
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperativity as quantification and optimization paradigm for nuclear receptor modulators†
Pim J.
de Vink‡
 a,
Auke A.
Koops‡
a,
Giulia
D'Arrigo‡
ab,
Gabriele
Cruciani
c,
Francesca
Spyrakis
*b and
Luc
Brunsveld
*a
a,
Auke A.
Koops‡
a,
Giulia
D'Arrigo‡
ab,
Gabriele
Cruciani
c,
Francesca
Spyrakis
*b and
Luc
Brunsveld
*a
aLaboratory of Chemical Biology, Department of Biomedical Engineering and Institute for Complex Molecular Systems, Eindhoven University of Technology, P. O. Box 513, 5600MB Eindhoven, The Netherlands. E-mail: l.brunsveld@tue.nl
bDepartment of Drug Science and Technology, University of Turin, via Giuria 9, 10125 Turin, Italy. E-mail: francesca.spyrakis@unito.it
cDepartment of Chemistry, Biology and Biotechnology, University of Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy
First published on 19th January 2022
Abstract
Nuclear Receptors (NRs) are highly relevant drug targets, for which small molecule modulation goes beyond a simple ligand/receptor interaction. NR–ligands modulate Protein–Protein Interactions (PPIs) with coregulator proteins. Here we bring forward a cooperativity mechanism for small molecule modulation of NR PPIs, using the Peroxisome Proliferator Activated Receptor γ (PPARγ), which describes NR–ligands as allosteric molecular glues. The cooperativity framework uses a thermodynamic model based on three-body binding events, to dissect and quantify reciprocal effects of NR–coregulator binding (KID) and NR–ligand binding (KIID), jointly recapitulated in the cooperativity factor (α) for each specific ternary ligand·NR·coregulator complex formation. These fundamental thermodynamic parameters allow for a conceptually new way of thinking about structure–activity-relationships for NR–ligands and can steer NR modulator discovery and optimization via a completely novel approach.
Introduction
The rapidly increasing field of small molecule stabilization of Protein–Protein Interactions (PPIs)1,2 is intrinsically connected to molecular cooperativity mechanisms, because of the simultaneous interactions between more than two molecular entities.3–5 The development of such cooperativity concepts is not only of relevance for so-called molecular glues2 and PROTACs,4 but also harbours great potential for more classical drug targets that also undergo PPIs.6 Nuclear Receptors (NRs) form a superfamily of ligand-mediated transcription factors that regulate genes involved in numerous cell processes such as metabolism and development. Their concurrent prominent role in related diseases makes them highly pursued as drug targets.7Despite a large number of studies on NRs and the significant number of drugs developed for this class of proteins, there are still many unknowns regarding the regulation of NR activity at the molecular level. Recent examples that highlight the identification of novel molecular regulatory mechanisms include studies on the conformational dynamics upon ligand binding,8 co-binding of multiple ligands,9,10 preferential cofactor recruitment,11 allosteric communication,12 ligands for orphan receptors,13 interplay with DNA binding,14 unexpected hetero-dimerization,15 and NR crosstalk.16 As an example of the continuing evolution of our thinking and understanding of NR activity regulation, the highly valuable mouse-trap model has now been additionally refined to include coregulator binding.17
A crucial reason why many of these molecular insights are still being discovered, despite the long-standing therapeutic applications of NR–ligands, relates to the fact that NR–ligands are not simple active site inhibitors, but actually modulate complex PPIs via an allosteric binding mechanism that transfers the molecular effects of ligand binding to the surface of the NR.18,19 The influence of ligand binding is as such indirect, modulating the equilibrium of conformations that NRs can address.20 Thus, NR modulation goes beyond a simple ligand/receptor interaction and should be viewed as a more complex three- or multi-body interplay.21 A single KD value is therefore not sufficient to describe the NR·ligand·coregulator complex formation in a valuable and quantitative manner. Fundamental insights and more uniform quantitative descriptions of the molecular mechanisms underlying NR/ligand binding and ensuing allosteric PPI regulation are urgently needed, along with robust experimental approaches that allow dissecting these different molecular binding events occuring simultaneously.
Inspired by recent progress in the field of PPI modulation,3 GPCRs,6 PROTACs,4 and molecular glues,2 we revisit NR modulation by means of cooperativity.5,22 Cooperativity is a common phenomenon in complex biological systems, where an initial binding event alters the affinity of subsequent ones.23,24 Though traditionally not viewed as such, NR–ligands basically act as allosteric molecular glues, enhancing NR/coregulator PPIs. Also, NR/ligand binding is frequently reported and compared using very different types of biochemical and cellular studies. In most cases, the reported affinity or EC50 values are the result of a combination of molecular events, including coregulator binding, and do not solely represent the ligand binding process and affinity.
To elucidate this underlying cooperativity mechanism concept of ligand mediated NR regulation, we used the Peroxisome Proliferator Activated Receptor γ (PPARγ) as a case study. PPARγ is a member of the NR1 subfamily and, being involved in fatty acid storage and glucose metabolism, is a target for different diseases such as inflammation, diabetes, obesity, neurodegenerative disorders, and cancer.25,26 The high number of diverse natural and synthetic ligands as well as coregulators known to bind PPARγ, make it an ideal NR model to explore a framework that dissects and describes the cooperative events of ligand and coregulator binding in a quantitative manner.27 Here, we report a cooperativity analysis of the NR·ligand·coregulator complex formation, utilizing an integrated biochemical, biophysical and computational approach. The framework uses a thermodynamic model based on the different binding events (Fig. 1), to dissect and quantify reciprocal effects of coregulator (KID) and ligand (KIID) binding on receptor affinity, jointly recapitulated in the cooperativity factor (α) for the ternary complex formation.3 These fundamental thermodynamic parameters allow for a conceptually new way of thinking about Structure–Activity-Relationships (SAR) for NR–ligands and their ensuing modulation of NR PPIs.
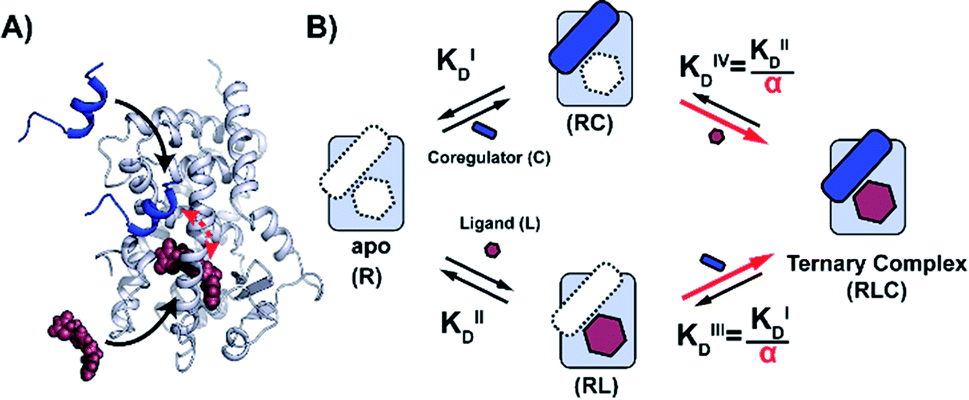 | ||
| Fig. 1 Cooperativity analysis square depicting the multiple binding events in NR–ligand mediated coregulator recruitment. (A) Structural depiction of an exemplary NR·ligand·coregulator complex composed of PPARy, rosiglitazone, shown as spheres, and MED1, shown as blue cartoon (PDB ID 5YCP28 and 6ONJ29). (B) Cooperativity scheme for ligand coregulator interplay involving sequential binding events of receptor (R), ligand (L) and coregulator (C). The coregulator binds to the target protein with KID and in the presence of a ligand this affinity is altered to KID/α. Similarly, the ligand binds with an intrinsic affinity KIID and an enhanced affinity KIID/α when the coregulator binding partner is already bound to the target protein. | ||
Results and discussion
2D-FA titrations deconvolute cooperative binding
NRs can be activated by lipophilic endogenous ligands such as fatty acids and steroid hormones, and by synthetic ligands that typically bind to a pocket in the NR–ligand binding domain (LBD). Ligand binding results in conformational changes of the LBD, in particular of helix 12 (H12), favouring the formation of PPIs with coregulator proteins that exert their effect on the genome.30,31 We adopted a cooperativity model (Fig. 1B) to interpret and deconvolute the various binding events and describe the interplay of ligand and coregulator binding more quantitatively. The coregulator (C in blue) and the ligand (L in red) both bind to the receptor (R in light blue), but do not bind to each other in the absence of R. The coregulator binds to the apo receptor with an affinity of KID. In the presence of a prebound ligand the affinity of the coregulator for the receptor is altered by a cooperativity factor (α) to KID/α. The α-factor represents the effect of the ligand-induced conformational rearrangement of the receptor on coregulator apparent affinity. An α-factor bigger than 1 represents a stimulatory effect of the ligand on the receptor/coregulator interaction (agonism), while an α-factor smaller than 1 represents a negative effect of the ligand (antagonism) and induces a lower affinity of the coregulator for the receptor. Similarly, the ligand binds with an intrinsic affinity KIID, generally enhanced upon coregulator binding to KIID/α. This dual interplay is frequently overlooked when describing NR/ligand binding, but ligand and coregulator influence each other to the exact same extend given the first law of thermodynamics. To provide a solution for such a ternary equilibrium system, we developed a semi-numerical thermodynamic model based on mass-action laws and mass-balance equations. These expressions can be rewritten into three interdependent master equations, which can be solved in a straightforward numerical recursion, without additional constraints or approximation on the free and total concentrations (see ESI† for derivation and detailed explanation).32We performed a series of 2D fluorescence anisotropy (FA) titrations, independently varying both PPARγ and ligand concentration. This allowed deconvoluting the synergistic interplay of ligand and coregulator binding to PPARγ. PPARγ–LBD was titrated to 10 nM FAM-labeled MED1 coregulator peptide in the presence of various concentrations of the ligand rosiglitazone, held constant over each titration (0–200 μM). The resulting 2D interaction profile shows the influence of rosiglitazone on the PPAR/MED1 interaction, as depicted in Fig. 2A. Without any rosiglitazone (grey line), the EC50 of PPARγ to MED1 was around 1 μM. In contrast, in the presence of an excess of rosiglitazone (200 μM, line in dark red) the EC50 of PPARγ to MED1 shifted to 16 nM (a 22-fold enhancement). Indeed, increasing doses of rosiglitazone induced a gradual shift of the titration curve towards the left. The increased occupancy of PPARγ by the ligand at higher concentrations results in an increased contribution of ligand-mediated binding of the coregulator, thereby enhancing the overall apparent affinity between PPARγ and MED1. However, from concentrations of 1 μM and higher, the occupancy of rosiglitazone maxes out and the PPARγ/MED1 affinity does not increase further. The maximum EC50-shift is influenced by the cooperativity factor, while the progression based on ligand concentration is determined by a combination of the intrinsic affinity of the ligand (KIID) and the cooperativity factor (ESI Fig. 1B and C†).
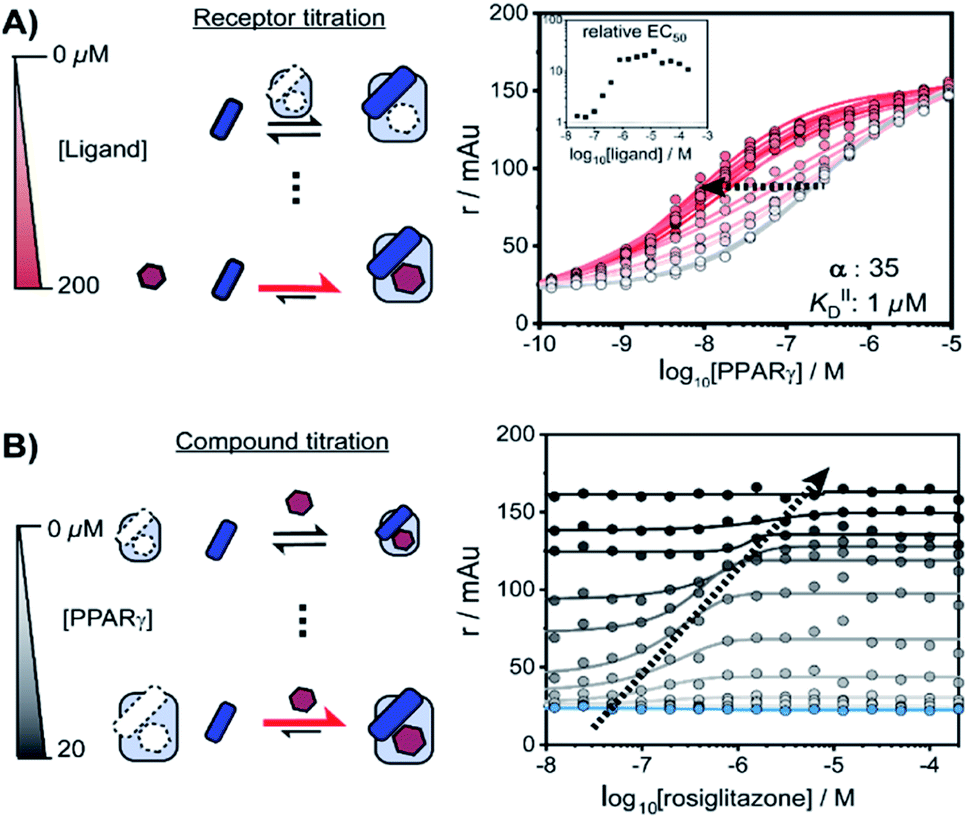 | ||
| Fig. 2 Interplay of rosiglitazone and MED1 binding to PPARγ–LBD measured by 2D fluorescence anisotropy. (A) 2D-FA protein-titration of PPARγ to 10 nM labeled MED1 at various rosiglitazone concentrations (0–200 μM). (B) 2D-FA compound titration of rosiglitazone to 10 nM labeled MED1 with various concentrations of PPARγ (0–20 μM; blue to black). | ||
By fitting 2D data profiles (vide infra) with the model, the values of α and KIID are obtained. The KID is obtained experimentally by the titration of PPARγ to MED1 without any ligand present (0.23 μM). Analysis of the experimental data depicted in Fig. 2A, resulted in an intrinsic affinity (KIID) of 1 μM for rosiglitazone to the apo receptor and an α-factor of around 35.
Intriguingly, at ligand concentrations below 500 nM, the ligand is not continuously in excess over the protein throughout the whole PPARγ titration. As a result, the overall titration becomes a composite of two distinct binding modes. Initially the binding of PPARγ to MED1 is ligand-mediated (most of the PPARγ is bound to rosiglitazone), but at higher PPARγ concentrations the apo state of the protein is in excess. These data highlight the complexity of the interdependent binding events involved within the receptor·ligand·coregulator system and the importance of the choice of assay conditions; i.e. concentrations of the different components.
In an alternative titration format, the ligand was titrated to a set concentration of MED1 and a discrete concentration range of PPARγ (Fig. 2B). In this assay format, the effect of the ligand on different starting points of the PPARγ/MED1 equilibrium is probed. As expected, without any PPARγ present, there is no effect from the addition of rosiglitazone (Fig. 2B, blue line). Likewise, at a high PPARγ concentration (above 20 μM), the binding of MED1 to PPARγ is already saturated and additional ligand cannot further shift this equilibrium (Fig. 2B, black line). However, at intermediate receptor concentrations, different dose response curves were obtained, reflecting the shift of the PPARγ/MED1 equilibrium to a more bound state by virtue of the ligand binding. The lower and upper end plateaus are influenced by the PPARγ concentration and illustrate that a large excess of rosiglitazone is not the only prerequisite for coregulator binding. In fact, the EC50 of the dose–response curve shifts to the right and to higher anisotropy values by virtue of the moving bottom and upper plateau as indicated by the dotted arrow. For this reason, receptor titrations (like those in Fig. 2A) are preferred for data fitting.
Dissecting PPAR agonists in terms of affinity and cooperativity
We thus applied the aforementioned approach to characterize in detail a library of various PPARγ ligands (Fig. 3A). The library contains both partial and full agonists from different compound classes including the glitazones33 and fibrates.34 PPARγ was titrated to 10 nM labeled MED1 coregulator in the presence of different doses of each ligand, resulting in 2D titration profiles of all compounds in the library (see Fig. 3B for an additional example and ESI Fig. S2† for all data). As previously observed with rosiglitazone, the affinity of PPARγ to MED1 gradually increased with increasing ligand concentrations (shades from light to dark). However, the extent of the shift and the concentration window in which the shift occurred, varied according to the ligand. While rosiglitazone and tesaglitazar (ESI Fig. S2A and E†) induced an EC50 shift of 22- and 24-fold, other ligands, such as pemafibrate and telmisartan, induced a smaller EC50 shift. We applied the cooperativity model to the 2D plots, to extract the cooperativity factor and intrinsic affinity (KIID) for each ligand (Fig. 3C and D).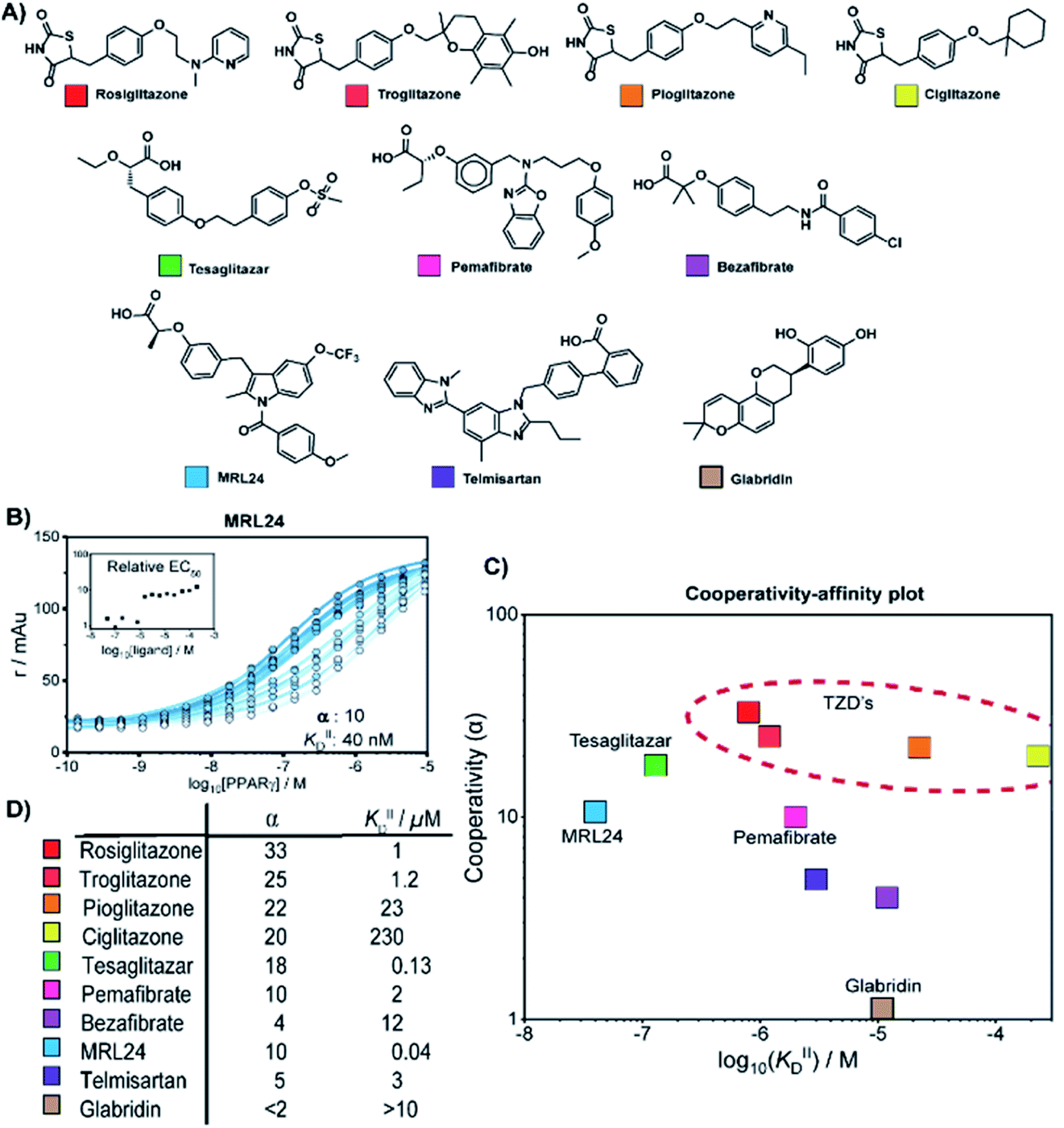 | ||
| Fig. 3 Cooperativity analyses of PPAR ligands on the interaction with the MED1 coregulator. (A) Molecular structure of PPARγ agonists. (B) Representative 2D titration. PPARγ–LBD is titrated to labelled MED1, at several constant concentrations of agonist (0–200 μM). Insets show the relative EC50 as function of the concentration ligand used (see ESI Fig. S2† for total overview). (C) Cooperativities and intrinsic affinities parameters. The cooperativity factor α, defined as the ratio between ligand bound affinity and the non-stabilized affinity of the cofactor for the receptor, and KIIID are obtained through data-fitting according to the model depicted in Fig. 1. (D) Overview of the distribution between cooperativity (α) and intrinsic affinity (KIID) parameters of the tested PPAR ligands. | ||
The cooperativity analysis revealed a wide diversity in cooperativity and affinity values for the different ligands (Fig. 3C). Classical full agonists such as rosiglitazone and tesaglitazar have high cooperativity factors, resulting in large EC50-shifts in the experimental profiles. Conversely, PPAR ligands typically classified as partial agonists induce a smaller EC50-shift, but with still notable cooperativity values. Of particular note is MRL24, known to strongly bind PPARγ but with limited effects in transcriptional assays,35,36 having a modest cooperativity factor of 10 but a very high KIID of around 40 nM. Au contraire, pemafibrate features a similar modest cooperativity as MRL24, but binds with a much weaker affinity of around 2 μM. Only in the case of ligands with very low cooperativity, such as the flavonoid glabridin (ESI Fig. S2J†), no exact cooperativity factor could be determined, in the absence of a large EC50 shift. The resulting KIID value fitted of ∼10 μM is in line with previous observations.37
Interestingly, all glitazone ligands (also known as thiazolidinediones (TZDs); ESI Fig. S2A–D†) showed similar cooperativity factors but quite different affinities (KIID), as in the following decreasing order: rosiglitazone > troglitazone > pioglitazone > ciglitazone (Fig. 3D). As a result, at high dose (200 μM) the cooperativity effect exerted by rosiglitazone and ciglitazone (showing the most distant values in terms of affinity) was quite the same, while at low dose only rosiglitazone was able to enhance the interaction between PPARγ and MED1. Presumably, the strength of the cooperativity effect of this class of ligands is dominated by the thiazolidinedione group, which is shared by all compounds and makes contact with PPARγ helix 12, thus favouring efficient binding of MED1.38 The variance in affinity for PPARγ amongst the TZDs can be attributed to the structural diversity of their hydrophobic tails (Fig. 3A and ESI Fig. S3†).
Thermodynamic analysis of the cooperativity effects
We employed isothermal titration calorimetry (ITC) as an orthogonal technique to verify the observed affinities from the 2D-FA titrations. ITC measurements detect the heat originating from the various binding events involved within the two or three components of the system, i.e., receptor, ligand, and coregulator, thus observing all the binding events simultaneously. This provides an interesting alternative perspective into the interplay of ligand and coregulator binding to the nuclear receptor; it complements the cooperativity analysis by 2D-FA that reports on the molecular binding events solely via the fluorescently labelled coregulator.First the intrinsic affinity (KIID) of the ligands for PPARγ was determined in a two-component study, in the absence of any coregulator (Fig. 4A). The binding affinities obtained by (1D) ITC and via 2D-FA followed the same global trend, though the absolute affinity values determined were, in general, slightly weaker with ITC than as obtained by the 2D-FA model. For example, for tesaglitazar we determined a KIID of 320 nM and 120 nM with ITC and 2D-FA, respectively, for rosiglitazone 1 μM vs. 15 μM values were obtained, while for telmisartan the same KIID of 6 μM and 5 μM was measured with the two approaches.
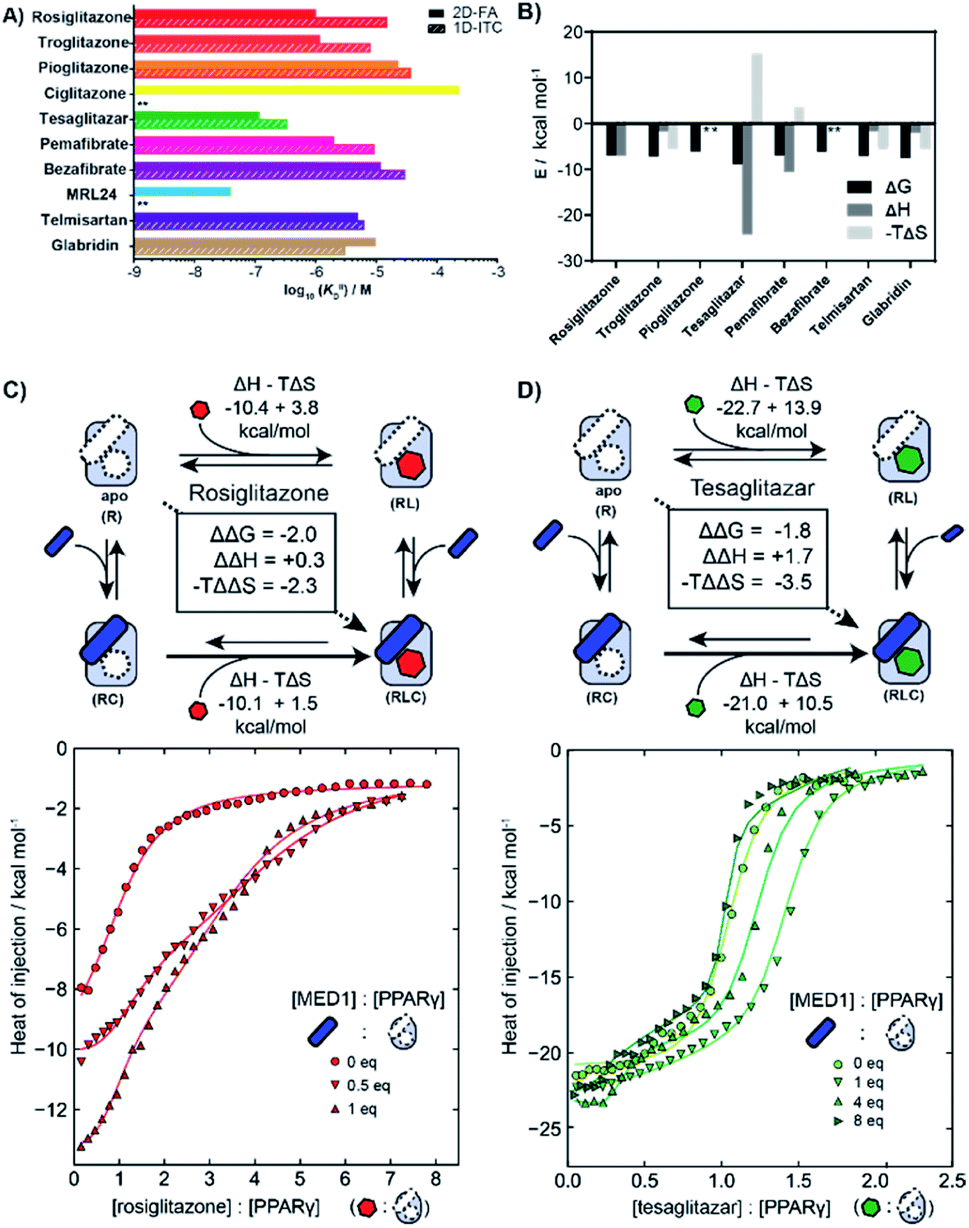 | ||
| Fig. 4 Isothermal titration calorimetry measurements (ITC) of PPAR ligands and synergy with the coregulator MED1. (A) Comparison of intrinsic affinities (KIID) of the various PPARy ligands as measured by 1D-ITC and 2D-FA (** = not determined). (B) Thermodynamic characteristics of binary interaction of ligand for PPARγ (* = not determined; see additional ITC data & ESI Table S1† for total overview). (C) 2D-ITC of rosiglitazone to PPARγ in the presence of various concentrations of MED1 added in cell and syringe. (D) 2D-ITC of tesaglitazar to PPARγ in the presence of various concentrations of MED1 added in cell and syringe. | ||
In addition to the dissociation constant of the ligands, ITC measurements can provide the thermodynamic binding characteristic of the ligand–receptor interaction (Fig. 4B). In general, all the tested ligands showed an enthalpy-driven binding, where the favourable enthalpic contribution is partially off-set by an unfavourable entropy. Intriguingly, the ΔH contribution for tesaglitazar is much larger than for the other full agonists such as rosiglitazone and troglitazone (−23 vs. −10 and −3 kcal mol−1). Also, pemafibrate, which has a similar carboxylic head group, has a high enthalpy driven interaction (−12 kcal mol−1). These two PPAR agonists are known to make polar interactions via their carboxylic acid with helices 3 and 12 in the PPARγ–LBD39 conferring rigidity to the complex; hence the unfavourable entropy. We were not able to determine the affinity of MRL24 due to a low signal to noise ratio, indicating a low net enthalpy contribution, while the interaction for ciglitazone was weaker than 200 μM (see ESI and Table S1† for a general overview).
2D-ITC experiments were conducted titrating either rosiglitazone or tesaglitazar to PPARγ, in the presence of MED1 in both cell and syringe. Representative binding curves and binding data are shown in Fig. 4C and D. In the presence of MED1, the isotherm consists of two distinct binding phases. Depending on the PPARγ:MED1 ratio, the titrated ligand initially predominately binds to the preformed binary PPARγ·MED1 complex, while at larger ligand equivalents the binding is predominately to the apo PPARγ. During the whole titration, as a result of the shifting equilibrium upon ligand binding to PPARγ, more MED1 starts to bind to PPARγ and the PPARγ·ligand complex.
To dissect the multitude of binding events, the three component ITC measurements were analysed with SEDPHAT.40–42 As an input, the thermodynamic parameters of the binary MED1/PPARγ interaction were determined separately (KD: 66.3 μM; ΔH: −28.3 kcal mol; −TΔS: 22.5 kcal mol; ESI Fig. S6†). Fitting of the ITC data, representative curves shown in Fig. 4C, resulted in a cooperativity value α = 28 for the PPARγ·rosiglitazone·MED1 complex with an enthalpy and entropy component of ΔΔH = +0.3 kcal mol−1 and −TΔΔS = −2.3 kcal mol−1 (ESI Fig. S9†). In contrast, the presence of MED1 causes an unfavourable enthalpic effect on the binding of tesaglitazar (Fig. 4D): the magnitude of the ΔH decreases from 22.7 to 21.0 kcal mol−1, with a net ΔΔH of +1.7 kcal mol−1. Overall, there is still a net positive cooperative effect (α = 18) caused by the −TΔΔS of −3.5 kcal mol−1 (ESI Fig. S8†).
To corroborate these results, a reverse titration of MED1 against PPARγ in the presence of tesaglitazar was also performed. Similarly in this case, we observed that the affinity of MED1 significantly increases in the presence of the ligand. This shows that the order of binding is reversible and corroborates the presented cooperativity square (Fig. 1). Furthermore, we observed a distinct two state binding mode, similar to the tesaglitazar titration (ESI Fig. S7†). The binding of coregulators to NRs is known to require a conformational change of H12, which is in itself unfavourable in terms of entropy.17 When this entropic penalty is already paid by the ligand binding event, it does not need to be overcome by the coregulator binding event anymore, thus enhancing the affinity.
Molecular dynamics reveals the underlying molecular mechanisms of cooperativity
Molecular Dynamics (MD) was employed to gain mechanistic insights in the interplay of the three components and underlying cooperativity mechanism. A ternary complex of receptor·ligand·coregulator was built by merging the coordinates of PPARγ bound to rosiglitazone (PDB: 5YCP), together with the coordinates of MED1 bound to PPARγ (PDB: 6ONJ) and adding the Ω loop absent in the crystal structures (Fig. 5A). Rosiglitazone was chosen as representative ligand for this investigation, being an extensively studied and approved drug and showing reliable results in the experimental assays (vide supra).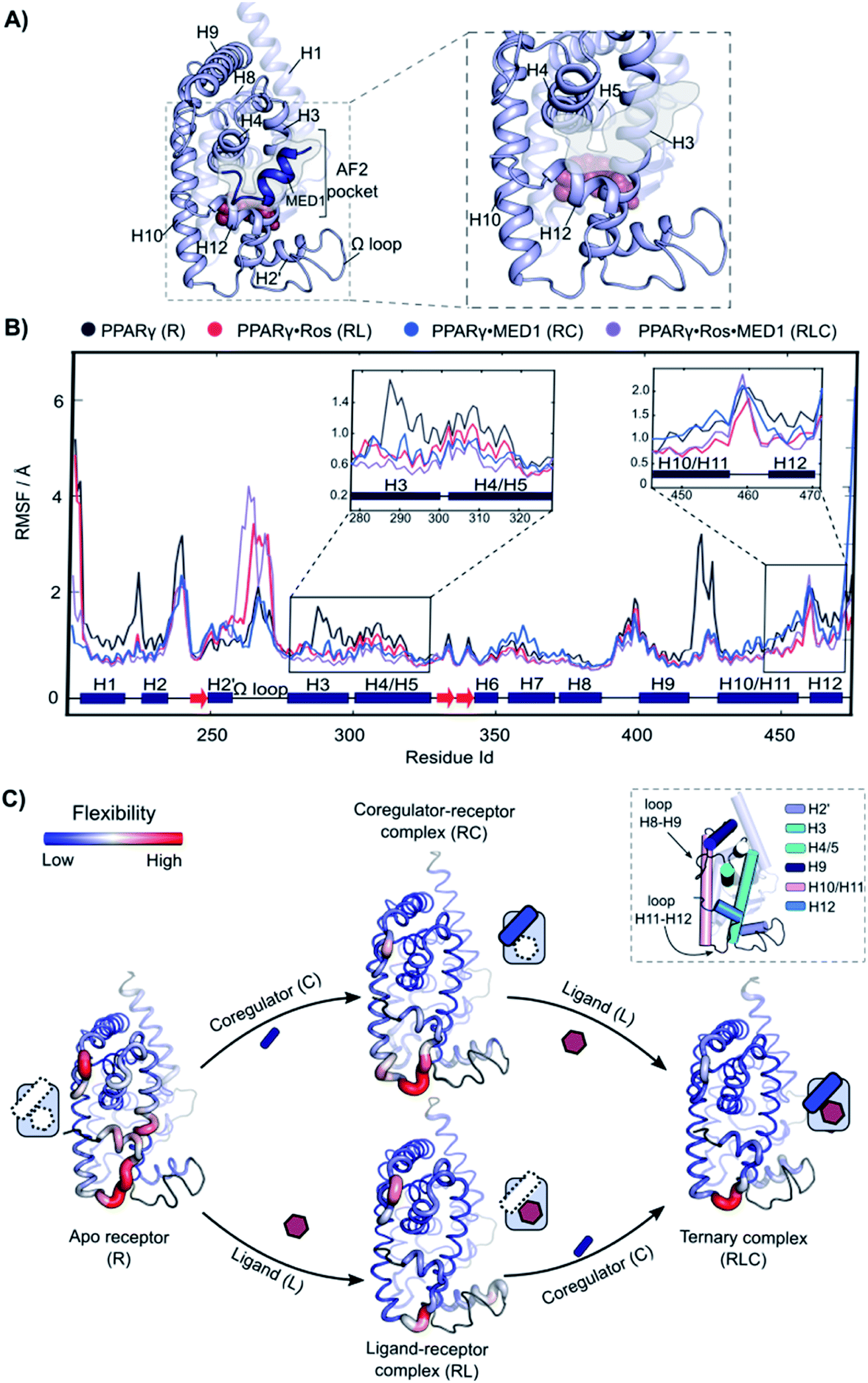 | ||
| Fig. 5 Increasing rigidity of PPARγ upon ligand and coregulator binding. (A) 3D representation of the model used for the analysis. Left: overall view of the structure with rosiglitazone shown as spheres within the ligand binding site and the MED1 coregulator shown as blue cartoon and highlighted. The coregulator binding groove (AF2-pocket) is indicated. Right: zoom of the structural portions interacting with the ligand and the coregulator (omitted for clarity). (B) RMSF of the four states of the cooperativity square. The secondary structure of the protein is show at the bottom of the graph. The regions involved in the interaction with both the ligand and the coregulator, namely the areas within H3, H4 and H5 and H10–H12 are zoomed. (C) 3D structures of the four states are represented as putty cartoons and inserted within the cooperativity scheme. The flexibility is shown with a gradient scale going from blue (low flexibility) to red (high flexibility). The thickness of the tube is also proportional to the magnitude of the flexibility. Flexibility values of loop areas and terminal residues are omitted (colored in dark grey in the cartoon). On the top right, a cartoon representation of the receptor with the most flexible portions highlighted and labelled is reported. | ||
The following four different states were simulated for 1 μs each: apo PPARγ (R), PPARγ·MED1 (RC), PPARγ·ROS (RL) and PPARγ·ROS·MED1 (RLC). The Root Mean Square Fluctuation (RMSF) analysis showed a stepwise reduction in the overall dynamicity from apo PPARγ to the binary structures (RL and RC) and to the ternary complex (Fig. 5B and C). Except for the H2 loop and the modelled Ω loop, the most flexible regions are those directly interacting with the ligand (H3, H12 and the bottom part of H10/11) and the coregulator (H3, H4/5 and H12, namely the AF2-pocket). The apo state fluctuations of H3 are reduced by the presence of both the coregulator and the ligand, while H4 flexibility is decreased mainly by coregulator binding (Fig. 5C). Less pronounced differences can be observed for the loop connecting H8 and H9, whose flexibility is lessened by the MED1 binding and further reduced upon subsequent rosiglitazone binding (Fig. 5C). In contrast, the mobility of H10/11 is mainly reduced by the ligand binding event, with little additional structuring by coregulator binding. Conversely, the loop connecting H10/11 and H12 is only marginally stabilized by the ligand and is maintained quite flexible in all four states. Importantly, the fluctuations of H12 are lowered both in the presence of the ligand or the coregulator and synergistically further reduced upon ternary complex formation (Fig. 5C).
To better understand how the ligand and coregulator mutually influence each other, we performed independent analyses on rosiglitazone and MED1 in the binary and ternary states. For rosiglitazone, the thiazolidinedione moiety is the only part of the molecule responsible for polar interactions. When bound only to PPARγ, one H-bond occurs between the thiazolidinedione moiety and Tyr473 (on H12), while in ternary complex a second H-bond interaction with His449 (H10) is also formed (Fig. 6A and S10†). This stronger anchoring to both H10 and H12 in the presence of the coregulator results in a higher rigidity of the ligand and PPARγ helices as observed in the RMSF analysis. Coregulator binding thus strengthens the interaction between the ligand and the receptor. This observation supports the experimental data (Fig. 2C) ascribing the polar head of PPARγ ligands as important element in dictating the cooperativity effect.
The MED1 coregulator also shows a higher mobility in complex with PPARγ alone than in the ternary complex (see the RMSD profile in Fig. S11†). Comparison of the essential modes reveals that MED1 experiences larger movements in the absence of the stabilizing, cooperative ligand (Fig. 6B). This is consistent with the observed formation of more stable interactions between the coregulator and the receptor upon addition of the ligand (Fig. 6C, Tables S2 and S3†). Rosiglitazone reduces the MED1 flexibility and induces longer lasting H-bonds between MED1 and PPARγ. In particular, polar interactions are confined within the N- and C-terminal loops, as the α-helical LXXLL motif43 is mainly hydrophobic. The H-bond between Lys301 (H3) and Leu648 (MED1), forming the so-called charge clamp,29 shows a higher occupancy in the ternary complex and anchors the coregulator C-terminus. Also, the flexibility of the MED1 N-terminus is lowered upon rosiglitazone binding, due to a key H-bond between Leu468 (H12) and His642 (MED1), featuring an occupancy > 90% and anchoring the coregulator in the AF-2 pocket. A superposition of the average structures of the simulations of the binary and ternary complexes, highlights the different positioning of both H12 and MED1 due to the ligand binding (Fig. 6D). In the absence of rosiglitazone, H12 is moved upwards and consequently MED1 is hindered and pushed out of the AF-2. This suggests that the stabilizing effect the ligand exerts on H12 in the ternary complex, ultimately translates cooperatively into a most favorable interaction between the coregulator and the receptor.
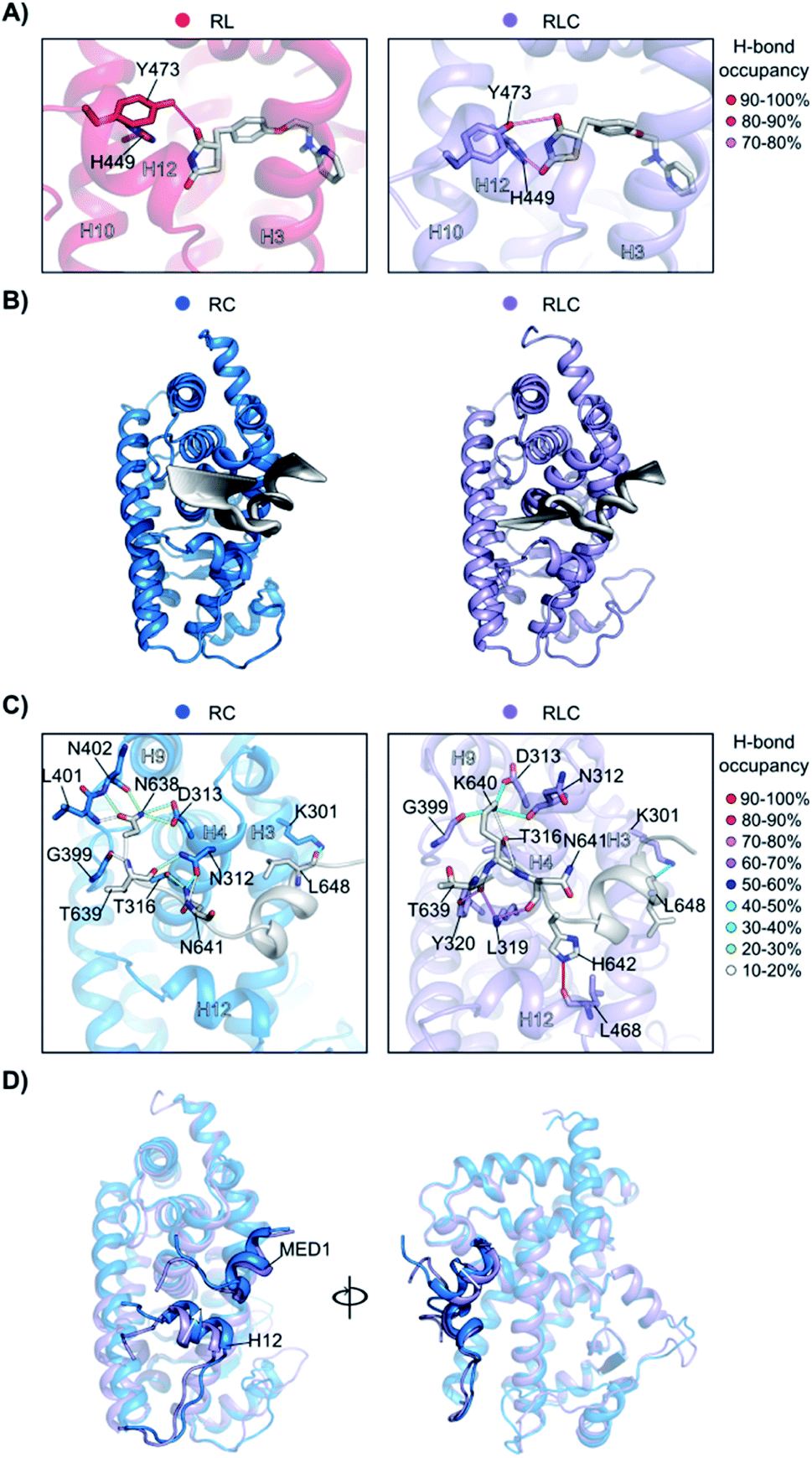 | ||
Fig. 6 Synergistic interplay of ligand and coregulator binding to PPARγ. (A) Hydrogen bonds between rosiglitazone and PPARγ. On the left, the comparison of the occupancy (%) of the hydrogen bonds in the presence ( ) and in the absence ( ) and in the absence ( ) of the coregulator. On the right, the representation of the interactions. Hydrogen bonds are shown as lines colored according to their occupancy over the simulation, as shown in the legend. Residues and helices are labelled. (B) Essential motions of MED1 in the two simulations. The amplitude of the cartoon is proportional to the magnitude of the motion and the gradient of colour indicates the direction of the motion (from black to white). (C) Hydrogen bonds occupancy between MED1 and PPARγ in the absence ( ) of the coregulator. On the right, the representation of the interactions. Hydrogen bonds are shown as lines colored according to their occupancy over the simulation, as shown in the legend. Residues and helices are labelled. (B) Essential motions of MED1 in the two simulations. The amplitude of the cartoon is proportional to the magnitude of the motion and the gradient of colour indicates the direction of the motion (from black to white). (C) Hydrogen bonds occupancy between MED1 and PPARγ in the absence ( ) and in the presence ( ) and in the presence ( ) of the ligand. The coregulator is colored in white and the interacting residues and helices are labelled. Hydrogen bonds are shown as lines colored according to their occupancy over the simulation, as shown in the legend. (D) Superposition of the average structures of PPARγ·MED1( ) of the ligand. The coregulator is colored in white and the interacting residues and helices are labelled. Hydrogen bonds are shown as lines colored according to their occupancy over the simulation, as shown in the legend. (D) Superposition of the average structures of PPARγ·MED1( ) and PPARγ·Ros·MED1( ) and PPARγ·Ros·MED1( ). The different positioning of H12 and MED1 is highlighted. ). The different positioning of H12 and MED1 is highlighted. | ||
Conclusions
There is a strong need for conceptually new ways to modulate NRs via modulators with a more differentiated molecular profile. Here we have provided a cooperativity framework for nuclear receptor/ligand/coregulator binding interpreting NR–ligands as allosteric molecular glues, using a combination of a computational and experimental methods. Our approach dissects the effect of ligands on the NR/coregulator interaction, by looking at their cooperativity factor (α), as well as their intrinsic affinity towards the apo-receptor (KIID). Both parameters could be obtained in a straightforward manner through 2D-FA titrations, in combination with a numerical model. ITC analyses corroborated these results and showed that the underlying cooperativity is entropy driven. The molecular dynamics simulations confirmed the bidirectionality between ligand and coregulator regarding the cooperative effect. The presence of the ligand stabilizes the AF2 surface of the NR which enhances the interaction with the coregulator, through preorganization. Vice versa, the presence of the coregulator increases the overall rigidification of the receptor, which in turn increases the duration of the hydrogen bond contacts with the ligand.The intrinsic affinities of the tested NR–ligands span a range of five orders of magnitude. The direct affinity of a NR–ligand can often be challenging to measure directly and is typically not reported, with some notable exceptions.44,45 In the past direct affinities were measured more often with radioactive displacement assay with steroids,46 but current indirect binding assays or transcriptional activation assays report on a combination of multiple binding effects and functional activity. 2D fluorescent anisotropy titration is a straightforward way of obtaining these NR/ligand affinities, concomitant with determining the cooperativity values for the ternary complex with the coregulator under study.
Intrinsic KD's and cooperativity factors are fundamental thermodynamic parameters and therefore allow for an objective comparison of different ligands, regardless of the assay conditions or type of ligand. MRL24 is exemplary here, having high binding affinity for PPARγ, but not strongly stabilizing the interaction with MED1. Similarly, other NR partial or silent agonists are potentially more quantitatively described via their KD's and cooperativity factors, than via a certain transcriptional activity in a cellular system.47 This quantitative framework for NR–ligands will furthermore facilitate NR drug discovery by attributing structural and activity changes to the underlying cooperativity and affinity parameters. We foresee that this framework will allow targeted optimization of NR–ligand properties via their cooperativity analysis. The cooperativity factor should also provide an attractive entry towards understanding and addressing preferential coregulator recruitment. The cooperativity framework describing the mechanisms of PPARγ·ligand·coregulator complex formation allows to understand NR modulation on a fundamental level and should not only be of importance for PPARγ, but be of equally high relevance for other NRs. Also, the equilibria studied in this manuscript will be further influenced by other interaction partners in a cellular context. These include for example competing cofactors, including of corepressor and coactivator nature,48 NR dimerization,49–51 or dual ligand binding.9,10,52 We are currently directing our approach towards such studies.
Data availability
Experimental data associated with the article can be found in the ESI.†Author contributions
Experimental contributions PJdV, AAK, GDA; conceptualization PJdV, AAK, GDA, FS, LB, data analysis PJdV, AAK, GDA, FS, LB; manuscript writing PJdV, AAK, GDA, GC, FS, LB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to express our gratitude towards Chad Brautigam for his help with NITPIC software, making it compatible with our ITC-setup. We also kindly acknowledge Molecular Discovery Ltd for supporting GDA, the Centro di Competenza sul Calcolo Scientifico (C3S) at the University of Turin (c3s.unito.it) for providing the computational time and resources and BiKi Technologies for providing the BiKi LiFe Sciences suite. This research was funded by the Netherlands Organization for Scientific Research (NWO) through Gravity program 024.001.035 and VICI grant 016.150.366, and by the University of Turin “Ricerca Locale” (SPYF_RILO_19_01). Part of the simulations were carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.Notes and references
- S. A. Andrei, E. Sijbesma, M. Hann, J. Davis, G. O'Mahony, M. W. D. Perry, A. Karawajczyk, J. Eickhoff, L. Brunsveld, R. G. Doveston, L.-G. Milroy and C. Ottmann, Expert Opin. Drug Discovery, 2017, 12, 925–940 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- P. J. de Vink, S. A. Andrei, Y. Higuchi, C. Ottmann, L.-G. Milroy and L. Brunsveld, Chem. Sci., 2019, 10, 2869–2874 RSC.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- A. Whitty, Nat. Chem. Biol., 2008, 4, 435–439 CrossRef CAS PubMed.
- T. Kenakin, Chem. Rev., 2017, 117, 4–20 CrossRef CAS PubMed.
- L. Zhao, S. Zhou and J.-Å. Gustafsson, Endo. Rev., 2019, 40, 1207–1249 Search PubMed.
- J. Zheng, M. R. Chang, R. E. Stites, Y. Wang, J. B. Bruning, B. D. Pascal, S. J. Novick, R. D. Garcia-Ordonez, K. R. Stayrook, M. J. Chalmers, J. A. Dodge and P. R. Griffin, Nat. Commun., 2017, 8, 923 CrossRef PubMed.
- J. Shang, R. Brust, S. A. Mosure, J. Bass, P. Munoz-Tello, H. Lin, T. S. Hughes, M. Tang, Q. Ge, T. M. Kamenekca and D. J. Kojetin, Elife, 2018, 7, e43320 CrossRef PubMed.
- R. M. J. M. de Vries, F. A. Meijer, R. G. Doveston, I. A. Leijten-van de Gevel and L. Brunsveld, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2021287118 CrossRef CAS PubMed.
- K. S. Bramlett, Y. Wu and T. P. Burris, Mol. Endo., 2001, 15, 909–922 CrossRef CAS PubMed.
- M. Scheepstra, S. Leysen, G. C. van Almen, J. R. Miller, J. Piesvaux, V. Kutilek, H. van Eenennaam, H. Zhang, K. Barr, S. Nagpal, S. M. Soisson, M. Kornienko, K. Wiley, N. Elsen, S. Sharma, C. C. Correll, B. W. Trotter, M. van der Stelt, A. Oubrie, C. Ottmann, G. Parthasarathy and L. Brunsveld, Nat. Commun., 2015, 6, 8833 CrossRef PubMed.
- Y. Jang, W. Kim, P. Leblanc, C.-H. Kim and K.-S. Kim, Exp. Mol. Med., 2021, 53, 19–29 CrossRef CAS PubMed.
- I. M. S. de Vera, J. Zheng, S. Novick, J. Shang, T. S. Hughes, R. Brust, P. Munoz-Tello, W. J. Gardner, D. P. Marciano, X. Kong, P. R. Griffin and D. J. Kojetin, Structure, 2017, 25, 1506–1518 CrossRef CAS PubMed.
- B.-D. K. Putcha, E. Wright, J. S. Brunzelle and E. J. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6084–6087 CrossRef CAS PubMed.
- K. De Bosscher, S. J. Desmet, D. Clarisse, E. Estébanez-Perpiña and L. Brunsveld, Nat. Rev. Endo., 2020, 16, 363–377 CrossRef CAS PubMed.
- F. Rastinejad, V. Ollendorff and I. Polikarpov, Trends Biochem. Sci., 2015, 40, 16–24 CrossRef CAS PubMed.
- J. Osz, Y. Brélivet, C. Peluso-Iltis, V. Cura, S. Eiler, M. Ruff, W. Bourguet, N. Rochel and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E588–E594 CrossRef PubMed.
- C. Köhler, G. Carlström, A. Gunnarsson, U. Weininger, S. Tångefjord, V. Ullah, M. Lepistö, U. Karlsson, T. Papavoine, K. Edman and M. Akke, Sci. Adv., 2020, 6, eabb5277 CrossRef PubMed.
- I. M. Chrisman, M. D. Nemetchek, I. M. S. de Vera, J. Shang, Z. Heidari, Y. Long, H. Reyes-Caballero, R. Galindo-Murillo, T. E. Cheatham, A.-L. Blayo, Y. Shin, J. Fuhrmann, P. R. Griffin, T. M. Kamenecka, D. J. Kojetin and T. S. Hughes, Nat. Commun., 2018, 9, 1794 CrossRef PubMed.
- S. J. Edelstein and N. Le Novère, J. Mol. Biol., 2013, 425, 1424–1432 CrossRef CAS PubMed.
- J. Shang, R. Brust, P. R. Griffin, T. M. Kamenecka and D. J. Kojetin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 22179–22188 CrossRef CAS PubMed.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- J.-P. Changeux, Annu. Rev. Biophys., 2012, 41, 103–133 CrossRef CAS PubMed.
- P. Tontonoz and B. M. Spiegelman, Annu. Rev. Biochem., 2008, 77, 289–312 CrossRef CAS PubMed.
- J. P. Berger, T. E. Akiyama and P. T. Meinke, Trends Pharmacol. Sci., 2005, 26, 244–251 CrossRef CAS PubMed.
- A. Farce, N. Renault and P. Chavatte, Curr. Med. Chem., 2009, 16, 1768–1789 CrossRef CAS PubMed.
- J. Y. Jang, H. Bae, Y. J. Lee, Y. I. Choi, H.-J. Kim, S. B. Park, S. W. Suh, S. W. Kim and B. W. Han, Sci. Rep., 2018, 8, 31 CrossRef PubMed.
- J. Shang, S. A. Mosure, J. Zheng, R. Brust, J. Bass, A. Nichols, L. A. Solt, P. R. Griffin and D. J. Kojetin, Nat. Commun., 2020, 11, 956 CrossRef CAS PubMed.
- D. M. Lonard and B. W. O'Malley, Mol. Cell, 2007, 27, 691–700 CrossRef CAS PubMed.
- A. Aranda and A. Pascual, Physiol. Rev., 2001, 81, 1269–1304 CrossRef CAS PubMed.
- E. F. Douglass, C. J. Miller, G. Sparer, H. Shapiro and D. A. Spiegel, J. Am. Chem. Soc., 2013, 135, 6092–6099 CrossRef CAS PubMed.
- R. E. Soccio, E. R. Chen and M. A. Lazar, Cell Metab., 2014, 20, 573–591 CrossRef CAS PubMed.
- B. Staels, M. Maes and A. Zambon, Nat. Clin. Pract. Cardiovasc. Med., 2008, 5, 542–553 CrossRef CAS PubMed.
- J. J. Acton, R. M. Black, A. B. Jones, D. E. Moller, L. Colwell, T. W. Doebber, K. L. MacNaul, J. Berger and H. B. Wood, Bioorg. Med. Chem. Lett., 2005, 15, 357–362 CrossRef CAS PubMed.
- J. H. Choi, A. S. Banks, T. M. Kamenecka, S. A. Busby, M. J. Chalmers, N. Kumar, D. S. Kuruvilla, Y. Shin, Y. He, J. B. Bruning, D. P. Marciano, M. D. Cameron, D. Laznik, M. J. Jurczak, S. C. Schürer, D. Vidović, G. I. Shulman, B. M. Spiegelman and P. R. Griffin, Nature, 2011, 477, 477–481 CrossRef CAS PubMed.
- J. F. Rebhun, K. M. Glynn and S. R. Missler, Fitoterapia, 2015, 106, 55–61 CrossRef CAS PubMed.
- R. T. Nolte, G. B. Wisely, S. Westin, J. E. Cobb, M. H. Lambert, R. Kurokawa, M. G. Rosenfeld, T. M. Willson, C. K. Glass and M. V. Milburn, Nature, 1998, 395, 137–143 CrossRef CAS PubMed.
- P. Cronet, J. F. W. Petersen, R. Folmer, N. Blomberg, K. Sjöblom, U. Karlsson, E.-L. Lindstedt and K. Bamberg, Structure, 2001, 9, 699–706 CrossRef CAS PubMed.
- S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, Anal. Chem., 2012, 84, 5066–5073 CrossRef CAS PubMed.
- J. C. D. Houtman, P. H. Brown, B. Bowden, H. Yamaguchi, E. Appella, L. E. Samelson and P. Schuck, Prot. Sci., 2007, 16, 30–42 CrossRef CAS PubMed.
- A. Velazquez-Campoy, G. Goñi, J. R. Peregrina and M. Medina, Biophys. J., 2006, 91, 1887–1904 CrossRef CAS PubMed.
- D. M. Heery, E. Kalkhoven, S. Hoare and M. G. Parker, Nature, 1997, 387, 733–736 CrossRef CAS PubMed.
- S. Raghuram, K. R. Stayrook, P. Huang, P. M. Rogers, A. K. Nosie, D. B. McClure, L. L. Burris, S. Khorasanizadeh, T. P. Burris and F. Rastinejad, Nat. Struct. Mol. Biol., 2007, 14, 1207–1213 CrossRef CAS PubMed.
- J. Kallen, R. Lattmann, R. Beerli, A. Blechschmidt, M. J. J. Blommers, M. Geiser, J. Ottl, J.-M. Schlaeppi, A. Strauss and B. Fournier, J. Biol. Chem., 2007, 282, 23231–23239 CrossRef CAS PubMed.
- A. Freyberger and H.-J. Ahr, Toxicology, 2004, 195, 113–126 CrossRef CAS PubMed.
- N. E. Bruno, J. C. Nwachukwu, S. Srinivasan, C. C. Nettles, T. Izard, Z. Jin, J. Nowak, M. D. Cameron, S. V. Boregowda, D. G. Phinney, O. Elemento, X. Liu, E. A. Ortlund, R. Houtman, D. A. Stavreva, G. L. Hager, T. M. Kamenecka, D. J. Kojetin and K. W. Nettles, Nat. Chem. Biol., 2021, 17, 307–316 CrossRef CAS PubMed.
- D. Merk, S. Sreeramulu, D. Kudlinzki, K. Saxena, V. Linhard, S. L. Gande, F. Hiller, C. Lamers, E. Nilsson, A. Aagaard, L. Wissler, N. Dekker, K. Bamberg, M. Schubert-Zsilavecz and H. Schwalbe, Nat. Commun., 2019, 10, 2915 CrossRef PubMed.
- D. S. Lala, R. Mukherjee, I. G. Schulman, S. S. Koch, L. J. Dardashti, A. M. Nadzan, G. E. Croston, R. M. Evans and R. A. Heyman, Nature, 1996, 383, 450–453 CrossRef CAS PubMed.
- L. Fadel, B. Rehó, J. Volkó, D. Bojcsuk, Z. Kolostyák, G. Nagy, G. Müller, Z. Simandi, É. Hegedüs, G. Szabó, K. Tóth, L. Nagy and G. Vámosi, J. Biol. Chem., 2020, 295, 10045–10061 CrossRef CAS PubMed.
- W. Kilu, D. Merk, D. Steinhilber, E. Proschak and J. Heering, J. Biol. Chem., 2021, 297, 100814 CrossRef CAS PubMed.
- S. Willems, L. Gellrich, A. Chaikuad, S. Kluge, O. Werz, J. Heering, S. Knapp, S. Lorkowski, M. Schubert-Zsilavecz and D. Merk, Cell Chem. Biol., 2021, 28, 1489–1500 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, supporting figures and tables. See DOI: 10.1039/d1sc06426f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |