
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast, solvent-free synthesis of ferrocene-containing organic cages via dynamic covalent chemistry in the solid state†
Tom
Kunde
 a,
Tobias
Pausch
a,
Piotr A.
Guńka
b,
Maurycy
Krzyżanowski
b,
Artur
Kasprzak
*b and
Bernd M.
Schmidt
*a
a,
Tobias
Pausch
a,
Piotr A.
Guńka
b,
Maurycy
Krzyżanowski
b,
Artur
Kasprzak
*b and
Bernd M.
Schmidt
*a
aInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Bernd.Schmidt@hhu.de; Web: http://www.bmschmidtlab.de
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego Str. 3, 00-664 Warsaw, Poland. E-mail: akasprzak@ch.pw.edu.pl
First published on 3rd February 2022
Abstract
A simple, solvent-free synthetic protocol towards the synthesis of organic self-assembled macromolecules has been established. By employing mechanochemistry using glassware readily available to every organic chemist, we were able to synthesise three novel organic cage compounds exemplarily and to speed up the synthesis of a ferrocene-containing macrocycle by a factor of 288 compared to the solution-based synthesis. The structural investigation of the newly synthesised cages revealed different modes of connectivity from using ferrocene-containing aldehydes caused by the free rotation of the cyclopentadienyl units against each other. By extending the facile solvent-free synthesis to ball-milling, even compounds that show lower reactivity could be employed in the dynamic covalent formation of organometallic cage compounds. The presented protocol gives access to otherwise inaccessible structures, speeds up general synthetic workflows, and simultaneously reduces the environmental impact of supramolecular syntheses.
Introduction
The fabrication of supramolecular structures using dynamic bonds is often accompanied by the use of metal ions to generate metal-containing superstructures formed by dative bonds, and/or the use of a large amount of solvents to facilitate the formation of those macro- or supramolecules via the dilution principle.1 Using dynamic covalent chemistry (DCC) for the synthesis of organic cage compounds circumvents the use of metals but is still accompanied by the use of large quantities of hazardous organic solvents.2 During the formation of these symmetrical cage molecules, large quantities of solvents are often needed to avoid precipitation of oligomeric intermediates, facilitate complete dissolving of sparely soluble reactants, or to allow stabilizing or templating effects of the solvent molecules throughout the formation of a single supramolecule by dynamic bond formation and breaking.3 In recent years, researchers have been eager to replace high-dilution DCC, which involves mixing two or more reactants in large volumes of solvents and stirring them for days, or even weeks,4 in search of less hazardous, green methods, which they discovered in mechanochemistry. Mechanochemistry was named as one of the most rapidly expanding fields in chemistry by the IUPAC in 2019![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and has been used to synthesise a wide array of organic, inorganic, metal–organic, and supramolecular compounds.6–9 Its ability to generate metal–organic frameworks, covalent organic frameworks and supramolecular frameworks in a matter of minutes instead of days, with only minimum amounts of solvent or completely solvent-free, has made it a well-established synthesis method for large framework structures.10,11 However, the use of mechanochemistry as a method for synthesising organic cages based on dynamic covalent bond formation has had limited success so far. To the best of our knowledge, only two groups succeeded in the formation of organic cage compounds by mechanochemical synthesis. The group of Severin reported the assembly of two cages, each consisting of 11 individual building blocks, synthesised by dynamic imine and boronic ester bond formation via ball-milling at room temperature in almost quantitative yields.12 The group was not only the first to report the solvent-free formation of a discrete supramolecular structure, but it remains the only one to have accomplished this task to this date, to the best of our knowledge. Crawford, Greenaway, and co-workers were the second group to report the synthesis of an organic cage molecule by liquid-assisted mechanochemical methods. They used twin screw extrusion (TSE) to facilitate the synthesis of their CC3 cage in high yields (up to 50 g scale), a short amount of time, and high purity.13 Both groups utilized equipment that is regularly not found in a common organic chemistry laboratory. We herein present the synthesis of three novel macromolecules via a straightforward, fast, solvent-free, single-step synthesis that only relies on laboratory glassware, present in every organic chemistry lab and even in most schools. By simply grinding the reactants, 1,1′-diformylferrocene (1), 1,1′-bis(4-formylphenyl)ferrocene (2), and different amines (3 and 4), using a glass rod and a glass reaction tube, we were able to selectively synthesise three different cage molecules in a matter of minutes without the use of any solvent. We were able to generate a bowl-shaped macrocycle14 using 1,2-(R,R)-diaminocyclohexane (R,R-DACH) in similar yields as previously reported, but in much less time.
5 and has been used to synthesise a wide array of organic, inorganic, metal–organic, and supramolecular compounds.6–9 Its ability to generate metal–organic frameworks, covalent organic frameworks and supramolecular frameworks in a matter of minutes instead of days, with only minimum amounts of solvent or completely solvent-free, has made it a well-established synthesis method for large framework structures.10,11 However, the use of mechanochemistry as a method for synthesising organic cages based on dynamic covalent bond formation has had limited success so far. To the best of our knowledge, only two groups succeeded in the formation of organic cage compounds by mechanochemical synthesis. The group of Severin reported the assembly of two cages, each consisting of 11 individual building blocks, synthesised by dynamic imine and boronic ester bond formation via ball-milling at room temperature in almost quantitative yields.12 The group was not only the first to report the solvent-free formation of a discrete supramolecular structure, but it remains the only one to have accomplished this task to this date, to the best of our knowledge. Crawford, Greenaway, and co-workers were the second group to report the synthesis of an organic cage molecule by liquid-assisted mechanochemical methods. They used twin screw extrusion (TSE) to facilitate the synthesis of their CC3 cage in high yields (up to 50 g scale), a short amount of time, and high purity.13 Both groups utilized equipment that is regularly not found in a common organic chemistry laboratory. We herein present the synthesis of three novel macromolecules via a straightforward, fast, solvent-free, single-step synthesis that only relies on laboratory glassware, present in every organic chemistry lab and even in most schools. By simply grinding the reactants, 1,1′-diformylferrocene (1), 1,1′-bis(4-formylphenyl)ferrocene (2), and different amines (3 and 4), using a glass rod and a glass reaction tube, we were able to selectively synthesise three different cage molecules in a matter of minutes without the use of any solvent. We were able to generate a bowl-shaped macrocycle14 using 1,2-(R,R)-diaminocyclohexane (R,R-DACH) in similar yields as previously reported, but in much less time.
Results and discussion
Ferrocene (Fc) offers an intriguing combination of rigidity and rotational flexibility while being chemically stable and easily functionalized, and also adding additional functionality to any self-assembled system.15,16 Fc-containing polymers have been extensively used in mechanochemical studies using ultrasound in solution.17 Although the Fe–cyclopentadienyl (Cp) bond shows remarkable stability towards homo- and heterolytic cleavage when the distance between the Cp-rings is increased along the normal of the Cp-plane, force applied parallel to the Cp-plane readily results in shearing of both Cp-units against each other and bond scission.17b This phenomenon could be employed with the use of Fc units inside a polymer backbone, creating a mechanophore in the process. The role of Fc in tribochemistry, however, has received far less attention.17,18 Wondering whether this method could be employed for the synthesis of cage molecules by DCC, we combined three equivalents of diformylferrocene 1 with two equivalents of tridentate amine 3 in an attempt to create a Tri2Di319 cage (FcC1, Fig. 1). During the grinding process, using a simple glass rod to grind the reactants inside of a glass reaction tube, the two dry powders quickly became a sticky mixture that was attached to the glass rod. By scraping off the sticky reaction mixture at regular intervals, complete mixing of all the reactants was assured. To our surprise, the MALDI-MS and ESI-HRMS of the grinding product (solid product mixed with dithranol and directly subjected to MALDI analysis) showed only a single intense peak of the cage FcC1 (Fig. S24 and S25†). When we subjected the mixture to immediate NMR-analysis (<10 min from the end of the grinding to the finished 1H NMR experiment), we observed only sharp signals that can be assigned to the quantitative formation of FcC1 (Fig. 2c). We investigated the formation of the cage during the grinding process by measuring ESI-MS spectra after 2, 5, 7, and 10 min to observe possible intermediates (Fig. S30†). After 2 min of grinding, already intense signals corresponding to FcC1 (A), alongside three different signals which can be attributed to an open form of FcC1 + H2O (B), and two oligomeric structures resulting from the [4 + 4] condensation reaction of 1 and 3 (C) and from the [5 + 5] condensation of 1 and 3 (D), can be observed. All signals not corresponding to the closed cage species decrease over time until, at 10 min, only the product peak remains. Therefore, we assume that the condensation reaction proceeds via initial oligomeric intermediates, releasing water and facilitating the very fast DCC formation of the apparently thermodynamically favoured cage product FcC1. By dissolving the mixture in dry CHCl3 followed by slow vapor diffusion of n-pentane, single-crystals suitable for X-ray diffraction (SC-XRD) could be obtained. The structure acquired from SC-XRD ultimately confirms the successful formation of the targeted Tri2Di3 cage FcC1 (Fig. 2a). Only two cage compounds using 1 as a building block have been reported in the literature to this date.16e,20 In both cases, the formyl groups are found to be stacked above one another (syn conformation), minimizing the distance between the two triamines that form the top and bottom parts of these small triangular prims. In the crystal structure of FcC1, the Cp-units are rotated 150° against each other (anti conformation). Additionally, the Fc units are also rotated by 90° compared to the formerly reported Tri2Di3 cages. This leads to an angle of 60° between the imine groups of the upper and bottom half of the molecule when looked at from the top (Fig. 2b).
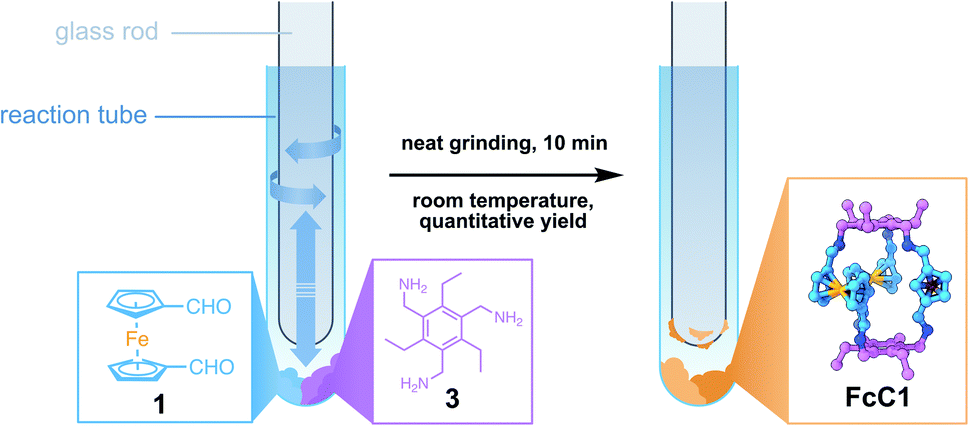 | ||
| Fig. 1 Solvent-free synthesis of FcC1 using a glass rod and a reaction tube to grind the starting materials 1 and 3 in stoichiometric equivalents. | ||
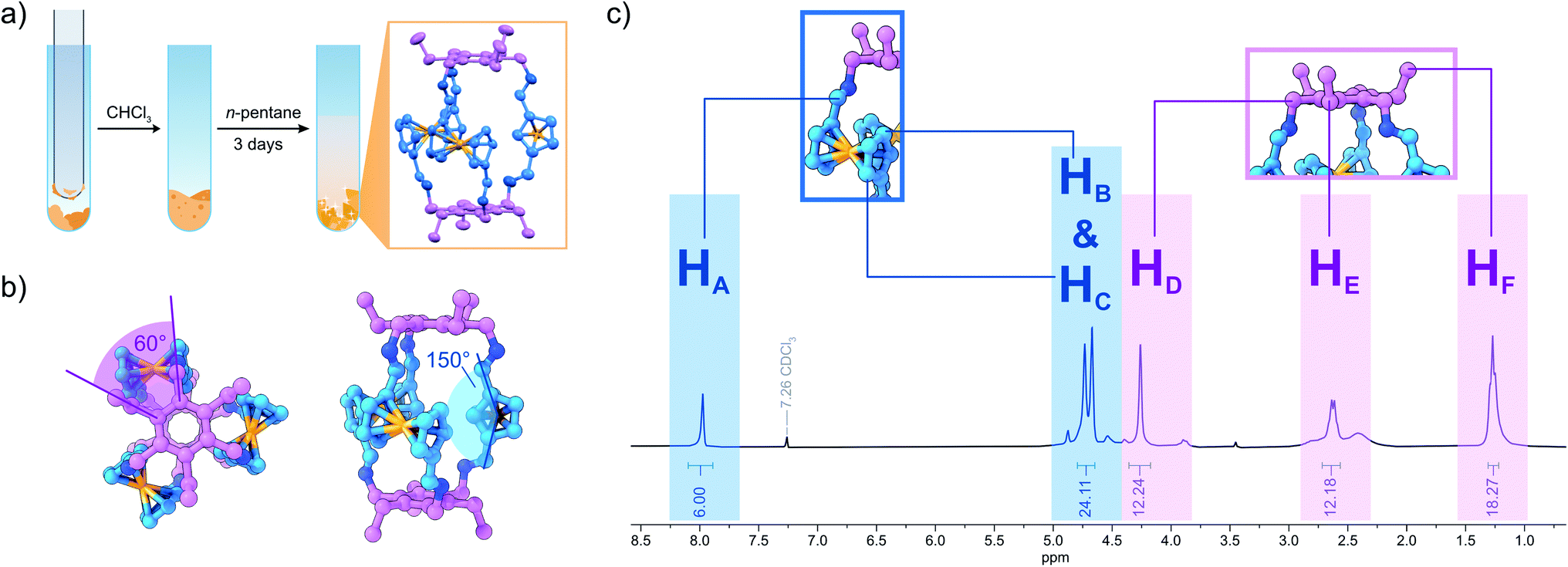 | ||
Fig. 2 (a) Preparation of single-crystals of FcC1 by dissolving the mixture obtained by grinding in CHCl3, followed by precipitation of FcC1 after slow vapour diffusion of n-pentane. The SC-XRD structure is depicted in the orange frame (solvent molecules and hydrogen atoms omitted for clarity; P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , R1 = 0.06, wR2 = 0.18, Rint = 0.07); (b) top view (left) and side view (right) of the crystal structure of FcC1. Important angles are highlighted in blue and purple; (c) 1H NMR obtained immediately after grinding the reactants for 10 min and dissolving the mixture in CDCl3. , R1 = 0.06, wR2 = 0.18, Rint = 0.07); (b) top view (left) and side view (right) of the crystal structure of FcC1. Important angles are highlighted in blue and purple; (c) 1H NMR obtained immediately after grinding the reactants for 10 min and dissolving the mixture in CDCl3. | ||
To rule out the possibility of a very fast formation of FcC1 in solution after dissolving the mixture in CDCl3, we investigated the time scale of the DCC reaction using 1H NMR analysis. We combined the two reactants inside an NMR tube (concentration 71 mM, representative for a sample prepared by grinding being dissolved for NMR measurement) and added 0.7 mL of dry CDCl3. Immediately after mixing, we observed that there are almost only starting materials and minute amounts of oligomeric products present, which are indicated by broad signals in the spectrum (Fig. S1†). After 2 h of no stirring or additional mixing, FcC1 accounts for roughly 70% of the dissolved species. Significant precipitation occurred, likely caused by large amounts of insoluble oligomers (redissolved aliquots show only traces FcC1 by MALDI, Fig. S32†). Although solution synthesis of FcC1 is possible, it necessitates additional purification steps due to the formation of significant amounts of (oligomeric) side products, takes longer, and employs hazardous solvents.
Scope of the method
With the knowledge of this fast, solvent-free method for generating FcC1 at hand, we wanted to expand this method to other compounds. We therefore combined 1 with either tris(4-aminophenyl)amine (4) or R,R-DACH, employing similar conditions (Fig. 3). When grinding three equivalents of 1 and two equivalents of 4 for 10 min at room temperature, starting materials are present, according to the 1H NMR. Increasing the reaction time to 30 min leads to full conversion of the reactants and the clean formation of the Tri2Di3 cage, FcC2. This is possibly due to aromatic amines being less reactive in dynamic imine condensations. 4 is also conformationally less flexible in direct comparison to 3. To compare the solvent-free synthesis of FcC2 with its synthesis from solution, we prepared a 71 mM solution in CDCl3 to monitor the reaction process by 1H NMR. To our surprise, the solution became turbid and precipitation occurred quickly. After 7 h, we could detect neither signals corresponding to FcC2 nor to the starting materials, concluding that only insoluble oligomeric species must have been formed. After careful optimization, we were able to generate FcC2 by an acid-catalysed reaction in dilute conditions from ethanol over the course of 24 h in very good yield (pages S5 and S6 in the ESI†). The solvent-free synthesis is 48 times faster than a comparable solution-based method. From a benzene/CHCl3 solution of FcC2, single-crystals suitable for SC-XRD analysis were obtained. In contrast to the structure of FcC1, the Fc units are stacked, similar to previously reported Tri2Di3 structures. This indicates that the Fc building blocks are able to adapt to the most stable geometry in the solid state, independent of the geometry in the solid starting material.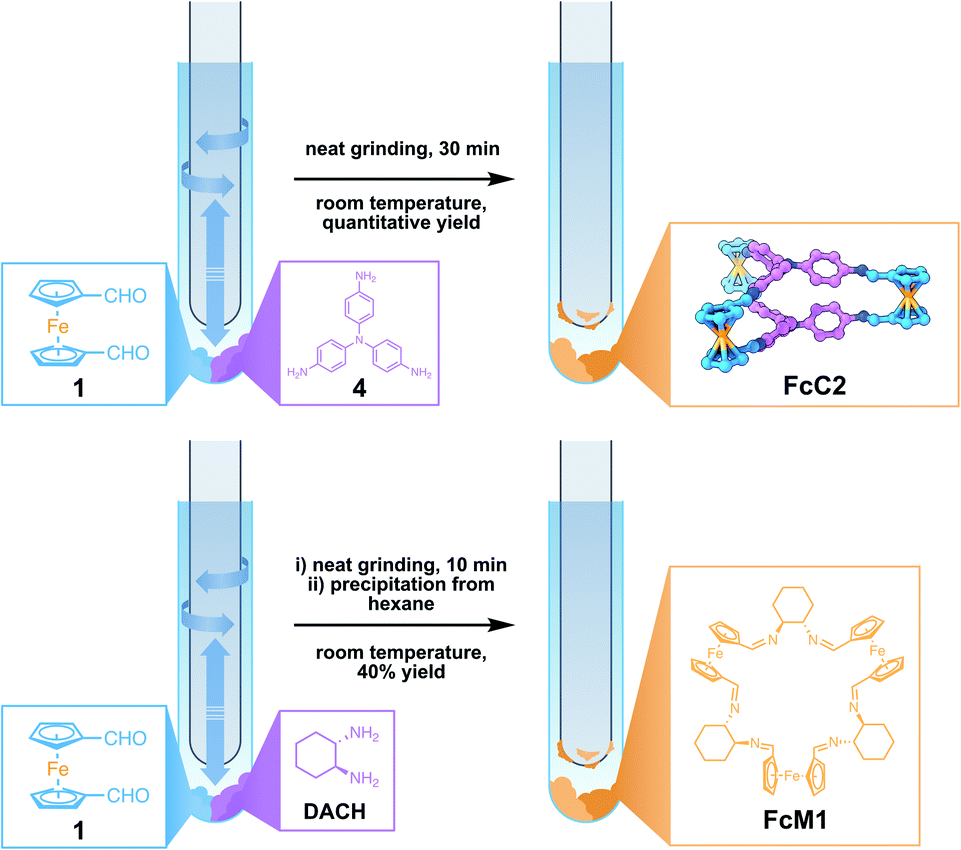 | ||
| Fig. 3 Synthesis of FcC2 from 1 and tris(4-aminophenyl)amine (4) (top) and synthesis of FcM1 from 1 and R,R-DACH (bottom). | ||
When preparing a Fc-containing macrocycle14FcM1 by reacting three equivalents of 1 with three equivalents of R,R-DACH, we observed the formation of oligomeric side products along with FcM1 in the 1H NMR and MS spectra even after elongated periods of grinding (Fig. S18 and S28†). We were able to isolate the pure FcM1 from the mixture by dissolving the sticky crude after 10 min of grinding in a minute amount of chloroform (0.1 mL/50 mg of product) and precipitation with n-hexane (1.0 mL/50 mg of product). Although the yield of this procedure is lower than the solution-based synthesis (40% by grinding vs. 80% from solution), the solvent-free reaction is faster by a factor of 288, showing the potential this method offers for the synthesis of Fc-containing macromolecules as well.
We additionally examined if other aldehydes could be used in this solvent-free synthesis.9 When using three equivalents of isophthalaldehyde and two equivalents of 3, we could only observe minimum amounts of oligomeric species and recover most of the unreacted starting materials (Fig. S22†). Since fluorinated building blocks often tend to react faster and drive the formation of otherwise inaccessible structures, we tried to synthesise a previously published Tri4Di6 cage using four equivalents of 3 and six equivalents of the highly reactive tetrafluoroterephthalaldehyde (Fig. S23†).21 Again, we could only identify small amounts of oligomeric compounds and the unreacted starting materials by both NMR and MS analysis.
Following these initial results, we examined the use of 1,1′-bis(4-formylphenyl)ferrocene (2) as an aldehyde building block in a solvent-free reaction with 3 to further understand the role of the Fc group during the mechanochemical reaction. Ferrocene 2 exhibits an eclipsed conformation of Cp and phenyl rings and an antiperiplanar configuration of aldehyde groups, as evidenced by its SC-XRD (Fig. S11†) measured at room temperature. Targeting a novel Tri2Di3 cage, FcC3, we reacted three equivalents of 2 and two equivalents of 3 by grinding the two compounds following the previous conditions (Fig. 4a). After 10 min, only a complex mixture of products along with unreacted starting materials can be identified by 1H NMR analysis. After grinding the compounds for 30 min, we observed the emergence of sharp peaks corresponding to the formation of FcC3. Compared to the successful and fast formation of related FcC1, FcC2 and FcM1, the relatively low abundance of product signals, even after 30 minutes of grinding, is indicative of a relationship between the position of the functional group inside the Fc-containing building block and the reactivity in a mechanochemical reaction. The much slower and less specific reaction of 2 can possibly be attributed to (i) a larger steric hindrance of the newly introduced phenylene spacer group on the cage formation and (ii) a possibly diminished reactivity from shifting the aldehyde group further away from the Fc core.22
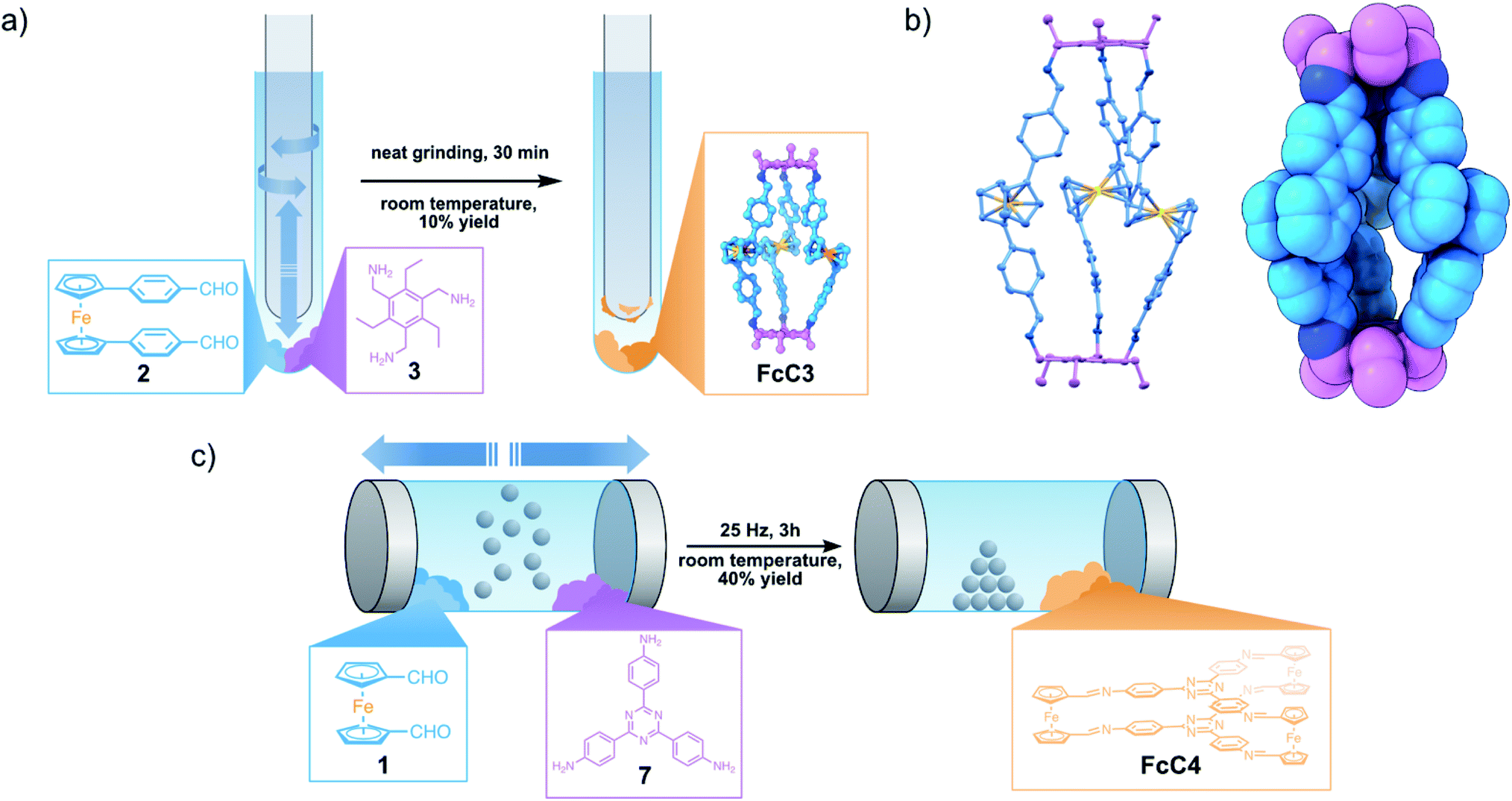 | ||
| Fig. 4 (a) Grinding reaction conditions for the synthesis of FcC3 from three equivalents of 2 and two equivalents of 3; (b) SC-XRD structure of FcC3 (left) with thermal ellipsoids at 50% probability (solvent molecules and hydrogen atoms were omitted for clarity, P, R1 = 0.11, wR2 = 0.33, Rint = 0.115) and space-filling model of FcC3 (right); (c) ball-milling procedure for solvent-free synthesis of FcC4 by using two equivalents of less reactive amine 7 and three equivalents of 1. | ||
If one assumes that shearing force is applied to the crystal lattice of 1 during the grinding process, which is then transferred to an individual molecule of 1, this could lead to ultrafast electron redistribution at the Cp-moiety, creating a highly electron-deficient and therefore reactive aldehyde group in the process. The local shift of electron density at the Cp-moiety can therefore only influence groups directly attached to the Cp-ring. In 2, the phenylene spacer limits the transfer of electron density to the formyl group, resulting in decreased reactivity as a result. To examine if the sharp signals observed in the 1H NMR can really be attributed to the initial formation of FcC3, we continued the reaction in the NMR tube for 24 h. The signal intensity of the sharp peaks increased only to a limited extent. When combining the starting materials 2 and 3 in solution, no signals corresponding to the formation of FcC3 were observed, rendering this cage inaccessible by solution-based methods (Fig. S21† for a formation study in CDCl3).
By slow vapour diffusion of n-pentane into a chloroform solution of the ground mixture of 2 and 3, we were able to isolate FcC3 as block-shaped crystals suitable for SC-XRD analysis (Fig. 4b). The structure reveals a very close proximity of the phenylene units with a dihedral angle of 0° between the phenylene unit and the Cp-ring. Indeed, this indicates a possible steric hindrance influencing the cage formation, since the coplanarity of the Cp-ring and the phenylene spacer group, caused by π-system maximization, limits the flexibility of the ligands, which is important during the assembly process of FcC3.
To investigate the feasibility of the solvent-free reaction between 2 and 3, we applied a stronger and continuous mechanical force by conducting the reaction inside of a ball mill. Surprisingly, even after 3 h at 25 Hz, we could not identify any peaks corresponding to the formation of FcC3, neither by NMR nor by MS analysis. This is possibly due to a much lower surface contact area when the reactants are adhering to the steel balls, and secondly, to the inherent lower reactivity of 2 compared to 1.
Lastly, we wanted to investigate the role of amine reactivity during the fast, solvent-free formation of organic cages containing Fc units. Therefore, we tried to synthesise the known Tri2Di3 cage FcC4, by using two equivalents of 2,4,6-tris(4-aminophenyl)-1,3,5-triazine (7) and three equivalents of 1.20 We were ultimately able to synthesise FcC4 in 40% yield, after ball-milling the starting materials for 3 h at 25 Hz (Fig. 4c). This indicates that the reactivity of the amine also plays a vital role in a successful solvent-free DCC reaction in the solid state. This represents an economically interesting alternative to the solvent-based previous synthesis of FcC4 using acetic acid, toluene and 1,4-dioxane, especially considering upscaling for potential applications.
Conclusions
We investigated the fast, solvent-free synthesis of organic cages using only readily available laboratory equipment. We discovered that Fc-containing reactants were able to react under the presented conditions, reliably resulting in an organic cage compound. To this extent, we were able to synthesise four different supramolecular compounds in a matter of minutes, of which cages FcC1–3 have not been reported previously. The structural analysis of FcC1–3 revealed differences in their connectivity, since FcC1 and FcC3 possess a ‘stair-like’ connection between the two former amines. FcC2 shows a ‘flat’ prismatic structure, highlighting the geometric diversity induced by the Fc aldehydes 1 and 2, even in the solid state. Additionally, we were able to synthesise the Fc-containing macrocycle FcM1 without the use of solvent during the synthesis, in good yields and by a factor of 288 faster than the solvent-based synthesis. Even compounds with low reactivity, like amine 7, can be used in a solvent-free synthesis of organic cages when a stronger and continuous mechanical force is applied. Our findings mark an important addition to not only the field of solvent-free, mechanochemical synthesis but also to the synthesis of organic cages and ferrocene chemistry and lay the foundation for further investigations towards sensors, porous, and electrochemically active materials.Data availability
All relevant data is presented in the paper and ESI.†Author contributions
T. K., T. P., A. K. and B. M. S. conceived the project; T. K., T. P. and B. M. S. designed the experiments; T. K. and T. P. performed the majority of the experiments, analysed the data and wrote the manuscript with B. M. S.; P. A. G. and B. M. S. did all SC-XRD measurements and refinements; M. K. and A. K. contributed additional solution screenings. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors state that there are no conflicts to declare.Acknowledgements
We would like to thank Esther Nieland for conducting the ball-milling and PXRD experiments. This work was supported by the Fonds der Chemischen Industrie by a Kekulé Fellowship (T. K.) by the North Rhine-Westphalian Academy of Sciences, Humanities and the Arts, funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SCHM 3101/5-1 (B. M. S.), Warsaw University of Technology within the NChem1 grant (504/04508/1020/43.120004) (A. K.) and supported by The Center for Structural Studies, which is funded by the Deutsche Forschungsgemeinschaft (DFG grant number 417919780) and INST 208/740-1 FUGG. SC-XRD measurements of compounds 2 and FcC2 were financed by Warsaw University of Technology (P. A. G.). M. K.’s contribution was within the “Szkoła Orłów” project, co-financed by the European Social Fund under the Knowledge-Education-Development Operational Programme, Axis III, Higher Education for the Economy and Development, Measure 3.1, Competences In Higher Education.Notes and references
-
(a) D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS
; (b) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC
-
(a) T. Kunde, T. Pausch and B. M. Schmidt, Eur. J. Org. Chem., 2021, 43, 5844–5856 CrossRef
-
(a) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC
- B. Teng, M. A. Little, T. Hasell, S. Y. Chong, K. E. Jelfs, R. Clowes, M. E. Briggs and A. I. Cooper, Cryst. Growth Des., 2019, 19, 3647–3651 CrossRef CAS PubMed
-
(a) W. Pickhardt, S. Grätz and L. Borchardt, Chem.–Eur. J., 2020, 26, 12903–12911 CrossRef CAS PubMed
-
(a) D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC
-
(a) B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed
-
(a) N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC
-
(a) L. Catalano, L. S. Germann, P. A. Julien, M. Arhangelskis, I. Halasz, K. Užarević, M. Etter, R. E. Dinnebier, M. Ursini, M. Cametti, J. Martí-Rujas, T. Friščić, P. Metrangolo, G. Resnati and G. Terraneo, Chem, 2021, 7, 146–154 CrossRef CAS
-
(a) I. Brekalo, W. Yuan, C. Mottillo, Y. Lu, Y. Zhang, J. Casaban, K. T. Holman, S. L. James, F. Duarte, P. A. Williams, K. D. M. Harris and T. Friščić, Chem. Sci., 2020, 11, 2141–2147 RSC
-
(a) E. Troschke, S. Grätz, T. Lübken and L. Borchardt, Angew. Chem., Int. Ed., 2017, 56, 6859–6863 CrossRef CAS PubMed
- B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154–3155 CrossRef PubMed
- B. D. Egleston, M. C. Brand, F. Greenwell, M. E. Briggs, S. L. James, A. I. Cooper, D. E. Crawford and R. L. Greenaway, Chem. Sci., 2020, 11, 6582–6589 RSC
- B. K. Hong, I. S. Lee, D. M. Shin and Y. K. Chung, Chem. Commun., 2004, 936–937 RSC
-
(a) R. A. S. Vasdev, J. A. Findlay, D. R. Turner and J. D. Crowley, Chem.–Asian J., 2021, 16, 39–43 CrossRef CAS PubMed
-
(a) A. Kasprzak, N. Kasprzak, A. Kowalczyk and A. M. Nowicka, Dalton Trans., 2021, 50, 8426–8433 RSC
-
(a) Y. Cha, T. Zhu, Y. Sha, H. Lin, J. Hwang, M. Seraydarian, S. L. Craig and C. Tang, J. Am. Chem. Soc., 2021, 143, 11871–11878 CrossRef CAS PubMed
-
(a) C. Imrie, P. Kleyi, V. O. Nyamori, T. I. A. Gerber, D. C. Levendis and J. Look, J. Organomet. Chem., 2007, 692, 3443–3453 CrossRef CAS
- For the nomenclature used herein, see: V. Santolini, M. Miklitz, E. Berardo and K. E. Jelfs, Nanoscale, 2017, 9, 5280–5298 RSC
- A. I. Kosińska, M. K. Nisiewicz, A. M. Nowicka and A. Kasprzak, ChemPlusChem, 2021, 86, 820–826 CrossRef PubMed
-
(a) T. Kunde, T. Pausch, G. J. Reiss and B. M. Schmidt, Synlett, 2022, 33, 161–165 CrossRef CAS
- The cage FcC3 comprises Cp-units in anti-conformation. This π–π stacking between phenylene units in the starting material might limit the ability of the cp-rings of 2 to rotate, thus, demands for a high activation energy to additionally compensate breaking of the, possibly attractive, π–π interaction. See also: J. D. Crowley, I. M. Steele and B. Bosnich, Chem.–Eur. J., 2006, 12, 88935–88951 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compounds characterization data and CCDC numbers. CCDC 2122280, 2085773, 2122279 and 2117653. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06372c |
| This journal is © The Royal Society of Chemistry 2022 |