAS PubMed.
- R. L. Hoffman, R. S. Kania, M. A. Brothers, J. F. Davies, R. A. Ferre, K. S. Gajiwala, M. He, R. J. Hogan, K. Kozminski, L. Y. Li, J. W. Lockner, J. Lou, M. T. Marra, L. J. Mitchell Jr, B. W. Murray, J. A. Nieman, S. Noell, S. P. Planken, T. Rowe, K. Ryan, G. J. Smith III, J. E. Solowiej, C. M. Steppan and B. Taggart, J. Med. Chem., 2020, 63, 12725–12747 CrossRef CAS PubMed.
- S. Vankadara, Y. X. Wong, B. Liu, Y. Y. See, L. H. Tan, Q. W. Tan, G. Wang, R. Karuna, X. Guo, S. T. Tan, J. Y. Fong, J. Joy and C. S. B. Chia, Bioorg. Med. Chem. Lett., 2021, 48, 128263 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- K. Vandyck and J. Deval, Curr. Opin. Virol., 2021, 49, 36–40 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X. M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L. K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- C. Ma, M. D. Sacco, B. Hurst, J. A. Townsend, Y. Hu, T. Szeto, X. Zhang, B. Tarbet, M. T. Marty, Y. Chen and J. Wang, Cell Res, 2020, 30, 678–692 CrossRef CAS PubMed.
- K. S. Yang, X. R. Ma, Y. Ma, Y. R. Alugubelli, D. A. Scott, E. C. Vatansever, A. K. Drelich, B. Sankaran, Z. Z. Geng and L. R. Blankenship, ChemMedChem, 2020, 16, 942–948 CrossRef PubMed.
- N. Kitamura, M. D. Sacco, C. Ma, Y. Hu, J. A. Townsend, X. Meng, F. Zhang, X. Zhang, M. Ba, T. Szeto, A. Kukuljac, M. T. Marty, D. Schultz, S. Cherry, Y. Xiang, Y. Chen and J. Wang, J. Med. Chem., 2022, 65(4), 2848–2865 CrossRef CAS PubMed.
- L. Fu, F. Ye, Y. Feng, F. Yu, Q. Wang, Y. Wu, C. Zhao, H. Sun, B. Huang, P. Niu, H. Song, Y. Shi, X. Li, W. Tan, J. Qi and G. F. Gao, Nat. Commun., 2020, 11, 4417 CrossRef CAS PubMed.
- M. M. Ghahremanpour, J. Tirado-Rives, M. Deshmukh, J. A. Ippolito, C. H. Zhang, I. Cabeza de Vaca, M. E. Liosi, K. S. Anderson and W. L. Jorgensen, ACS Med. Chem. Lett., 2020, 11, 2526–2533 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- H. Peacock and H. Suga, Trends Pharmacol. Sci., 2021, 42, 385–397 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- Y. Goto, A. Ohta, Y. Sako, Y. Yamagishi, H. Murakami and H. Suga, ACS Chem. Biol., 2008, 3, 120–129 CrossRef CAS PubMed.
- T. Fujino, Y. Goto, H. Suga and H. Murakami, J. Am. Chem. Soc., 2013, 135, 1830–1837 CrossRef CAS PubMed.
- L. Zhu, S. George, M. F. Schmidt, S. I. Al-Gharabli, J. Rademann and R. Hilgenfeld, Antiviral Res., 2011, 92, 204–212 CrossRef CAS PubMed.
- J. Shi and J. Song, FEBS J., 2006, 273, 1035–1045 CrossRef CAS PubMed.
- W. A. Hendrickson, Trends Biochem. Sci., 2000, 25, 637–643 CrossRef CAS PubMed.
- J. Lee, L. J. Worrall, M. Vuckovic, F. I. Rosell, F. Gentile, A. T. Ton, N. A. Caveney, F. Ban, A. Cherkasov, M. Paetzel and N. C. J. Strynadka, Nat. Commun., 2020, 11, 5877 CrossRef CAS PubMed.
- Schrödinger Release 2019-1: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef CAS PubMed.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CrossRef CAS.
- E. J. Nielsen, S. Yoshida, N. Kamei, R. Iwamae, S. Khafagy el, J. Olsen, U. L. Rahbek, B. L. Pedersen, K. Takayama and M. Takeda-Morishita, J. Control. Release, 2014, 189, 19–24 CrossRef CAS PubMed.
- K. Muto, N. Kamei, M. Yoshida, K. Takayama and M. Takeda-Morishita, J. Pharm. Sci., 2016, 105, 2014–2017 CrossRef CAS PubMed.
- Z. Qian, J. R. LaRochelle, B. Jiang, W. Lian, R. L. Hard, N. G. Selner, R. Luechapanichkul, A. M. Barrios and D. Pei, Biochemistry, 2014, 53, 4034–4046 CrossRef CAS PubMed.
- L. L. Bennink, D. J. Smith, C. A. Foss, M. G. Pomper, Y. Li and S. M. Yu, Mol. Pharm., 2017, 14, 1906–1915 CrossRef CAS PubMed.
- J. M. Rogers, T. Passioura and H. Suga, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 10959–10964 CrossRef CAS PubMed.
- N. K. Bashiruddin, M. Hayashi, M. Nagano, Y. Wu, Y. Matsunaga, J. Takagi, T. Nakashima and H. Suga, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 31070–31077 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06750h |
| This journal is © The Royal Society of Chemistry 2022 |