DOI:
10.1039/D1SC06346D
(Edge Article)
Chem. Sci., 2022,
13, 2391-2404
Ligand and solvent effects on CO2 insertion into group 10 metal alkyl bonds†
Received
16th November 2021
, Accepted 31st January 2022
First published on 9th February 2022
Abstract
The insertion of carbon dioxide into metal element σ-bonds is an important elementary step in many catalytic reactions for carbon dioxide valorization. Here, the insertion of carbon dioxide into a family of group 10 alkyl complexes of the type (RPBP)M(CH3) (RPBP = B(NCH2PR2)2C6H4−; R = Cy or tBu; M = Ni or Pd) to generate κ1-acetate complexes of the form (RPBP)M{OC(O)CH3} is investigated. This involved the preparation and characterization of a number of new complexes supported by the unusual RPBP ligand, which features a central boryl donor that exerts a strong trans-influence, and the identification of a new decomposition pathway that results in C–B bond formation. In contrast to other group 10 methyl complexes supported by pincer ligands, carbon dioxide insertion into (RPBP)M(CH3) is facile and occurs at room temperature because of the high trans-influence of the boryl donor. Given the mild conditions for carbon dioxide insertion, we perform a rare kinetic study on carbon dioxide insertion into a late-transition metal alkyl species using (tBuPBP)Pd(CH3). These studies demonstrate that the Dimroth–Reichardt parameter for a solvent correlates with the rate of carbon dioxide insertion and that Lewis acids do not promote insertion. DFT calculations indicate that insertion into (tBuPBP)M(CH3) (M = Ni or Pd) proceeds via an SE2 mechanism and we compare the reaction pathway for carbon dioxide insertion into group 10 methyl complexes with insertion into group 10 hydrides. Overall, this work provides fundamental insight that will be valuable for the development of improved and new catalysts for carbon dioxide utilization.
Introduction
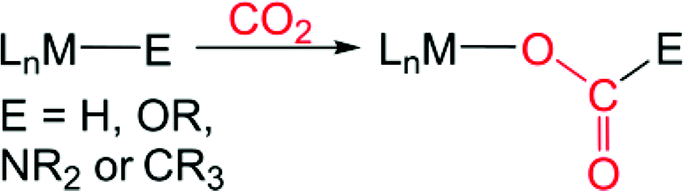
Carbon dioxide (CO2) is an attractive target as a chemical feedstock due to its low cost, non-toxic nature, and abundance.1 Although there are several chemicals currently industrially prepared from CO2, such as urea and carbonates,2 only a small fraction of our available CO2 is used as a source of carbon for either commodity1h or fine3 chemical synthesis. The full exploitation of CO2 as a feedstock is limited by its kinetic and thermodynamic stability, which poses a scientific challenge for developing synthetic routes to make more valuable chemicals from CO2. Utilizing homogeneous transition metal catalysts is one attractive approach for solving kinetic problems associated with the conversion of CO2 into more valuable chemicals and a variety of systems have been developed for the thermal, electrochemical, and photochemical conversion of CO2.1,3 An elementary step which is proposed in many of these catalytic reactions is CO2 insertion into an M–E σ-bond (for example E = H, OR, NR2, or CR3) to form a product with a M–O bond (eqn (1)).4 To date, the majority of catalysts feature late transition metals,1,3 likely due to the relative weakness of the M–O bonds that are formed,4 which makes cleavage of the M–O bond and subsequent transformation more facile.| | 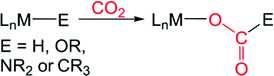 | (1) |
The crucial role of CO2 insertion into late transition metal element σ-bonds in catalysis means that understanding the pathway for insertion is important for rationally designing improved catalytic systems.4 In particular, understanding the impact of the ancillary ligand, solvent, and additives that are commonly used in catalysis, such as Lewis acids, is valuable for the development of optimized catalysts and reaction conditions. Although the influence of these factors on CO2 insertion into metal hydrides has been studied,5 there is limited information on how these variables affect CO2 insertion into metal alkyl bonds.6–13 These reactions are particularly important because they can result in the generation of products containing C–C bonds from CO2, which has been identified as a high priority research target by the United States National Academies of Science.2 For example, group 10 catalysts have been used for the reductive carboxylation of a variety of alkyl halides and pseudo halides.3h,i In these reactions, C–C bonds are proposed to form between CO2 and the alkyl electrophile via the insertion of CO2 into a metal alkyl bond.
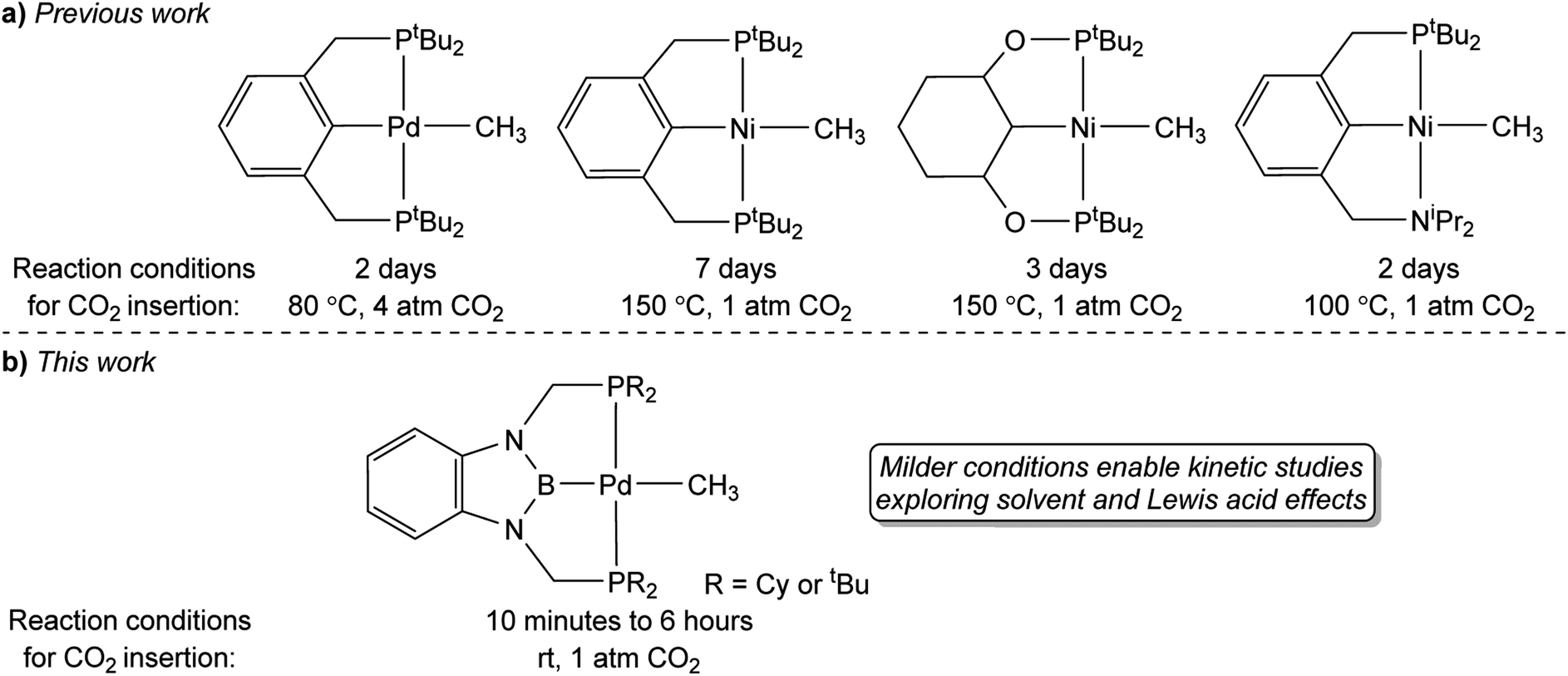
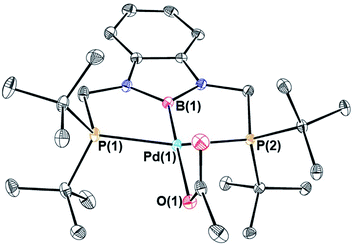
A major limitation in studying CO2 insertion into group 10 metal alkyl bonds is the paucity of systems that are stable and react under mild conditions.14 In fact, the majority of well-defined examples of CO2 insertion reactions into group 10 metal alkyls require elevated temperatures, long reaction times, and do not give quantitative yields of products, which makes it difficult to perform kinetic studies (Fig. 1a). Due to the need for high temperatures, there has been a focus on complexes supported by pincer ligands, which often result in complexes with high thermal stability. For example, Wendt and co-workers demonstrated that treatment of (tBuPCP)Pd(CH3) (tBuPCP = 2,6-C6H3(CH2PtBu2)2) with 4 atm of CO2 at 80 °C results in complete conversion to the κ1-acetate complex (tBuPCP)Pd{OC(O)CH3}, but the reaction takes two days.15 Similarly, we reported that the reaction of 1 atm of CO2 with the related compound (tBuPCP)Ni(CH3) requires heating to 150 °C to form the acetate complex (tBuPCP)Ni{OC(O)CH3}, but only approximately 75% of the acetate product is formed.16 Changing the pincer ligand from tBuPCP to a system containing an sp3-hybridized carbon donor or replacing one of the phosphine arms of the pincer ligand with a hemi-labile nitrogen donor only leads to small changes in the rate of reaction.17 A notable example of a group 10 methyl complex that does not contain a pincer ligand and inserts CO2 at room temperature is an unusual anionic palladium dimethyl complex, with a pendant Lewis acid, prepared by Jordan and co-workers.7 Although this system inserts CO2 rapidly it requires careful handling as it is unstable at room temperature. Overall, in order to gain detailed information on the factors that influence the rate of CO2 insertion there is a need for stable alkyl complexes that cleanly react with CO2 under mild conditions.
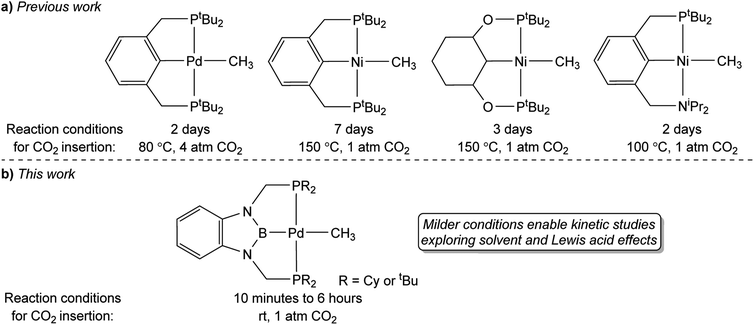 |
| | Fig. 1 (a) Previous examples of pincer-supported group 10 methyl complexes that undergo CO2 insertion to form κ1-acetate complexes. (b) RPBP supported palladium complexes studied in this work, which undergo CO2 insertion under milder conditions. | |
In this work, we describe the insertion of CO2 into palladium and nickel methyl complexes supported by RPBP (RPBP = B(NCH2PR2)2C6H4−; R = Cy or tBu) pincer ligands, which contain a strongly donating central boryl donor (Fig. 1b). As a result of the strong trans-influence of the boryl donor, these complexes insert CO2 at significantly milder conditions (room temperature) than previously reported pincer systems. This enables us to measure the kinetics of CO2 insertion and study the effect of modulating the pincer ligand and the solvent on CO2 insertion. We demonstrate that the Dimroth–Reichardt ET(30) parameter18 for a solvent correlates with the rate of CO2 insertion and show that Lewis acids, which are often present in catalysis, do not promote CO2 insertion. Our experimental studies are complemented by computational investigations to understand the pathway for CO2 insertion and identify the structure of key transition states and intermediates. Overall, our work is one of the first quantitative investigations of the impact of the ancillary ligand, solvent, and LAs on CO2 insertion into metal alkyl bonds and will assist in the development of improved and new catalysts for CO2 utilization.
Results and discussion
Synthesis of RPBP supported nickel and palladium methyl complexes and reactivity with CO2
Previous studies on CO2 insertion into pincer supported group 10 metal hydrides demonstrate that the rate of CO2 insertion increases as the trans-influence of the donor opposite to the hydride ligand increases.19 As a result, nickel and palladium hydride complexes supported by CyPSiP (CyPSiP = Si(Me)(2-PCy2-C6H4)2) ligands, which contain a strongly donating silyl ligand trans to the hydride insert CO2 more rapidly than the corresponding species containing RPCP ligands. We hypothesized that a similar trend may also occur for nickel and palladium methyl species. Unfortunately, the previously described complexes (CyPSiP)M(CH3) (M = Ni or Pd) undergo an intramolecular ligand rearrangement,20 as does (tBuPSiP)Pd(CH3) (tBuPSiP = Si(Me)(2-PtBu2-C6H4)2, see ESI†) which complicates studies of CO2 insertion. Therefore, we prepared nickel and palladium methyl complexes supported by RPBP ligands (RPBP = B(NCH2PR2)2C6H4; R = Cy or tBu) that feature a boryl donor, which has previously been shown to exert a large trans-influence,21 opposite the methyl ligand.
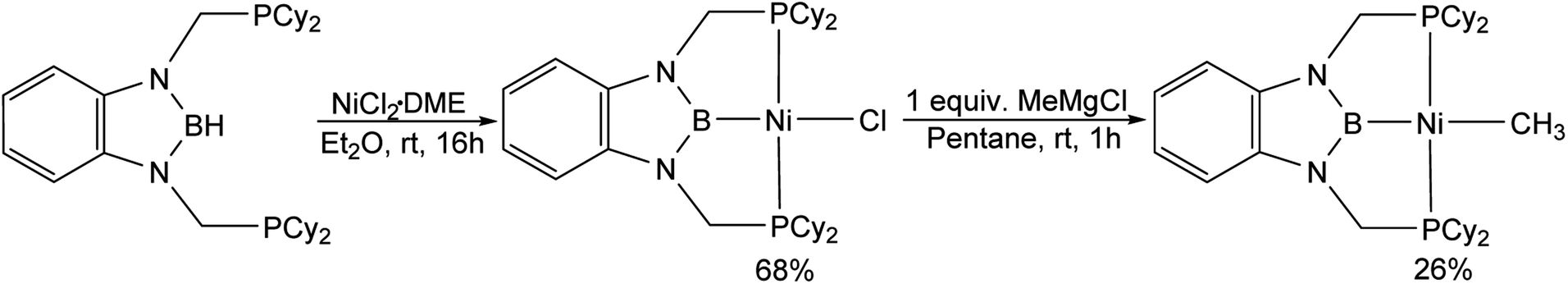
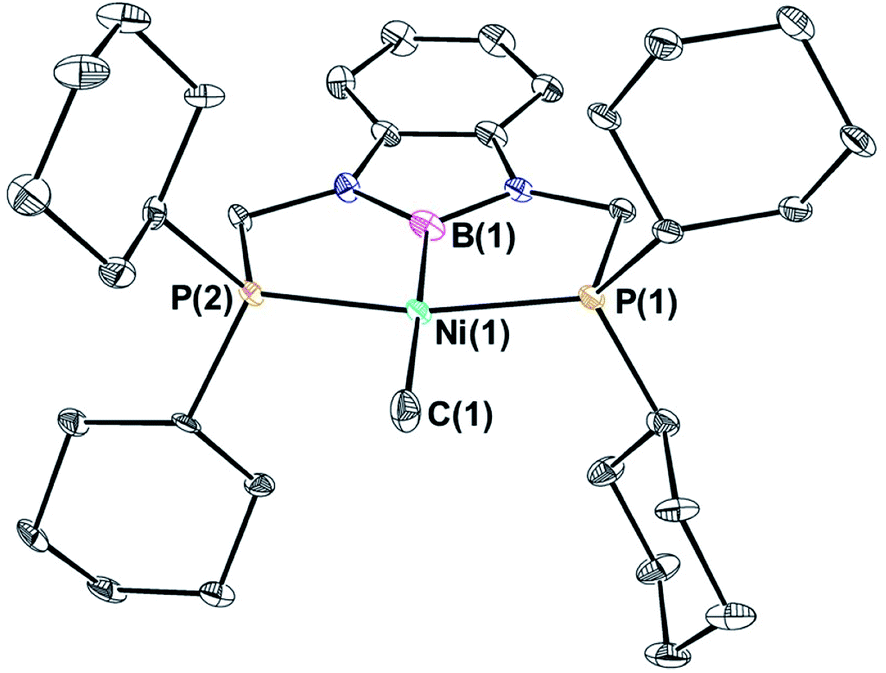
(CyPBP)Ni(CH3) was synthesized via an analogous route to that previously described for (tBuPBP)Ni(CH3).21h Initially, NiCl2·DME was treated with CyPBHP to generate (CyPBP)NiCl in 68% yield (Scheme 1). The subsequent reaction of (CyPBP)NiCl with MeMgCl generates (CyPBP)Ni(CH3) in 26% yield. The major cause of the low yield of (CyPBP)Ni(CH3) is the need to perform sequential recrystallizations to remove magnesium salts. Specifically, considerable care is required to remove MgCl2 by-products from the Grignard reagent, which cause the methyl species to convert back to the chloride complex. (CyPBP)Ni(CH3) was characterized in the solid-state using X-ray crystallography (Fig. 2) and has an analogous distorted square planar geometry to (tBuPBP)Ni(CH3).21h The P(1)–Ni(1)–P(2) bond angle is 158.67(10)°, which is similar to that observed in (tBuPBP)Ni(CH3) (157.58(2)°). The B(1)–Ni(1)–C(1) bond angle is almost linear (179.1(4)°), which presumably results in the boryl ligand exerting the maximum possible trans-influence on the methyl ligand. The Ni(1)–B(1) bond length in (CyPBP)Ni(CH3) is 1.927(9) Å, which is comparable to other species with a Ni–boryl bond.21h,22 The nickel methyl bond length is 2.019(9) Å, which is significantly shorter than the nickel methyl bond length in (tBuPBP)Ni(CH3), which is 2.059(2) Å. The longer bond length in (tBuPBP)Ni(CH3) is likely due to steric factors, as the larger steric properties of the tert-butyl substituents in (tBuPBP)Ni(CH3) may cause the nickel methyl bond to elongate. The increased steric demand of the tBuPBP ligand was confirmed using buried volume calculations (SambVca 2.1 (ref. 23)) using the crystal structures of (RPBP)Ni(CH3), which show that the buried volume of (tBuPBP)Ni(CH3) is 92.1% compared to 89.3% for (CyPBP)Ni(CH3) (see ESI†).
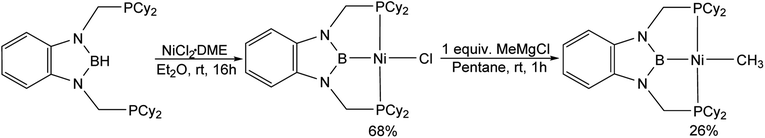 |
| | Scheme 1 Synthesis of (CyPBP)Ni(CH3). | |
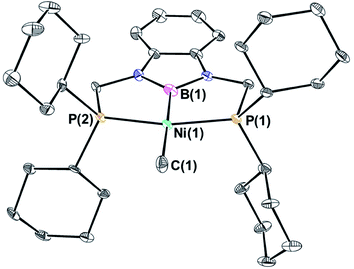 |
| | Fig. 2 Solid-state structure of (CyPBP)Ni(CH3) with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (°): Ni(1)–B(1) 1.927(9), Ni(1)–C(1) 2.019(9), Ni(1)–P(1) 2.148(2), Ni(1)–P(2) 2.138(2), B(1)–Ni(1)–C(1) 179.1(4), B(1)–Ni(1)–P(1) 80.1(3), C(1)–Ni(1)–P(1) 100.6(3), B(1)–Ni(1)–P(2) 78.7(3), C(1)–Ni(1)–P(2) 100.6(3), P(1)–Ni(1)–P(2) 158.67(10). | |
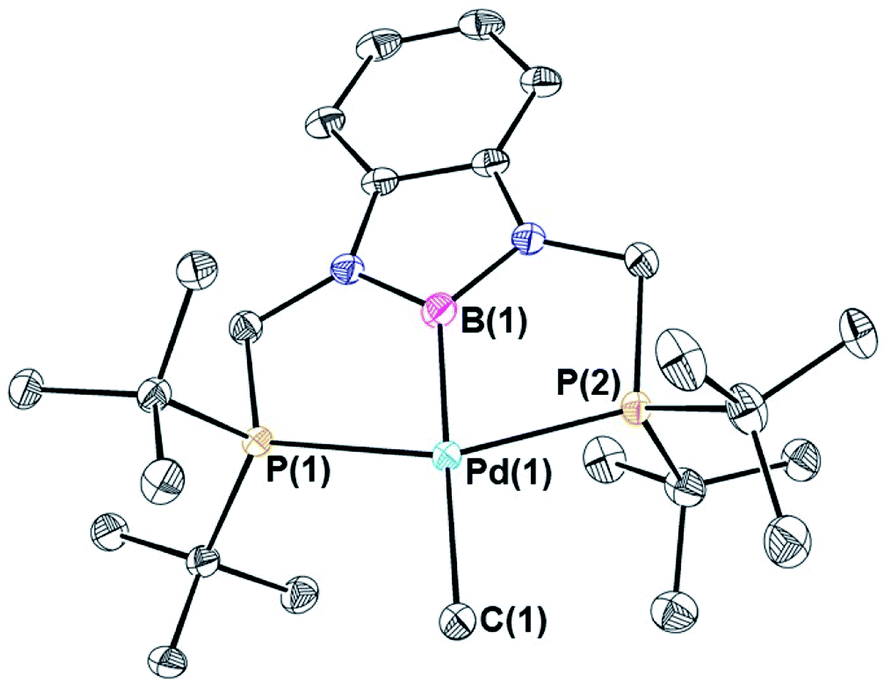
Palladium chloride complexes supported by RPBP (R = Cy or tBu) ligands were synthesized through the reaction of the protonated RPBHP ligand with commercially available [(η3-allyl)PdCl]2 in yields of 69% and 63%, respectively. This is different to a previous route utilized to metallate the tBuPBP ligand, which used the more expensive Pd(COD)Cl2 as the palladium source.21k Treatment of (RPBP)PdCl with MeLi results in the formation of the (RPBP)Pd(CH3) in yields of 25% for (CyPBP)Pd(CH3) and 60% for (tBuPBP)Pd(CH3) (Scheme 2). In an analogous fashion to (CyPBP)Ni(CH3), it is important to remove alkali halide by-products as they can cause the palladium methyl species to be converted back to the palladium chloride precursor and the multiple recrystallizations required for (CyPBP)Pd(CH3) lowers the yield. (tBuPBP)Pd(CH3) was characterized using X-ray crystallography (Fig. 3). The geometry around palladium is distorted square planar. The P(1)–Pd(1)–P(2) bond angle is 153.74(3)°, which is a greater deviation from linearity than that observed in a related complex with a tBuPCP ligand. Specifically in (tBuPCP)PdCl P–Pd–P is 165.79(2)°.24 As a consequence, the fourth coordination site in pincer complexes supported by tBuPBP is likely more sterically accessible than in species supported by tBuPCP ligands. The Pd(1)–B(1) bond length in (tBuPBP)Pd(CH3) is 2.027(4) Å, which is significantly longer than the Pd–B bond length in the only other crystallographically characterized palladium complex with a tBuPBP ligand, (tBuPBP)PdCl (Pd–B is 1.972(4) Å). This is consistent with the methyl ligand exerting a significantly stronger trans-influence than the chloride ligand.
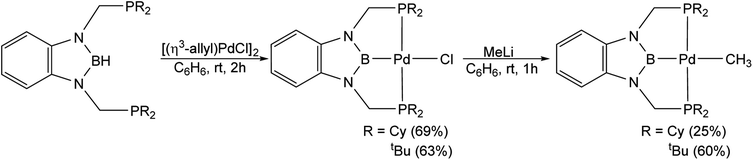 |
| | Scheme 2 Synthesis of (RPBP)Pd(CH3) (R = Cy or tBu). | |
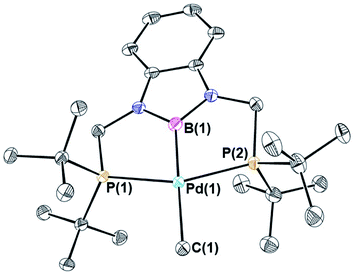 |
| | Fig. 3 Solid-state structure of (tBuPBP)Pd(CH3) with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (°): Pd(1)–B(1) 2.027(4), Pd(1)–C(1) 2.185(4), Pd(1)–P(1) 2.3030(8), Pd(1)–P(2) 2.3035(8), B(1)–Pd(1)–C(1) 178.09(15), B(1)–Pd(1)–P(1) 77.36(12), C(1)–Pd(1)–P(1) 103.49(10), B(1)–Pd(1)–P(2) 77.33(12), C(1)–Pd(1)–P(2) 102.02(10), P(1)–Pd(1)–P(2) 153.74(3). | |
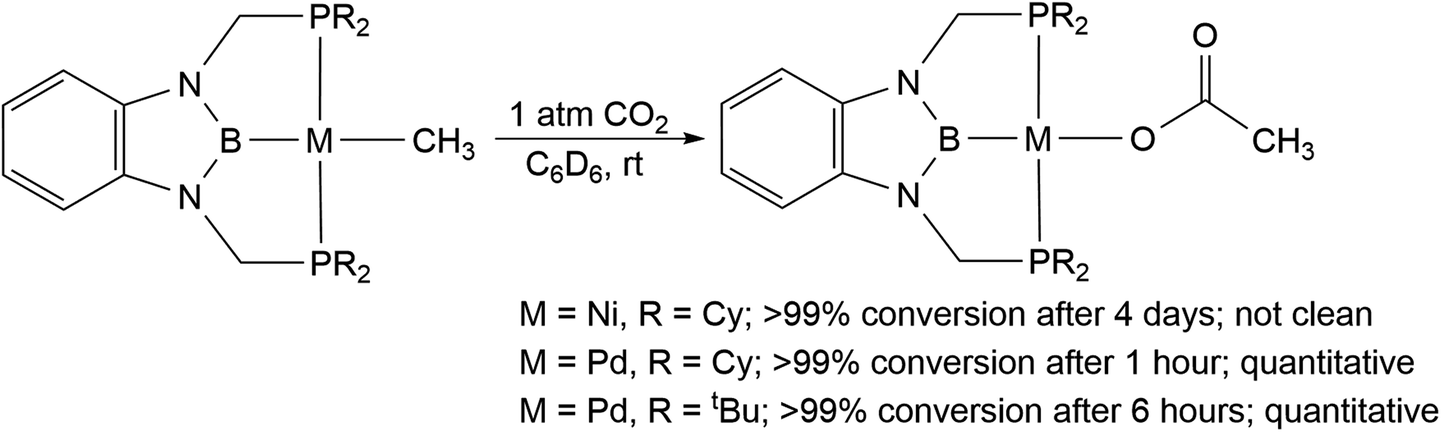
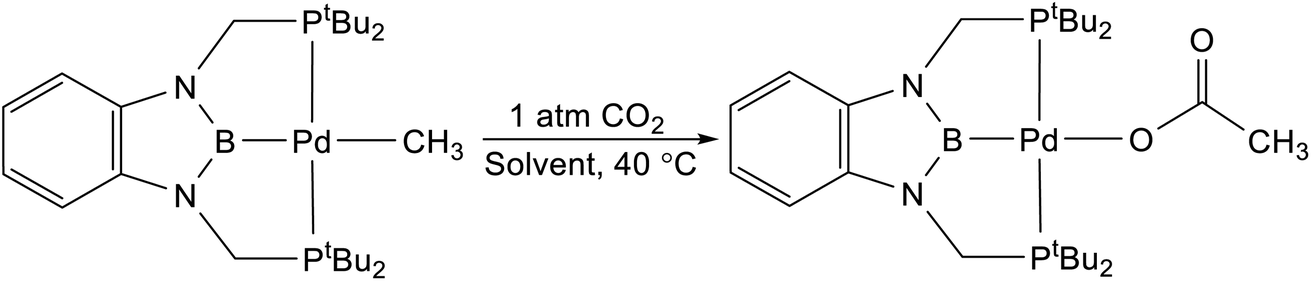
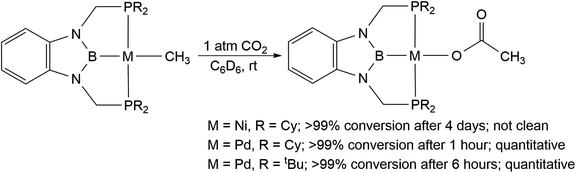
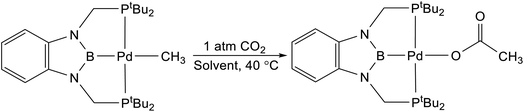
We treated complexes of the type (RPBP)M(CH3) (M = Ni or Pd, R = Cy or tBu) with 1 atm of CO2 in C6D6 at room temperature (Scheme 3). In the cases of (RPBP)Pd(CH3) (R = Cy or tBu) NMR spectroscopy indicated clean and essentially quantitative CO2 insertion to the corresponding acetate complexes at room temperature. The reactions took 1 and 6 hours to reach completion for (CyPBP)Pd(CH3) and (tBuPBP)Pd(CH3), respectively. These are the most rapid insertions of CO2 into pincer-supported group 10 metal alkyl complexes observed to date. The acetate products were isolated and (tBuPBP)Pd{OC(O)CH3} was characterized using X-ray crystallography (Fig. 4). The solid-state structure of (tBuPBP)Pd{OC(O)CH3} has the same distorted square planar geometry as (tBuPBP)Pd(CH3) and confirms κ1-binding of the acetate. As expected, the Pd(1)–B(1) bond length (1.975(2) Å) in (tBuPBP)Pd{OC(O)CH3} is significantly shorter than the Pd–B bond length in (tBuPBP)Pd(CH3) (2.027(4) Å) due to the weaker trans-influence of the acetate ligand. Further, the increased Pd–O bond length in (tBuPBP)Pd{OC(O)CH3} (2.1861(14) Å) in comparison to (tBuPCP)Pd{OC(O)CH3} (2.1279(19) Å)15 is consistent with the increased trans-influence of the tBuPBP ligand in comparison to the related tBuPCP ligand. In contrast, the insertion of CO2 into (CyPBP)Ni(CH3) was not clean. Although NMR spectroscopy suggested that (CyPBP)Ni{OC(O)CH3}, which was not isolated, was the major product, free ligand and several unidentified decomposition products were present (see ESI†). Additionally, it took approximately 4 days at room temperature for all of the (CyPBP)Ni(CH3) to disappear. Overall, from these initial qualitative examples of CO2 insertion into (CyPBP)M(CH3) (M = Ni or Pd) and (tBuPBP)Pd(CH3) we can make the following conclusions: (i) the insertion of CO2 into (CyPBP)Pd(CH3) is faster than the corresponding insertion into (CyPBP)Ni(CH3). This is in agreement with results for CO2 insertion into (tBuPCP)M(CH3) (M = Ni or Pd)15,16 but stands in contrast to computational studies of CO2 insertion into (tBuPCP)MH (M = Ni or Pd), where insertion is proposed to be faster for nickel.19 (ii) The insertion of CO2 into (tBuPBP)Pd(CH3) is significantly faster than the corresponding insertion into (tBuPCP)Pd(CH3).15 This suggests that the rate of CO2 insertion can be increased by having a stronger trans-influence donor opposite the methyl, similar to what has been observed for CO2 insertion into group 10 metal hydrides.19 (iii) The faster insertion of CO2 into (CyPBP)Pd(CH3) compared to (tBuPBP)Pd(CH3) suggests that steric factors are important.
 |
| | Scheme 3 Generation of (RPBP)M{OC(O)CH3} (M = Ni, R = Cy; M = Pd, R = Cy or tBu) through CO2 insertion into (RPBP)M(CH3). | |
 |
| | Fig. 4 Solid-state structure of (tBuPBP)Pd{OC(O)CH3} with thermal ellipsoids at 30% probability. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (°): Pd(1)–B(1) 1.975(2), Pd(1)–O(1) 2.1861(14), Pd(1)–P(1) 2.3256(5), Pd(1)–P(2) 2.3336(5), B(1)–Pd(1)–O(1) 174.99(7), B(1)–Pd(1)–P(1) 77.32(7), O(1)–Pd(1)–P(1) 100.17(4), B(1)–Pd(1)–P(2) 79.00(7), O(1)–Pd(1)–P(2) 103.05(4), P(1)–Pd(1)–P(2) 155.805(18). | |
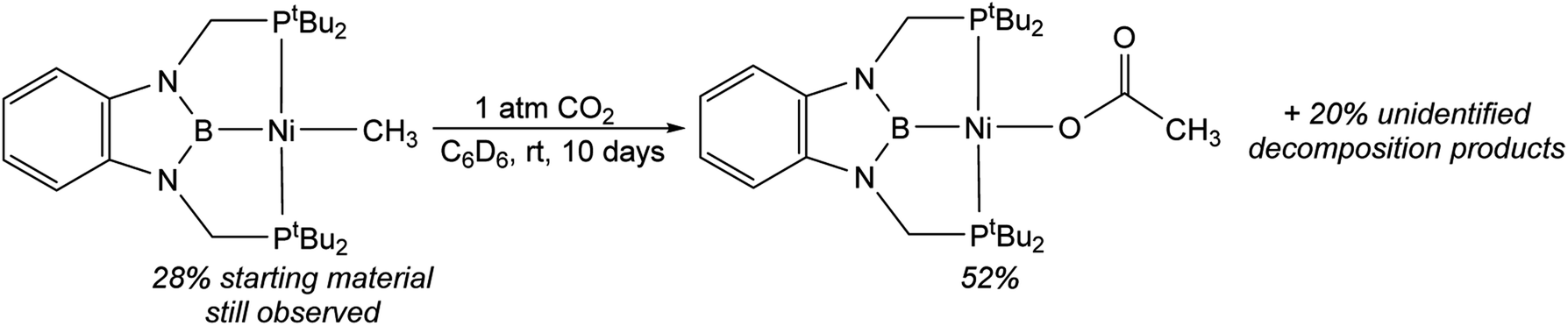
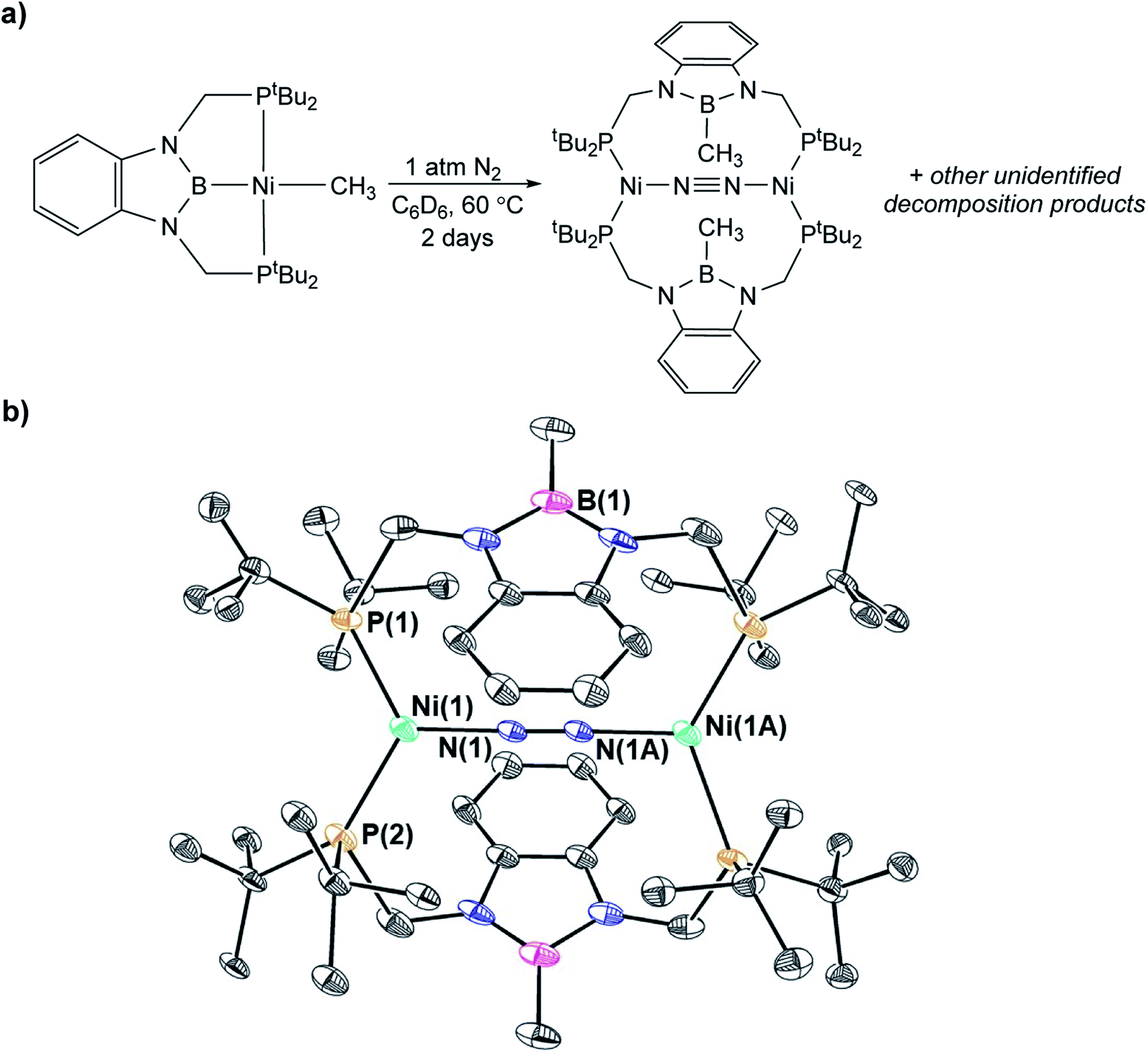
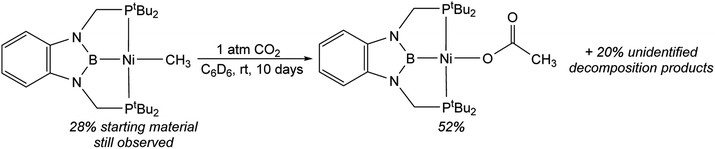
In contrast to the quantitative or relatively clean insertion of CO2 observed for (CyPBP)Pd(CH3), (tBuPBP)Pd(CH3), and (CyPBP)Ni(CH3), insertion into (tBuPBP)Ni(CH3) was not clean and was significantly slower than for the other methyl compounds. After 10 days at room temperature, approximately 30% of (tBuPBP)Ni(CH3) was still observed, along with 50% of a product we propose to be (tBuPBP)Ni{OC(O)CH3} on the basis of its similar 31P NMR chemical shift to (CyPBP)Ni{OC(O)CH3}, and 20% of unidentified decomposition products (Scheme 4). Attempts to independently synthesize (tBuPBP)Ni{OC(O)CH3} via reactions between silver acetate and (tBuPBP)NiCl or sodium acetate and (tBuPBP)Ni(OTf) were unsuccessful. Although, initially a new resonance is observed in the 31P NMR spectra of these reactions with the same chemical shift as the proposed insertion product with CO2 (84.9 ppm), this resonance disappears over time, indicating that (tBuPBP)Ni{OC(O)CH3} is likely unstable in solution. This decomposition prohibited isolation and full characterization of (tBuPBP)Ni{OC(O)CH3}. Nevertheless, since the rate of CO2 insertion into (tBuPBP)Ni(CH3) is presumably independent of the decomposition of the corresponding insertion product, we can make qualitative conclusions about the rate of insertion. We hypothesize that the slower rate of CO2 insertion is related to the increased steric bulk of (tBuPBP)Ni(CH3), which makes it harder for CO2 to interact with the complex. When (tBuPBP)Ni(CH3) was treated with CO2 at higher temperatures even larger amounts of decomposition products were observed. Control experiments indicate that (tBuPBP)Ni(CH3) is not stable even when heated under an N2 atmosphere. Although we were unable to identify all of the decomposition products of (tBuPBP)Ni(CH3), an unusual dinuclear species (tBuPBMeP)2Ni2(μ-N2) (tBuPBMeP = MeB(NCH2PtBu2)2C6H4), containing two nickel(0) centers and a bridging dinitrogen ligand crystallized out of solution (Fig. 5). In this complex, the nickel methyl ligand has undergone reductive coupling with the boryl ligand to form a new C–B bond. The boron atom no longer coordinates to the nickel center and the two phosphorus donors of the new bidentate tBuPBMeP ligands do not coordinate to the same nickel center but instead coordinate to two different nickel atoms. The geometry around the nickel centers is distorted trigonal planar and the dinitrogen ligand coordinates in an end-on fashion. The N–N bond distance of 1.145(8) Å is slightly elongated compared to that of free N2 (1.098 Å), consistent with weak activation of N2 and what has been observed in related nickel dinitrogen complexes.25 The decomposition of (tBuPBP)Ni(CH3) into (tBuPBMeP)2Ni2(μ-N2) demonstrates a new reductive pathway for decomposition of complexes supported by RPBP ligands. However, this is likely a particular problem for the tBuPBP ligand when it is coordinated to Ni because of steric constraints associated with the bulky tert-butyl substituents and small size of nickel.
 |
| | Scheme 4 CO2 insertion into (tBuPBP)Ni(CH3). | |
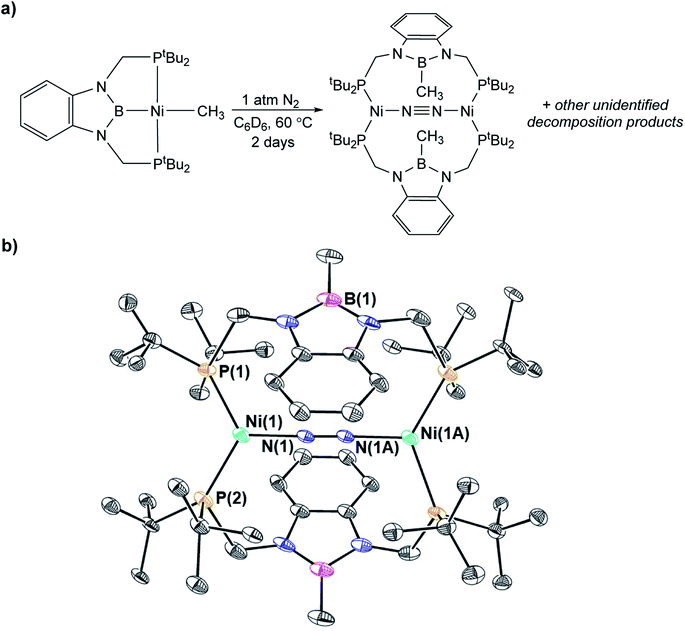 |
| | Fig. 5 a) Decomposition of (tBuPBP)Ni(CH3) under N2 at 60 °C. (b) Solid-state structure of (tBuPBMeP)2Ni2(μ-N2) with thermal ellipsoids at 30% probability. The model is near the crystallographic inversion center; half of the structure is generated by symmetry. Hydrogen atoms are omitted for clarity. Selected distances (Å) and angles (°): Ni(1)–N(1) 1.854(4), Ni(1)–P(1) 2.293(11), Ni(1)–P(2) 2.164(11), N(1)–N(1A) 1.145(8), P(1)–Ni(1)–P(2) 122.0(5), N(1)–Ni(1)–P(1) 118.9, N(1)–Ni(1)–P(2) 110.8(3), Ni(1)–N(1)–N(1A) 178.7(5). | |
Kinetic studies of CO2 insertion into (tBuPBP)Pd(CH3).
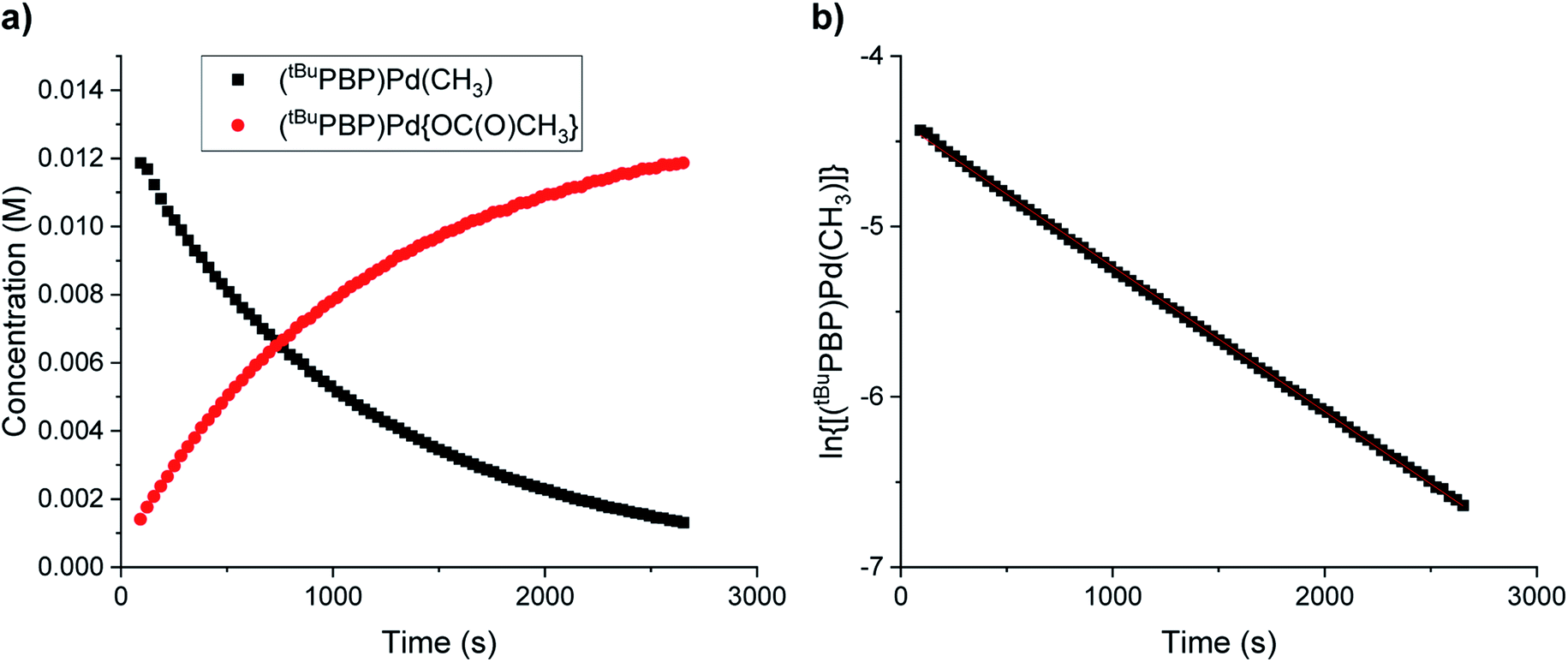
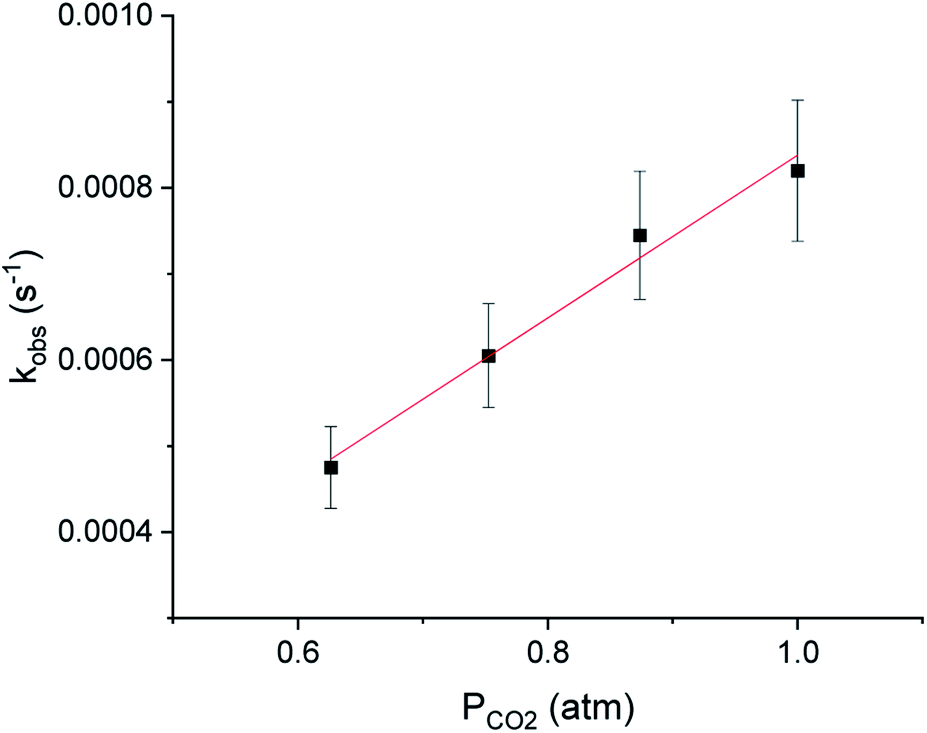
It has traditionally been challenging to perform kinetics experiments on CO2 insertion into group 10 alkyl complexes either because of the instability of the complexes14 or the harsh conditions required for insertion.16,17 In contrast, the insertion of CO2 into (tBuPBP)Pd(CH3) under mild conditions enables kinetics experiments to be performed. Initially, 1H or 31P NMR spectroscopy was used to monitor the reaction of (tBuPBP)Pd(CH3) with CO2 as a function of time (Fig. 6a). It was possible to measure the concentrations of both the (tBuPBP)Pd(CH3) starting material and the (tBuPBP)Pd{OC(O)CH3} product, which enabled us to confirm the reaction is clean and quantitative. An excess of CO2 was used to ensure that the reactions were performed under pseudo-first order conditions with the concentration of CO2 measured using quantitative 13C NMR spectroscopy (see SI).26 Initial experiments in C6D6 indicate that the reaction is first order in both [(tBuPBP)Pd(CH3)] and [CO2], demonstrating that the overall rate law is k1[(tBuPBP)Pd(CH3)][CO2] (Fig. 6b and 7). The kobs values obtained from a plot of ln([(tBuPBP)Pd(CH3)]) versus time were divided by the concentration of CO2 to obtain values of k1 (see ESI†). Monitoring the rate of the reaction in C6D6 at various temperatures allowed for the activation parameters to be determined through Eyring analysis (see ESI†). The enthalpy of activation, ΔH‡, 10.9 ± 1.1 kcal mol−1, for insertion into (tBuPBP)Pd(CH3) is significantly lower than that for (tBuPCP)Pd(CH3) (ΔH‡ = 17.4 ± 1.7 kcal mol−1),26 which is the most closely related system for which an experimental activation energy for CO2 insertion has been measured. This is consistent with the hypothesis that the strong trans-influence of the tBuPBP ligand weakens the palladium methyl bond and destabilizes the ground state. The entropies of activation, ΔS‡, for (tBuPBP)Pd(CH3) and (tBuPCP)Pd(CH3) are within error of each other at −32.2 ± 3.2 and −28.2 ± 4.54 cal mol−1 K−1, respectively. The similar negative values suggest that the nature of the rate-limiting transition state in both reactions are analogous and is in agreement with two molecules combining to form one molecule in the transition state (vide infra). The overall difference in ΔG‡ at 298 K between (tBuPBP)Pd(CH3) (20.5 ± 2.1 kcal mol−1) and (tBuPCP)Pd(CH3) (25.8 ± 2.6 kcal mol−1) is consistent with the much faster rate of insertion observed for the tBuPBP system.
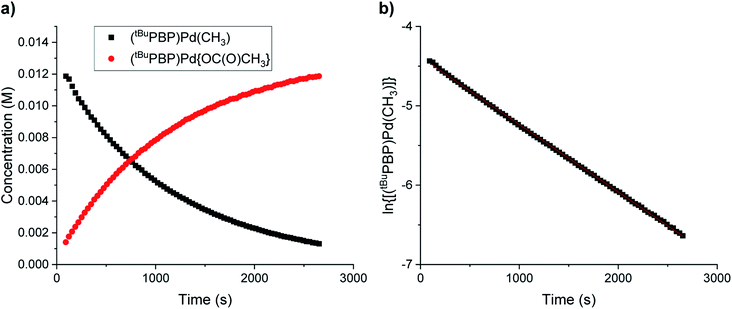 |
| | Fig. 6 Representative traces for the insertion of CO2 into (tBuPBP)Pd(CH3) at 40 °C in C6D6 with 1 atm of CO2 showing (a) the concentrations of (tBuPBP)Pd(CH3) and (tBuPBP)Pd{OC(O)CH3} as a function of time and (b) the ln of the concentration of (tBuPBP)Pd(CH3) as a function of time. | |
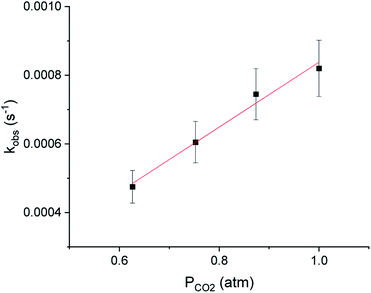 |
| | Fig. 7 Reaction order in [CO2] of the insertion of CO2 into (tBuPBP)Pd(CH3) in C6D6 at 40 °C. | |
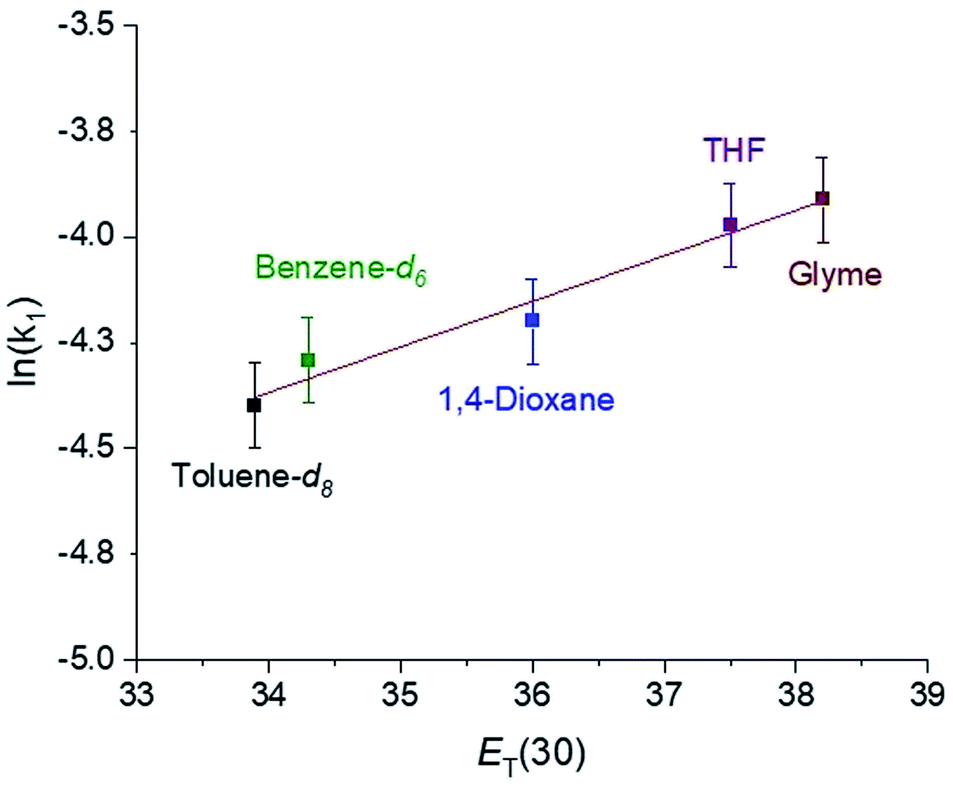
In CO2 insertion reactions into metal hydrides, it has been demonstrated that the solvent can have a major influence on the rate of insertion.5 To date there are no studies exploring how the rate of CO2 insertion into metal alkyls varies as the solvent is changed. We determined the rate of CO2 insertion (k1) into (tBuPBP)Pd(CH3) in toluene-d8, benzene-d6, 1,4-dioxane, THF, and glyme (Table 1). There was only a small solvent effect with approximately a 1.9-fold difference in k1 between the slowest (toluene-d8) and fastest (glyme) solvent (Table 1). This compares with a 30-fold increase in the rate of CO2 insertion into an iridium hydride in a similar set of solvents.5a We propose that the smaller impact of solvent in this case is because there is less build-up of charge in the rate-determining transition state for CO2 insertion (vide infra) compared to the corresponding transition state for metal hydrides. To determine if the variation in the rate of insertion correlated with any solvent parameters, ln(k1) was plotted against the dielectric constant, Gutmann Acceptor Number (AN),27 or Dimroth–Reichardt ET(30) parameter18 of the solvent. No significant correlation was observed between the rate of CO2 insertion and the dielectric constant or AN of a solvent (see ESI†). In contrast, an excellent correlation was observed between the ET(30) value of a solvent and the rate of CO2 insertion (Fig. 8), suggesting that ET(30) is a good predictor of the rate of CO2 insertion. A similar relationship was observed for CO2 insertion into metal hydrides and hydroxides,5b raising the possibility that this correlation is general across a variety of distinct metal-element σ-bonds. It is likely that ET(30) is a better predictor of CO2 insertion rates because it explicitly considers specific solute/solvent interactions, which are not accounted for when the solvent is modelled using either dielectric constant or AN.5b Unfortunately, we were unable to extend the correlation between the rate of CO2 insertion into (tBuPBP)Pd(CH3) and ET(30) to more polar solvents. This is because these solvents led to the decomposition of (tBuPBP)Pd(CH3) or resulted in rates that were too fast to measure using NMR spectroscopy. For example, CO2 insertion into (tBuPBP)Pd(CH3) is complete in less than 10 minutes at room temperature in pyridine and benzonitrile, which is too fast to obtain reliable kinetics by NMR spectroscopy. Nevertheless, given the slow rates of CO2 insertion previously observed into pincer-supported group 10 alkyls, the completion of reactions in less than 10 minutes at room temperature is unprecedented.
Table 1 Effect of solvent on rate of CO2 insertion into (tBuPBP)Pd(CH3) at 40 °C and 1 atm of CO2
|
|
| Entry |
Solvent |
k
1
(M−1 s−1 × 10−2) |
Rate enhancement |
|
These values are the average of two trials and the errors are ±10%.
|
| 1 |
Toluene-d8 |
1.1 |
1 |
| 2 |
Benzene-d6 |
1.3 |
1.2 |
| 3 |
1,4-Dioxane |
1.5 |
1.4 |
| 4 |
Tetrahydrofuran |
1.8 |
1.6 |
| 5 |
Glyme |
2.1 |
1.9 |
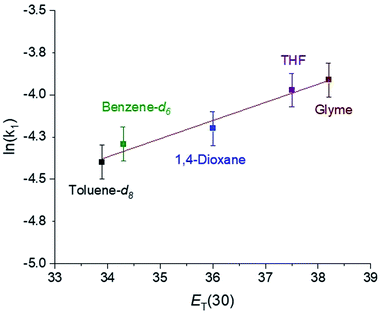 |
| | Fig. 8 Plot of ln(k1) for the insertion of CO2 into (tBuPBP)Pd(CH3) versus the Dimroth–Reichardt ET(30) solvent parameter for various solvents at 40 °C and 1 atm of CO2. | |
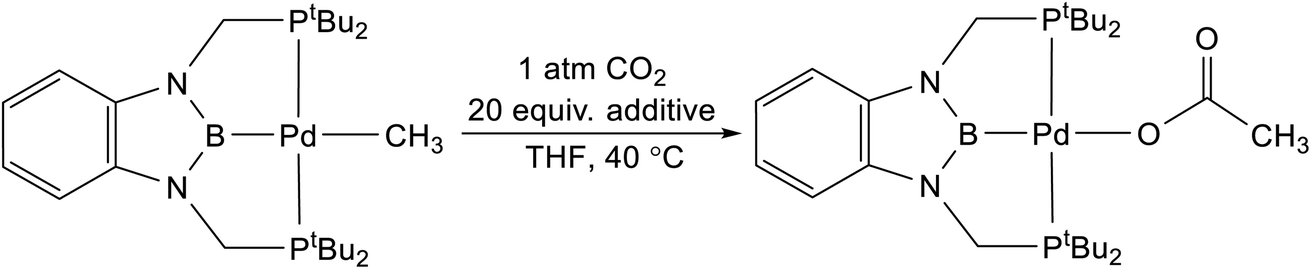
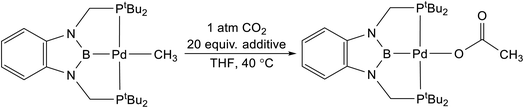
Lewis acids have been proposed to increase the rate of CO2 insertion into a variety of different metal-element σ-bonds, but these effects are often dependent on the pathway of insertion and the reaction conditions.5,7,8,9b,28 As a result, it is difficult to predict when a Lewis acid will increase the rate of a CO2 insertion reaction. We evaluated the impact of a variety of different Lewis acids on the rate of CO2 insertion into (tBuPCP)Pd(CH3) using THF as the solvent (Table 2).29 Although there is an increase in rate when the reaction was performed in the presence of 20 equivalents of a variety of Lewis acids, a control experiment using 20 equivalents of (nBu4)PF6, which does not contain a Lewis acid, gives the same enhancement. This indicates that there is no increase in rate caused by Lewis acids and instead it is likely the case that increasing the ionic strength of the solution results in a slight enhancement in the rate of CO2 insertion. Consistent with this hypothesis, all of the different Lewis acids evaluated give approximately the same increase in the rate of CO2 insertion. At this stage, it is unclear why no Lewis acid effect is observed in the insertion of CO2 into (tBuPBP)Pd(CH3), given that Li+ has been demonstrated to increase the rate of CO2 insertion into (CO)5W(CH3).8 However, in the next section, DFT calculations are performed to understand the exact nature of the transition states involved in CO2 insertion into (tBuPBP)Pd(CH3).
Table 2 Effect of Lewis acids on rate of CO2 insertion into (tBuPBP)Pd(CH3) in THF at 40 °C and 1 atm of CO2
|
|
| Entry |
Additive (20 equiv.) |
k
obs
(s−1 × 10−3) |
Rate enhancement |
|
NTf = triflimide.
These values are the average of two trials and the errors are ±10%.
|
| 1 |
None |
1.6 |
1 |
| 2 |
(NBu4)PF6 |
2.3 |
1.4 |
| 3 |
LiNTf2a |
2.2 |
1.4 |
| 4 |
NaNTf2a |
2.1 |
1.3 |
| 5 |
KNTf2a |
2.3 |
1.4 |
| 6 |
LiPF6 |
2.4 |
1.5 |
| 7 |
LiBPh4·3DME |
2.5 |
1.6 |
| 8 |
LiOTf |
2.1 |
1.3 |
Computational studies of CO2 insertion into (RPBP)Pd(CH3) and related species.
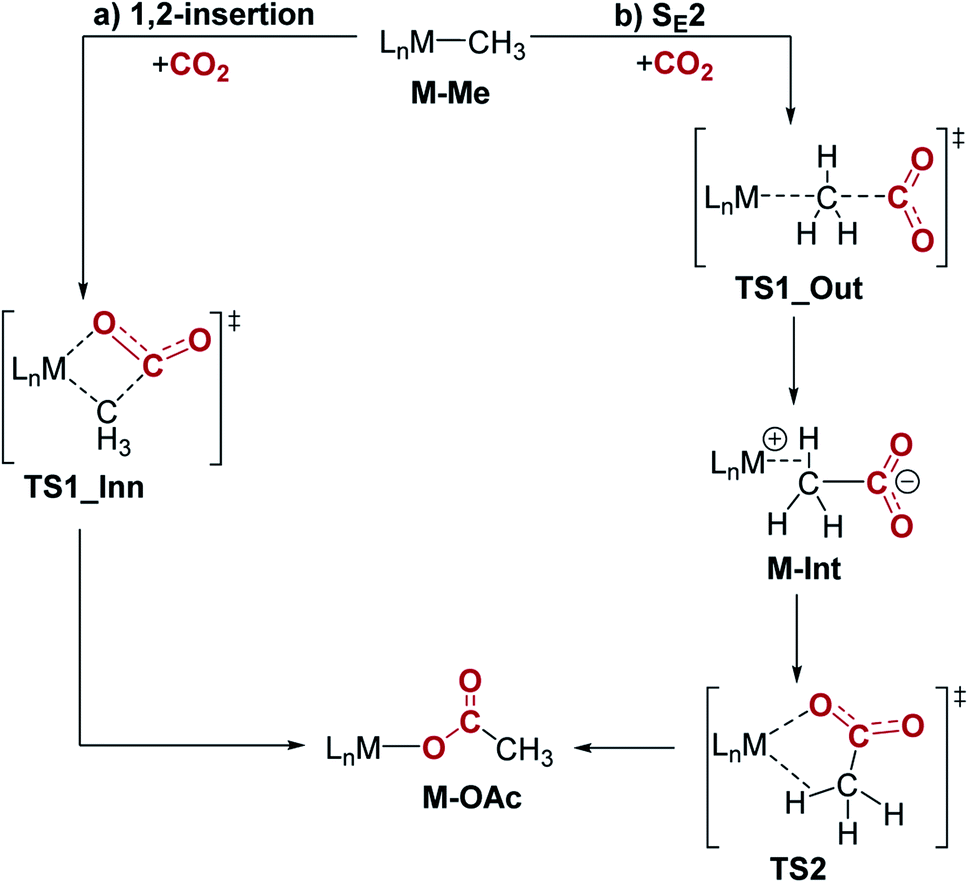
Previous studies investigating CO2 insertion into PCP-supported palladium and nickel methyl complexes have proposed two plausible pathways for insertion: (i) a 1,2-insertion (inner sphere) or (ii) an SE2 pathway (outer sphere).16,17c,26 In the 1,2-insertion pathway, the O-bound acetate product is formed in one step and CO2 interacts with the metal in the key transition state (Fig. 9a). This means that both the C–C and M–O bonds are formed at the same time. In contrast, the SE2 pathway involves two steps and starts with electrophilic attack of CO2 on the coordinated methyl, without any direct interaction between CO2 and the metal (Fig. 9b). This results in the formation of the new C–C bond. In the second step, the intermediate containing an acetate-metal ion-pair rearranges via TS2 to form the O-bound acetate adduct. Here, we used DFT methods to understand the pathway for CO2 insertion into (tBuPBP)M(CH3) (M = Ni or Pd). We also independently calculated CO2 insertion into (tBuPCP)Pd(CH3) in order to directly compare it with the tBuPBP systems.
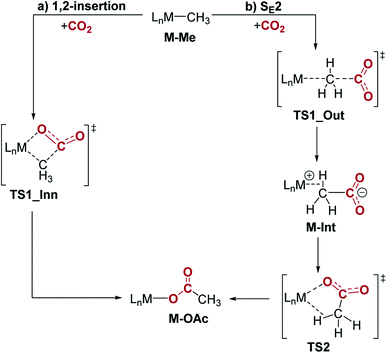 |
| | Fig. 9 Two plausible mechanisms for CO2 insertion into pincer-supported group 10 methyls: (a) 1,2-insertion and (b) SE2. | |
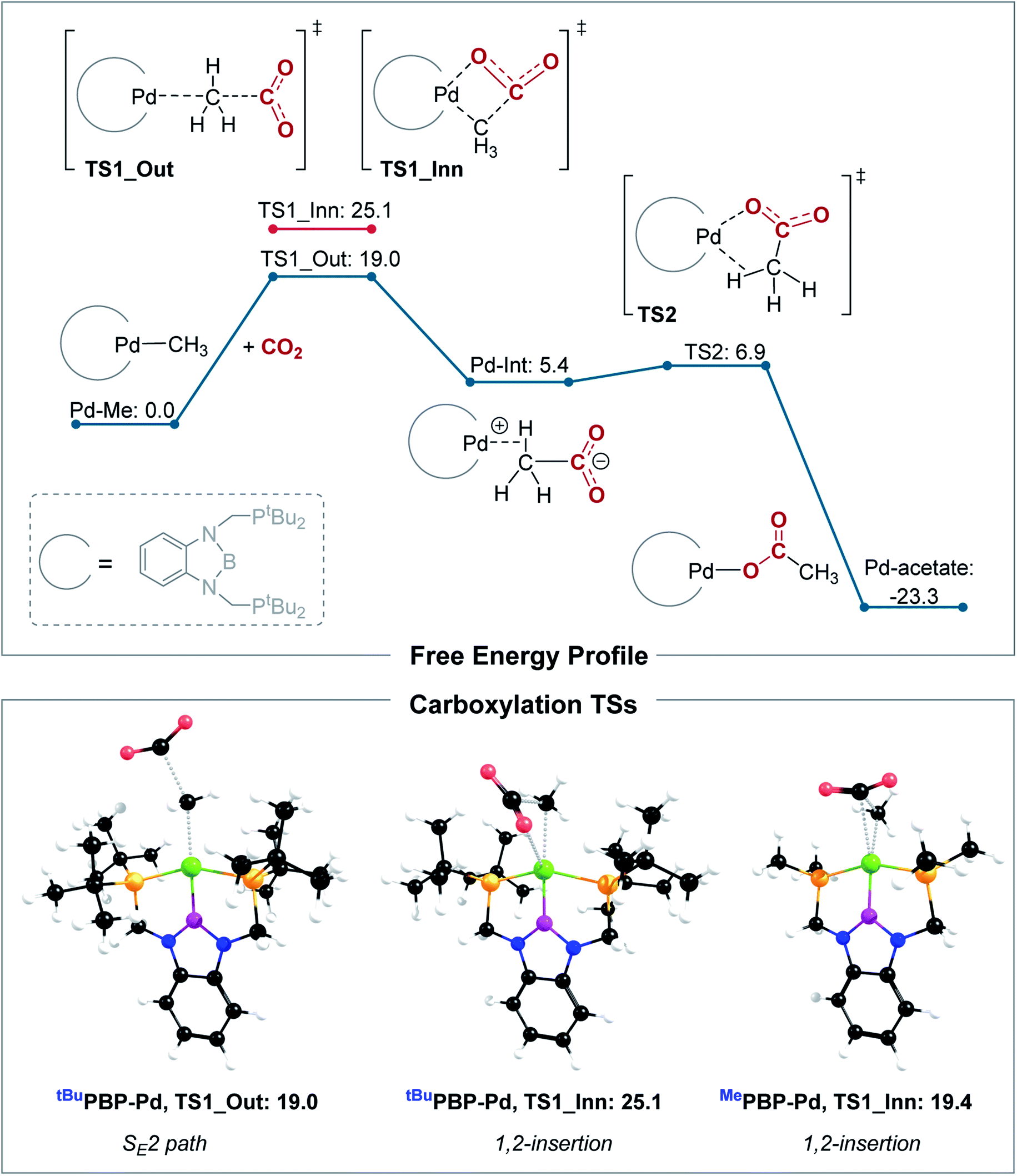
Our calculations indicate that CO2 insertion into (tBuPBP)Pd(CH3) proceeds via an SE2 pathway (Fig. 10, S43, S44† & Table 3). The rate-limiting step is the initial attack of CO2 on the palladium methyl group (TS1_Out), which has a computed barrier of 19.0 kcal mol−1 (298 K). This is within error of the activation barrier measured experimentally (vide supra). Although there is no interaction between CO2 and the metal in TS1_Out, we note that there are two C–H…OCO interactions between the tert-butyl substituents of the tBuPBP ligand and CO2 (2.43 and 2.74 Å, respectively), which provide charge stabilization. This may explain why the addition of Lewis acids to the system does not result in a rate enhancement (vide supra). The second step of the SE2 pathway has a very small barrier and involves rotation of the acetate-palladium ion pair to form the O-bound acetate product (Fig. 10). In contrast, the barrier for the alternative 1,2-insertion is significantly higher at 25.1 kcal mol−1 (TS1_Inn, Fig. 10 and S45†). The high barrier is likely caused by the bulky tert-butyl substituents of the tBuPBP ligand, which make it difficult for CO2 to approach palladium without steric repulsion. In order to investigate this hypothesis, we calculated CO2 insertion into (MePBP)Pd(CH3), where the tert-butyl substituents on the tBuPBP ligand have been replaced with methyl substituents. For CO2 insertion into (MePBP)Pd(CH3) the 1,2-insertion pathway is preferred by 3.1 kcal mol−1 over the SE2 mechanism (Fig. 10, S46 and S47†). Thus, the size of the pincer ligand has a decisive role in promoting either a 1,2-insertion (small phosphine substituents) or an SE2 pathway (large phosphine substituents). Similar ligand effects were previously observed for NHC–Cu complexes.30
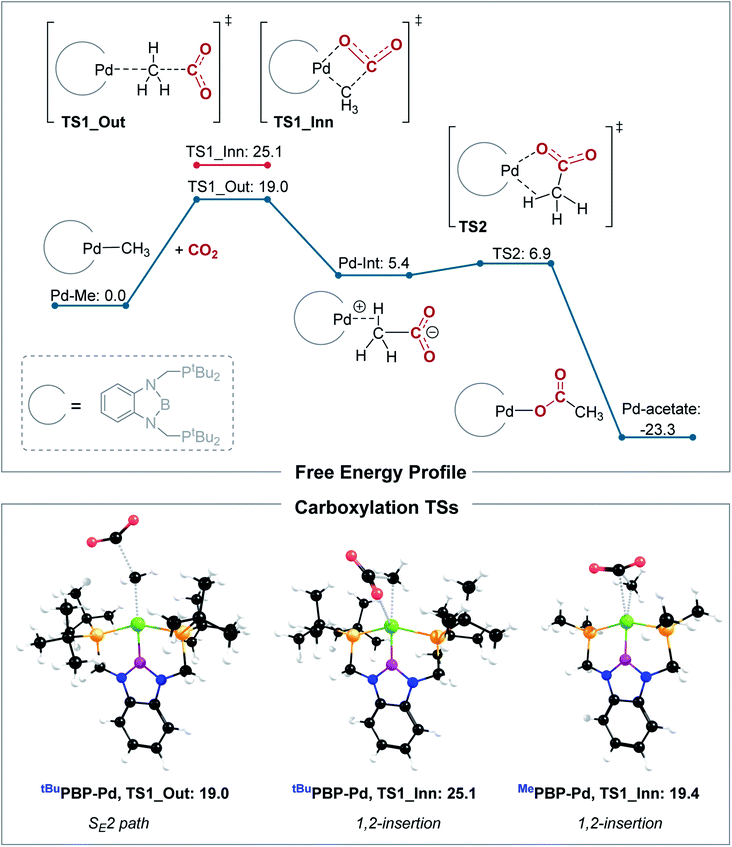 |
| | Fig. 10 Top: computed free energy profile (B3LYP-D3/def2-TZVPPD[CPCM]//B3LYP-D3/def2-SVP[CPCM], 298 K) for CO2 insertion into (tBuPBP)Pd(CH3), bottom: optimized transition state structures for CO2 insertion into (RPBP)Pd(CH3), with R = tBu or Me. All energies are in kcal mol−1. | |
Table 3 Calculated free energy barriers for inner and outer sphere C–CO2 bond formation with different pincer-supported palladium and nickel complexes (kcal mol−1, B3LYP-D3/def2-TZVPPD[CPCM]//B3LYP-D3/def2-SVP[CPCM], 298 K)a
| Complex |
TS1_Out (kcal mol−1) |
TS1_Inn (kcal mol−1) |
|
For comparison, results for the ωB97XD functional are given in parenthesis, showing the same mechanistic trends.
|
| (tBuPBP)Pd(CH3) |
19.0 (23.9) |
25.1 (30.6) |
| (MePBP)Pd(CH3) |
22.5 (23.4) |
19.4 (21.0) |
| (tBuPBP)Ni(CH3) |
24.5 (27.8) |
31.9 (33.7) |
| (tBuPCP)Pd(CH3) |
24.2 (29.2) |
36.7 (41.1) |
| (MePCP)Pd(CH3) |
26.8 (30.0) |
25.0 (27.8) |
The tBuPCP-supported complex, (tBuPCP)Pd(CH3), also shows a preference for the SE2 pathway, with a computed barrier of 24.2 kcal mol−1 (TS_Out, Fig. S48 and S49†). Consistent with previous results,26 the 1,2-insertion mechanism is unlikely for (tBuPCP)Pd(CH3) due to the high barrier of 36.7 kcal mol−1 (TS_Inn, Fig. S50†).31 Thus, both (tBuPBP)Pd(CH3) and (tBuPCP)Pd(CH3) show a clear preference for an SE2 mechanism. In the rate limiting transition state (TS_Out), (tBuPBP)Pd(CH3) displays a longer C–CO2 bond (by 0.09 Å) and a smaller CO2 angle (by 1.7°) compared to (tBuPCP)Pd(CH3), which indicates an earlier TS. The lower barrier for (tBuPBP)Pd(CH3) (by 5.2 kcal mol−1) is in agreement with our kinetic studies, showing that CO2 insertion is much faster for (tBuPBP)Pd(CH3). The decrease in the barrier for CO2 insertion is also matched by the greater thermodynamic driving force for CO2 insertion into (tBuPBP)Pd(CH3) compared with (tBuPCP)Pd(CH3) (−23.3 kcal mol−1versus −15.8 kcal mol−1). These results highlight how a donor atom with strong trans-influence opposite to the methyl can lower the barrier for insertion and suggest that the enhancement is due to destabilization of the ground state as opposed to a kinetic effect associated with stabilization of the transition state.
The slower qualitative rate of CO2 insertion into (tBuPBP)Ni(CH3) compared to (tBuPBP)Pd(CH3) (vide supra) is surprising given that CO2 insertion into nickel hydrides is typically faster than palladium hydrides.19 Calculations on CO2 insertion into (tBuPBP)Ni(CH3) confirm our experimental results as the calculated barrier is 24.5 kcal mol−1 at 298 K, more than 5 kcal mol−1 higher than for (tBuPBP)Pd(CH3) (B3LYP-D3, Table 3). The lowest energy pathway for CO2 insertion into (tBuPBP)Ni(CH3) also follows an SE2 mechanism (Fig. S51 and 52†). Geometrical analysis of the rate-determining transition states for CO2 insertion into (tBuPBP)Ni(CH3) and (tBuPBP)Pd(CH3) shows that the Ni–P bonds are shorter by around 0.15 Å compared with the corresponding Pd–P bonds due to the smaller atomic radius of nickel. We propose that the increased steric congestion around nickel results in the higher barrier for CO2 insertion relative to the palladium system.32 This is in agreement with previous results showing that CO2 insertion reactions into nickel hydrides are highly sensitive to steric factors.5a It is likely, however, that there is no inherent electronic reason for CO2 insertion into nickel methyl complexes to be slower than palladium methyl complexes, and if a nickel complex could be prepared where steric factors were not relevant,33 we expect to see comparable rates of insertion.
Conclusions
In this work, we have demonstrated that nickel and palladium methyl complexes supported by RPBP ligands undergo CO2 insertion under significantly milder conditions than any previously reported pincer-ligated group 10 alkyl species. This is because of the strong trans-influence of the central boryl donor in RPBP type ligands, which destabilizes the M–CH3 bond. Qualitatively, we show that CO2 insertion into (RPBP)Pd(CH3) is faster than CO2 insertion into (RPBP)Ni(CH3), which is likely due to steric factors. In contrast, we propose that steric factors are less significant in CO2 insertion into group 10 hydrides, and as a result nickel hydrides typically undergo faster insertion than palladium hydrides.19 Given the rapid rate of insertion into RPBP-ligated complexes, we were able to complete a rare kinetic study on CO2 insertion into a metal alkyl complex. Our results demonstrate that the solvent parameter that is best for predicting the rate of CO2 insertion is the Dimroth–Reichardt ET(30) parameter. This is analogous to what we have observed for transition metal hydrides,5b but the dependence of the insertion rate on the solvent is significantly less pronounced in the case of the metal alkyl complex. We hypothesize that this is because there is less charge build-up in the transition state for carbon dioxide insertion into a metal alkyl complex compared to related metal hydrides. Consistent with there being only a small build-up of charge in the transition state, we did not observe any enhancement from Lewis acids in CO2 insertion reactions into (tBuPBP)Pd(CH3). DFT calculations indicate that the proposed mechanism for insertion into (tBuPBP)M(CH3) (M = Ni or Pd) follows an SE2 pathway, but this is dependent on steric factors and compounds with less bulky substituents on the phosphine donors are more likely to undergo insertion via a 1,2-insertion mechanism. Overall, our results indicate that the fastest systems for CO2 insertion into a group 10 methyl will have a strong trans-influence donor opposite the methyl, be able to undergo insertion in solvents with high ET(30) values, and not have sterically bulky ancillary ligands that prevent the approach of CO2. Future research in our group will aim to extend these principles to other metal-element σ-bonds and apply them to improve catalytic reactions.
Data availability
Additional information about selected experiments, NMR spectra, and other details are availabe via the internet.
Author contributions
Project conception: AJD, MRE, DJC, NH; experimental work: AJD, MRE, DJC; X-ray crystallography: BQM; computational analysis: LjP, KHH. All authors contributed to writing the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
NH acknowledges support from National Science Foundation through Grant CHE-1953708. We thank Dr Hee Won Suh for preliminary work on this project and Dr Fabian Menges for assistance obtaining high resolution mass spectrometry data with equipment funded by NSF MRI Grant CHE-1828190. KHH and LjP thank the Research Council of Norway (Grant No. 300769), the Tromsø Research Foundation (Grant No. TFS2016KHH), NordForsk (Grant No. 85378) and Sigma2 - the National Infrastructure for High Performance Computing and Data Storage in Norway (No. nn9330k).
References
-
(a) E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, Carbon Dioxide Recycling: Emerging Large-Scale Technologies with Industrial Potential, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed;
(b) J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction Catalysts, Chem. Soc. Rev., 2012, 41, 2036–2051 RSC;
(c) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed;
(d) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed;
(e)
M. Limbach, 'Chapter Four - Acrylates from Alkenes and CO2, the Stuff That Dreams Are Made of'. in Advances in Organometallic Chemistry, ed. Pérez P. J., Academic Press, 2015, pp. 175–202 Search PubMed;
(f) W. H. Bernskoetter and N. Hazari, Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts, Acc. Chem. Res., 2017, 50, 1049–1058 CrossRef CAS PubMed;
(g) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed;
(h) M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, Opportunities and Challenges for Catalysis in Carbon Dioxide Utilization, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS;
(i) C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, The Technological and Economic Prospects for CO2 Utilization and Removal, Nature, 2019, 575, 87–97 CrossRef CAS PubMed;
(j) Z. Zhang, S.-Y. Pan, H. Li, J. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Recent Advances in Carbon Dioxide Utilization, Renewable Sustainable Energy Rev., 2020, 125, 109799 CrossRef CAS;
(k) W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Industrial Carbon Dioxide Capture and Utilization: State of the Art and Future Challenges, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
-
National Academies of Sciences, Engineering, and Medicine, Gaseous Carbon Waste Streams Utilization: Status and Research Needs, The National Academies Press, Washington DC, 2019 Search PubMed.
-
(a) K. Huang, C.-L. Sun and Z.-J. Shi, Transition-Metal Catalyzed C-C Bond Formation Through the Fixation of Carbon Dioxide, Chem. Soc. Rev., 2011, 40, 2435–2452 RSC;
(b) I. Omae, Recent Developments in Carbon Dioxide Utilization for the Production of Organic Chemicals, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS;
(c) Y. Tsuji and T. Fujihara, Carbon Dioxide as a Carbon Source in Organic Transformation: Carbon-Carbon Bond Forming Reactions by Transition-Metal Catalysts, Chem. Commun., 2012, 48, 9956–9964 RSC;
(d) C. S. Yeung and V. M. Dong, Making C-C Bonds from Carbon Dioxide via Transition-Metal Catalysis, Top. Catal., 2014, 57, 1342–1350 CrossRef CAS;
(e) Q. Liu, L. Wu, R. Jackstell and M. Beller, Using Carbon Dioxide as a Building Block in Organic Synthesis, Nat. Commun., 2015, 6, 5933 CrossRef PubMed;
(f) D. Yu, S. P. Teong and Y. Zhang, Transition Metal Complex Catalyzed Carboxylation Reactions with CO2, Coord. Chem. Rev., 2015, 293–294, 279–291 CrossRef CAS;
(g) A.-H. Liu, B. Yu and L.-N. He, Catalytic Conversion of Carbon Dioxide to Carboxylic Acid Derivatives, Greenhouse Gases: Sci. Technol., 2015, 5, 17–33 CrossRef CAS;
(h) M. Börjesson, T. Moragas, D. Gallego and R. Martin, Metal-Catalyzed Carboxylation of Organic (Pseudo)halides with CO2, ACS Catal., 2016, 6, 6739–6749 CrossRef PubMed;
(i) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed;
(j) M. Obst, L. Pavlovic and K. H. Hopmann, Carbon-Carbon Bonds with CO2: Insights from Computational Studies, J. Organomet. Chem., 2018, 864, 115–127 CrossRef CAS;
(k) Y. Yang and J.-W. Lee, Toward Ideal Carbon Dioxide Functionalization, Chem. Sci., 2019, 10, 3905–3926 RSC;
(l) J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, C−H Bond Carboxylation with Carbon Dioxide, ChemSusChem, 2019, 12, 6–39 CrossRef CAS PubMed;
(m) C. S. Yeung, Photoredox Catalysis as a Strategy for CO2 Incorporation: Direct Access to Carboxylic Acids from a Renewable Feedstock, Angew. Chem., Int. Ed., 2019, 58, 5492–5502 CrossRef CAS PubMed.
- N. Hazari and J. E. Heimann, Carbon Dioxide Insertion into Group 9 and 10 Metal-Element σ-Bonds, Inorg. Chem., 2017, 56, 13655–13678 CrossRef CAS PubMed.
-
(a) J. E. Heimann, W. H. Bernskoetter, N. Hazari and J. M. Mayer, Acceleration of CO2 Insertion into Metal Hydrides: Ligand, Lewis Acid, and Solvent Effects on Reaction Kinetics, Chem. Sci., 2018, 8, 6629–6638 RSC;
(b) J. E. Heimann, W. H. Bernskoetter and N. Hazari, Understanding the Individual and Combined Effects of Solvent and Lewis Acid on CO2 Insertion into a Metal Hydride, J. Am. Chem. Soc., 2019, 141, 10520–10529 CrossRef CAS.
- For an example of CO2 insertion into a Sc alkyl bond see: F. A. LeBlanc, A. Berkefeld, W. E. Piers and M. Parvez, Reactivity of Scandium β-Diketiminate Alkyl Complexes with Carbon Dioxide, Organometallics, 2012, 31, 810–818 CrossRef CAS.
- For an example of CO2 insertion into a Zr alkyl bond see: K.-C. Lau, B. J. Petro, S. Bontemps and R. F. Jordan, Comparative Reactivity of Zr–and Pd–Alkyl Complexes with Carbon Dioxide, Organometallics, 2013, 32, 6895–6898 CrossRef CAS.
- For an example of CO2 insertion into a Cr alkyl bond see: D. J. Darensbourg and A. Rokicki, Reduction of Carbon Dioxide and Carbonyl Sulfide by Anionic Group VIB Metal Hydrides and Alkyls. Carbon-Hydrogen and Carbon-Carbon Bond Formation Processes and the Structure of [PNP][Cr(CO)5SC(O)H], J. Am. Chem. Soc., 1982, 104, 349–350 CrossRef CAS.
- For examples of CO2 insertion into W-alkyl bonds see:
(a) D. J. Darensbourg, R. K. Hanckel, C. G. Bauch, M. Pala, D. Simmons and J. N. White, A Kinetic Investigation of Carbon Dioxide Insertion Processes Involving Anionic Tungsten-Alkyl and -Aryl Derivatives: Effects of Carbon Dioxide Pressure, Counterions, and Ancillary Ligands. Comparisons with Migratory Carbon Monoxide Insertion Processes, J. Am. Chem. Soc., 1985, 107, 7463–7473 CrossRef CAS;
(b) D. J. Darensbourg and M. Pala, Cation-Anion Interaction in the [Na-kryptofix-221][W(CO)5O2CH] Derivative and its Relevance in Carbon Dioxide Reduction Processes, J. Am. Chem. Soc., 1985, 107, 5687–5693 CrossRef CAS;
(c) D. J. Darensbourg and G. Grotsch, Stereochemical Studies of the Carbon Dioxide Insertion Reactions into the Tungsten-Alkyl Bond, J. Am. Chem. Soc., 1985, 107, 7473–7476 CrossRef CAS;
(d) D. J. Darensbourg and R. Kudaroski, Metal-Induced Transformations of Carbon Dioxide. Carbon-Carbon Bond-Forming Processes Involving Anionic Group VIB Metal Derivatives, and the X-ray structure of [PNP][cis-MeW(CO)4PMe3], J. Am. Chem. Soc., 1984, 106, 3672–3673 CrossRef CAS.
- For examples of CO2 insertion into Fe-alkyl bonds see:
(a) S. Ittel, C. Tolman, A. English and J. Jesson, The Chemistry of 2-Naphthyl bis[bis(dimethylphosphino)ethane] Hydride Complexes of Iron, Ruthenium, and Osmium. 2. Cleavage of sp and sp3 Carbon-Hydrogen, Carbon-Oxygen, and Carbon-Halogen bonds. Coupling of Carbon Dioxide and Acetonitrile, J. Am. Chem. Soc., 1978, 100, 7577–7585 CrossRef CAS;
(b) I. M. Arafa, K. Shin and H. M. Goff, Carbon Monoxide and Carbon Dioxide Carbon-Metal Bond Insertion Chemistry of Alkyliron(III) Porphyrin Complexes, J. Am. Chem. Soc., 1988, 110, 5228–5229 CrossRef CAS;
(c) O. R. Allen, S. J. Dalgarno, L. D. Field, P. Jensen, A. J. Turnbull and A. C. Willis, Addition of CO2 to Alkyl Iron complexes, Fe(PP)2Me2, Organometallics, 2008, 27, 2092–2098 CrossRef CAS;
(d) K.-C. Lau and R. F. Jordan, Reactivity of (Pyridine-Diimine) Fe Alkyl Complexes with Carbon Dioxide, Organometallics, 2016, 35, 3658–3666 CrossRef CAS.
- For examples of CO2 insertion into Ru-alkyl bonds see reference 10a and:
(a) O. R. Allen, S. J. Dalgarno, L. D. Field, P. Jensen and A. C. Willis, Insertion of CO2 into the Ru−C Bonds of cis-and trans-Ru(dmpe)2Me2 (dmpe = Me2PCH2CH2PMe2), Organometallics, 2009, 28, 2385–2390 CrossRef CAS;
(b) J. F. Hartwig, R. G. Bergman and R. A. Andersen, Insertion Reactions of Carbon Monoxide and Carbon Dioxide with Ruthenium Benzyl, Arylamido, and Aryloxide complexes: A Comparison of the Reactivity of Ruthenium-Carbon, Ruthenium-Nitrogen, and Ruthenium-Oxygen Bonds, J. Am. Chem. Soc., 1991, 113, 6499–6508 CrossRef CAS.
- For examples of CO2 insertion into Rh-alkyl bonds see:
(a) D. J. Darensbourg, G. Groetsch, P. Wiegreffe and A. L. Rheingold, Insertion Reactions of Carbon Dioxide with Square-Planar Rhodium Alkyl and Aryl Complexes, Inorg. Chem., 1987, 26, 3827–3830 CrossRef CAS;
(b) T. G. Ostapowicz, M. Hölscher and W. Leitner, CO2 Insertion into Metal–Carbon Bonds: A Computational Study of RhI Pincer Complexes, Chem. – Eur. J., 2011, 17, 10329–10338 CrossRef CAS;
(c) T. Suga, T. Saitou, J. Takaya and N. Iwasawa, Mechanistic Study of the Rhodium-Catalyzed Carboxylation of Simple Aromatic Compounds with Carbon Dioxide, Chem. Sci., 2017, 8, 1454–1462 RSC;
(d) L. Pavlovic, J. Vaitla, A. Bayer and K. H. Hopmann, Rhodium-Catalyzed Hydrocarboxylation: Mechanistic Analysis Reveals Unusual Transition State for Carbon–Carbon Bond Formation, Organometallics, 2018, 37, 941–948 CrossRef CAS.
- For examples of CO2 insertion into Cu-alkyl bonds see:
(a) T. Ikariya and A. Yamamoto, Preparation and Properties of Ligand-Free Methylcopper and of Copper Alkyls Coordinated with 2,2′-Bipyridyl and Tricyclohexylphosphine, J. Organomet. Chem., 1974, 72, 145–151 CrossRef;
(b) N. P. Mankad, T. G. Gray, D. S. Laitar and J. P. Sadighi, Synthesis, Structure, and CO2 Reactivity of a Two-Coordinate (Carbene)copper(I) Methyl Complex, Organometallics, 2004, 23, 1191–1193 CrossRef CAS;
(c) S. Sakaki and K. Ohkubo, Ab initio MO Study of Carbon Dioxide Insertion into a Methyl-Copper(I) Bond. Critical Difference from CO2 Insertion into a Hydrogen-Copper(I) Bond, Organometallics, 1989, 8, 2970–2973 CrossRef CAS.
- The following examples describe CO2 insertion into Ni(I) alkyl species. In both cases, the starting materials and products are highly unstable, which precludes detailed kinetic studies.
(a) J. B. Diccianni, C. T. Hu and T. Diao, Insertion of CO2 Mediated by a (Xantphos)NiI–Alkyl Species, Angew. Chem., Int. Ed., 2019, 58, 13865–13868 CrossRef CAS PubMed;
(b) R. J. Somerville, C. Odena, M. F. Obst, N. Hazari, K. H. Hopmann and R. Martin, Ni(I)–Alkyl Complexes Bearing Phenanthroline Ligands: Experimental Evidence for CO2 Insertion at Ni(I) Centers, J. Am. Chem. Soc., 2020, 142, 10936–10941 CrossRef CAS PubMed.
- R. Johansson, M. Jarenmark and O. F. Wendt, Insertion of Carbon Dioxide into (PCP)PdII-Me Bonds, Organometallics, 2005, 24, 4500–4502 CrossRef CAS.
- T. J. Schmeier, N. Hazari, C. D. Incarvito and J. R. Raskatov, Exploring the Reactions of CO2 with PCP Supported Nickel Complexes, Chem. Commun., 2011, 47, 1824–1826 RSC.
-
(a) K. J. Jonasson and O. F. Wendt, Synthesis and Characterization of a Family of POCOP Pincer Complexes with Nickel: Reactivity Towards CO2 and Phenylacetylene, Chem. – Eur. J., 2014, 20, 11894–11902 CrossRef CAS PubMed;
(b) A. H. Mousa, J. Bendix and O. F. Wendt, Synthesis, Characterization, and Reactivity of PCN Pincer Nickel Complexes, Organometallics, 2018, 37, 2581–2593 CrossRef CAS;
(c) A. H. Mousa, A. V. Polukeev, J. Hansson and O. F. Wendt, Carboxylation of the Ni–Me Bond in an Electron-Rich Unsymmetrical PCN Pincer Nickel Complex, Organometallics, 2020, 39, 1553–1560 CrossRef CAS.
-
(a) C. Reichardt, Empirical Parameters of the Polarity of Solvents, Angew. Chem., Int. Ed., 1965, 4, 29–40 CrossRef;
(b) C. Reichardt, Solvatochromic Dyes as Solvent Polarity Indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Experimental and Computational Studies of the Reaction of Carbon Dioxide with Pincer-Supported Nickel and Palladium Hydrides, Organometallics, 2012, 31, 8225–8236 CrossRef CAS.
- S. J. Mitton, R. McDonald and L. Turculet, Nickel and Palladium Silyl Pincer Complexes: Unusual Structural Rearrangements that Involve Reversible Si-C(sp3) and Si-C(sp2) Bond Activation, Angew. Chem., Int. Ed., 2009, 48, 8568–8571 CrossRef CAS PubMed.
-
(a) Y. Segawa, M. Yamashita and K. Nozaki, Syntheses of PBP Pincer Iridium Complexes: A Supporting Boryl Ligand, J. Am. Chem. Soc., 2009, 131, 9201–9203 CrossRef CAS PubMed;
(b) Y. Segawa, M. Yamashita and K. Nozaki, Diphenylphosphino- or Dicyclohexylphosphino-Tethered Boryl Pincer Ligands: Syntheses of PBP Iridium(III) Complexes and Their Conversion to Iridium−Ethylene Complexes, Organometallics, 2009, 28, 6234–6242 CrossRef CAS;
(c) A. F. Hill, S. B. Lee, J. Park, R. Shang and A. C. Willis, Analogies between Metallaboratranes, Triboronates, and Boron Pincer Ligand Complexes, Organometallics, 2010, 29, 5661–5669 CrossRef CAS;
(d) M. Hasegawa, Y. Segawa, M. Yamashita and K. Nozaki, Isolation of a PBP-Pincer Rhodium Complex Stabilized by an Intermolecular C-H σCoordinationas the Fourth Ligand, Angew. Chem., Int. Ed., 2012, 51, 6956–6960 CrossRef CAS;
(e) H. Ogawa and M. Yamashita, Platinum Complexes Bearing a Boron-Based PBP Pincer Ligand: Synthesis, Structure, and Application as a Catalyst for Hydrosilylation of 1-Decene, Dalton Trans., 2013, 42, 625–629 RSC;
(f) T. Miyada and M. Yamashita, Oxygenation of a Ruthenium Complex Bearing a PBP-Pincer Ligand Inducing the Formation of a Boronato Ligand with a Weak Ru–O Bond, Organometallics, 2013, 32, 5281–5284 CrossRef CAS;
(g) T.-P. Lin and J. C. Peters, Boryl–Metal Bonds Facilitate Cobalt/Nickel-Catalyzed Olefin Hydrogenation, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed;
(h) N. Curado, C. Maya, J. López-Serrano and A. Rodríguez, Boryl-Assisted Hydrogenolysis of a Nickel–Methyl Bond, Chem. Commun., 2014, 50, 15718–15721 RSC;
(i) P. Ríos, N. Curado, J. López-Serrano and A. Rodríguez, Selective Reduction of Carbon Dioxide to Bis(silyl)acetal Catalyzed by a PBP-Supported Nickel Complex, Chem. Commun., 2016, 52, 2114–2117 RSC;
(j) P. Rios, A. Rodriguez and J. Lopez-Serrano, Mechanistic Studies on the Selective Reduction of CO2 to the Aldehyde Level by a Bis(phosphino)boryl (PBP)-Supported Nickel Complex, ACS Catal., 2016, 6, 5715–5723 CrossRef CAS;
(k) Y. Ding, Q.-Q. Ma, J. Kang, J. Zhang, S. Li and X. Chen, Palladium(II) Complexes Supported by PBP and POCOP Pincer Ligands: A Comparison of their Structure, Properties and Catalytic Activity, Dalton Trans., 2019, 48, 17633–17643 RSC.
- D. Adhikari, J. C. Huffman and D. J. Mindiola, Structural Elucidation of a Nickel Boryl Complex. A Recyclable Borylation Ni(II) Reagent of Bromobenzene, Chem. Commun., 2007, 4489–4491 RSC.
-
(a) H. Clavier and S. P. Nolan, Percent Buried Volume for Phosphine and N-Heterocyclic Carbene Ligands: Steric Properties in Organometallic Chemistry, Chem. Commun., 2010, 46, 841–861 RSC;
(b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- B. F. M. Kimmich, W. J. Marshall, P. J. Fagan, E. Hauptman and R. M. Bullock, Palladium Complexes with PCP Ligands: Preparation and Structural Studies of Two Forms of [(PCP)Pd(OH2)]+BF4−, Inorg. Chim. Acta, 2002, 330, 52–58 CrossRef CAS.
-
(a) R. Beck, M. Shoshani, J. Krasinkiewicz, J. A. Hatnean and S. A. Johnson, Synthesis and Chemistry of Bis(triisopropylphosphine) Nickel(I) and Nickel(0) Precursors, Dalton Trans., 2013, 42, 1461–1475 RSC;
(b) W. H. Harman, T.-P. Lin and J. C. A. Peters, d10 Ni–(H2) Adduct as an Intermediate in H-H Oxidative Addition across a Ni-B Bond, Angew. Chem., Int. Ed., 2014, 53, 1081–1086 CrossRef CAS PubMed;
(c) Y.-E. Kim, J. Kim and Y. Lee, Formation of a Nickel Carbon Dioxide Adduct and its Transformation Mediated by a Lewis Acid, Chem. Commun., 2014, 50, 11458–11461 RSC;
(d) Y.-E. Kim, S. Oh, S. Kim, O. Kim, J. Kim, S. W. Han and Y. Lee, Phosphinite-Ni(0) Mediated Formation of a Phosphide-Ni(II)-OCOOMe Species via Uncommon Metal–Ligand Cooperation, J. Am. Chem. Soc., 2015, 137, 4280–4283 CrossRef CAS PubMed;
(e) D. J. Charboneau, D. Balcells, N. Hazari, H. M. C. Lant, J. M. Mayer, P. R. Melvin, B. Q. Mercado, W. D. Morris, M. Repisky and H.-W. Suh, Dinitrogen-Facilitated Reversible Formation of a Si–H Bond in a Pincer-Supported Ni Complex, Organometallics, 2016, 35, 3154–3162 CrossRef CAS.
- M. T. Johnson, R. Johansson, M. V. Kondrashov, G. Steyl, M. S. G. Ahlquist, A. Roodt and O. F. Wendt, Mechanisms of the CO2 Insertion into (PCP) Palladium Allyl and Methyl σ-Bonds. A Kinetic and Computational Study, Organometallics, 2010, 29, 3521–3529 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, The Acceptor Number — A Quantitative Empirical Parameter for the Electrophilic Properties of Solvents, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- D. J. Charboneau, G. W. Brudvig, N. Hazari, H. M. Lant and A. K. Saydjari, Development of an Improved System for the Carboxylation of Aryl Halides Through Mechanistic Studies, ACS Catal., 2019, 9, 3228–3241 CrossRef CAS PubMed.
- THF was selected as the solvent due to the improved solubility of many Lewis acids in THF compared to other ethereal solvents.
- D. García-López, L. Pavlovic and K. H. Hopmann, To Bind or Not to Bind: Mechanistic Insights into C–CO2 Bond Formation with Late Transition Metals, Organometallics, 2020, 39, 1339–1347 CrossRef.
- We also calculated CO2 insertion into (MePCP)Pd(CH3), where the tert-butyl substituents on the tBuPCP ligand have been replaced with methyl substituents. The calculations indicate that for (MePCP)Pd(CH3), the 1,2-insertion pathway is preferred over the SE2 mechanism by 1.8 kcal mol−1 (Table 3, Fig. S53 and S54†). This is consistent with the hypothesis that the steric properties of the pincer ligand play a large role in determining the pathway for CO2 insertion.
- We note that other computational protocols (ωB97XD, PBE0-D3BJ) provide the same reactivity trends as B3LYP-D3 (Table S3†).
- Our results do not explain Wendt's observation (see ref. 17b and 17c) that CO2 insertion into (tBuPCiPrN)Ni(CH3) (tBuPCiPrN = 1-(3-((ditert-butylphosphino)methyl)phenyl)-N,N-diisopropyl-methanamine)) is faster than insertion into (tBuPCMeN)Ni(CH3) (tBuPCMeN = 1-(3-((ditert-butylphosphino)methyl)phenyl)-N,N-methyl-methanamine)). In this case, ligand hemilability may play a role and it is possible that the more sterically bulky tBuPCiPrN is more likely to decoordinate.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
David J.
Charboneau
a,
Nilay
Hazari
b,
David J.
Charboneau
a,
Nilay
Hazari