
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSingle-atom cobalt-hydroxyl modification of polymeric carbon nitride for highly enhanced photocatalytic water oxidation: ball milling increased single atom loading†
Fei
Yu
,
Tingting
Huo
,
Quanhua
Deng
,
Guoan
Wang
,
Yuguo
Xia
 ,
Haiping
Li
* and
Wanguo
Hou
,
Haiping
Li
* and
Wanguo
Hou
National Engineering Research Center for Colloidal Materials, School of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, China. E-mail: hpli@sdu.edu.cn
First published on 15th December 2021
Abstract
Expediting the oxygen evolution reaction (OER) is the key to achieving efficient photocatalytic overall water splitting. Herein, single-atom Co–OH modified polymeric carbon nitride (Co-PCN) was synthesized with single-atom loading increased by ∼37 times with the assistance of ball milling that formed ultrathin nanosheets. The single-atom Co-N4OH structure was confirmed experimentally and theoretically and was verified to enhance optical absorption and charge separation and work as the active site for the OER. Co-PCN exhibits the highest OER rate of 37.3 μmol h−1 under visible light irradiation, ∼28-fold higher than that of common PCN/CoOx, with the highest apparent quantum yields reaching 4.69, 2.06, and 0.46% at 400, 420, and 500 nm, respectively, and is among the best OER photocatalysts reported so far. This work provides an effective way to synthesize efficient OER photocatalysts.
Massive fuel energy consumption induced environmental and ecological problems, especially the greenhouse effect, and the resultant extreme climates and rise in sea level are threatening human life.1 As a potential substitution for fuel energy, hydrogen energy conversion from solar energy via photocatalytic water splitting attracts great attention from scientists.2–5 However, the photocatalytic hydrogen evolution efficiency from overall water splitting is still restricted by the sluggish oxygen evolution reaction (OER) that involves energy absorption, four-electron transfer, breakage of O–H bonds, and formation of O–O bonds,6,7 and thus efficient OER photocatalysts become the key to achieving efficient overall water splitting. Though numerous hydrogen evolution photocatalysts have been reported, research on OER photocatalysts is mainly around a few semiconductors including BiVO4, WO3, Ag3PO4, α-Fe2O3, etc.8–11 and their activity is not high enough yet for practical applications. Therefore, exploring high-efficiency OER photocatalysts is still necessary.
Polymeric carbon nitride (PCN) was first reported in 2009 (ref. 12) as a photocatalyst with a layered melon-type orthorhombic structure,13 and thereafter quickly became a “star” photocatalyst thanks to its advantages of being visible-light responsive and metal-free, non-toxic, and low cost, and its relatively high chemical stability.14 Because of several self-deficiencies including fast photogenerated charge recombination and a narrow optical absorption spectrum, PCN exhibits relatively low photocatalytic activity.15 Then, a series of strategies were put forward successively to enhance the photoactivity of PCN, such as enhancement of crystallinity,16 morphological control,17 structural modification18 (including extensively researched single atom modification in recent years19,20), exfoliation,21 construction of hetero-(homo-)junctions,22 and loading of noble metals.23 Though photocatalytic water splitting on PCN was extensively researched in the past, the research was mainly around the hydrogen evolution half-reaction used for exploring properties and the catalytic mechanism of photocatalysts, and little research was focused on the industrially useable overall water splitting process owing to the sluggish OER.15 Therefore, enhancing the photocatalytic OER activity of PCN becomes the key to practical applications.
To increase OER rates of PCN, several kinds of methods were proposed, such as rational design of compound cocatalysts (e.g., CoOx, IrO2, CoP, CoPi, RhOx, RuOx, PtOx, MnOx, Co(OH)2, Ni(OH)2, and CoAl2O4 (ref. 24–30)), modification of carbon dots and carbon rings,31,32 fabrication of special architectures of PCN (e.g., PCN quantum dot stacked nanowires33), and single-atom (e.g., B, Co, and Mn34–36) modification. For instance, Zhao and coauthors prepared B and N-vacancy comodified PCN that exhibits the highest OER rate of ∼28 μmol h−1 (ref. 36) and recently their group further used these B doped PCN ultrathin nanosheets to fabricate a Z-scheme heterojunction for overall water splitting with a solar-to-hydrogen efficiency reaching ∼1.2%.37 Comparatively, PCN loaded with compound cocatalysts can only enhance OER activity to a limited degree and there are finite methods for carbon modification and special architecture fabrication. Single-atom modification shows a bright prospect, on account of metal atoms capable of being inserted into the framework of PCN and effectively increasing the OER activity. However, reported single metal atom modification routes are all based on direct ion adsorption on PCN or calcination of mixtures of metal salts and PCN feedstocks.34,35,38 New routes need be explored to increase effective loading of single atoms in PCN. Besides, the metal-OH structure is considered efficient for the OER,30,39,40 and a single metal atom-OH structure has never been reported for modification of PCN, though Mn–OH was thought to play a key role in the OER process.34
Ball milling is an extensively used versatile and scalable way for preparation of heterogeneous catalysts and even single-atom catalysts,41,42 but was rarely used in synthesis of PCN-based single-atom photocatalysts. In this work, we synthesized single-atom Co–OH modified PCN (Co-PCN) with the single-atom content in PCN highly increased with the assistance of ball milling. The simple synthetic route is shown in Fig. 1a. PCN was ball-milled to obtain BM-PCN that then adsorbed Co2+ till saturation to form BM-PCN/Co which was calcined to obtain BM-PCN/Co-c (Co-PCN). For comparison, PCN was directly used to adsorb Co2+ till saturation to form PCN/Co which was calcined to obtain PCN/Co-c. PCN mainly comprises large blocks with the size of several micrometers (Fig. S1†), while BM-PCN contains massive irregular particles with the size reduced to several hundreds of nanometers (Fig. S2†), indicative of high efficacy of ball milling. BM-PCN/Co-c exhibits a similar morphology as BM-PCN (Fig. 1b and S3†) and PCN/Co-c exhibits a similar morphology to PCN (Fig. S4†), but the surface area and mesopore volume of BM-PCN and BM-PCN/Co-c are not higher than those of PCN and PCN/Co-c (Fig. S5†), manifesting that ball-milling and subsequent calcination did not form massive mesopores, which accords well with the particle size variation from several micrometers (before ball milling) to several hundreds of nanometers (after ball milling). However, the Co content in BM-PCN/Co-c, BM-PCN/Co, PCN/Co-c, and PCN/Co was measured to be 0.75, 0.50, 0.02, and ∼0.02 wt%, respectively, by inductively coupled plasma mass spectrometry (ICP-MS). The ∼37 times higher Co content in BM-PCN/Co-c than in PCN/Co-c suggests the ball-milling enhanced adsorption of Co2+ on surfaces of BM-PCN, which should arise mainly from the ball-milling induced increase of surface energy and adsorption sites.43
 | ||
| Fig. 1 (a) Schematic illustration for synthesis of single-atom CoII-OH modified PCN (BM-PCN/Co-c); and (b) SEM, (inset in b) TEM, (c) AFM, (d) EDS elemental mapping, and (e) HAADF-STEM images of BM-PCN/Co-c. | ||
The TEM image shows the existence of small and ultrathin nanosheets in BM-PCN/Co-c (inset in Fig. 1b) which can also be observed in the atomic force microscopy (AFM) image with a thickness of ∼7–10 nm and lateral size of <70 nm (Fig. 1c), and formation of these ultrathin nanosheets results from the ball milling of PCN.44 It should be noted that most formed ultrathin nanosheets with high surface energy may stack into compact particles upon ball milling, leading to no increase of the total surface area. Energy dispersive X-ray spectroscopy (EDS) elemental mapping images of BM-PCN/Co-c indicate homogeneous distribution of C, N, O, and Co elements in the sample (Fig. 1d). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of BM-PCN/Co-c shows massive white spots (marked by circles) with a mean size of <1 Å dispersed in the sample (Fig. 1e and S6†), which should correspond to single-atom Co.
To further verify the single-atom Co structure in BM-PCN/Co-c, Co K-edge X-ray absorption near-edge structure spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) analysis were performed. As shown in Fig. 2a, the absorption-edge position of BM-PCN/Co-c is quite close to that of CoO and their peak positions are similar and far from those of other reference samples, indicating that the valence of Co in BM-PCN/Co-c is about +2. The bonding structure around Co was determined by Fourier transformed (FT) k3-weighted EXAFS analysis. As shown in Fig. 2b, a distinct single Co-ligand peak at ∼1.6 Å for BM-PCN/Co-c is observed, which prominently differs from the Co–Co coordination peak at ∼2.2 Å for Co foil and the CoII–O coordination peak at ∼1.7 Å for CoO. The wavelet transform (WT) contour plot of BM-PCN/Co-c shows only one intensity maximum (Fig. S7†), and the Cl 2p core-level XPS spectrum of BM-PCN/Co-c reveals no residue of Cl (Fig. S8†). These further indicate the single-atom dispersion of Co species. Apparently, the Co-ligand peak is almost consistent with the CoII–N peak for Co porphyrin, suggesting that the single-atom Co in BM-PCN/Co-c mainly coordinates with N. Least-square EXAFS curve fitting was performed to confirm quantitative structural parameters of CoII in BM-PCN/Co-c, as shown in Fig. 2c, S9, and S10 and Table S1.† Simple Co–N single-shell fitting of BM-PCN/Co-c (Fig. S10†) gave a coordination number of 5.6 ± 0.4 (Table S1†), that is, CoII coordinates with five atoms. Considering that the PCN monolayer provides four appropriate N coordination sites at most,45 CoII likely coordinates with four N atoms and one OH atom. Thus, we further performed Co–N4/Co–O double-shell fitting (Fig. 2c) and the obtained R-factor (0.0011) remarkably reduces relative to that from Co–N single-shell fitting (0.0035), indicative of rationality of the proposed CoII–N4OH structure. Confirmed Co–N and Co–O bond lengths are 2.04 and 2.15 Å, respectively (Table S1†).
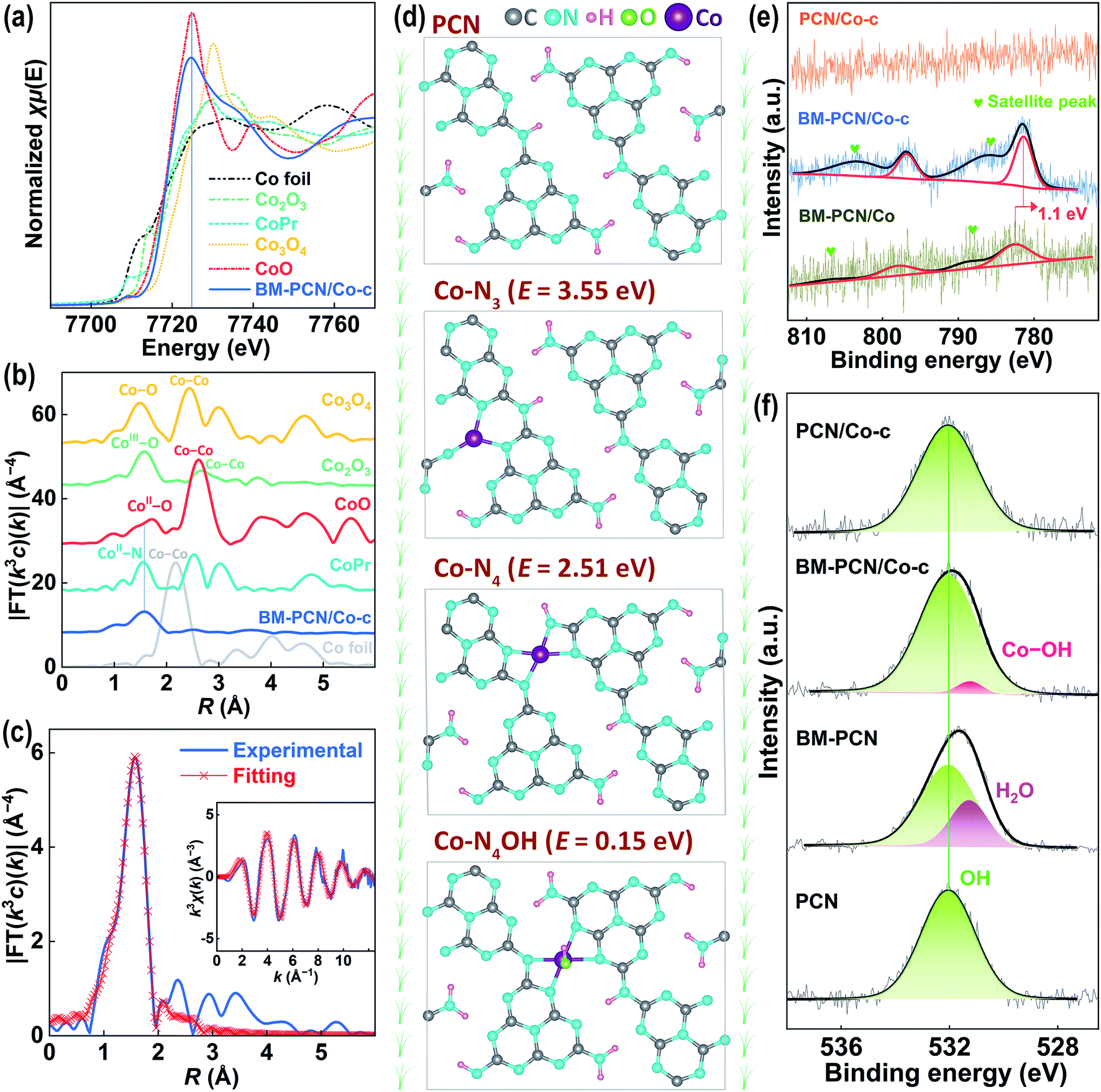 | ||
| Fig. 2 (a) Co K-edge XANES and (b) EXAFS spectra of Co foil, Co porphyrin (Copr), CoO, Co3O4, Co2O3, and BM-PCN/Co-c; EXAFS (c) R space-fitting and (inset in c) K space-fitting curves of BM-PCN/Co-c; (d) optimized structure of PCN and Co-doped PCN with different doping configurations and calculated formation energies (e) of Co doped PCN; and (e) Co 2p and (f) O 1s core-level XPS spectra of samples. | ||
To further confirm rationality of the Co–N4OH coordination structure, density functional theory (DFT) calculations were conducted. As shown in Fig. 2d, three possible CoII coordination structures in the PCN monolayer were explored. The Co–N4OH structure without removal of H from PCN exhibits a much lower formation energy (∼0.15 eV) than Co–N4 and Co–N3 structures with removal of two H atoms from PCN (∼2.51 and 3.55 eV), demonstrating a high probability of existence of the Co–N4OH structure in BM-PCN/Co-c. This structure can also be evidenced by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2e, the Co 2p core-level XPS spectrum of BM-PCN/Co-c shows two distinct peaks at binding energies of 796.8 and 781.4 eV beside satellite peaks, corresponding to Co 2p1/2 and 2p3/2 of CoII ions.46 The spectrum of BM-PCN/Co also shows two Co 2p peaks but at a binding energy ∼1.1 eV higher, suggesting variation of the Co coordination structure from BM-PCN/Co to BM-PCN/Co-c. PCN/Co-c exhibits no peaks because of its low Co content. Fig. 2f shows O 1s core-level spectra of PCN, BM-PCN, BM-PCN/Co-c, and PCN/Co-c. All the samples exhibit one peak at a binding energy of ∼532.0 eV, ascribed to surface hydroxyl species,47 but an additional peak could be obtained for BM-PCN or BM-PCN/Co-c after deconvolution. The peak at a binding energy of ∼531.3 eV for BM-PCN should be assigned to adsorbed H2O at new active adsorption sites generated by ball milling. This peak can also be observed in the spectrum of BM-PCN/Co, but with a ∼0.1 eV shift to a higher binding energy (Fig. S11†) owing to the influence of adsorbed CoII ions. The peak at ∼531.2 eV for BM-PCN/Co-c should be assigned to Co–OH,48 given that there is only one O 1s peak for BM-PCN-c (synthesized by direct calcination of BM-PCN) (Fig. S11†). The calculated Co/O(–Co) molar ratio, based on the XPS data, is ∼1.07 (Table S2†), close to 1, consistent with the Co–N4OH coordination structure.
In C 1s and N 1s core-level XPS spectra, BM-PCN, BM-PCN/Co-c, PCN/Co-c, and BM-PCN/Co exhibit similar peaks to PCN (Fig. S12a–d†), indicative of their similar framework structure which can also be evidenced by their similar N/C molar ratios, 1.53 (Table S3†), but the N–H peak of BM-PCN shifts ∼0.2 eV to a lower binding energy relative to that of PCN, likely arising from the ball-milling induced destruction of intralayer hydrogen bonds (Fig. S13†). The Co content in BM-PCN/Co, BM-PCN/Co-c, and PCN/Co-c is too low to cause detectable variation of C 1s and N 1s peaks. Similar FT-IR absorption bands of the samples (Fig. S14a and b†) also indicate their basic frame structure, but in enlarged spectra (Fig. S14c†), ν(C–N) and ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) absorption bands of BM-PCN shift 16 cm−1 to a higher wavenumber and 19 cm−1 to a lower wavenumber, respectively, relative to those of PCN at 1242 and 1640 cm−1,49 likely resulting from the ball-milling induced hydrogen bond destruction, and the shift of these two absorption bands turns smaller for BM-PCN/Co-c, suggesting calcination-induced reforming of the destroyed hydrogen bonds, which is consistent with the XPS results (Fig. S12c†). Besides, BM-PCN exhibits a wider and relatively stronger ν(N–H)/ν(O–H) absorption band than PCN (Fig. S14a†), probably owing to the hydrogen bond destruction and new adsorbed H2O, while this absorption band for BM-PCN/Co-c becomes much weaker, suggesting hydrogen bond reforming and loss of new adsorbed H2O (Fig. 2f). Zeta potentials of the samples dispersed in water reflect variation of surface adsorbed hydroxyl species. As shown in Fig. S15a,† all the samples exhibit negative zeta potentials because of dissociation of surface hydroxyl species. The zeta potentials, following the order PCN (−24 mV) > BM-PCN (−41 mV) < BM-PCN/Co-c (−30 mV) ≈ PCN/Co-c (−28 mV), suggest the ball milling-induced increase of surface hydroxyls in BM-PCN and calcination-induced decrease in BM-PCN/Co-c, consistent with the FT-IR results.
N) absorption bands of BM-PCN shift 16 cm−1 to a higher wavenumber and 19 cm−1 to a lower wavenumber, respectively, relative to those of PCN at 1242 and 1640 cm−1,49 likely resulting from the ball-milling induced hydrogen bond destruction, and the shift of these two absorption bands turns smaller for BM-PCN/Co-c, suggesting calcination-induced reforming of the destroyed hydrogen bonds, which is consistent with the XPS results (Fig. S12c†). Besides, BM-PCN exhibits a wider and relatively stronger ν(N–H)/ν(O–H) absorption band than PCN (Fig. S14a†), probably owing to the hydrogen bond destruction and new adsorbed H2O, while this absorption band for BM-PCN/Co-c becomes much weaker, suggesting hydrogen bond reforming and loss of new adsorbed H2O (Fig. 2f). Zeta potentials of the samples dispersed in water reflect variation of surface adsorbed hydroxyl species. As shown in Fig. S15a,† all the samples exhibit negative zeta potentials because of dissociation of surface hydroxyl species. The zeta potentials, following the order PCN (−24 mV) > BM-PCN (−41 mV) < BM-PCN/Co-c (−30 mV) ≈ PCN/Co-c (−28 mV), suggest the ball milling-induced increase of surface hydroxyls in BM-PCN and calcination-induced decrease in BM-PCN/Co-c, consistent with the FT-IR results.
Solid-state 13C magic-angle-spinning nuclear magnetic resonance (NMR) spectra of PCN, BM-PCN, BM-PCN/Co-c, and PCN/Co-c show two similar peaks at chemical shifts of ∼164 and 156 ppm (Fig. S15b†), ascribed to C−NHx and NC–N, respectively,50 indicating their similar molecular framework, but in enlarged spectra, BM-PCN exhibits ∼0.3° movement of the NC–N peak to a lower chemical shift compared with PCN, because of the ball-milling induced hydrogen bond destruction, and the C−NHx peak of BM-PCN/Co-c moves ∼0.2° to a lower chemical shift, likely owing to formation of the C–N–Co structure whose peak lies close to the C−NHx peak.51 The XRD patterns of the samples are shown in Fig. S15c.† PCN and PCN/Co-c exhibit typical diffraction peaks of melon-type carbon nitride with a layered orthorhombic structure and peaks at 13.1° and 27.6° correspond to (210) and (002) facets, respectively,13,52 but BM-PCN reveals remarkably decreased peak intensity and ∼0.2° shift of the (002) peak to a lower 2θ (indicative of the increased interlayer distance) relative to PCN, demonstrating the ball-milling induced hydrogen bond destruction and substantial decrease of crystallinity. The remarkable decrease of crystallinity and almost no change of the surface area of BM-PCN, compared with those of PCN, further suggest that ball milling may form massive thin nanosheets (Fig. 1c) most of which stack into compact particles (Fig. 1b) owing to their high surface energy. In comparison with BM-PCN, BM-PCN/Co-c exhibits a narrower (002) peak, suggesting enhanced crystallinity owing to the calcination-induced hydrogen bond reforming, consistent with the FT-IR results. On the whole, it is likely the ball-milling induced destruction of hydrogen bonds that contributes largely to the increase of surface energy and new active adsorption centers and thus Co2+ adsorption on BM-PCN.
Optical absorption capability of samples was investigated by UV-vis diffuse reflectance spectroscopy (DRS). As shown in Fig. 3a, BM-PCN/Co-c, BM-PCN, and PCN/Co-c exhibit considerably higher, lower, and similar optical absorption than/to PCN, respectively. For BM-PCN/Co-c, the optical absorption enhancement at a wavelength of <400 nm may benefit from the electron-rich Co that enhances π–π* transitions in heptazine rings,53 and the Urbach tail absorption should arise from the Co–OH doping.54,55 Bandgaps (Eg) of PCN, BM-PCN, BM-PCN/Co-c, and PCN/Co-c were roughly confirmed as 2.70, 2.81, 2.56, and 2.73 eV, respectively, via the formula Eg/eV = 1240/(λed/nm)56 where λed is the absorption edge determined by solid lines in the spectra. The wider Eg of BM-PCN probably results from the quantum size effect of massive ultrathin crystal nanosheets (Fig. 1c) formed by ball milling, and the narrower Eg of BM-PCN/Co-c arises from the Co–OH doping that was then verified by DFT calculations. As shown in Fig. S16,† the calculated Eg of BM-PCN/Co-c, ∼1.90 eV, is much smaller than that of PCN (2.57 eV), in accordance with the experimental results. For PCN, the conduction band (CB) is contributed by C 2p and N 2p orbitals and the valence band (VB) mainly by N 2p orbitals, while for BM-PCN/Co-c, the CB is contributed by Co 3d, C 2p, and N 2p orbitals and the VB mainly by Co 3d and N 2p orbitals (Fig. S16c and d†), effectively manifesting that the narrowing of Eg of BM-PCN/Co-c results from the Co–OH doping. In addition, there are prominent doping levels (Ed) in the bandgap of BM-PCN/Co-c, mainly contributed by Co 3d and O 2p orbitals (Fig. S16d†), effectively proving the Co–OH doping effect in BM-PCN/Co-c. Similar calculation results have been reported for Pt–OH modified carbon nitride.57 Given that the experimental Co content (0.75 wt%) is much lower than the theoretical (6.71 wt%), practical doping levels in the bandgap may approach more to the VB. CB edges of the samples (ECB) could be roughly determined by using Mott-Schottky plots (Fig. S17†) and their Fermi levels (Ef) were subsequently confirmed based on VB-XPS spectra (Fig. S18†). Energy band levels of the samples are shown in Fig. 3b, and it seems that ball milling causes a slight downshift of the VB edge (EVB) of BM-PCN, favorable for photocatalytic water splitting, but the Co–OH doping causes a slight downshift of ECB and upshift of EVB of BM-PCN/Co-c. It is noteworthy that the Ed close to the VB edge (EVB) can capture photogenerated holes58 and thus the single-atom Co–OH works as the active site for the OER (Fig. 3b).
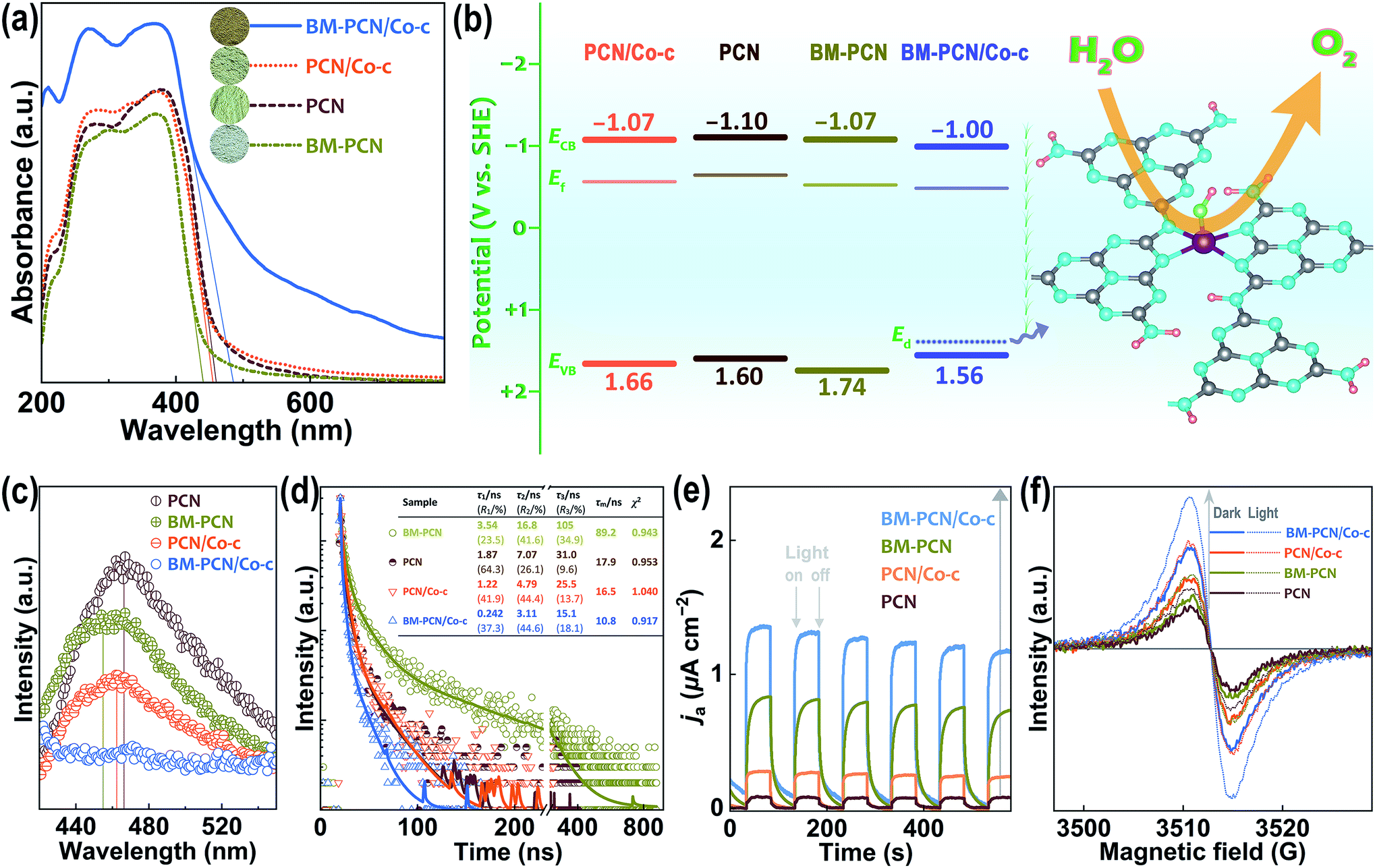 | ||
| Fig. 3 (a) UV-vis diffuse reflectance spectra of PCN, BM-PCN, BM-PCN/Co-c, and PCN/Co-c; (b) energy band levels of the samples and schematic illustration for water oxidation on BM-PCN/Co-c; (c) photoluminescence spectra, (d) time-resolved fluorescence spectra, and (e) anodic photocurrent (Ja) response of the samples; and (f) EPR spectra of the samples in the dark and under visible light irradiation. Data in (d) are the results of fitting decay curves to a tri-exponential model. Dark Ja in (e) was set as zero for distinct comparison. | ||
Spectroscopy and photoelectrochemical tests were conducted to evaluate photogenerated charge separation and transfer performance. As shown in Fig. 3c, photoluminescence (PL) spectra of all the samples show one emission peak, basically corresponding to their bandgap emission. BM-PCN exhibits weaker PL intensity than PCN, revealing a decreased photogenerated charge recombination efficiency, which originates from faster charge transfer from the inside to the surface of ultrathin nanosheets (Fig. S19†) and trapped by surface states.59 BM-PCN/Co-c exhibits the lowest PL intensity and the PL intensity of PCN/Co-c is lower than that of PCN, which arises from the Ed capturing photogenerated holes to reduce their direct recombination with electrons beside the ultrathin nanosheet effect in BM-PCN/Co-c. Fig. 3d shows time-resolved fluorescence spectra of the samples. Decay curves were well fitted to a tri-exponential model (S3) and the obtained results are shown in Fig. 3d. Three lifetimes (τ1–τ3) and their mean lifetime (τm, 89.2 ns) of BM-PCN are all much longer than those of PCN (τm = 17.9 ns), further suggesting the faster charge transfer from the inside to the surface of ultrathin nanosheets in BM-PCN, decreasing the direct charge recombination efficiency, but with subsequent surface radiative recombination.60 Interestingly, the τ1–τ3 and τm (10.8 ns) of BM-PCN/Co-c are much shorter than those of PCN, which should result from faster transfer of holes to Ed that effectively decreases the charge recombination efficiency, with subsequent nonradiative energy transformation.61 The Co–OH doping effect also makes PCN/Co-c exhibit shorter τ1–τ3 and τm (16.5 ns) than PCN. Fig. 3e shows the photocurrent response of the samples. Their anodic photocurrent density follows the order PCN < PCN/Co-c < BM-PCN < BM-PCN/Co-c, indicating gradually increased photogenerated charge separation efficiencies,62 basically consistent with the PL results. The relatively high photocurrent response of BM-PCN benefits from the applied bias that effectively inhibits surface recombination of photogenerated charge carriers.
To assess charge mobility of the samples, their electrochemical impedance spectroscopy (EIS) spectra were tested with high-frequency data simply fitted to an equivalent circuit (Fig. S20†). The obtained charge transfer resistance (Rct) follows the order PCN (26 Ω) > BM-PCN (18 Ω) ≈ PCN/Co-c (19 Ω) > BM-PCN/Co-c (13 Ω). Apparently, BM-PCN/Co-c exhibits smaller Rct than BM-PCN and PCN/Co-c, and PCN/Co-c exhibits smaller Rct than PCN, indicating the highest charge transfer performance of BM-PCN/Co-c63 which originates from the single-atom Co modification64 that may increase the electron density to facilitate charge transport. The smaller Rct of BM-PCN than that of PCN indicates the additional favorable effect of ultrathin nanosheets.65Fig. 3f shows electron paramagnetic resonance (EPR) spectra of the samples. All reveal one single Lorentzian line centered at a g of 2.0039, attributed to unpaired electrons in heptazine rings.66 In the dark, the EPR signal intensity follows the order PCN < BM-PCN < PCN/Co-c < BM-PCN/Co-c, and the stronger signal of BM-PCN than that of PCN results from formation of ultrathin nanosheets that enhances delocalization of unpaired electrons, while the stronger signal of BM-PCN/Co-c and PCN/Co-c mainly benefits from the Co doping that increases the delocalized electron density.67 Under visible light irradiation, the samples exhibit remarkable signal enhancement, following the sequence PCN < BM-PCN < PCN/Co-c < BM-PCN/Co-c, similar to that of the signal intensity in the dark, suggesting that the increase in the delocalized electron density facilitates charge photoexcitation. The high delocalized electron density favors charge transport, consistent with the EIS results, and the high photoexcited charge density benefits enhancement of photocatalytic activity.
Photocatalytic OER activity of various samples was well evaluated using Ag+ as the sacrificial agent (Fig. S21†). The Co content in BM-PCN/Co-c was optimized according to the photocatalytic OER rates and BM-PCN-c exhibits no detectable OER activity (Fig. S22†), indicating indispensability of the Co–OH structure for the OER. The influence of the calcination temperature (Tc °C) of BM-PCN/Co on OER rates of BM-PCN/Co-c (Tc = 460) and BM-PCN/Co-cTc was investigated and BM-PCN/Co-c exhibits the highest photoactivity (Fig. 4a), manifesting that the optimal calcination temperature is 460 °C. Under both simulated solar light and visible light irradiation (λ ≥ 420 nm), BM-PCN/Co-c exhibits substantially higher OER activity than PCN/Co-c (Fig. 4b), further suggesting the significance of the single-atom Co loading amount, and remarkably higher activity than common PCN/CoOx (with 0.75 wt% Co, obtained via photodeposition) and BM-PCN-c/Co(OH)2 (with 0.75 wt% Co), demonstrating the high efficacy of the single-atom distribution of Co–OH in BM-PCN/Co-c. Besides, urea was used as the feedstock to synthesize carbon nitride (marked as PCN-urea) with a larger surface area (76 m2 g−1 (ref. 68)) than PCN, and PCN-urea was further used to synthesize PCN-urea/Co-c similar to the synthesis of BM-PCN/Co-c. The OER activity of BM-PCN/Co-c is prominently higher than that of PCN-urea/Co-c (with the optimized Co content and Co single atom distribution, Fig. S23†), suggesting the significant role of ball milling in fabricating the single-atom Co–N4OH structure. To quantitively compare photoactivity of the samples, their mean OER rates under visible light illumination for 2 h are shown in Fig. 4c. The OER rate of BM-PCN/Co-c can reach ∼37.3 μmol h−1, about 13.8, 28.7, 2.6, and 2.0 times those of PCN/Co-c, PCN/CoOx, BM-PCN-c/Co(OH)2, and PCN-urea/Co-c, respectively. Comparatively, less N2 was generated for BM-PCN/Co-c (Fig. S24†), further demonstrating the significance of single-atom Co–OH modification.
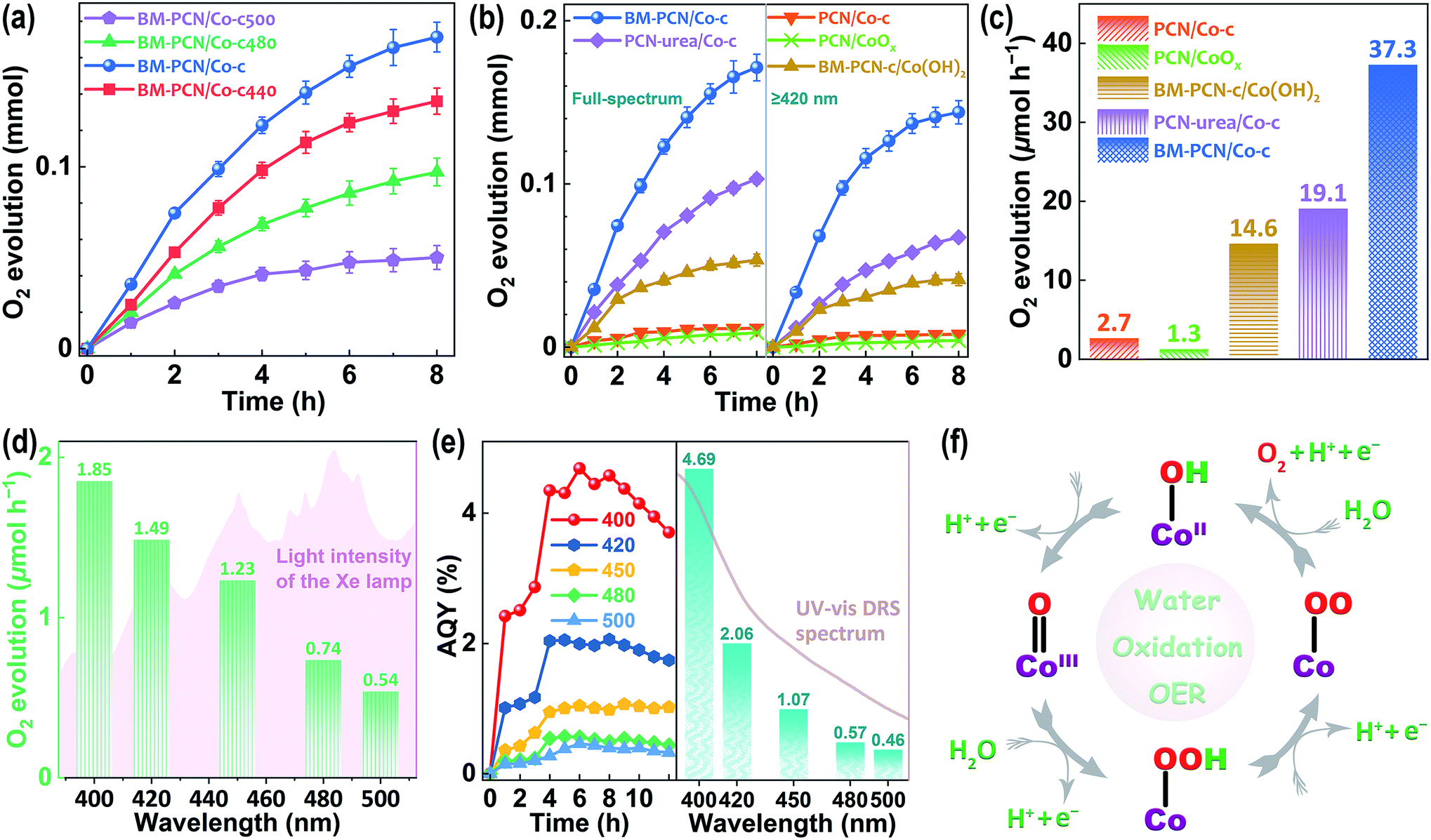 | ||
| Fig. 4 (a) The influence of the calcination temperature (Tc °C) of BM-PCN/Co on photocatalytic OER activity of BM-PCN/Co-c (Tc = 460) and BM-PCN/Co-cTc, under Xe-lamp illumination, with AgNo3 as the sacrificial agent; (b) photocatalytic oxygen evolution on various samples under Xe-lamp illumination with or without using a 420-nm filter; (c) corresponding OER rates of the samples in 2 h; (d) photocatalytic OER rates of BM-PCN/Co-c under irradiation with various monochromatic light sources for 12 h; (e) apparent quantum yields (AQYs) of BM-PCN/Co-c at different wavelengths and reaction times and the highest AQY at every wavelength, along with the UV-DRS spectrum; and (f) proposed mechanism for photocatalytic water oxidation on the single-atom CoII-OH structure. | ||
Photocatalytic oxygen evolution on BM-PCN/Co-c was also tested under monochromatic light irradiation (Fig. S25†). Apparently, BM-PCN/Co-c can exhibit OER activity even at a wavelength of 500 nm. The mean OER rate in 12 h decreases from 1.85 to 0.54 μmol h−1 with increasing wavelengths from 400 to 500 nm (Fig. 4d), independent of light intensity of the Xe lamp and is mainly dependent on optical absorption capability of BM-PCN/Co-c at various wavelengths (Fig. 3a). Fig. 4e shows apparent quantum yields (AQYs) of BM-PCN/Co-c at different reaction times and wavelengths. Basically, there are maxima of AQYs with increasing reaction time at every wavelength, suggesting the adverse effect of excessive photodeposited Ag on surfaces of samples. These maxima are shown in Fig. 4e and accord well with the UV-vis DRS spectrum with increasing wavelengths. The maxima of AQYs at 400, 420, 450, and 500 nm can reach 4.69, 2.06, 1.07, and 0.46%, respectively. Compared with the reported photocatalytic OER results for PCN (Table S4†), BM-PCN/Co-c exhibits the top-class performance.
To investigate chemical stability of BM-PCN/Co-c, the cyclic OER experiment was conducted. After five consecutive runs, OER rates of BM-PCN/Co-c decrease less (Fig. S26a†), with the morphology similar to the original (Fig. S26b†). Co single atoms in the sample could still be distinctly observed by HAADF-STEM (Fig. S26c and d†). In addition, N 1s core-level XPS spectra of BM-PCN/Co-c are almost similar before and after the cyclic experiment (Fig. S26e†). These indicate the high stability of the basic framework structure of the sample. However, Co 2p core-level spectra show remarkable differences before and after the experiment, not only the CoII peak shift, probably owing to ion (e.g., IO4−) adsorption, but also formation of a large amount of CoIII (Fig. S26f†). Coexistence of CoII/CoIII may suggest the photocatalytic OER mechanism.
The proposed OER mechanism based on the Co–OH structure is shown in Fig. 4f, according to the reported results in Mn doped PCN.34 Four holes are needed to complete four oxidation steps and obtain one O2 molecule. The first step starting with one hole may involve formation of the CoIIIO bond. The Co–N4OH structure should facilitate the water oxidation more compared with that of Co–N4 without OH coordination, by leaving out the initial adsorption process of H2O molecules.34 On the whole, the high photocatalytic OER activity of Co-PCN benefits from the Co–N4OH structure that not only effectively enhances optical absorption, and charge separation and transport, but also works as the highly active site for the OER.
Conclusions
Single-atom CoII-OH modified PCN with an optimized Co content of 0.75 wt% (Co-PCN) was simply prepared via a ball milling-adsorption-calcination process and the Co content increases ∼37 times thanks to the assistance of ball-milling that formed ultrathin nanosheets. The single-atom CoII–N4OH structure in Co-PCN was confirmed and evidenced to work as the active center for the OER. The CoII-OH modification was considered to endow Co-PCN with enhanced optical absorption, and photogenerated charge separation and transport capability. Co-PCN exhibits a highly enhanced OER rate (∼37.3 μmol h−1 at the highest), compared with those of common PCN/CoOx (∼28 times lower) and the control sample prepared without ball milling (∼13 times lower) under visible light irradiation, with the highest apparent quantum yields reaching 4.69, 2.06, 1.07, and 0.46% at 400, 420, 450, and 500 nm, respectively, and is among the best OER photocatalysts reported so far. This work provides an effective method to prepare efficient OER photocatalysts and may guide successive synthesis of high-performance photocatalysts (e.g., Z-scheme heterojunctions) for overall water splitting.Data availability
All experimental data are provided in the ESI.† Other data are available from the corresponding author upon reasonable request.Author contributions
F. Y. synthesized and characterized the samples and wrote this manuscript. Q. D., G. W. and Y. X. characterized the materials. H. L. conceived the experiments and revised the manuscript. W. H. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21872082) and the Young Scholars Program of Shandong University in China (No. 2018WLJH39). We are grateful for the assistance of Shandong University Structural Constituent and Physical Property Research Facilities (SDU SCPPRF) and Shiyanjia Lab (www.shiyanjia.com).Notes and references
- D. Kirk-Davidoff, in Green Chemistry, ed. B. Török and T. Dransfield, Elsevier, Netherlands, 2018, p. 211 Search PubMed.
- H. Zhang, S. Zuo, M. Qiu, S. Wang, Y. Zhang, J. Zhang and X. W. Lou, Sci. Adv., 2020, 6, eabb9823 CrossRef CAS.
- J. Liu, Y. Li, X. Zhou, H. Jiang, H. G. Yang and C. Li, J. Mater. Chem. A, 2020, 8, 17 RSC.
- Y. Bai, K. Hippalgaonkar and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 16222 RSC.
- S. Wang, Y. Wang, S.-Q. Zang and X. W. Lou, Small Methods, 2020, 4, 1900586 CrossRef CAS.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559 CrossRef CAS.
- G. Zhang, S. Zang, Z.-A. Lan, C. Huang, G. Li and X. Wang, J. Mater. Chem. A, 2015, 3, 17946 RSC.
- P. Li, X. Chen, H. He, X. Zhou, Y. Zhou and Z. Zou, Adv. Mater., 2018, 30, 1703119 CrossRef.
- J. Yan, T. Wang, G. Wu, W. Dai, N. Guan, L. Li and J. Gong, Adv. Mater., 2015, 27, 1580 CrossRef CAS.
- G. He, W. Yang, W. Zheng, L. Gong, X. Wang, Y. An and M. Tian, RSC Adv., 2019, 9, 18222 RSC.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2020, 8, 2002458 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612 CrossRef CAS.
- Y. Wang, S. Z. F. Phua, G. Dong, X. Liu, B. He, Q. Zhai, Y. Li, C. Zheng, H. Quan, Z. Li and Y. Zhao, Chem, 2019, 5, 2775 CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS.
- Z. Liang, X. Zhuang, Z. Tang, H. Li, L. Liu and W. Kang, J. Mater. Chem. A, 2021, 9, 6805 RSC.
- Q. Han, Z. Cheng, B. Wang, H. Zhang and L. Qu, ACS Nano, 2018, 12, 5221 CrossRef CAS.
- S. Yu, J. Li, Y. Zhang, M. Li, F. Dong, T. Zhang and H. Huang, Nano Energy, 2018, 50, 383 CrossRef CAS.
- G. Liu, H. Lv, Y. Zeng, M. Yuan, Q. Meng, Y. Wang and C. Wang, Trans. Tianjin Univ., 2021, 27, 139 CrossRef CAS.
- P. Zhou, Y. Chao, F. Lv, J. Lai, K. Wang and S. Guo, Sci. Bull., 2020, 65, 720 CrossRef CAS.
- P. Yang, H. Ou, Y. Fang and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 3992 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- C. Han, L. Du, M. Konarova, D.-C. Qi, D. L. Phillips and J. Xu, ACS Catal., 2020, 10, 9227 CrossRef CAS.
- G. Zhang, Z. A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261 RSC.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644 CrossRef CAS.
- Z. Pan, Y. Zheng, F. Guo, P. Niu and X. Wang, ChemSusChem, 2017, 10, 87 CrossRef CAS.
- L. Ge, C. Han, X. Xiao and L. Guo, Appl. Catal., B, 2013, 142–143, 414 CrossRef CAS.
- F. Raziq, L. Sun, Y. Wang, X. Zhang, M. Humayun, S. Ali, L. Bai, Y. Qu, H. Yu and L. Jing, Adv. Energy Mater., 2018, 8, 1701580 CrossRef.
- T. Kanazawa, K. Kato, R. Yamaguchi, T. Uchiyama, D. Lu, S. Nozawa, A. Yamakata, Y. Uchimoto and K. Maeda, ACS Catal., 2020, 10, 4960 CrossRef CAS.
- G. Zhang, S. Zang and X. Wang, ACS Catal., 2015, 5, 941 CrossRef CAS.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970 CrossRef CAS.
- W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139, 3021 CrossRef CAS.
- K. Zhang, L. Wang, X. Sheng, M. Ma, M. S. Jung, W. Kim, H. Lee and J. H. Park, Adv. Energy Mater., 2016, 6, 1502352 CrossRef.
- S. Sun, G. Shen, J. Jiang, W. Mi, X. Liu, L. Pan, X. Zhang and J. J. Zou, Adv. Energy Mater., 2019, 9, 1901505 CrossRef.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312 CrossRef CAS.
- D. Zhao, C. L. Dong, B. Wang, C. Chen, Y. C. Huang, Z. Diao, S. Li, L. Guo and S. Shen, Adv. Mater., 2019, 31, e1903545 CrossRef.
- D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen, F. Xue, S. Shen and L. Guo, Nat. Energy, 2021, 6, 388 CrossRef CAS.
- Y. Li, Y. Wang, C. L. Dong, Y. C. Huang, J. Chen, Z. Zhang, F. Meng, Q. Zhang, Y. Huangfu, D. Zhao, L. Gu and S. Shen, Chem. Sci., 2021, 12, 3633 RSC.
- S.-M. Xu, T. Pan, Y.-B. Dou, H. Yan, S.-T. Zhang, F.-Y. Ning, W.-Y. Shi and M. Wei, J. Phys. Chem. C, 2015, 119, 18823 CrossRef CAS.
- J. Yan, H. Wu, H. Chen, L. Pang, Y. Zhang, R. Jiang, L. Li and S. Liu, Appl. Catal., B, 2016, 194, 74 CrossRef CAS.
- H. Schreyer, R. Eckert, S. Immohr, J. de Bellis, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2019, 58, 11262 CrossRef CAS.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pan and X. Bao, Sci. Adv., 2015, 1, e1500462 CrossRef.
- S. Maleki and A. Karimi-Jashni, Appl. Clay Sci., 2017, 137, 213 CrossRef CAS.
- Q. Han, F. Zhao, C. Hu, L. Lv, Z. Zhang, N. Chen and L. Qu, Nano Res., 2015, 8, 1718 CrossRef CAS.
- Z. Guo, Y. Xie, J. Xiao, Z. J. Zhao, Y. Wang, Z. Xu, Y. Zhang, L. Yin, H. Cao and J. Gong, J. Am. Chem. Soc., 2019, 141, 12005 CrossRef CAS.
- L. Qian and Y. Miao, Polyhedron, 2019, 160, 213 CrossRef CAS.
- T.-Q. Zi, X.-R. Zhao, C. Liu, Y.-Q. Cao and A.-D. Li, J. Alloys Compd., 2021, 855, 157446 CrossRef CAS.
- J.-K. Chang, C.-M. Wu and I. W. Sun, J. Mater. Chem., 2010, 20, 3729 RSC.
- J. Xu, L. W. Zhang, R. Shi and Y. F. Zhu, J. Mater. Chem. A, 2013, 1, 14766 RSC.
- Q. Deng, G. Ba, T. Huo, H. Li, G. Wang, F. Yu and W. Hou, Appl. Surf. Sci., 2020, 148427 Search PubMed.
- Z. Liang, Y. Xia, G. Ba, H. Li, Q. Deng and W. Hou, J. Mater. Chem. A, 2019, 7, 25824 RSC.
- J. Wu, X. Ji, X. Yuan, Z. Zhao, Y. Li, B. Wen, H. Zhang, D. Yu, Y. Zhao and Y. Tian, Chem. Mater., 2019, 31, 9188 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178 CrossRef CAS.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708 RSC.
- W. Zhang, Q. Peng, L. Shi, Q. Yao, X. Wang, A. Yu, Z. Chen and Y. Fu, Small, 2019, 15, 1905166 CrossRef CAS.
- C. Zhu, L. Meng, J. Zhang, S. Qin, W. Lai, B. Qiu, J. Yuan, Y. Wan, W. Huang and Y. Li, Adv. Mater., 2021, 33, 2100474 CrossRef CAS.
- X. Li, P. Cui, W. Zhong, J. Li, X. Wang, Z. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233 RSC.
- K. Watanabe, A. Iwase, S. Nozawa, S.-i. Adachi and A. Kudo, ACS Sustainable Chem. Eng., 2019, 7, 9881 CrossRef CAS.
- K. Omika, K. Takahashi, A. Yasui, T. Ohkochi, H. Osawa, T. Kouchi, Y. Tateno, M. Suemitsu and H. Fukidome, Appl. Phys. Lett., 2020, 117, 171605 CrossRef CAS.
- L. Cavigli, F. Bogani, A. Vinattieri, V. Faso and G. Baldi, J. Appl. Phys., 2009, 106, 053516 CrossRef.
- T. Huo, G. Ba, Q. Deng, F. Yu, G. Wang, H. Li and W. Hou, Appl. Catal., B, 2021, 287, 119995 CrossRef CAS.
- P. Zhang, X. F. Lu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 8128 CrossRef CAS.
- X. Zhang and G. Lu, Carbon, 2016, 108, 215 CrossRef CAS.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- L. Zhou, J. Feng, B. Qiu, Y. Zhou, J. Lei, M. Xing, L. Wang, Y. Zhou, Y. Liu and J. Zhang, Appl. Catal., B, 2020, 267, 118396 CrossRef CAS.
- G. Dong, W. Ho and C. Wang, J. Mater. Chem. A, 2015, 3, 23435 RSC.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X.-M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826 CrossRef CAS.
- F. Yang, G. Ba, Z. Wang and H. Li, J. Colloid Interface Sci., 2021, 594, 64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of samples, details of characterization, partial SEM and TEM images, XANES and EXAFS spectra, XPS spectra, photocatalytic OER data, XRD patterns, XANES and EXAFS fitting data, VB-XPS and FT-IR spectra, nitrogen sorption data, zeta potential and Mott–Schottky plots, DFT calculation results of energy bands and DOS, and elemental analysis data (PDF). See DOI: 10.1039/d1sc06555f |
| This journal is © The Royal Society of Chemistry 2022 |