
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDimerizing cascades of enallenamides reveal the visible-light-promoted activation of cumulated C–C double bonds†
Andrea
Serafino
a,
Maurizio
Chiminelli
a,
Davide
Balestri
a,
Luciano
Marchiò
a,
Franca
Bigi
ab,
Rai-mondo
Maggi
 a,
Max
Malacria
c and
Giovanni
Maestri
*a
a,
Max
Malacria
c and
Giovanni
Maestri
*a
aDepartment of Chemistry, Life Sciences and Environmental Sustainability, Università di Parma, Parco Area Delle Scienze 17/A, 43124 Parma, Italy. E-mail: giovanni.maestri@unipr.it
bIMEM-CNR, Parco Area Delle Scienze 37/A, 43124 Parma, Italy
cSorbonne Université, Faculty of Science and Engineering, IPCM (UMR CNRS 8232), 4 Place Jussieu, 75252 Paris Cedex 05, France
First published on 26th January 2022
Abstract
The visible-light-promoted activation of conjugated C–C double bonds is well developed, while that of cumulated systems is underexplored. We present the feasibility of this challenging approach. The localization of a triplet on an allenamide arm can be favored over that on a conjugated alkene. Allenamides with an arylacryloyl arm dimerize at room temperature in the presence of visible light and an iridium(III) photocatalyst. Two orthogonal polycyclizations took place and their outcome is entirely dictated by the substitution of the alkene partner. Both cascades afford complex molecular architectures with high selectivity. Products form through the ordered rearrangement of twelve π electrons, providing a [3.2.0] bicyclic unit tethered to a fused tricycle, whose formation included an aryl C–H functionalization step, using disubstituted alkenes. The outcome was reverted with trisubstituted ones, which gave rise to taxane-like bridged tricycles that had two six-membered lactams flanking a cyclooctane ring, which was established through the creation of four alternate C–C bonds.
Introduction
The activation of C–C double bonds allows the synthesis of several elegant products upon either an electron transfer (ET) or an energy transfer (eT) from a species excited by visible light (Scheme 1).1 However, isolated, unbiased alkenes could not be directly activated. This issue can be addressed thanks to a conjugated π-unit. The role of this group has been thoroughly studied for many fragments, including styrenes, dienes and various cinnamic acid derivatives among others.2 The auxiliary is crucial to activate C–C double bonds by stabilizing biradical triplets via benzylic or allylic resonance. In contrast, eT to cumulated double bonds is more challenging because it forms an unstable vinyl radical arm. As a result, allene activation is at present limited to the use of thermal conditions.3 | ||
| Scheme 1 Activation of conjugated vs. cumulated C–C double bonds. | ||
We report herein the first dimerization of enallenamides.4 The term enallenamides has been used to describe the two functional groups, namely alkenes and allenamides, which are crucial for present cycloisomerization sequences, in analogy to those involving enynes and enallenes that are more frequently described in the literature. These visible-light promoted intermolecular cascades give highly complex molecular architectures through an original rearrangement of π-type electrons, including a novel route to cyclooctanes5 through the creation of four alternate C–C bonds.
Moreover, modeling and experimental data present the proof of principle of the activation by eT of cumulated, rather than conjugated, C–C double bonds, thus opening a new avenue for photochemical synthesis.
Following an interest in atom-economical cascades,6 we recently converted en-sulfonylallenamides6c into [3.2.0] bicycles. The [2+2] cycloaddition was coupled with the 1,3-shift of a sulfonyl radical,7 which was transferred to the bridgehead position of the product. We tried to elicit a C–C forming variant by preparing enallenamide 1 with the aim of generating a benzyl radical and induce its 1,3-migration. Experiments to test this hypothesis failed but afforded traces of a polar dimer. We did not find analogous reports in the literature,2,3 and we thus characterized the unexpected product.
The structure of 2 had a [3.2.0] bicyclic unit tethered to a fused tricycle. The formation of 2 involved the complete rearrangement of the π-bonding network of two substrate molecules, creating five new contiguous stereocenters through the assembly of five new C–C bonds and the functionalization of an aromatic C–H group. The photocatalytic cascade enabled the formation of four new cycles in an intermolecular fashion at room temperature and with a complete atom economy.
Deuterium labeling experiments performed to rationalize the hydrogen atom transfers (HATs)8 at work in the sequence (vide infra) led us to prepare 3 that had a trisubstituted alkene. However, the reagent provided a different dimer. Polycycle 4 had a cyclooctane decorated by two bridged lactams. It presented five stereocenters, including two synthetically challenging tetrasubstituted headbridging carbons that reminded us of the B ring of Taxol.9 To the best of our knowledge, the domino synthesis of a cyclooctane via creation of four alternate C–C bonds has not been previously reported.5
The outcome of the present cascades is entirely dictated by the nature of the R group that differentiates 1 from 3, while a variety of functional groups did not affect the domino process. This is remarkable in light of the intrinsic chemoselectivity issues connected with the use of enallenes. Their relatively simple intramolecular cycloaddition could indeed provide up to three different products10 and a mixture of isomers has been observed.3a,11 Moreover, the heat increases further for intermolecular events. The dimerization of methyl allene delivers a mixture of seven different isomers.12 These precedents show that the design of an ordered series of events controlling the regio- and diastereo-chemical demands of a dimerization sequence between two enallene molecules has major inherent challenges. However, solving these issues grants access to a spectacular degree of molecular complexity.
Results and discussion
In initial experiments, 0.2 mmol of 1a and 1 mol% Ir(III) photocatalyst were dissolved in DMF. The mixture was injected using a syringe in a 5 mm NMR tube and degassed by freeze–thaw. The tube was put in a silicon oil bath kept at 25 °C and irradiated using a 14 W household white LED strip for 18 hours (Table 1).|
|
|||
|---|---|---|---|
| Entrya | Catalyst (1 mol%) | Solvent | Yield of 2ab (%) |
| a Reaction conditions: 0.2 mmol of 1a, 1 mol% cat., 2 mL of degassed DMF in a 5 mm NMR tube kept at 25 °C and irradiated with an RGB 14 W LED strip overnight. b 1H NMR yield using 1,2,4,5-tetrachloro-3-nitrobenzene as the internal standard. c Isolated yield. d With 2 mol% cat. e Under air. f Without light. | |||
| 1 | Ir(p-F-ppy)3 | DMF | 28% |
| 2 | (Ir[dF(CF3)ppy]2(dtbpy))PF6 | DMF | 45% |
| 3c | Ir(ppy)3 | DMF | 54% |
| 4d | Ir(ppy)3 | DMF | 47% |
| 5 | Ru(bpy)3(PF6)2 | DMF | — |
| 6 | Eosin Y | DMF | — |
| 7 | Ir(ppy)3 | Dioxane | 32% |
| 8 | Ir(ppy)3 | MeCN | 48% |
| 9e | Ir(ppy)3 | DMF | 31% |
| 10f | Ir(ppy)3 | DMF | — |

The use of Ir(p-F-ppy)3 provided 28% 2a (entry 1, full optimization in the ESI†). The yield of 2a rose to 45% upon using the complex (Ir[dF(CF3)ppy]2(dtbpy))PF6 (entry 2). The best result was achieved with Ir(ppy)3 as the catalyst, which gave 2a in 54% yield (entry 3). A higher catalyst loading proved futile (entry 4). Several other organic and organometallic photocatalysts, such as Ru(bpy)32+ or eosin Y (entries 5 and 6), were unable to trigger the dimerization. A polar solvent was necessary (entries 7 and 8), and the product formed only in traces in apolar media. The presence of dioxygen was detrimental (31%, entry 9). Finally, no trace of 2a formed in the absence of either the Ir complex or light (entry 10). In all cases, tricycle 2′a was observed as a byproduct together with traces of another fused polycycle (vide infra). Enallenamide 1a slowly decomposes in solution and the best results were achieved using freshly prepared batches.
With the best conditions in hand,13 we tested the generality of the polycyclization (Fig. 1). Substrates 1 were prepared by isomerization of the corresponding 1,6-enynes. The latter were generally obtained by sequential Heck coupling, hydrolysis, chlorination and treatment with a secondary amine.
 | ||
| Fig. 1 Synthesis of polycycles 2 from enallenamides 1. | ||
The reaction of 1a on the 1 mmol scale performed in a 2 cm wide Schlenk tube led to a diminished yield of 2a (39%), suggesting that a high surface/volume ratio is beneficial to the cascade. We thus performed the experiment using 1.04 g of substrate hosting the resulting solution in the 5 mm wide internal coil of a refrigerant system, which was surrounded by an LED strip (pictures in the ESI†). The temperature of the apparatus was kept at 25 °C thanks to a water flux circulating in the outer chamber. The experiment allowed us to isolate 2a in 43% yield (447 mg). The dimerization was tolerant to various substituents on the nitrogen atom and products 2a–c were retrieved in moderate to good yields (36–54%, see the ESI† for substrate and scope limitations). The reagent incorporating an aniline unit provided a single diastereoisomer of 2d, although with a diminished yield. In contrast, an N-cyclopropyl arm worked well, affording 2e in 54% yield. Switching to functionalization of the styryl fragment, we started testing para-substituted ones. Strongly electron-withdrawing groups, such as fluorides and trifluoromethyl, could be tolerated, although the products were recovered in low yields. A better result was achieved with a chloride substituent (2f, 32%). The C(sp2)–Br group was untouched by the sequence, offering a convenient handle for further functionalization (2g, 44%). A similar outcome was observed with electron-donating groups (2h, 47%). A substrate with an ortho-substituted aryl gave a better yield (2i, 61%). This result showed that the C(sp2)–H functionalization could smoothly take place on aryls with a single ortho-C–H group. Reagents bearing a meta-substituted aryl led to the recovery of two products in ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Combined yields were very good, particularly using electron-rich substrates (2j–k, 54–63%). These results showed that the aryl C–H functionalization is unaffected by steric factors, suggesting that it is likely an intramolecular homolytic substitution.14 The use of a reagent with an electron-rich protected catechol arm led to a single regioisomer of the dimer in good yield (2l, 61%), which has a fused tetracyclic arm often encountered in bioactive molecules, such as the popular topoisomerase inhibitors etoposide and podophyllotoxin.15
:1 ratio. Combined yields were very good, particularly using electron-rich substrates (2j–k, 54–63%). These results showed that the aryl C–H functionalization is unaffected by steric factors, suggesting that it is likely an intramolecular homolytic substitution.14 The use of a reagent with an electron-rich protected catechol arm led to a single regioisomer of the dimer in good yield (2l, 61%), which has a fused tetracyclic arm often encountered in bioactive molecules, such as the popular topoisomerase inhibitors etoposide and podophyllotoxin.15
Two diastereoisomers of 2 were observed. The relative configuration of the stereocenters of the first one was assigned by X-ray analysis. Comparison of correlation NMR spectra allowed us to assign the relative configuration of the second one too. The [3.2.0] subunit showed identical cross-resonances. The stereotopic C(sp3)–H of the tricyclic unit gave strikingly different NOE correlations, indicating that the relative arrangement of the two subunits of 2 is scrambled. This is likely due to the limited stereocontrol in the intermolecular step.
The structure of 2 showed that several HATs took place throughout the sequence.8 We performed reactions with deuterium-labelled reagents (Scheme 2) to rationalize them.
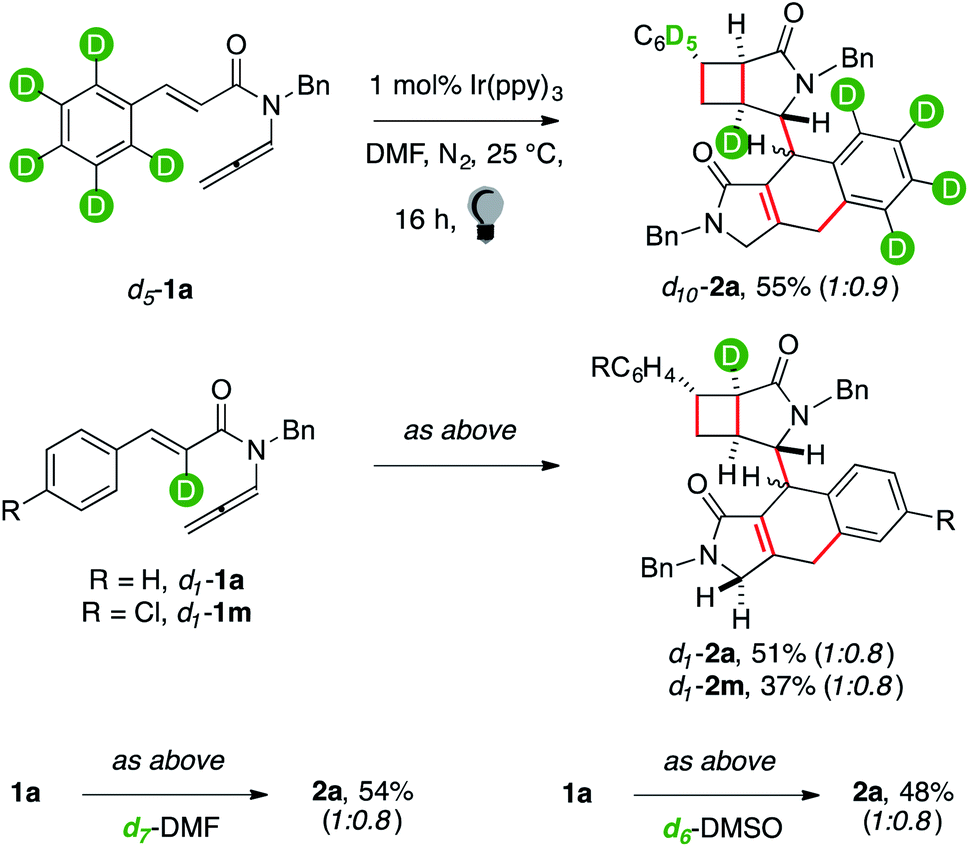 | ||
| Scheme 2 Deuterium labelling experiments. | ||
Enallenamide d5-1a had a deuterated aryl unit. It was tested to observe the position that the hydrogen nucleus, involved in the aryl C–H functionalization, occupied in the product (Scheme 2). The reaction provided d10-2a in 55% yield. NMR analyses coherently indicated that the headbridging position of the [3.2.0] unit away from the carbonyl group was extensively labelled. Deuteration of the C(sp2)–H α to the carbonyl was tested because this position became a quaternary C(sp2) in the fused tricyclic arm of 2. The reaction of d1-1a provided d1-2a in 51% yield. Surprisingly, a single deuterium atom was retrieved in 2. We performed the reaction in deuterated DMF, but no labeling occurred (2a, 54%). The same outcome was observed using deuterated DMSO (2a, 48%). These results suggest that no HAT between the substrate and the solvent occurred. We recovered the photocatalyst at the end of the reaction and checked if its ligands underwent H/D scrambling.16 None was observed. Puzzled by these observations, we prepared d1-1m to confirm the result. The reaction gave mono-deuterated dimer d1-2m (37%). NMR analyses of reaction mixtures quenched at low conversion of d1-1a still showed that the substrate was extensively labelled, strongly suggesting that the H/D exchange took place within the mechanism of the dimerization cascade. MS analyses of the main byproducts of the reaction (2′ and 2′′′) did not show the incorporation in their structure of additional deuterium nuclei. This seems to indicate that the 2H nucleus that is not retrieved in d1-2a and d1-2m has been transferred to those minor unidentified products that account for mass balance in these reactions.
Indeed, 2H NMR analyses of crude mixtures showed several minor deuterium resonances spread across the aliphatic region. This indicated that unselective HATs established the acryloyl unit of 2. We thus prepared 3a, which had no hydrogen atom α to its carbonyl group, to avoid the issue.
The reaction of 3a proceeded with a rate comparable to that of 1a. A dimer was recovered in 64% yield as a 1:0.8 mixture of two diastereoisomers (Fig. 2). X-ray analysis showed that the product was not 2′′a but bridged tricycle 4a. The two diastereomers of 4a differed by the relative configuration of one lactam with respect to the central cyclooctane. We then tested the generality of the new cascade to ascertain if it was a general tool for the preparation of taxane-like5,9 tricyclic cyclooctanes in a single step from two acyclic molecules.
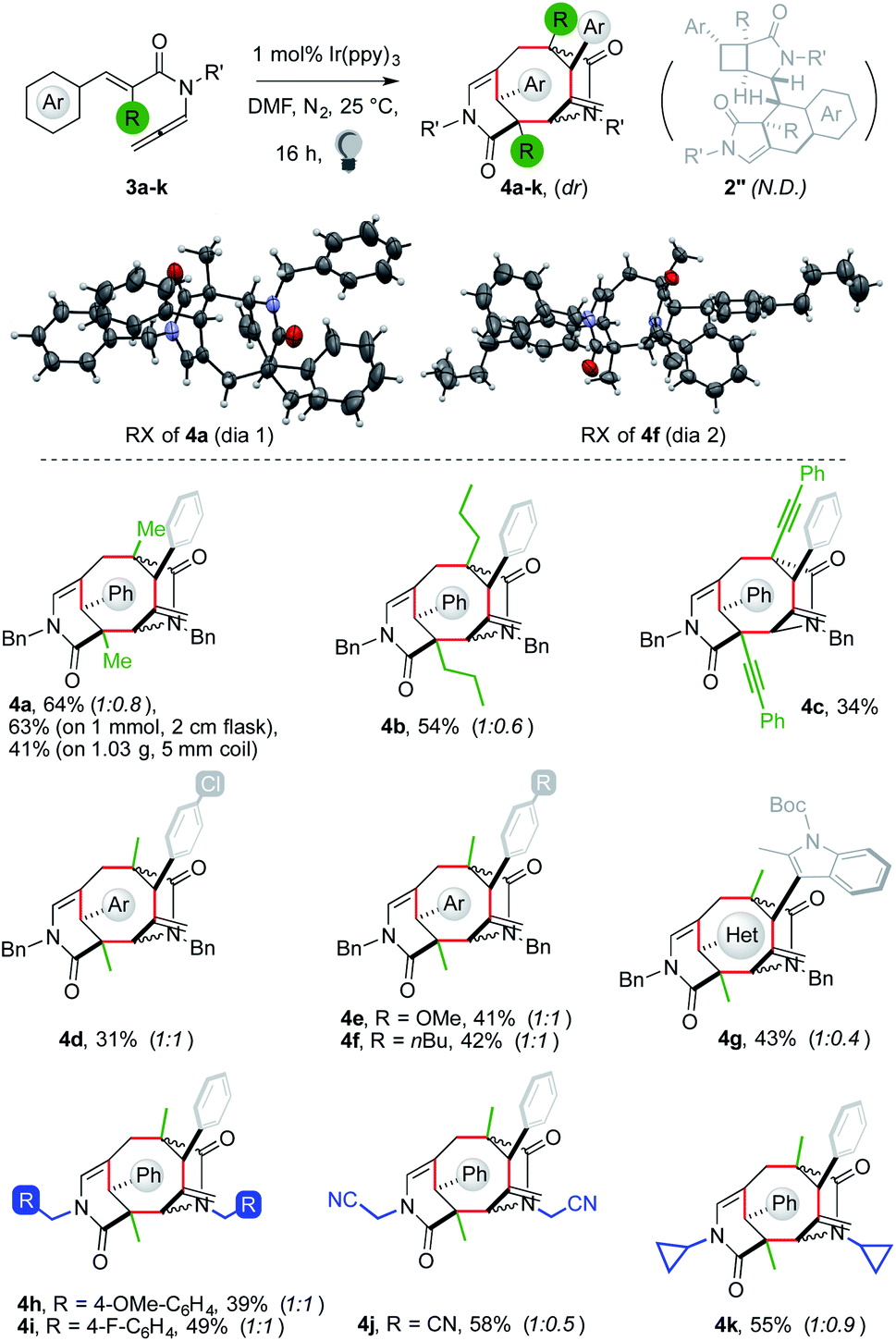 | ||
| Fig. 2 Synthesis of polycycles 4 from enallenamides 3. | ||
Substrates 3 were mostly prepared via malonate functionalization, decarboxylation and aldol reaction with an arylaldehyde. The resulting species were converted into 3 following the route devised for 1. The dimerization of 3a on a 1 mmol scale proved less sensitive to the shape of the reaction flask than that of 1a (vide supra), allowing one to recover 4a in 63% yield. The reaction performed in a coil-type reactor using 1.03 g of 3a led to the isolation of 422 mg of 4a (41%). An n-propyl R substituent led to the formation of 4b in 54% yield. Attempts to replace the R group with an allyl or benzyl group failed because of their unwillingness to undergo alkyne isomerization (limitations in the ESI†). An alkyne motif was tolerated, giving diyne 4c in a synthetically useful yield (34%).17 Switching to substitution of the aryl unit, an electron-withdrawing chloride group gave a moderate yield (4d, 31%). Better results were achieved with a methoxy group and an alkyl chain (4e–f, 41–42%). Remarkably, N-protected indoles could be employed, affording 4g in 43% yield. This result showed that sterically hindered ortho, ortho′-disubstituted (hetero)aryls could be tolerated by the dimerization. Finally, N-substitution on 3 was investigated. The use of a para-methoxybenzyl fragment led to the formation of 4h in 39% yield. A better outcome was achieved with para-fluorobenzyl and cyanomethylene arms (4i–j, 49–58%). N-Cyclopropyl allene 3k gave the corresponding product in 55% yield.18
The activation of 1 and 3 should occur through an eT pathway2 since these reagents did not have redox potentials matching those of the photocatalyst. This was confirmed by positive calculated ΔGs for these redox reactions at the M06/Def2-TZVP level6 (details in the ESI†). The calculated triplet energies of 31α and 33α were +48.7 and +44.1 kcal mol−1 (Scheme 3), matching that of 3Ir(ppy)3 (+56.3 and +55.1 kcal mol−1 for its experimental and calculated triplet energy, respectively).1 The geometries and spin-densities of 31α and 33α paralleled the literature.2 However, lower-lying triplets 31β and 33β were surprisingly found (+41.4 and +40.2 kcal mol−1, respectively). The spin density was spread over the three allenyl carbons in 3β, leaving the cinnamoyl units untouched. The 3β of N-acyl allenamides was more stable than 3α thanks to the presence of the α heteroatom and to a partial bis-allylic stabilization of one spin, which compensated the destabilizing vinyl character of the second one. The higher stability of 3β than 3α is likely further increased in substrates with hindered aryl rings because their ortho substituents impose a lower degree of conjugation to their cinnamoyl fragment (1i, 3g).
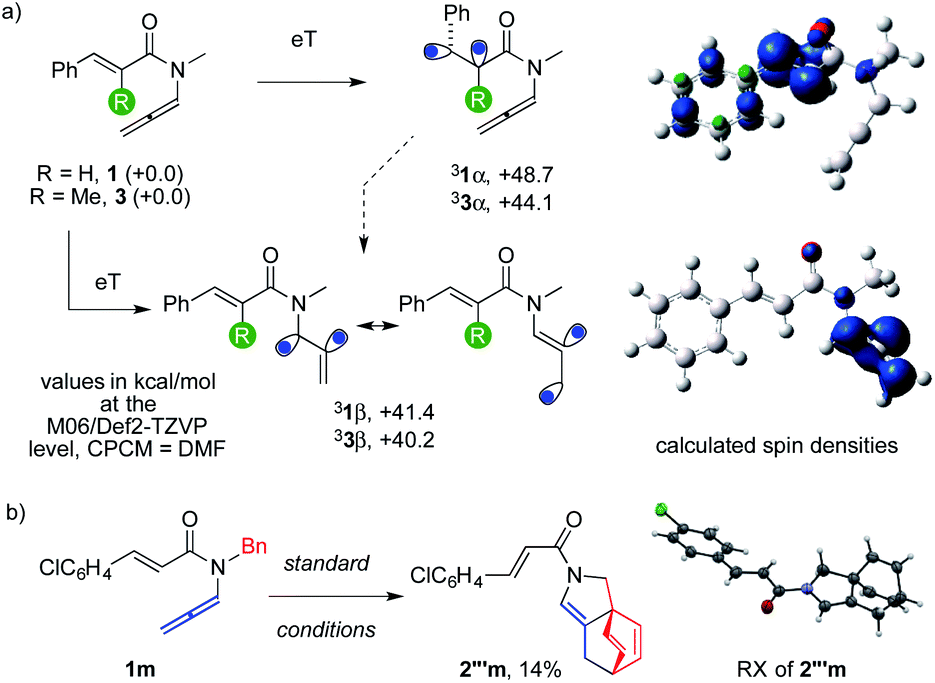 | ||
| Scheme 3 Triplet energies of substrates and experimental proof of allene activation. | ||
These observations suggested the feasibility of activating the cumulated rather than the conjugated C–C double bond of reagents. The experimental proof of this original event1,2 came by the characterization of byproduct 2′′′, for which we got crystals from the reaction of 1m. The structure showed that an exotic para-cycloaddition to the phenyl ring occurred,3b,19 leaving the conjugated alkene unscathed.
The allenamide activation can rationalize the outcome of the cascade employing either 1 or 3 (Fig. 3).
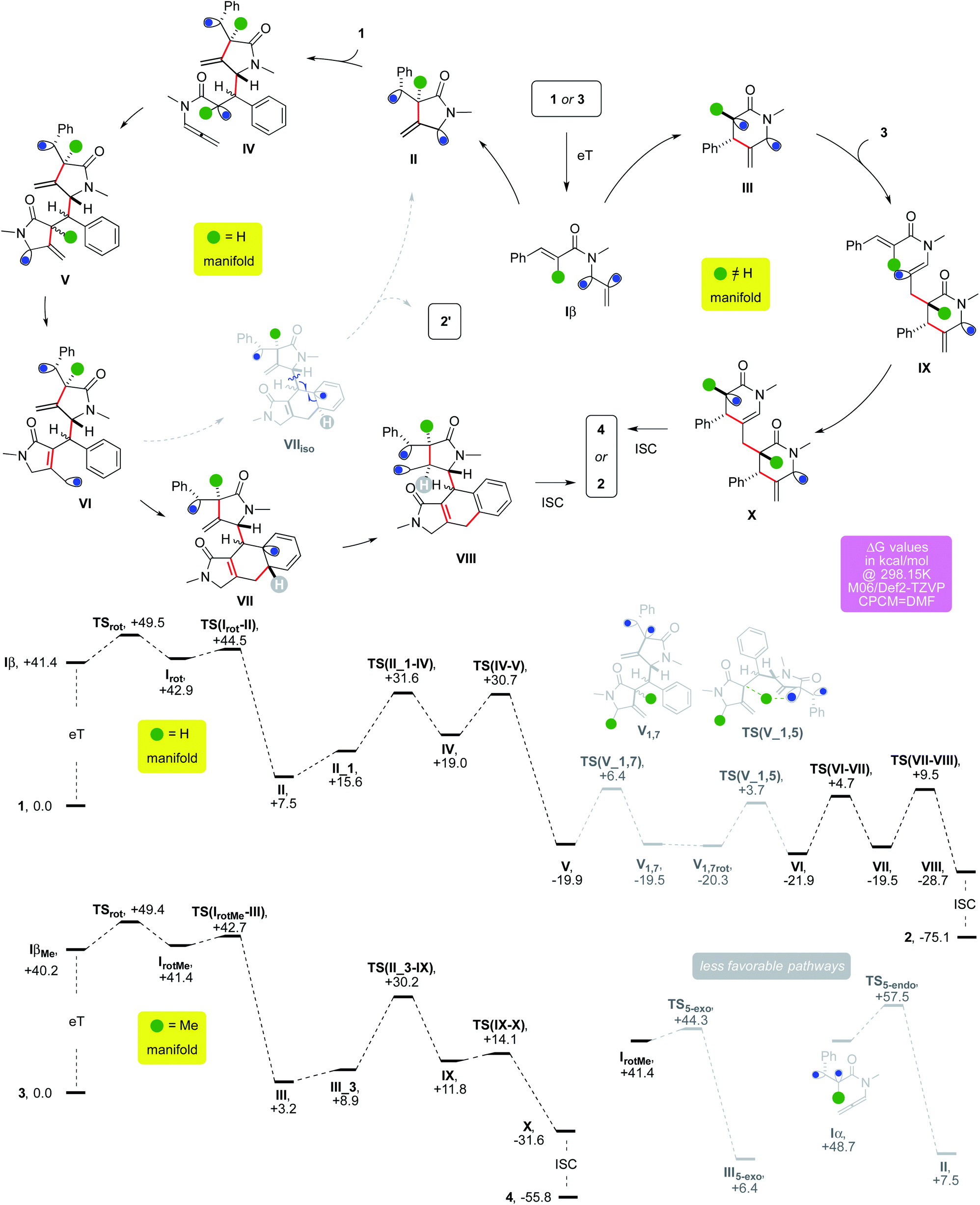 | ||
| Fig. 3 Possible mechanistic rationale and DFT modelling results. | ||
The former would undergo an initial 5-exo-trig cyclization, while a 6-endo-trig one would be favored for the latter because of its trisubstituted alkene.20 Triplet II would add onto a molecule of 1 thanks to polarity reversal.21 The α-acyl radical of IV could undergo 5-exo/endo-dig cyclization yielding V, which would evolve into VI through two HATs. A 6-exo/endo-trig cyclization could provide VII and the latter would give VIII through a favorable 1,5-HAT,8 eventually providing 2 upon ISC. Alternatively, III would add onto the least hindered terminus of 3 exploiting polarity reversal. A steric-driven second 6-endo-trig cyclization from IX would afford X, which seals the cyclooctane via ISC.
This scenario was backed by DFT modeling, which was carried out at the M06/Def2-TZVP level using DMF as implicit solvent. Upon eT, triplet Iβ could twist its former allenyl unit through TSrot by overcoming a barrier of +8.1 kcal mol−1 in ΔG. The resulting intermediate Irot has the unsaturated partners that are significantly closer, paving the way for a low cyclization barrier. Indeed, TS(Irot–II) requires just 1.6 kcal mol−1 in ΔG to occur. This step provides cyclic triplet II, which is considerably more stable thanks to the creation of a new C–C bond (ΔG = +7.5 kcal mol−1). Association of a second substrate molecule comes with a positive enthalpic contribution which is offset by a negative entropic variation (II_1, −1.2 and +15.6 kcal mol−1 for ΔH and ΔG, respectively). The intermediate could then form IVviaTS(II_1–IV) and this barrier requires 16.0 kcal mol−1 in ΔG. The usually challenging intermolecular radical addition is helped in this case by polarity reversal. The secondary allyl radical α to the nitrogen atom of II, which has a nucleophilic character, becomes an electrophilic one in IV because of the neighboring carbonyl. Intermediate IV (ΔG = +19.0 kcal mol−1) can undergo the subsequent cyclization, which could occur viaTS(IV–V) that presents a barrier of +11.7 kcal mol−1 in ΔG. The resulting triplet V lies below the entry channel because of the formation of an additional sigma C–C bond (ΔG = −19.9 kcal mol−1). The two HATs that establish the acryloyl unit of the final product are most likely unselective based on the results of deuterium labelling experiments (d1-2a–m, Scheme 2). It was thus not possible to model a precise pathway for these steps. In order to gain insights into their relative easiness, we modeled an intramolecular alternative based on sequential 1,7 and 1,5-HATs, which are usually favorable processes.8 The first one might in principle occur viaTS(V-1,7), which would require a barrier of +26.5 kcal mol−1. The resulting triplet could then provide intermediate VI through a slightly lower barrier (+24.0 kcal mol−1 in ΔG). These relatively difficult steps show the energetic challenges connected with these two putative HATs. Since no d2-2a–m was recovered, the actual pathway of the cascade, which is responsible for the formal 1,3-HAT that took place between VI and V, should involve more labile hydrogen atoms present in the reaction mixture. Intermediate VI is in the correct spatial arrangement to induce the attack of its allyl radical arm onto the aryl ring, providing the cyclohexadienyl radical fragment of VIIviaTS(VI–VII) (ΔG = +4.7 kcal mol−1). This step might rationalize the energetic convenience of the system to undergo the two elusive and experimentally unselective HATs mentioned above. Indeed, the 6-endo/exo-trig cyclization that attacks the aryl ring modelled from intermediate V resulted in a higher barrier (+33.1 kcal mol−1 in ΔG). This could be ascribed to a Thorpe–Ingold effect for which the lower flexibility of the tether between the aryl ring and the allyl radical in VI than that in V favored the cyclization of the former. An aromatization-driven 1,5-HAT on intermediate VII could then provide exergonic triplet VIII. This step can occur viaTS(VII–VIII) (ΔG = +9.5 kcal mol−1) and it is consistent with the outcome observed in the reaction of d5-1a (Scheme 2). Upon ISC, radical recombination eventually provides 2, which lies expectedly well below the entry channel (ΔG = −75.1 kcal mol−1). This route could explain the formation of byproduct 2′ (Table 1). If the 6-endo/exo-trig cyclization forms intermediate VIIiso, the relative arrangement of the hydrogen atom of the cyclohexadienyl fragment forbids any intramolecular 1,5-HAT. The intermediate could then evolve via β-fragmentation to regenerate II, affording 2′ upon an aromatization-driven ene-rearrangement.
The alternative route, modeled for R = Me, can involve a similar activation of the allenyl unit. This step is followed by a rotation to ensure the proximity of the two unsaturated arms, which occurs viaTSrotMe which requires a barrier of +9.2 kcal mol−1 in ΔG. The resulting triplet IrotMe is 1.2 kcal mol−1 less stable than its parent, thus paralleling the outcome observed for the unsubstituted substrate 1. The presence of the R group disfavors the 5-exo cyclization mode for 3, making the usually less efficient 6-endo one the most convenient route in this case (ΔΔG = −1.6 kcal mol−1). This step forms intermediate III, which lies slightly above the entry channel (ΔG = +3.2 kcal mol−1). The association of an additional substrate molecule can provide III_3 (ΔG = +8.9 kcal mol−1), in analogy to the cascade of 1. The most favorable intermolecular addition involves the reaction of the α-acyl radical of III onto the terminal end of the allenyl unit of 3. This step can occur through a barrier of +21.3 kcal mol−1 in ΔG. Alternatively, the formation of 4 could in principle occur through the reaction of the allyl radical arm of III on the most hindered side of the alkene arm of 3. This intermolecular reaction is, however, more energy costly, and seems therefore less likely (ΔΔG = +8.0 kcal mol−1). Intermediate IX could undergo a second 6-endo-trig cyclization, providing bicyclic triplet X by overcoming the low barrier TS(IX–X) (ΔΔG = +2.3 kcal mol−1). Finally, upon ISC the radical recombination yields exergonic bridged tricycle 4 (ΔG = −55.8 kcal mol−1).
Conclusions
We reported the first dimerization of enallenamides. These catalytic intermolecular cascades are among the longest reported to date for organic reactive intermediates, involving up to eleven elementary steps. These domino reactions occur with a complete atom economy, under ambient conditions, and display remarkable selectivity, which is even more striking given their complexity. These sequential reactions show a noticeably broad functional group tolerance and, nonetheless, their outcome is entirely dictated by the presence on their cinnamoyl arm of either a hydrogen atom or an R group. Up to five new C–C bonds are formed through the ordered rearrangement of the π-bonding network of two linear substrate molecules and an original synthesis of highly congested cyclooctanes is disclosed. Cumulated C–C double bonds can be activated in the presence of visible light, holding ample synthetic potential. Further mechanistic and synthetic studies are ongoing.Data availability
All data presented in this manuscript are available in the ESI.†Author contributions
GM conceived the work with contributions from all authors; AS and MC performed synthetic experiments; DB and LM performed crystallographic analyses; GM performed computational studies; all authors reviewed the data; GM composed the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has benefited from the equipment and framework of the COMP-HUB Initiative, funded by the ‘Departments of Excellence’ program of the Italian Ministry for Education, University and Research (MIUR, 2018–2022). We are also thankful for the support from UniPR. Modeling facilities were kindly provided by the ICSN-CNRS (Gif s/Yvette, France) and the HPC cluster of UniPR.Notes and references
- For selected reviews, see: (a) J. Groβkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626 Search PubMed; (b) F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888 CrossRef CAS; (c) D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org. Chem., 2020, 2020, 1310 CrossRef CAS; (d) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed; (e) M. Wang and P. Lu, Org. Chem. Front., 2018, 5, 254 RSC; (f) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (g) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (h) K. A. Margray and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997 CrossRef PubMed.
- For selected recent examples and seminal studies, see: (a) M. Plaza, J. Großkopf, S. Breitenlechner, C. Bannwarth and T. Bach, J. Am. Chem. Soc., 2021, 143, 11209 CrossRef CAS PubMed; (b) H. Jung, M. Hong, M. Marchini, M. Villa, P. S. Steinlandt, X. Huang, M. Hemmings, E. Meggers, P. Ceroni, J. Park and M.-H. Baik, Chem. Sci., 2021, 12, 9673 RSC; (c) M. Zhu, X.-L. Huang, S. Sun, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2021, 143, 13441 CrossRef PubMed; (d) T. Hostmann, T. Nevesely and R. Gilmour, Chem. Sci., 2021, 12, 10643 RSC; (e) E. N. Hancock, E. L. Kuker, D. J. Tantillo and M. K. Brown, Angew. Chem., Int. Ed., 2020, 59, 436 CrossRef CAS PubMed; (f) M. S. Oderinde, J. Kempson, D. Smith, N. A. Meanwell, E. Mao, J. Pawluczyk, M. Vetrichelvan, M. Pitchai, A. Karmakar, R. Rampulla, J. Li, T. G. M. Dhar and A. Mathur, Eur. J. Org. Chem., 2020, 41 CrossRef CAS; (g) J. C. Beck, C. R. Lacker, L. M. Chapman and S. E. Reisman, Chem. Sci., 2018, 10, 2315 RSC; (h) N. Munster, N. A. Parker, L. van Dijk, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 9468 CrossRef PubMed; (i) M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 7661 CrossRef CAS PubMed; (j) T. Lei, C. Zhou, M.-Y. Huang, L.-M. Zao, B. Yang, C. Ye, H. Xiao, Q.-Y. Meng, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 15407 CrossRef CAS PubMed; (k) A. E. Hurtley, Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 8991 CrossRef CAS PubMed; (l) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329 CrossRef CAS PubMed.
- (a) L. Henn and G. Himbert, Angew. Chem., Int. Ed. Engl., 1982, 21, 620 CrossRef; (b) A. Padwa, M. Meske, S. S. Murphree, S. H. Watterson and Z. Ni, J. Am. Chem. Soc., 1995, 117, 7071 CrossRef CAS ; for a review, see: ; (c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC.
- For selected reviews on the transformation of allenamides, see: (a) R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545 CrossRef CAS PubMed; (b) X. Li, Y. Liu, N. Ding, X. Tan and Z. Zhao, RSC Adv., 2020, 10, 36818 RSC; (c) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465 CrossRef PubMed; (d) C. Praveen, Coord. Chem. Rev., 2019, 392, 1 CrossRef CAS; (e) J. Le Bras and J. Muzart, Chem. Soc. Rev., 2014, 43, 4003 RSC; (f) T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862 CrossRef CAS PubMed; (g) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- For selected strategies, see: (a) S. Kulyk, B. B. Khatri and S. McN. Sieburth, Angew. Chem., Int. Ed., 2017, 56, 319 CrossRef CAS PubMed; (b) R. Brimioulle and T. Bach, Angew. Chem., Int. Ed., 2014, 53, 12921 CrossRef CAS PubMed; (c) C. Zhu, X. Zhang, X. Lian and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 7817 CrossRef CAS PubMed; (d) L. G. Monovich, Y. Hue, Y. Le Huérou, M. Rönn and G. A. Molander, J. Am. Chem. Soc., 2000, 122, 52 CrossRef CAS; (e) S. A. Hitchcock and G. Pattenden, Tetrahedron Lett., 1992, 33, 4843 CrossRef CAS ; a review, see: ; (f) Y.-J. Hu, L.-X. Li, J.-C. Han, L. Min and C. C. Li, Chem. Rev., 2020, 120, 5910 CrossRef CAS PubMed.
- (a) M. Lanzi, V. Santacroce, D. Balestri, L. Marchiò, F. Bigi, R. Maggi, M. Malacria and G. Maestri, Angew. Chem., Int. Ed., 2019, 58, 6703 CrossRef CAS PubMed; (b) G. Cera, N. Della Ca' and G. Maestri, Chem. Sci., 2019, 10, 10297 RSC; (c) A. Serafino, D. Balestri, L. Marchiò, M. Malacria, E. Derat and G. Maestri, Org. Lett., 2020, 22, 6354 CrossRef CAS PubMed; (d) N. Camedda, M. Lanzi, F. Bigi, R. Maggi and G. Maestri, Org. Lett., 2021, 23, 6536 CrossRef CAS PubMed.
- H. Zhang, B. E. Hay, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 16610 CrossRef CAS PubMed.
- For recent examples using visible light, see: (a) Y. Shen, I. Funez-Ardoiz, F. Schoenebeck and T. Rovis, J. Am. Chem. Soc., 2021, 134, 18952 CrossRef PubMed; (b) W. Shu and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1881 CrossRef CAS PubMed; (c) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef CAS PubMed ; for a review, see: ; (d) W. Guo, Q. Wang and J. Zhu, Chem. Soc. Rev., 2021, 50, 7359 RSC.
- For classics, see: (a) K. C. Nicolaou, Z. Yang, J. J. Liu, H. Ueno, P. G. Nantermet, R. K. Guy, C. F. Claiborne, J. Renaud, E. A. Couladouros, K. Paulvannan and E. J. Sorensen, Nature, 1994, 367, 630 CrossRef CAS PubMed; (b) R. A. Holton, C. Somoza, H.-B. Kim, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith, S. Kim, H. Nadizadeh, Y. Suzuki, C. Tao, P. Vu, S. Tang, P. Zhang, K. K. Murthi, L. N. Gentile and J. H. Liu, J. Am. Chem. Soc., 1994, 116, 1597 CrossRef CAS; (c) J. J. Masters, J. T. Link, L. B. Snyder, W. B. Young and S. J. Danishefsky, Angew. Chem., Int. Ed. Engl., 1995, 34, 1723 CrossRef CAS; (d) P. A. Wender, N. F. Badham, S. P. Conway, P. E. Floreancig, T. E. Glass, C. Granicher, J. B. Houze, J. Janichen, D. Lee, D. J. Marquess, P. L. McGrane, W. Meng, T. P. Mucciaro, M. Muhlebach, M. G. Natchus, H. Paulsen, D. B. Rawlins, J. Satkofsky, A. J. Shuker, J. C. Sutton, R. E. Taylor and K. Tomooka, J. Am. Chem. Soc., 1997, 119, 2755 CrossRef CAS ; for a recent approach, see: ; (e) Y. Kanda, H. Nakamura, S. Umemiya, R. K. Puthukanoori, V. R. M. Appala, G. K. Gaddamanugu, B. R. Paraselli and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 10526 CrossRef CAS PubMed.
- (a) J. M. Saya, K. Vos, R. A. Kleinnijenhuis, J. H. van Maarseveen, S. Ingemann and H. Hiemstra, Org. Lett., 2015, 17, 3892 CrossRef CAS PubMed; (b) J. D. Winkler and J. R. Ragains, Org. Lett., 2006, 8, 4031 CrossRef CAS PubMed; (c) M. S. Shepard and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 2597 CrossRef CAS.
- R. Miaou, S. G. Gramani and M. J. Lear, Tetrahedron Lett., 2009, 50, 1731 CrossRef.
- This thermal process is proposed to proceed through biradical intermediates, thus paralleling many photochemical ones, see: (a) J. J. Gajewski and C. N. Shih, J. Org. Chem., 1972, 37, 64 CrossRef CAS ; for a computational study, see: ; (b) S. L. Skraba and R. P. Johnson, J. Org. Chem., 2012, 77, 11096 CrossRef CAS PubMed.
- The average efficiency of each new C–C forming event, assuming quantitative HATs and ignoring any rotational barrier, is above 88% for 2a.
- For reviews, see: (a) C.-S. Wang, P. H. Dixneuf and J. F. Soulé, Chem. Rev., 2018, 118, 7532 CrossRef CAS PubMed; (b) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed.
- (a) W. Zhao, Y. Cong, H.-M. Li, S. Li, Y. Shen, Q. Qi, Y. Zhang, Y.-Z. Li and Y.-J. Tang, Nat. Prod. Rep., 2021, 38, 470 RSC; (b) Y.-Q. Liu, J. Tian, K. Qian, X.-B. Zhao, S. L. Morris-Natschke, L. Yang, X. Nan, X. Tian and K.-H. Lee, Med. Res. Rev., 2015, 35, 1 CrossRef PubMed.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Sully, D. E. Gomez, N. W. Barnett, A. Polyzosa and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646 CrossRef CAS PubMed.
- A single diastereoisomer of 4c was recovered, possibly for a stabilizing stacking between aryl and phenylacetylene arms.
- Attempts to synthesize substrates 3 and 1 with a polysubstituted allenyl unit, either by isomerization, direct alkylation or cross-coupling methods, have been so far fruitless, hampering the possibility of testing them in the present catalytic dimerizations.
- J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338 CrossRef CAS PubMed.
- Intermediates II and III might in principle form via 5-exo/endo-dig cyclization of the least stable 3α, in analogy to ref. 2d. Because of their partial endo character, these cyclization barriers provided higher energies than those of 3β. Moreover, 3α is unable to account for the formation of 2′′′.
- For a review, see: F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed optimization study, computational details, scope limitations, preparation of substrates, characterization of products, copies of NMR spectra, XYZ coordinates, and a comprehensive table in AU. CCDC 2110024, 2110025, 2120965 and 2120966. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06719b |
| This journal is © The Royal Society of Chemistry 2022 |