
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFully fused boron-doped polycyclic aromatic hydrocarbons: their synthesis, structure–property relationships, and self-assembly behavior in aqueous media†
Hiroki
Narita‡
 a,
Heekyoung
Choi‡
b,
Masato
Ito
a,
Naoki
Ando
a,
Soichiro
Ogi
a and
Shigehiro
Yamaguchi
*ab
a,
Heekyoung
Choi‡
b,
Masato
Ito
a,
Naoki
Ando
a,
Soichiro
Ogi
a and
Shigehiro
Yamaguchi
*ab
aDepartment of Chemistry, Graduate School of Science, Integrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan. E-mail: yamaguchi@chem.nagoya-u.ac.jp
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya 464-8601, Japan
First published on 6th January 2022
Abstract
Planarized triarylboranes are attracting increasing attention not only as models of boron-doped graphenes, but also as promising materials for organic optoelectronics. In particular, polycyclic aromatic hydrocarbon (PAH) skeletons with embedded boron atom(s) in the inner positions are of importance in light of their high chemical stability and π-stacking ability derived from their planar geometries. Herein, we disclose a robust synthesis of such fully fused boron-doped PAHs and their self-assembly behavior in aqueous media to explore their potential utility in biological applications. The synthesis using in situ-generated planar diarylboranes as a key precursor afforded a series of fully fused boron-doped PAHs, even including an amphiphilic derivative with hydrophilic side chains. These compounds exhibited red emission in solution, and slight structural modification resulted in increased fluorescence brightness. While these compounds showed relatively low Lewis acidity compared to their partially ring-fused counterparts, their Lewis acidities were slightly increased in polar solvents compared to those in nonpolar solvents. In addition, their B–N Lewis acid–base adducts, even those with a strong, charge-neutral Lewis base such as N,N-dimethylaminopyridine (DMAP), exhibited photo-dissociation behavior in the excited state. The amphiphilic derivative showed significant spectral changes with increased water content in DMSO/H2O mixed media and formed sheet-like aggregates. The disassembly and assembly processes of the aggregates were externally controlled by the addition of DMAP and an acid, accompanied by a change in the fluorescence intensity.
Introduction
The incorporation of a tricoordinate boron atom into π-conjugated skeletons produces promising π-electron materials with various attractive properties, such as intense fluorescence, reversible redox properties, two-photon absorption properties, anion-sensing ability, and photo-reactivity.1 While the conventional strategy to stabilize this compound class is the kinetic protection of the boron center with bulky aryl groups, the structural constraint of the triarylborane scaffold into a planar geometry has been introduced as an alternative approach (Fig. 1).2,3 The less sterically crowded and planar geometries of the thus-produced compounds endow them with not only effective π-conjugation, but also π-stacking ability and sufficient Lewis acidity to form labile adducts with charge-neutral Lewis bases such as pyridine or phosphine derivatives.4–8 These additional characteristics give rise to several attractive phenomena or utilities, and have thereby opened new application avenues. Specifically, a wide range of fluorescent materials have been developed using planarized triarylborane scaffolds and extensively utilized for organic light-emitting diodes (OLEDs).9 The π-stacking ability of the planarized scaffolds also furnishes self-assembly properties and thereby makes these compounds useful components for columnar liquid crystalline materials10 and supramolecular polymers.11 Moreover, their labile complexation ability with Lewis bases has not only led to the emergence of intriguing thermochromism and dual-emission properties,4,6b but has also been utilized for solution-processing thin-film fabrication of insoluble π-electron materials.12 These findings have increased the potential utility of this compound class and therefore promoted the exploration of their chemical space.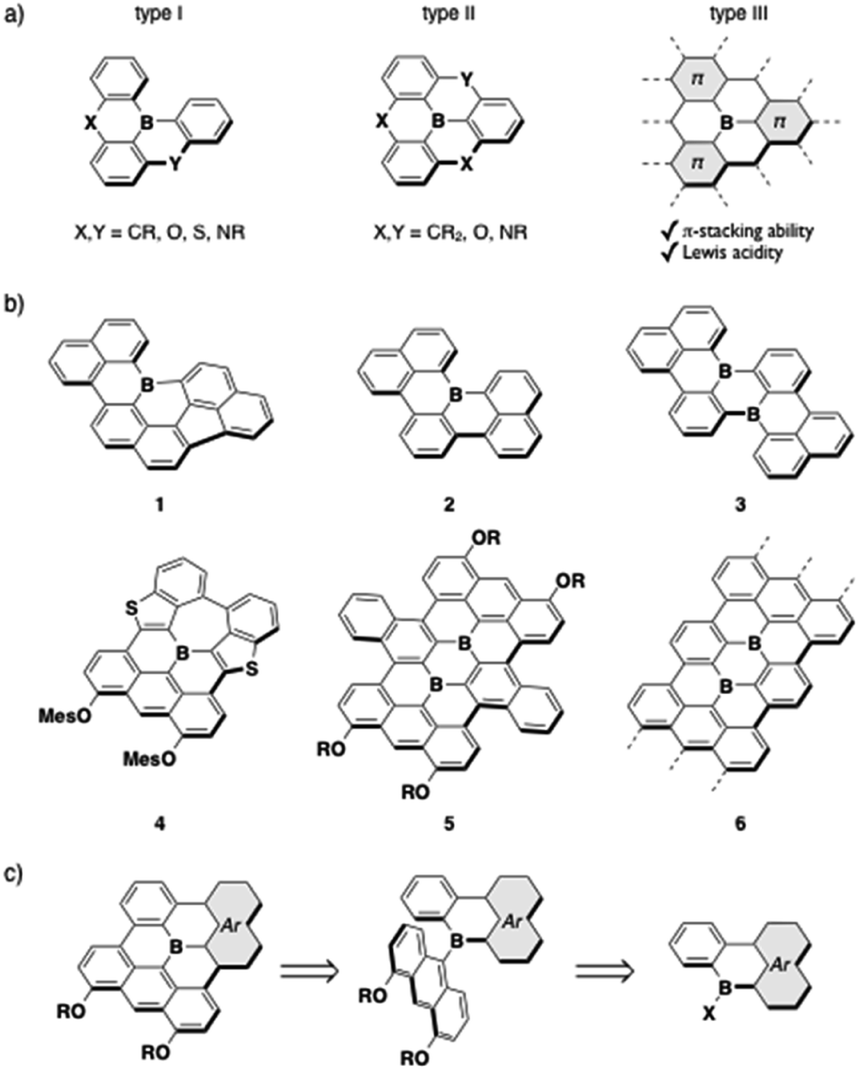 | ||
| Fig. 1 (a) Classification of planarized triarylboranes, (b) examples of previously reported boron-doped PAHs (1–6), and (c) retrosynthetic analysis of type-III fully fused boron-doped PAHs. | ||
The planarized triarylboranes that have been synthesized to date are categorized into three types: (I) partially and (II) fully bridged triarylboranes with carbon or heteroatom bridges, and (III) all sp2-carbon atom bridged analogues (Fig. 1a). Recent progress in synthetic methods has allowed access to a variety of type-I and -II compounds,13 including even type-I compounds with sp2-carbon bridges such as 1–3 (Fig. 1b).4b,5,14 However, the synthesis of type-III fully fused boron-doped polycyclic aromatic hydrocarbons (PAHs) is still limited to a few examples, despite their importance as models of boron-doped graphenes. Two entities of this compound type, 4 and 5, have been reported.4a,15 Both showed characteristic absorption and fluorescence properties in the visible/near-IR region, and their chemisorption ability and utility as battery electrodes, which reflects their reversible redox properties, were demonstrated. Boron-doped graphene nanoribbons (GNRs) 6 with well-defined boron-doping positions were also synthesized via metal-surface reactions, and their electronic and magnetic properties have been investigated.16
Several challenges remain in the further pursuit of functional type-III compounds. The primary one is widening the chemical space through the establishment of synthetic methods. The common key step for the synthesis of 4–6 involves the dehydrogenative oxidative cyclization of anthryl-substituted borane precursors, in which 4,5-diaryloxy-9-anthryl groups particularly play a crucial role in facilitating the cyclization in the synthesis of the boron-doped PAHs 4 and 5.17 In other words, to make this method more robust, strategies to introduce various planarized diarylboryl groups to the anthracene moiety must be developed (Fig. 1c). The other challenge is the exploration of the potential utility of this compound class. Recent successes in utilizing triarylborane-based fluorescent dyes in bioimaging18,19 have encouraged the use of boron-doped PAHs in aqueous media, although previous studies focused on their properties in organic solvents or in the solid state due to their hydrophobic structures. To this end, the type-III compounds are suitable scaffolds in terms of their high chemical stability owing to their rigid structures, while the introduction of hydrophilic side chains is necessary. In addition, their ability to self-assemble into nanostructures taking advantage of their planar geometries should further expand the application range of this compound class.
To tackle these challenges, herein, we report a robust synthesis for a series of type-III boron-doped PAHs. The study of their structure–property relationships showed that the fully fused structures affect not only their photophysical properties but also their Lewis acidity, the latter of which was assessed even in polar solvents. Their B–N Lewis acid–base adducts with pyridine derivatives underwent intriguing photo-dissociation in the excited state. Their self-assembly behavior to form sheet-like aggregates in aqueous media was also revealed, and the disassembly and assembly processes could be controlled by the external addition of Lewis bases and acids. The details of these results are discussed in this article.
Results and discussion
Synthesis of a series of fully fused boron-doped PAHs
A series of fully fused boron-doped PAHs was synthesized based on the generation of planar diarylborane precursors through intramolecular electrophilic C–H borylation13,14a followed by the introduction of an anthryl group and its oxidative dehydrocyclization (Scheme 1a). Thus, starting from o-bromobiaryl 7, lithiation with sec-BuLi followed by treatment with boron tribromide produced bromoboracycle intermediate 8. The subsequent treatment of 8 with lithiated 4,5-dimesityloxyanthracene afforded 9a in a moderate yield. The intramolecular dehydrocyclization of 9a using FeCl3 produced fully fused boron-doped PAH 10a, which was isolated in 48% yield as a purple solid via chromatography on silica gel. In this synthesis, two aryloxy groups were introduced on the anthryl groups not only to increase the solubility of the products, but also to facilitate the oxidative dehydrocyclization.17 The versatility of this method was demonstrated by the synthesis of π-extended analogues 11 and 12, which have phenanthrene and fluoranthene substructures, respectively. Moreover, when 1-bromobenzo[c]phenanthrene was used as a starting material, fully fused boron-doped PAH 13 with a symmetric structure was also obtained. These results demonstrate the robustness of this synthetic method for the preparation of various fully fused boron-doped PAHs.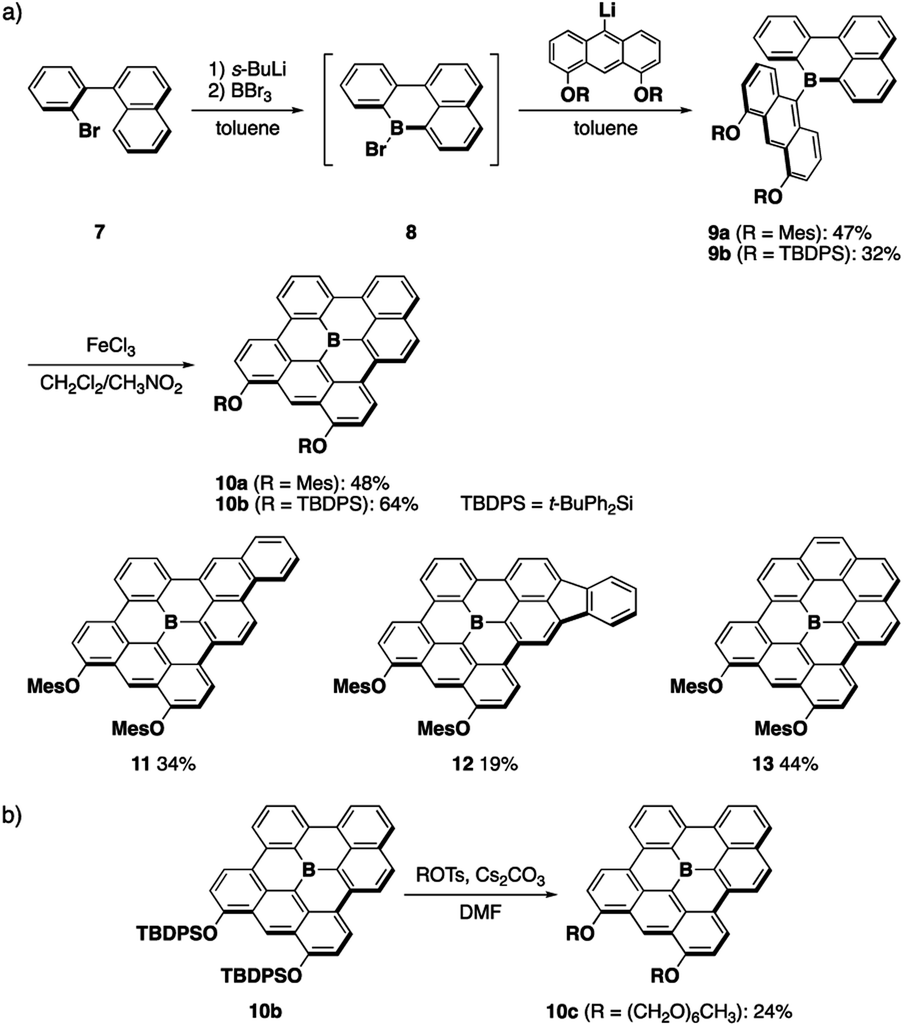 | ||
| Scheme 1 (a) Synthesis of fully fused boron-doped PAHs and (b) late-stage modification of a boron-doped PAH. | ||
The introduction of appropriate side chains onto the PAH scaffolds is crucial not only for increasing their solubility, but also for control of the crystal packing or aggregation modes. For example, we previously introduced several kinds of side chains on 5 to control the packing structure in the crystalline state.15 However, it was necessary to introduce these side chains at the anthryl groups in the initial stage of the synthesis, which made the procedure tedious and required multi-step processes. To solve this issue, we also synthesized t-butyldiphenylsilyl (TBDPS)-protected boron-doped PAH 10b in this study, which allowed us to conduct the late-stage introduction of other side chains (Scheme 1b). Thus, 10b was treated with hexaethylene glycol monomethyl ether tosylate in the presence of cesium carbonate in DMF under heating at 100 °C. In this reaction, deprotection of the TBDPS groups and subsequent alkylation proceeded in situ, without the precipitation of insoluble dihydroxy-substituted products, to afford amphiphilic derivative 10c, which enabled us to study the aggregation behavior of the boron-doped PAH in aqueous media (vide infra).
X-ray crystallographic analysis
The structures of the fully fused boron-doped PAHs 10a, 12, and 13 were determined via single-crystal X-ray diffraction analysis. As a representative example, Fig. 2 shows the structure of 12, which has a highly planar π-skeleton with C–B–C angles that sum to 360° and small torsion angles of 2.18–6.84° between the three hexagon rings directly linked to the boron atom. Notably, 12 has short B–C bond lengths of 1.519(3), 1.506(4), and 1.498(3) Å for B–C1, B–C2, and B–C3, respectively, which are significantly shorter than those of triphenylborane (1.571(3)–1.589(5) Å)20 and slightly shorter or comparable to those of 4 (1.508(2)–1.539(2) Å).4a The crystal packing of 12 forms a π-stacked dimeric motif with an upside-down arrangement and an interplanar distance of 3.34 Å (Fig. 2b and c).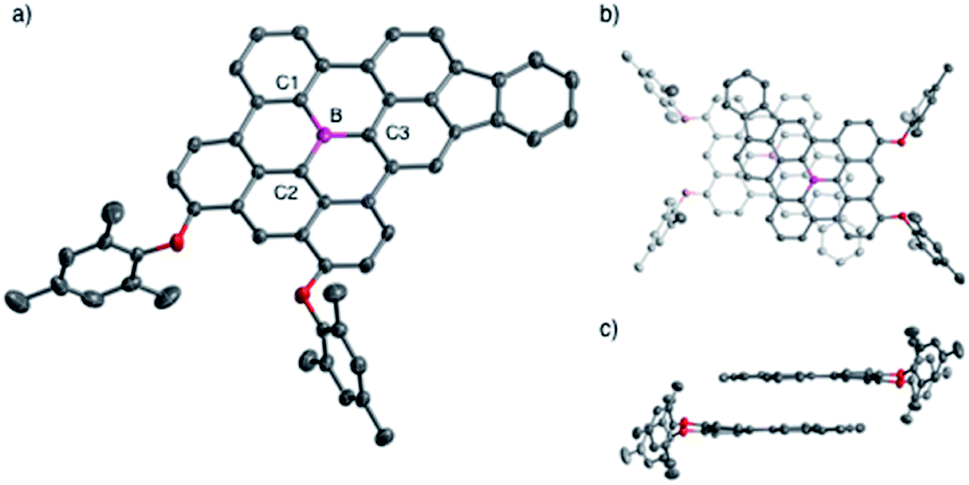 | ||
| Fig. 2 (a) Crystal structure of 12 with thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. (b) Top view and (c) side view of the π-stacked dimeric motif in the crystal packing of 12. | ||
UV-vis absorption and emission properties
Despite its all-hexagon ring-fused π-skeleton without strong electron-donating groups, compound 10a exhibited red emission in toluene. 10a showed a broad absorption band with a maximum wavelength (λabs) of 577 nm and an emission band with a maximum (λem) of 606 nm, with a fluorescence quantum yield (ΦF) of 0.41. These characteristic properties were not significantly affected even by the expansion of the π-skeleton in 11 and 12. Their λabs and λem values (11: λabs = 586 nm, λem = 599 nm; 12: λabs = 595 nm, λem = 614 nm) are comparable to those of 10a (Fig. 3). It should be noted, however, that the π-expansion resulted in an increase in the molar absorption coefficients (ε) and consequently gave rise to increased brightness of the fluorescence (ΦF × ε). For instance, the incorporation of one additional hexagon ring from 10a to 11 increased the fluorescence brightness more than two-fold. In contrast, the variation from 10a to 13 with an additional benzene ring in a symmetric fashion resulted in a slightly blue-shifted absorption band (λabs = 565 nm), whilst their λem and ΦF values are comparable. Taking into account the less-emissive characters of the previously reported fully fused boron-doped PAHs 4 (ΦF = 0.016)4a and 5 (ΦF = 0.04),15a the red-emissive characters of the present compounds 10a–13 with ΦF values of 0.36–0.56, irrespective of their structural symmetry, makes them useful core scaffolds for pursuing functional boron-doped PAHs.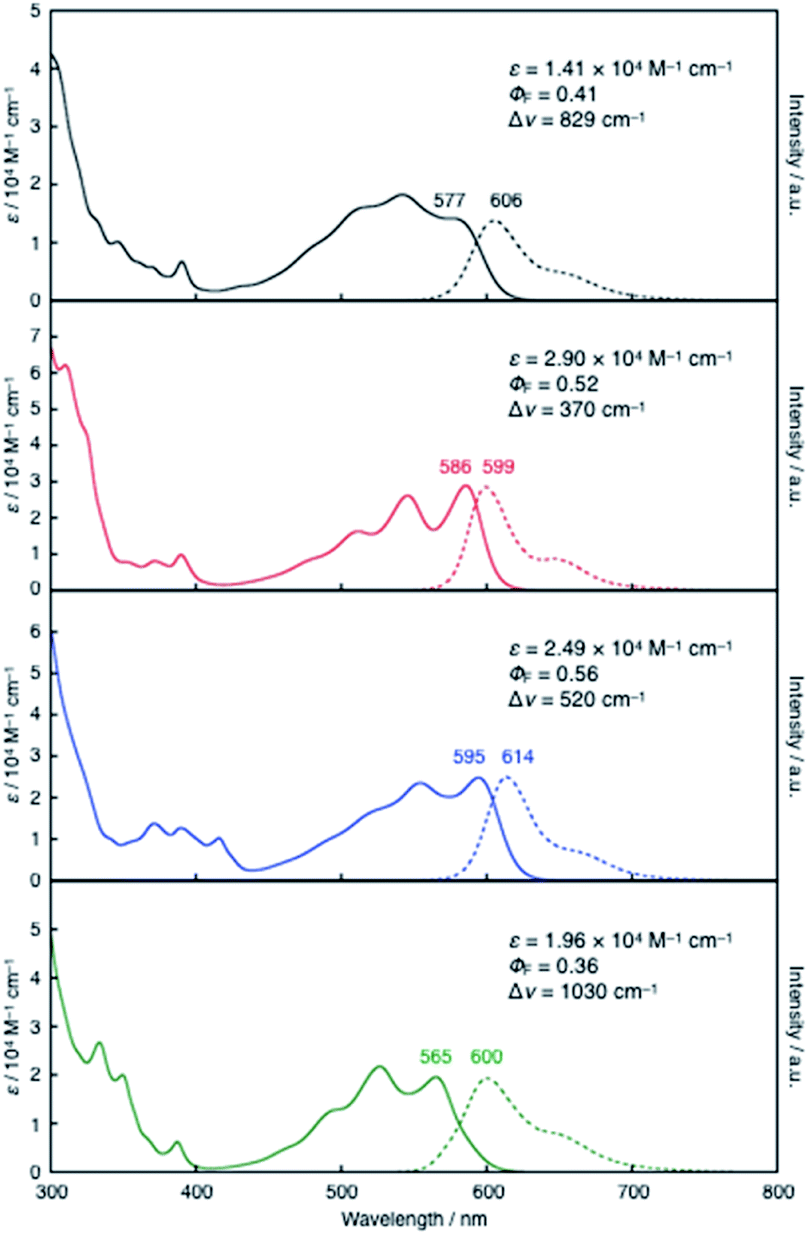 | ||
| Fig. 3 UV-vis absorption (solid lines) and fluorescence spectra (dashed lines) of 10a (black), 11 (red), 12 (blue), and 13 (green) in toluene. | ||
Effects of the planarization on the electronic structure
To understand the inherent electronic features of the present π-systems 10–13, their electronic structures were elucidated using time-dependent density-functional theory (TD-DFT) calculations (B3LYP/6-31G(d) level) and compared with those of non-planarized and partially planarized (type I) model analogues 14 and 15, respectively (Fig. 4; for the data of 11–13, see Fig. S15†). In the comparison among 14, 15, and 10a, the variation in the HOMO level is relatively small, indicating that the dimesityloxyanthracene moiety predominantly determines the HOMO levels, and that delocalization of the HOMO over the π-skeleton does not increase their energy levels. In contrast, the ring-closure modifications from 14 to 15 to 10a effectively decrease the LUMO level, resulting in smaller electronic-transition energies accompanied by increased oscillator strength (f) values. Further expansion of the π-skeleton from 10a to 11 to 12 increases the f values without significantly affecting the transition energy (Fig. S15†). Thus, the combination of the electron-donating diaryloxyanthracene moiety and the fully ring-fused triarylborane moiety is crucial for achieving the red-shifted absorption and emission with high intensities. However, the extent of the intramolecular charge-transfer character in the excited state should be small due to the effective delocalization of the HOMO and LUMO over the π-skeleton. Indeed, the red-shifts observed in the absorption and emission spectra of these compounds with changing the solvent from toluene to CH2Cl2 are only moderate (Table S2 and Fig. S4, S5†). Comparison between 10a and 13 showed that the one additional fused ring results in a decrease in the HOMO level rather than the LUMO level, which is responsible for the slightly blue-shifted λabs and λem of 13. The calculation of 13 at the CAM-B3LYP/6-31G(d) level of theory gave the lowest-energy electronic transition with an allowed character, which is consistent with the experimentally observed result (Fig. S16†).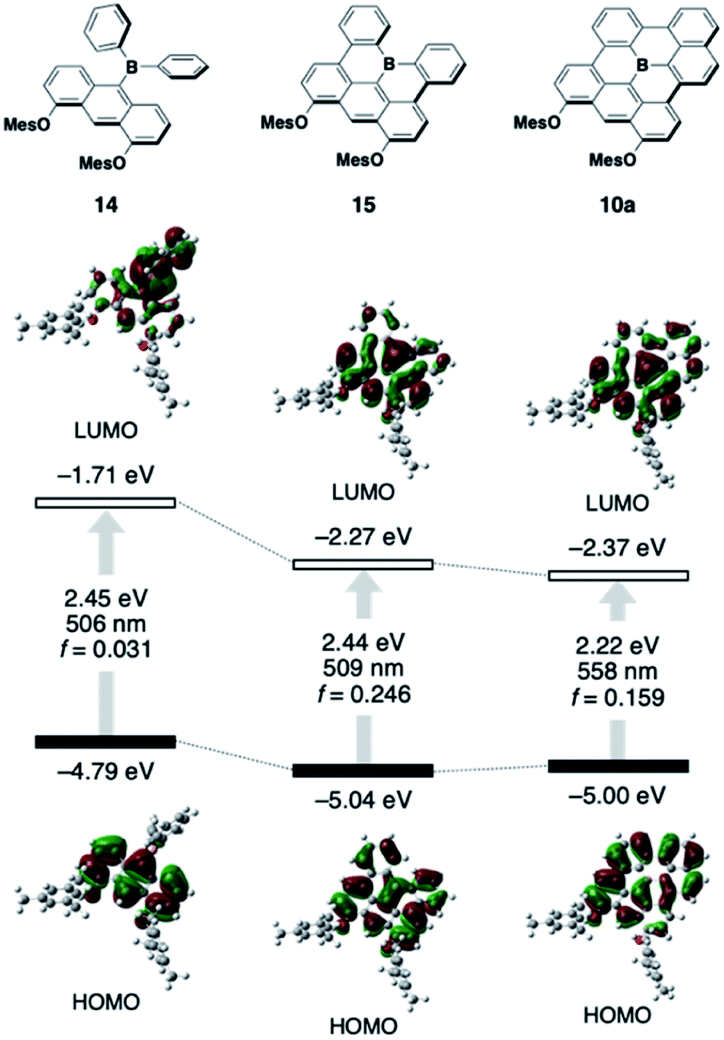 | ||
| Fig. 4 Kohn–Sham molecular orbitals for 10a, 14, and 15, together with their corresponding S0 → S1 excitation energies, wavelengths, and oscillator strengths obtained from TD-DFT calculations at the B3LYP/6-31G(d) level of theory. | ||
Labile Lewis acid–base complexation with DMAP
One attractive feature of the boron-doped PAHs is the Lewis acidity of the boron center, which is sufficient for the formation of a Lewis acid–base complex with charge-neutral bases such as pyridine derivatives. Compared to the type-I partially fused counterparts, the type-III fully fused boron-doped PAHs have lower Lewis acidity. A titration via UV-vis absorption spectroscopy in toluene determined the binding constant (Kpy) of 10a toward pyridine to be 0.66 M−1 (Fig. S6†). This value is comparable to that of 4 (Kpy = 0.35 M−1 in toluene),21 but four orders of magnitude smaller than those of 1 (Kpy = 5.1 × 103 M−1 in toluene)4b and 2 (Kpy = 8.2 × 102 M−1 in toluene).4c This fact suggests that the fully fused boron-doped PAHs would be suitable Lewis acids to study their labile complexation behavior with stronger Lewis bases such as N,N-dimethylaminopyridine (DMAP). Indeed, the binding constant (KDMAP) of 10a toward DMAP was determined to be 5.2 × 102 M−1 in toluene (Table 1). It is worth noting that the KDMAP values vary to some extent depending on the π-expansion modes. Namely, more π-expanded 11 and 12 have KDMAP values that are more than 2- or 3-fold higher, respectively, than those of 10a, while symmetric 13 has a much lower KDMAP value than 10a. The more rigid structure of fully hexagon-ring-fused 13 compared to the other derivatives due to the additional fused hexagon ring likely destabilizes the tetracoordinate species formed by the complexation, thereby decreasing the KDMAP value.| Compound | Solvent | K DMAP/M−1b |
|---|---|---|
| a Determined by titrations with the addition of DMAP monitored using UV-vis absorption spectroscopy. b Determined by the fitting within the error of ±0.1. | ||
| 10a | Toluene | 5.2 × 102 |
| 11 | Toluene | 1.5 × 103 |
| 12 | Toluene | 2.0 × 103 |
| 13 | Toluene | 1.8 × 102 |
| 10c | Toluene | 2.2 × 102 |
| DMSO | 5.3 × 102 | |
DMSO/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) :1, v/v) |
4.6 × 102 | |
Although the Lewis acidity of previously reported boron-doped PAHs has been evaluated in nonpolar solvents such as toluene, their Lewis acidity in polar solvents or aqueous media is crucial for their utilization in biological applications. The availability of amphiphilic boron-doped PAH 10c allowed us to determine the KDMAP values of the boron-doped PAHs in polar media. Titration experiments revealed that the KDMAP of 10c in DMSO increased to 5.3 × 102 M−1, which was more than twice that in toluene (2.2 × 102 M−1). The tetracoordinate borane–DMAP complex with a larger dipole moment is likely stabilized in such polar media, thereby increasing the KDMAP values. Calculations of the model compound 10d (R = Me) and DMAP-adduct 10d·DMAP at the M06-2X/6-311G(d) level of theory supported this rationalization, as the dipole moment increased from 4.33 D for 10d to 10.0 D for 10d·DMAP (Fig. S17†). On the other hand, the KDMAP value of 10c decreased slightly to 4.6 × 102 M−1 in a 9:1 DMSO/H2O mixed solvent, where 10c molecules were present in a monomeric form.
Photo-dissociation of B–N Lewis acid–base adducts
Notably, the Lewis adducts between the fully fused boron-doped PAH skeletons and strong Lewis base DMAP are still labile enough to exhibit photo-responsive behavior in the excited state. Thus, upon the addition of an excess amount of DMAP (9.5 × 10−2 M) to a DMSO solution of 10c (3.2 × 10−5 M), the absorption spectra showed complete conversion to 10c·DMAP (Fig. 5a). However, the fluorescence spectrum of the thus-prepared solution exhibited an emission with a λem of 640 nm, which corresponds to the emission from tricoordinate 10c (Fig. 5b). These results demonstrate that 10c·DAMP undergoes photo-dissociation in the excited state. Namely, in the lowest excited singlet state (S1), DMAP dissociates from [10c·DMAP]* to generate the excited state of tricoordinate 10c*, which produces the red fluorescence (Fig. 5c). While similar photo-dissociation behavior has been reported for several boron-doped PAHs,4,6 this is the first example to show photo-dissociation behavior in the presence of DMAP, a strong, charge-neutral Lewis base, in a polar solvent.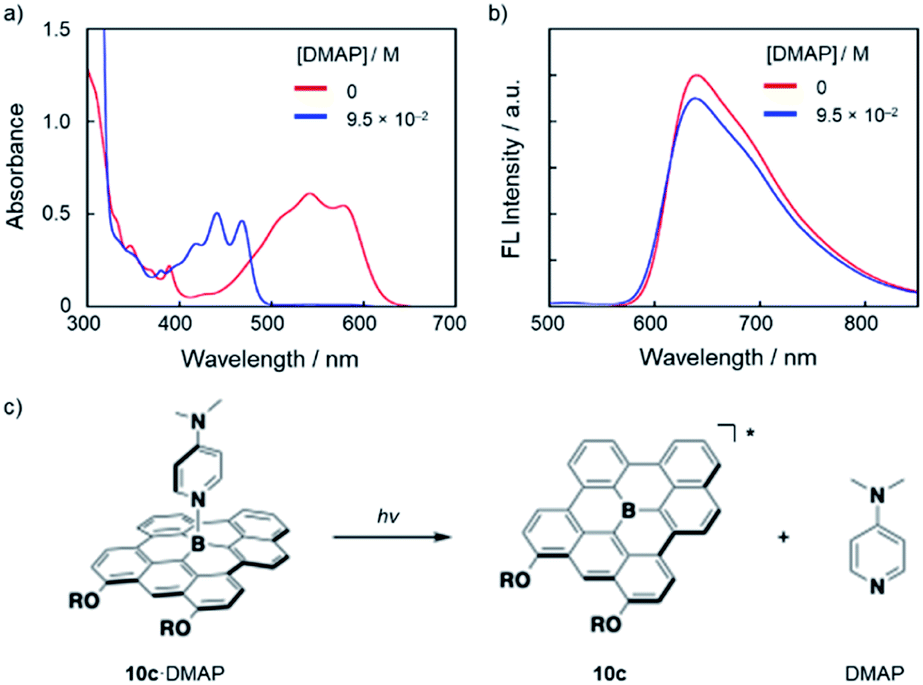 | ||
| Fig. 5 (a) UV-vis absorption and (b) fluorescence (λex = 477 nm) spectral changes upon addition of DMAP to a DMSO solution of 10c (3.2 × 10−5 M). (c) Schematic illustration of the photo-dissociation of 10c·DMAP. | ||
Self-assembly in aqueous media
Taking advantage of high stability toward water and planar structure of the boron-doped PAHs, the self-assembly behavior of amphiphilic derivative 10c with hydrophilic side chains was investigated in aqueous media. When the water content was increased from 5% to 30% in a DMSO/H2O mixed solvent system, the absorbance of the monomeric state at 500–600 nm gradually reduced in intensity, and the absorption band concomitantly showed a hypsochromic shift to λabs = 510 nm (Fig. 6a). Additionally, the intensity of the red emission band of 10c decreased with increasing water content, which is indicative of the formation of H-type aggregates of 10c in DMSO/H2O (Fig. S18a†).22 No noticeable changes in the absorption or fluorescence bands were observed for water contents over 30% (Fig. 6a and S18b†).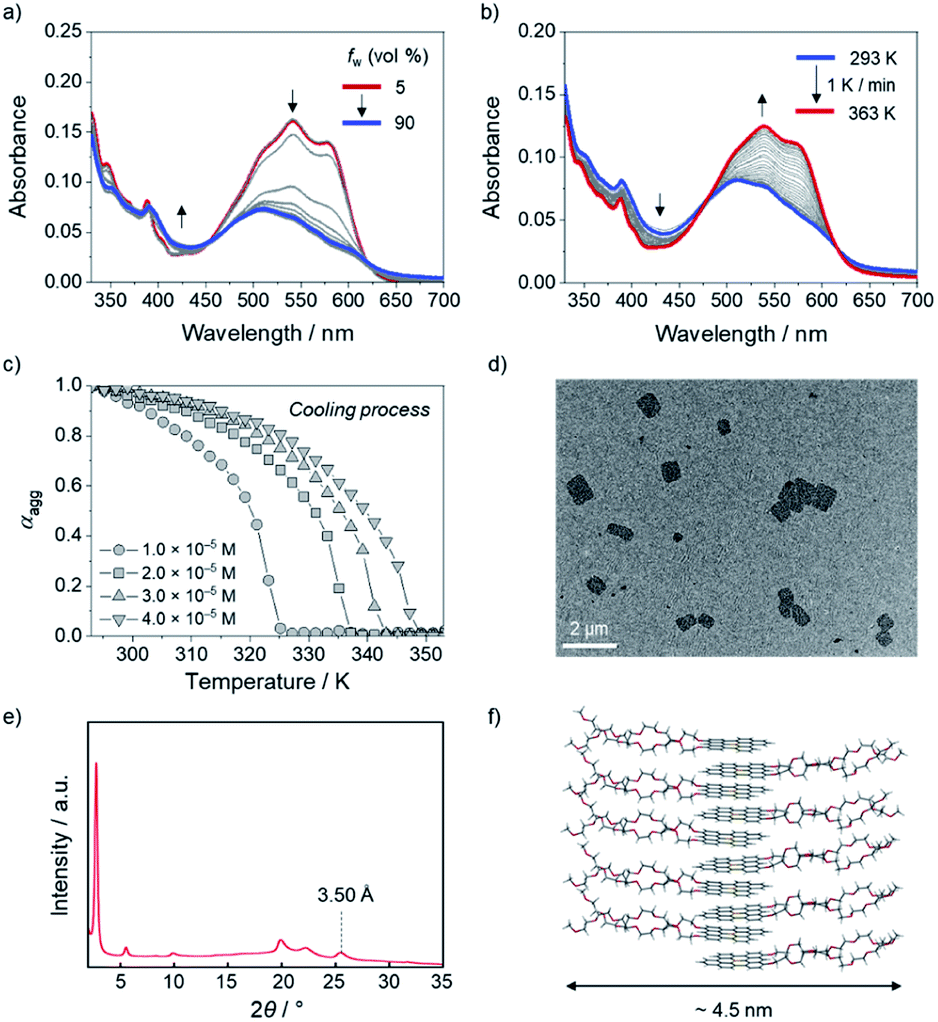 | ||
| Fig. 6 UV-vis absorption spectral changes of 10c (2.0 × 10−5 M) in DMSO/H2O (a) with increasing H2O fraction from 5 to 90% at room temperature and (b) during heating from 293 to 363 K at 1 K min−1 in DMSO/H2O (7:3, v/v). (c) Plots of the variation in the degree of aggregation (αagg) of 10c during cooling from 353 to 293 K at 1 K min−1 in DMSO/H2O (7:3, v/v), monitored at 577 nm. (d) TEM image and (e) XRPD pattern of aggregated 10c and (f) schematic illustration of assumed molecular alignment for the aggregates of 10c. | ||
The disassembly process of the aggregates of 10c (2.0 × 10−5 M) was monitored using temperature-dependent UV-vis absorption spectroscopy. In DMSO/H2O (5:5, v/v), no clear disassembly of the aggregates was observed, even upon heating to 363 K, because of the strong hydrophobic effect among 10c in the high-water-fraction medium (Fig. S19†).23 In contrast, the generation of the monomeric state of 10c was observed at 363 K in DMSO/H2O (7:3, v/v). Specifically, during the heating of 10c (2.0 × 10−5 M) from 293 to 363 K at a rate of 1 K min−1, the absorption band with maxima at 538 nm and 577 nm, which is characteristic of monomeric 10c, gradually increased (Fig. 6b).
The assembly process from the monomeric state was also monitored during cooling of the solution from 353 K to 293 K. The plot of the absorbance change of 10c (4.0 × 10−5 M) at 577 nm showed a non-sigmoidal transition (Fig. 6c) with a critical elongation temperature (Te) at 349 K (Fig. 6c and S20†).24 This result indicates that amphiphilic 10c follows a cooperative aggregation mechanism.25 The thermodynamic parameters governing the cooperative aggregation were determined using a van't Hoff plot. As the total concentration (cT) of 10c was decreased from 4.0 × 10−5 M to 1.0 × 10−5 M, the critical elongation temperature (Te) in the cooling process decreased (Fig. 6c). A linear relationship was observed in the van't Hoff plot (Fig. S21†), from which a standard enthalpy (ΔH°) of −56.9 kJ mol−1 and a standard entropy (ΔS°) of −79.0 J mol−1 K−1 were determined. The negative values of ΔH° and ΔS° imply that the aggregation process is enthalpically driven. Using the equation ΔG° = ΔH° − TΔS°, the Gibbs free energy (ΔG°) at 298 K was calculated to be −33.4 kJ mol−1, from which an elongation equilibrium constant (Ke) of 7.2 × 105 M−1 was estimated for the aggregation of 10c (Table S15†).
For further analysis of the aggregated 10c, its morphology was observed using transmission electron microscopy (TEM) and atomic force microscopy (AFM). The TEM image revealed the formation of sheet-like aggregates with a length of approximately 1 μm (Fig. 6d). The AFM image confirmed the presence of sheet-like aggregates with a thickness of 4 nm (Fig. S23†). In addition, X-ray powder diffraction (XRPD) analysis showed a peak at 2θ = 25.5°, which corresponds to a π-stacking arrangement of molecules with an interfacial distance of 3.50 Å (Fig. 6e). Taking the strong hydrophobic effect in aqueous media into account, we assumed a bilayer-like molecular arrangement that orients the hydrophobic boron-doped PAH moiety inward and the hydrophilic side chains outward while retaining a π-stacked array (Fig. 6f). This molecular arrangement corresponds to a width of ca. 4.5 nm, which is consistent with the results obtained from AFM (Fig. S23†).
Stimuli-responsive self-assembly of 10c in aqueous media
Given the dynamic behavior of 10c in aqueous media, we turned our attention to controlling the self-assembly process of 10c by switching the coordination number of the Lewis acidic boron center using an external stimulus. To this end, the disassembly process of aggregated 10c upon the addition of a Lewis base was studied. When an excess of DMAP was added to a solution of 10c in DMSO/H2O (7:3, v/v), aggregated 10c was transformed into the axially coordinated adduct 10c·DMAP, as evidenced by the emergence of an absorption band with a λabs of 467 nm (Fig. 7a and d), which is blue-shifted compared to that of monomeric tricoordinate 10c. The steric hindrance derived from the axially coordinated DMAP in the tetracoordinate complex is responsible for the disassembly of the aggregates, as it prevents π-stacking. Notably, the fluorescence spectrum of the solution exhibited a red emission band with a λem of 640 nm, even under conditions that mainly produce the tetracoordinate 10c·DMAP (Fig. 7b and d). This fact demonstrates that the Lewis acid–base adduct 10c·DMAP undergoes photo-dissociation in the excited state even in aqueous solution.
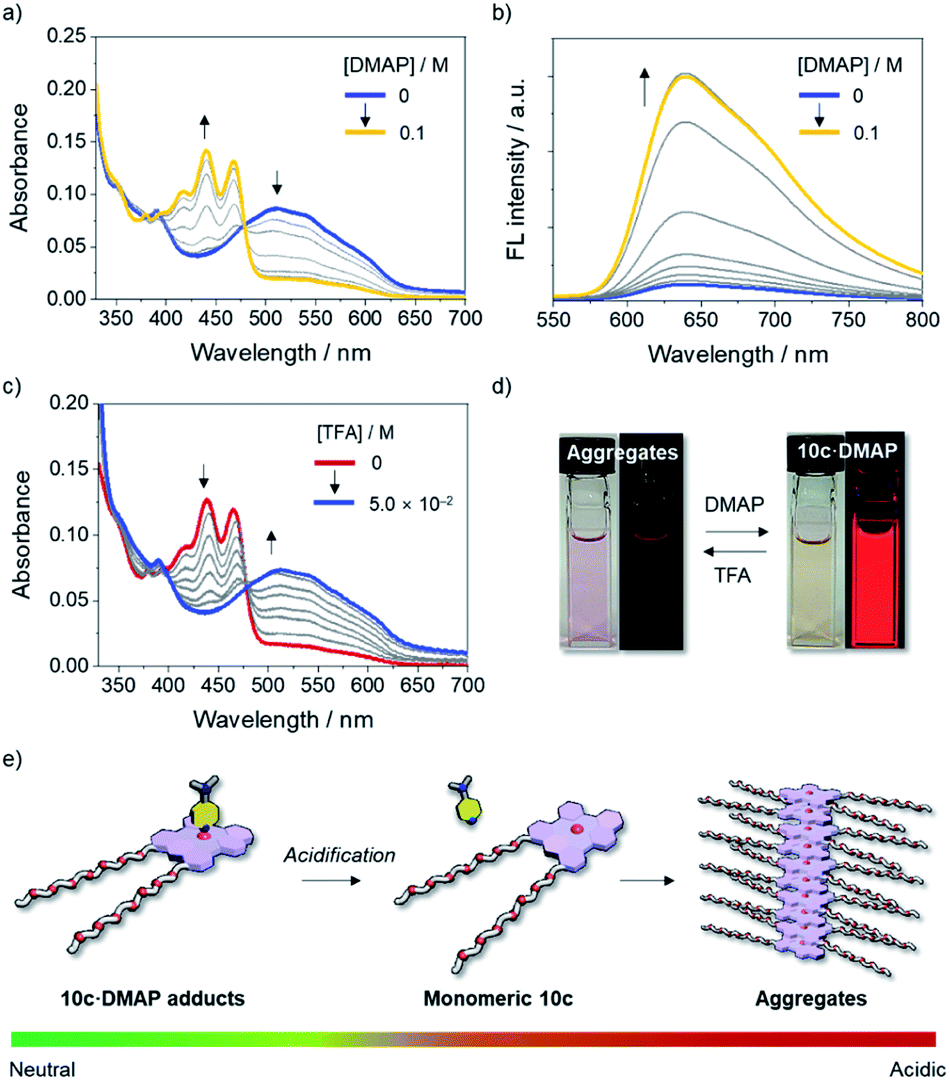 | ||
| Fig. 7 (a) UV-vis absorption and (b) fluorescence spectral changes (λex = 477 nm) of aggregated 10c (2.0 × 10−5 M) in DMSO/H2O (7:3, v/v) at room temperature with increasing concentration of DMAP and (c) UV-vis absorption spectral change of 10c in the presence of DMAP (6.0 × 10−2 M) in DMSO/H2O (7:3, v/v) with an increasing concentration of TFA, along with (d) photographs of these changes. (e) Schematic representation of the pH-responsive self-assembly of the boron-doped PAH 10c·DMAP adduct in aqueous media. | ||
Importantly, the assembly-incompetent Lewis adduct 10c·DMAP can be reverted to the aggregated state by tuning the acidity of the medium. Upon the addition of trifluoroacetic acid (TFA) to a solution of 10c·DMAP in DMSO/H2O (7:3, v/v), the absorption band of 10c·DMAP gradually decreased in intensity with the concomitant growth of an absorption band corresponding to the aggregates with an λabs of 510 nm (Fig. 7c and d). Thus, the protonation of DMAP led to a shift in the equilibrium toward the dissociation of 10c·DMAP, and, ultimately, the self-assembly process became preferable under the acidified conditions (Fig. 7e). This result was also corroborated by the quenching of the red emission attributed to the formation of H-type aggregates (Fig. 7d and S22†). The stimuli-responsive geometric transformation between tri- vs. tetracoordinate species is a crucial factor to control the (dis)assembly process, and can be easily recognized by the “on–off” fluorescence change (Fig. 7d and e).
Conclusions
Since the discovery of the effective stabilization of triarylboranes in a planar fashion via structural constraint, planarized triarylboranes have attracted increasing attention as promising scaffolds for π-electron materials. Among the various types of planarized triarylboranes, fully fused boron-doped PAHs are of particular importance for their potential applications in aqueous media in light of their high stability toward water and oxygen. To explore their utilities along this line, a series of fully fused boron-doped PAHs has been synthesized in this study, and their structure–property relationships have been studied. The thus-obtained boron-doped PAHs have planar geometries with shortened B–C bond distances and exhibit red fluorescence with moderate fluorescence quantum yields. Late-stage modification via in situ TBDPS deprotection afforded an amphiphilic derivative with hydrophilic side chains, which allowed assessment of the Lewis acidity in polar solvents. Their B–N Lewis acid–base adducts also exhibited photo-dissociation behavior with even strong, charge-neutral Lewis bases such as DMAP. With the amphiphilic derivative in hand, the self-assembly of the boron-doped PAH in aqueous media was demonstrated for the first time in DMSO/H2O mixed solvents. Making use of the labile complexation ability of the Lewis-acidic boron center, the disassembly and assembly processes from the aggregates were also controlled via the successive addition of DMAP and TFA in aqueous media. These results shed light on a new aspect of the boron-doped PAHs. The combined use of their π-stacking ability and Lewis acidity gives them a crucial advantage over all-carbon-based PAHs. The present findings should form the basis to open a new avenue for their use in biological applications.Author contributions
H. N., H. C., M. I., and S. Y. conceived the idea. H. N., M. I., and N. A. synthesized the compounds and evaluated their properties. H. C. and S. O. studied the self-assembly properties. H. N., H. C., and S. Y. wrote the manuscript, and all authors discussed and commented on the manuscript. S. Y. directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by KAKENHI Grants JP18H05261 from the Japan Society for the Promotion of Science (JSPS) and Japan Science and Technology Agency (JST; JPMJCR2105). H. N. and M. I. thank the JSPS for a Research Fellowship for Young Scientists and the “Graduate Program of Transformative Chem-Bio Research” at Nagoya University, supported by MEXT (WISE Program). The synchrotron X-ray crystallographic analyses were performed at the BL02B1 beamline of SPring-8 with the approval of the Japanese Synchrotron Radiation Research Institute (JASRI; proposal 2021B1833).Notes and references
- (a) C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS; (b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (c) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (d) Z. M. Hudson and S. Wang, Acc. Chem. Res., 2009, 42, 1584–1596 CrossRef CAS PubMed; (e) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (f) F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed; (g) Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC; (h) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS; (i) A. Wakamiya and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1357–1377 CrossRef CAS.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- (a) A. Escande and M. J. Ingleson, Chem. Commun., 2015, 51, 6257–6274 RSC; (b) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (c) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (d) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- (a) S. Saito, K. Matsuo and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 9130–9133 CrossRef CAS PubMed; (b) K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 CrossRef CAS PubMed; (c) N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed.
- K. Schickedanz, T. Trageser, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2015, 51, 15808–15810 RSC.
- (a) Y. Kitamoto, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Chem. Commun., 2016, 52, 7098–7101 RSC; (b) Y. Kitamoto, F. Kobayashi, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Dalton Trans., 2019, 48, 2118–2127 RSC.
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS PubMed.
- G. Meng, T. Peng, Y. Shi, H. Li, X. Wang, X. Yin, D.-T. Yang, S. Wang and N. Wang, J. Mater. Chem. C, 2020, 8, 7749–7754 RSC.
- (a) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed; (b) K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195–1198 CrossRef CAS PubMed; (c) X. Liang, Z.-P. Yan, H.-B. Han, Z.-G. Wu, Y.-X. Zheng, H. Meng, J.-L. Zuo and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 11316–11320 CrossRef CAS PubMed; (d) A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed; (e) S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 3082–3089 RSC; (f) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS; (g) Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 CrossRef CAS PubMed; (h) S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311–9314 CrossRef CAS PubMed; (i) M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed; (j) M. Nagata, H. Min, E. Watanabe, H. Fukumoto, Y. Mizuhata, N. Tokitoh, T. Agou and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 20280–20285 CrossRef CAS PubMed; (k) M. Yang, S. Shikita, H. Min, I. S. Park, H. Shibata, N. Amanokura and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 23142–23147 CrossRef CAS PubMed; (l) P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2021, 2106954 CrossRef PubMed.
- T. Kushida, A. Shuto, M. Yoshio, T. Kato and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 6922–6925 CrossRef CAS PubMed.
- (a) B. Adelizzi, P. Chidchob, N. Tanaka, B. A. G. Lamers, S. C. J. Meskers, S. Ogi, A. R. A. Palmans, S. Yamaguchi and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 16681–16689 CrossRef CAS PubMed; (b) H. Choi, S. Ogi, N. Ando and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 2953–2961 CrossRef CAS PubMed.
- K. Matsuo, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 11984–11988 CrossRef CAS PubMed.
- (a) S. A. Iqbal, J. Pahl, K. Yuan and M. J. Ingleson, Chem. Soc. Rev., 2020, 49, 4564–4591 RSC; (b) S. Oda and T. Hatakeyama, Bull. Chem. Soc. Jpn., 2021, 94, 950–960 CrossRef CAS; (c) A. Escande, D. L. Crossley, J. Cid, I. A. Cade, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 17160–17167 RSC; (d) J. M. Farrell, C. Mützel, D. Bialas, M. Rudolf, K. Menekse, A.-M. Krause, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2019, 141, 9096–9104 CrossRef CAS PubMed; (e) J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed; (f) J.-J. Zhang, L. Yang, F. Liu, Y. Fu, J. Liu, A. A. Popov, J. Ma and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 25695–25700 CrossRef CAS PubMed.
- (a) F. Miyamoto, S. Nakatsuka, K. Yamada, K. Nakayama and T. Hatakeyama, Org. Lett., 2015, 17, 6158–6161 CrossRef CAS PubMed; (b) J. Radtke, K. Schickedanz, M. Bamberg, L. Menduti, D. Schollmeyer, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2019, 10, 9017–9027 RSC.
- (a) C. Dou, S. Saito, K. Matsuo, I. Hisaki and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 12206–12210 CrossRef CAS PubMed; (b) S. Osumi, S. Saito, C. Dou, K. Matsuo, K. Kume, H. Yoshikawa, K. Awaga and S. Yamaguchi, Chem. Sci., 2016, 7, 219–227 RSC.
- (a) S. Kawai, S. Saito, S. Osumi, S. Yamaguchi, A. S. Foster, P. Spijker and E. Meyer, Nat. Commun., 2015, 6, 8098 CrossRef CAS PubMed; (b) E. Carbonell-Sanromà, A. Garcia-Lekue, M. Corso, G. Vasseur, P. Brandimarte, J. Lobo-Checa, D. G. de Oteyza, J. Li, S. Kawai, S. Saito, S. Yamaguchi, J. E. Ortega, D. Sánchez-Portal and J. I. Pascual, J. Phys. Chem. C, 2018, 122, 16092–16099 CrossRef; (c) N. Friedrich, P. Brandimarte, J. Li, S. Saito, S. Yamaguchi, I. Pozo, D. Peña, T. Frederiksen, A. Garcia-Lekue, D. Sánchez-Portal and J. I. Pascual, Phys. Rev. Lett., 2020, 125, 146801 CrossRef CAS PubMed.
- (a) N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945–3947 CrossRef CAS PubMed; (b) N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124–2127 CrossRef CAS PubMed; (c) N. K. S. Davis, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 30–31 CrossRef CAS PubMed; (d) L. Zeng, C. Jiao, X. Huang, K.-W. Huang, W.-S. Chin and J. Wu, Org. Lett., 2011, 13, 6026–6029 CrossRef CAS PubMed.
- (a) X. Li, X. Guo, L. Cao, Z. Xun, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2014, 53, 7809–7813 CrossRef CAS PubMed; (b) J. Liu, X. Guo, R. Hu, X. Liu, S. Wang, S. Li, Y. Li and G. Yang, Anal. Chem., 2016, 88, 1052–1057 CrossRef CAS PubMed; (c) J. Liu, S. Zhang, C. Zhang, J. Dong, C. Shen, J. Zhu, H. Xu, M. Fu, G. Yang and X. Zhang, Chem. Commun., 2017, 53, 11476–11479 RSC.
- (a) S. Griesbeck, Z. Zhang, M. Gutmann, T. Lühmann, R. M. Edkins, G. Clermont, A. N. Lazar, M. Haehnel, K. Edkins, A. Eichhorn, M. Blanchard-Desce, L. Meinel and T. B. Marder, Chem.–Eur. J., 2016, 22, 14701–14706 CrossRef CAS PubMed; (b) S. Griesbeck, M. Ferger, C. Czernetzi, C. Wang, R. Bertermann, A. Friedrich, M. Haehnel, D. Sieh, M. Taki, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 7679–7688 CrossRef CAS; (c) S. Griesbeck, E. Michail, C. Wang, H. Ogasawara, S. Lorenzen, L. Gerstner, T. Zang, J. Nitsch, Y. Sato, R. Bertermann, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem. Sci., 2019, 10, 5405–5422 RSC; (d) S. Griesbeck, E. Michail, F. Rauch, H. Ogasawara, C. Wang, Y. Sato, R. M. Edkins, Z. Zhang, M. Taki, C. Lambert, S. Yamaguchi and T. B. Marder, Chem.–Eur. J., 2019, 25, 13164–13175 CrossRef CAS PubMed; (e) Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 RSC; (f) S. M. Berger, J. Rühe, J. Schwarzmann, A. Phillipps, A.-K. Richard, M. Ferger, I. Krummenacher, L.-M. Tumir, Ž. Ban, I. Crnolatac, D. Majhen, I. Barišić, I. Piantanida, D. Schleier, S. Griesbeck, A. Friedrich, H. Braunschweig and T. B. Marder, Chem.–Eur. J., 2021, 27, 14057–14072 CrossRef CAS PubMed; (g) S. M. Berger and T. B. Marder, Mater. Horiz., 2021, 9, 112–120 Search PubMed.
- F. Zettler, H. D. Hausen and H. Hess, J. Organomet. Chem., 1974, 72, 157–162 CrossRef CAS.
- N. Ando, T. Kushida and S. Yamaguchi, Chem. Commun., 2018, 54, 5213–5216 RSC.
- (a) F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS PubMed; (b) N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef.
- (a) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS; (b) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS; (c) M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- (a) M. Hecht, P. Leowanawat, T. Gerlach, V. Stepanenko, M. Stolte, M. Lehmann and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 17084–17090 CrossRef CAS PubMed; (b) B. Lei, H. Pan, Y. Zhang, X.-K. Ren and Z. Chen, Org. Biomol. Chem., 2021, 19, 6108–6114 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral data, details of the computational studies, analysis of self-assembly, and crystallographic data for 10a, 10b, 12, and 13. CCDC 2124573–2124576. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06710a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |