
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent directed chemically divergent synthesis of β-lactams and α-amino acid derivatives with chiral isothiourea†
Dong-Sheng
Ji
a,
Hui
Liang
a,
Kai-Xuan
Yang
a,
Zhi-Tao
Feng
c,
Yong-Chun
Luo
 *a,
Guo-Qiang
Xu
a,
Yucheng
Gu
d and
Peng-Fei
Xu
*ab
*a,
Guo-Qiang
Xu
a,
Yucheng
Gu
d and
Peng-Fei
Xu
*ab
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: xupf@lzu.edu.cn; luoych@lzu.edu.cn
bState Key Laboratory of Veterinary Etiological Biology, College of Veterinary Medicine, Lanzhou University, Lanzhou, 730000, P. R. China
cDepartment of Chemistry, University of California Davis, One Shields Avenue, Davis, California 95616, USA
dSyngenta Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 18th January 2022
Abstract
A protocol for the chemically divergent synthesis of β-lactams and α-amino acid derivatives with isothiourea (ITU) catalysis by switching solvents was developed. The stereospecific Mannich reaction occurring between imine and C(1)-ammonium enolate generated zwitterionic intermediates, which underwent intramolecular lactamization and afforded β-lactam derivatives when DCM and CH3CN were used as solvents. However, when EtOH was used as the solvent, the intermediates underwent an intermolecular esterification reaction, and α-amino acid derivatives were produced. Detailed mechanistic experiments were conducted to prove that these two kinds of products came from the same intermediates. Furthermore, chemically diversified transformations of β-lactam and α-amino acid derivatives were achieved.
Introduction
Synthetic chemists have especially devoted themselves to developing concise, green and efficient methodologies for accessing target molecules.1 Divergent synthesis as an intriguing and efficient strategy has made synthesis of complex molecules possible, which has attracted much attention from chemists to construct complex natural products starting from simple materials by regulating and adjusting reaction conditions. Therefore, different target products are obtained by different reaction pathways (Scheme 1).2 This strategy was inspired by natural biosynthesis, in which many natural products are derived from the same intermediates by different biosynthesis routes. It is widely used not only by synthetic chemists in the field of total synthesis,3 but also in the field of developing new synthetic methodologies.4 | ||
| Scheme 1 The strategy of divergent synthesis. | ||
β-lactams and α-amino acid derivatives play important roles in life events.5 β-lactams refer to a large class of antibiotics with a β-lactam ring in their chemical structure, including the most commonly used penicillins and cephalosporins in clinical practice, as well as newly developed cephamycins and thiobacillus antibiotics, monocyclic β-lactams and other atypical β-lactam antibiotics, such as aztreonam and zetia.6 Amino acids are the basic units of proteins, which are mainly used as therapeutic drugs and as basic units of synthetic peptide drugs (Fig. 1).7 Therefore, it is essential to develop concise and efficient synthetic methods for the synthesis of chiral β-lactams and α-amino acid derivatives.8 Over the past few decades, the synthesis of β-lactams mediated by chiral Lewis base nucleophilic catalysts has progressed very well. Lectka developed cinchona alkaloid participating reactions for accessing chiral β-lactams.9 Fu reported planar-chiral DMAP involving protocols for obtaining chiral β-lactams.10 Birman and Smith have carried out pioneering studies in this field via C1 ammonium generated from chiral isothiourea11 (Scheme 2A). Although there are some reports on the construction of chiral β-lactams or amino acids, all these reactions were developed for the synthesis of only one kind of them.8c,11c In terms of chemistry, these two kinds of compounds are structurally closely linked to each other, and we envisioned that they could be synthesized from the same intermediates as in biosynthesis. Therefore, we attempted to synthesize these two important kinds of molecules exploiting the strategy of divergent synthesis, which will contribute to enriching potential candidates for screening and discovering drugs.
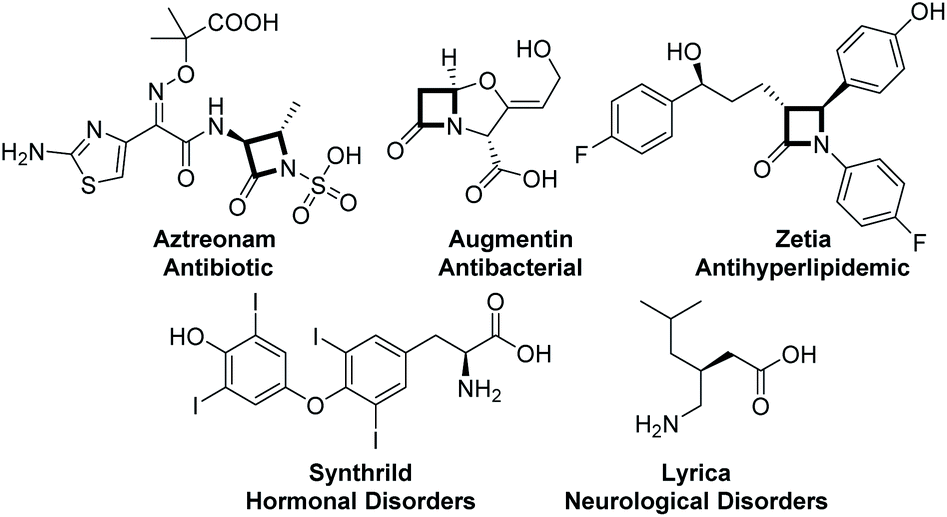 | ||
| Fig. 1 Pharmaceuticals containing β-lactam and amino acid structures. | ||
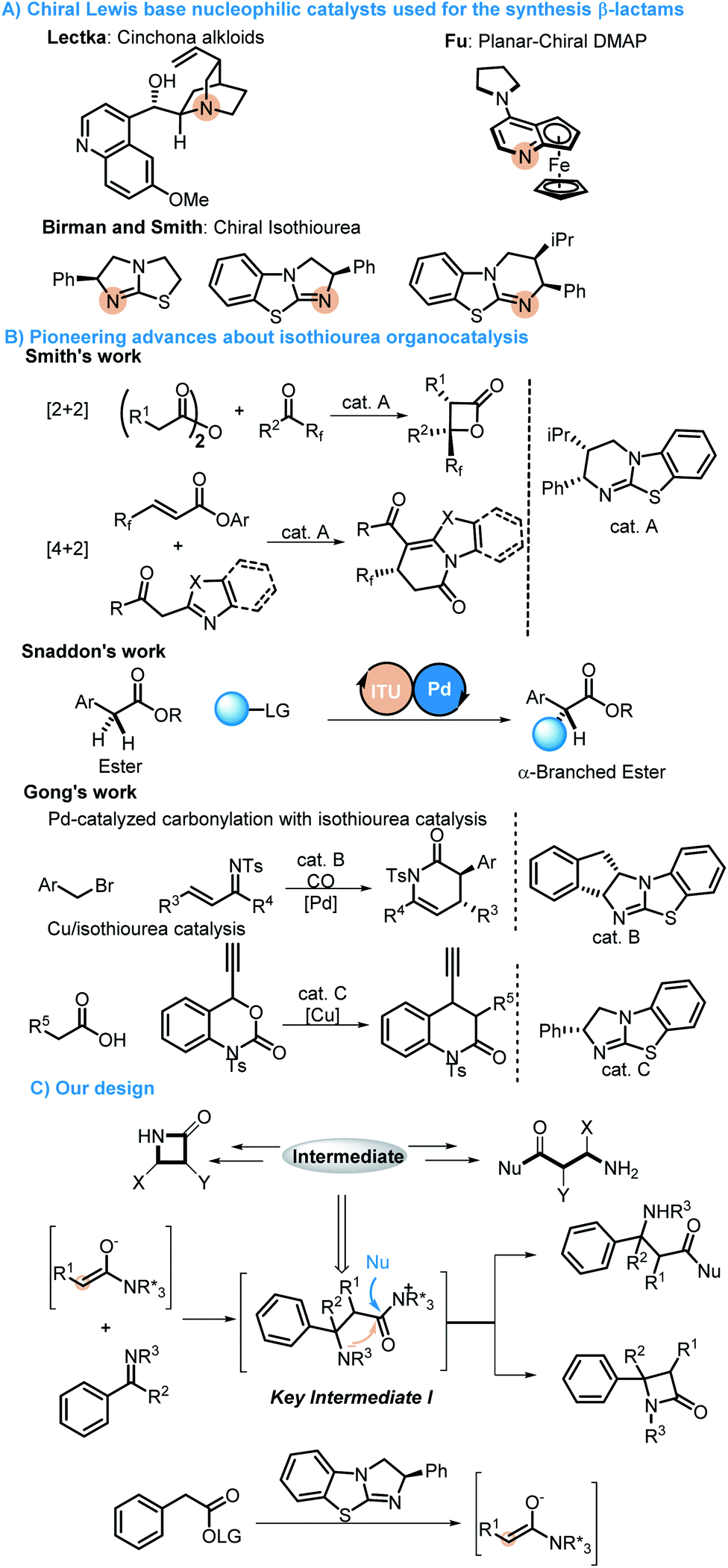 | ||
| Scheme 2 (A) Chiral Lewis base nucleophilic catalysts used for the synthesis β-lactams. (B) Pioneering advances about isothiourea organocatalysis. (C) The proposed protocol. | ||
In recent years, isothiourea (ITU) catalysis, as a new kind of organocatalysis, has attracted tremendous attention from organic chemists,12 and not only shows outstanding performance in controlling product stereoselectivity but can also be combined with transition metal,13 photo14 and amine catalysis.15 Smith, as a pioneer in this field, developed a new homobenzotetramisole (HBTM) catalyst and achieved many [2 + 2] and [4 + 2] cycloaddition processes, with which many useful molecules have been constructed.16 Snaddon developed a method for the enantioselective α-functionalization of acyclic esters through a synergistic combination of ITU catalysis and transition metal catalysis.17 In 2017, Gong reported an asymmetric [4 + 2] annulation of C1 ammonium enolates generated from chiral isothiourea catalysts and copper–allenylidene complexes. Later, a new strategy combining the palladium-catalyzed carbonylation and chiral ITU catalysis was developed to access dihydropyridones and β-lactams by the same group (Scheme 2B).18 There are also other chemists who have devoted their efforts to this field and promoted the development of ITU catalysis.19 Based on the principle of divergent synthesis, ITU catalysis, the longstanding exploration in organocatalysis20 and the hypothesis that β-lactams and α-amino acid derivatives may be produced from the same intermediates, a strategy for the divergent synthesis of β-lactams and α-amino acid derivatives by switching reaction solvents was proposed. We envisioned that a stereospecific Mannich reaction between C(1)-ammonium enolates and imines would produce key intermediate I (Scheme 2C). Then, there would be two different pathways to regenerate the catalyst from intermediate I: one is the process of the intramolecular lactamization, and the other is the process of the intermolecular esterification when ethanol is used as the solvent. Therefore, β-lactams and α-amino acid derivatives would be easily obtained from using different solvents. There are two obvious challenges in the protocol: one is the competitive intramolecular and intermolecular reactions of intermediates I, which will determine whether or not we can access either only β-lactams or only α-amino acid derivative products; the other is the diastereoselectivity issue because imines might have two pathways to approach enolates.
Results and discussion
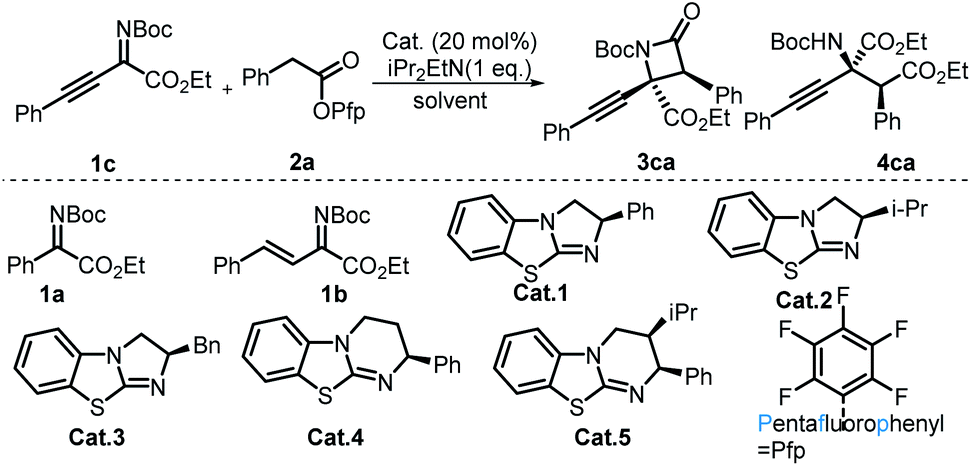
Initial experiments were started with imine 1a and aryl acetic acid ester 2a in 1 mL of CH2Cl2 with 20 mol% Cat.1 as well as 1 equivalent i-Pr2EtN. Unfortunately, no product formation was detected, and all the substrates were totally recovered. Considering the higher electron cloud density and larger steric hindrance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond of imine 1a, alkenyl and alkynyl groups were introduced into imine 1a,21 and we believed that this tactic not only increased the reactivity of the carbon atom of the imine bond but also introduced two useful functional groups which can be easily transformed into other useful molecules. To our delight, imine 1c22 produced the desirable β-lactam, which contained a quaternary carbon stereocenter. Then, imine 1c and aryl acetic acid ester 2a were chosen as model substrates to optimize the reaction conditions. First, catalysts with either a [5,5] fused ring system or a [5,6] fused ring system were tested. It was found that catalysts with [5,5] fused ring systems were superior for this reaction, and the classical Cat.1 was the optimal catalyst. Next, we turned our attention to solvents. After the evaluation of non-nucleophilic solvents, acetonitrile was proven to be the optimal solvent for the synthesis of β-lactams (Table 1, entry 8). Then, alcohols were used as the solvent for the divergent synthesis of α-amino acid derivatives by switching the solvents. The acquisition of the corresponding amino acid derivative 4ca showed that solvent-directed divergent synthesis of β-lactams and α-amino acid derivatives was successfully achieved (Table 1, entry 9). To improve the yields and stereoselectivities of the reactions, the bases and the reaction temperatures were surveyed (see the ESI for details†). The results showed that high stereoselectivities and yields were obtained when either acetonitrile or ethanol was used as the solvent when the reactions were performed at a lower temperature. Taking into consideration the case that the poor diastereoselectivity of 3ca might originate from the process of the base-promoted enolate epimerization of 3ca, lower temperatures might repress this process, and the best results were obtained when the reaction was performed at −50 °C. After the evaluation of other parameters, including the ratios of substrates and additives, the optimal reaction conditions for the synthesis of β-lactam (Table 1, entry 15) and the amino acid derivative (Table 1, entry 14) were obtained.
N double bond of imine 1a, alkenyl and alkynyl groups were introduced into imine 1a,21 and we believed that this tactic not only increased the reactivity of the carbon atom of the imine bond but also introduced two useful functional groups which can be easily transformed into other useful molecules. To our delight, imine 1c22 produced the desirable β-lactam, which contained a quaternary carbon stereocenter. Then, imine 1c and aryl acetic acid ester 2a were chosen as model substrates to optimize the reaction conditions. First, catalysts with either a [5,5] fused ring system or a [5,6] fused ring system were tested. It was found that catalysts with [5,5] fused ring systems were superior for this reaction, and the classical Cat.1 was the optimal catalyst. Next, we turned our attention to solvents. After the evaluation of non-nucleophilic solvents, acetonitrile was proven to be the optimal solvent for the synthesis of β-lactams (Table 1, entry 8). Then, alcohols were used as the solvent for the divergent synthesis of α-amino acid derivatives by switching the solvents. The acquisition of the corresponding amino acid derivative 4ca showed that solvent-directed divergent synthesis of β-lactams and α-amino acid derivatives was successfully achieved (Table 1, entry 9). To improve the yields and stereoselectivities of the reactions, the bases and the reaction temperatures were surveyed (see the ESI for details†). The results showed that high stereoselectivities and yields were obtained when either acetonitrile or ethanol was used as the solvent when the reactions were performed at a lower temperature. Taking into consideration the case that the poor diastereoselectivity of 3ca might originate from the process of the base-promoted enolate epimerization of 3ca, lower temperatures might repress this process, and the best results were obtained when the reaction was performed at −50 °C. After the evaluation of other parameters, including the ratios of substrates and additives, the optimal reaction conditions for the synthesis of β-lactam (Table 1, entry 15) and the amino acid derivative (Table 1, entry 14) were obtained.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Cat. | Solvent | Yield 3cab | Yield 4cab | drc | eed |
a Reactions performed with 0.1 mmol 1a, 0.1 mmol 2a, 0.1 mmol i-Pr2NEt, catalyst (20 mol%), in solvent (1 mL) at 15 °C for 24 hours.
b Isolated yield of the major isomer.
c Determined by 1H NMR analysis of the crude products.
d Determined by chiral-phase HPLC.
e The reaction was performed at 0 °C for 40 hours.
f The reaction was performed at −40 °C.
g The reaction was performed at −50 °C for 48 hours, CH3CN/DCM = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
h 0.12 mmol 2a was used. :1.
h 0.12 mmol 2a was used.
|
||||||
| 1 | Cat.1 | CH2Cl2 | 53 | — | 3:1 |
90 |
| 2 | Cat.2 | CH2Cl2 | 56 | — | 3:1 |
85 |
| 3 | Cat.3 | CH2Cl2 | 51 | — | 3:1 |
78 |
| 4 | Cat.4 | CH2Cl2 | 42 | — | 2:1 |
74 |
| 5 | Cat.5 | CH2Cl2 | 38 | — | 3:1 |
91 |
| 6 | Cat.1 | CHCl3 | 56 | — | 3:1 |
88 |
| 7 | Cat.1 | Toluene | Trace | — | — | — |
| 8 | Cat.1 | CH3CN | 70 | — | 3:1 |
90 |
| 9 | Cat.1 | EtOH | — | 72 | 5:1 |
92 |
| 10e | Cat.1 | CH3CN | 72 | — | 3:1 |
91 |
| 11e | Cat.1 | EtOH | — | 78 | 20:1 |
99 |
| 12f | Cat.1 | CH3CN | 80 | — | 8:1 |
99 |
| 13g | Cat.1 | CH3CN | 78 | — | 8:1 |
98 |
| 14g,h | Cat.1 | CH3CN | 85 | — | 8:1 |
99 |
| 15e,h | Cat.1 | EtOH | — | 81 | 20:1 |
99 |
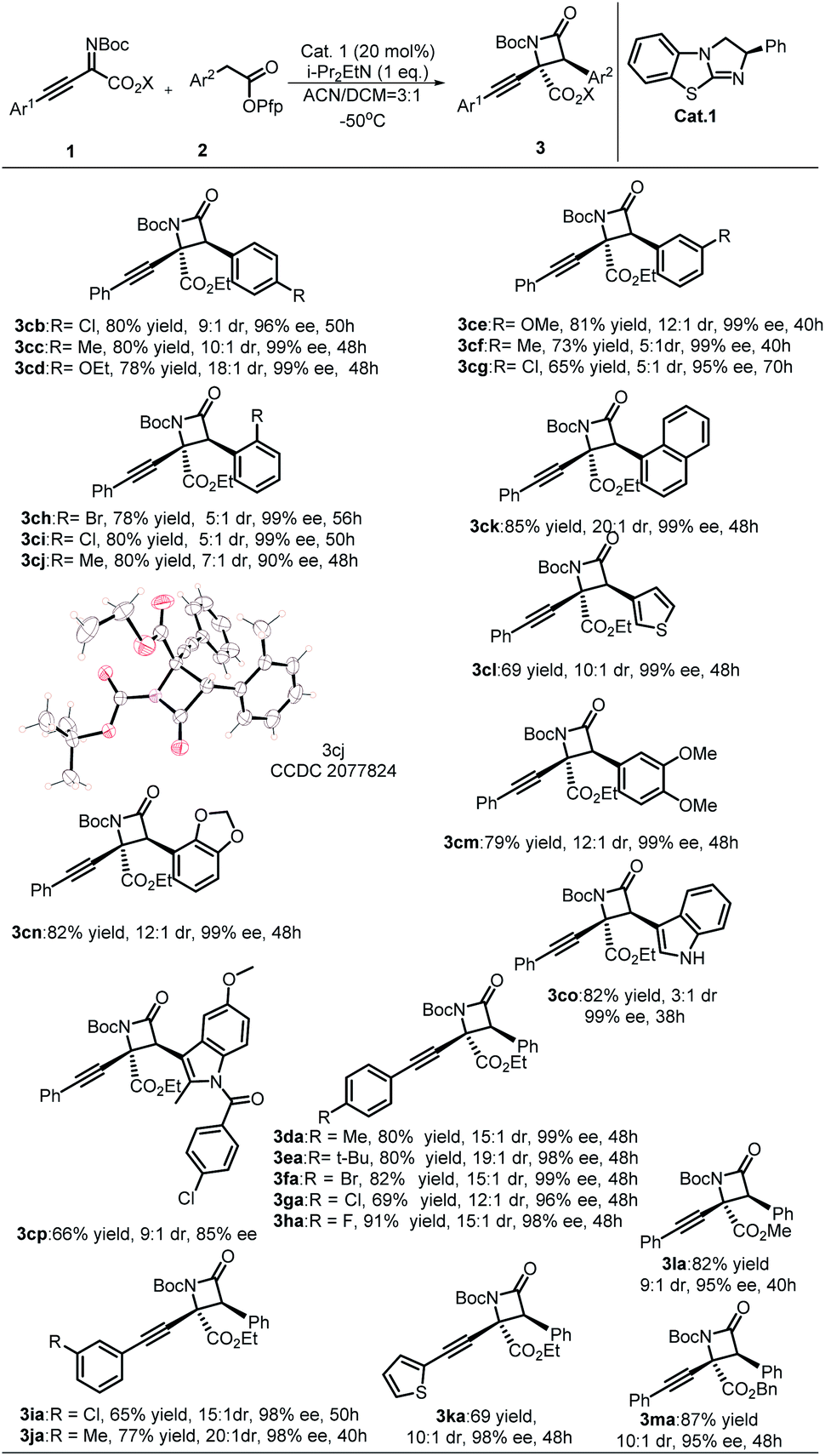
With the optimal reaction conditions established, the substrate scope to synthesize β-lactams was explored (Table 2). First, the electronic and steric effects on aryl acetic acid esters were explored. Acetic acid esters containing para- and meta-substituted phenyls all gave desirable products with excellent enantioselectivities and diastereoselectivities (3cb–3cg), while substrates containing electron withdrawing groups (EWG) needed more reaction time than substrates containing electron donating groups (3cb, 3cg). By comparison, an obvious steric effect was observed when ortho-substituted phenylacetic acid ester was used, which produced 3ch and 3ci as a 5:1 mixture of diastereomers with good yields and excellent enantioselectivity (99% ee). Ortho-methyl substituted substrate 2j with less steric hindrance gave ideal results with higher enantioselectivity (99% ee) and diastereoselectivity than ortho-chlorine- or bromine-substituted substrates. Next, both naphthyl and thienyl acetic acid esters gave desirable products (3ck, 3cl) with satisfying results. Substrates 2 possessing multiple substituents were also compatible with the protocol, which produced the corresponding products (3cm, 3cn) with excellent stereoselectivities and yields. To demonstrate the value of this protocol for the synthesis of β-lactams, commercial pharmaceutical indomethacin-derived β-lactam 3cp was constructed with moderate stereoselectivities and yield. In general, imines 1 containing either EWG- or EDG-substituted Ar1 afforded products with satisfactory results. When the phenyl group of imines was changed to a thienyl group, the desired products were obtained with excellent stereoselectivities in modest yields. When the ester group of imines 1 was turned into either methyl ester or benzyl acetate, the expected products were obtained with similar stereoselectivities and yields. The absolute configuration of product 3cj (CCDC 2077824) was determined to be (2S,3S) by single-crystal X-ray diffraction analysis, and the structures of other products were assigned by analogy.23
| a All reactions were performed with 20 mol% of Cat.1, 0.1 mmol of 1, 0.12 mmol of 2, and 0.1 mmol i-Pr2EtN in 0.75 mL CH3CN and 0.25 mL CH2Cl2 at −50 °C for t h; isolated yields of major isomers are provided. |
|---|
|
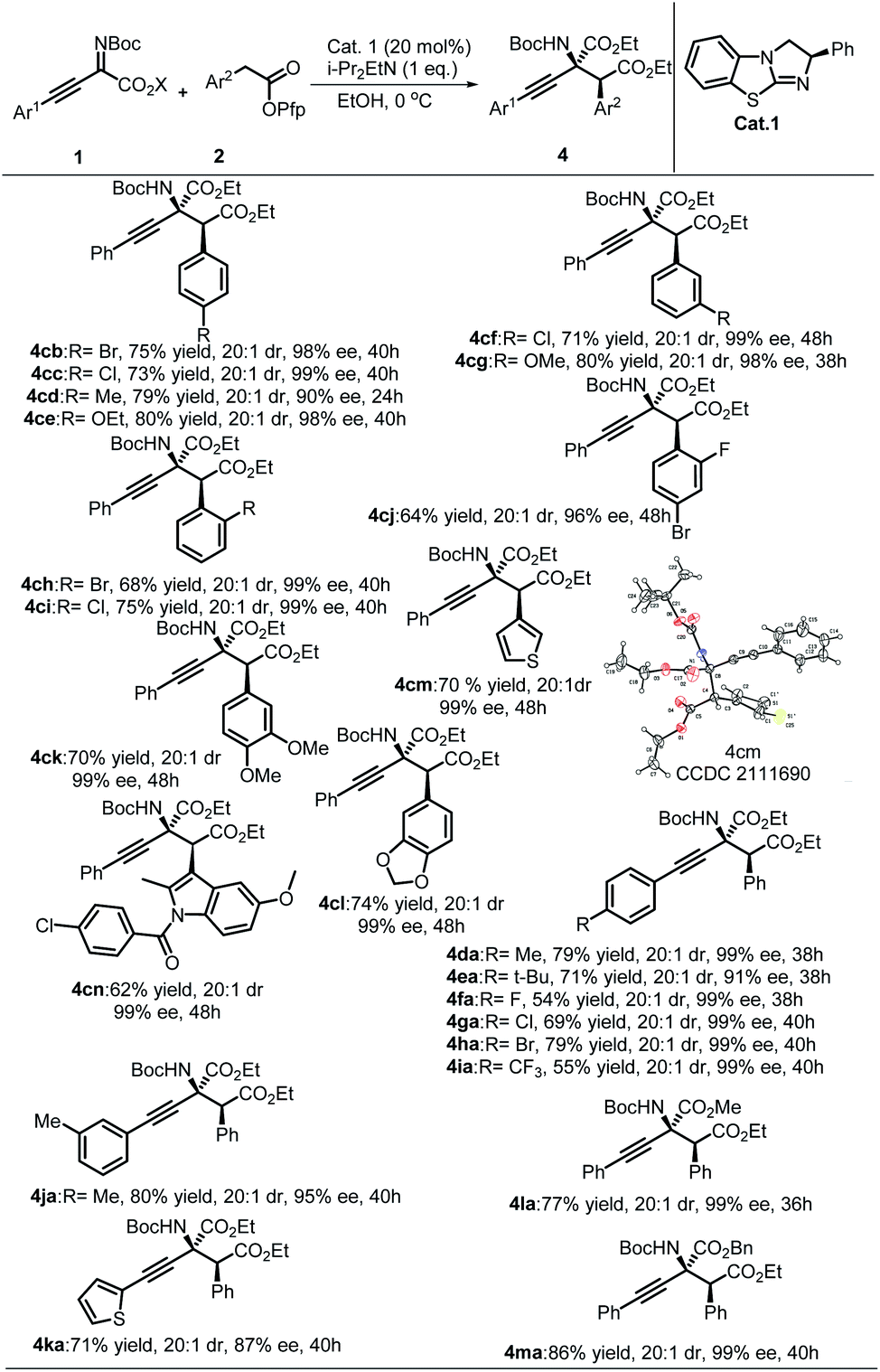
Next, the universality of EtOH-directed synthesis of α-amino acid derivatives was studied (Table 3). Generally, this reaction showed better diastereoselectivity than the synthesis of β-lactams, and all substrates afforded products with excellent stereoselectivities. Substrates 2 with EWG produced the expected products with lower yields than substrates with EDG (4cb, 4cd to 4cc, 4ce). Substrates containing meta-position-substituted Ar2 all afforded desirable products in excellent results without apparent substituent effects. Vicinal larger steric substituents resulted in lower yields and stereoselectivities (4ci and 4cj). Satisfying results were observed for substrates containing Ar2 with multiple substituents (4ck and 4cl). The substrate with an aryl group replaced by a 3-thienyl group yielded the corresponding product with an acceptable outcome (4cm). Furthermore, the commercial antiphlogistic drug indomethacin was also compatible with this protocol, and the corresponding amino acid derivative 4cn was obtained with a modest yield and stereoselectivities. The generality of substrates of imines 1 was also inspected. As shown in Table 3, the reaction results showed excellent stereoselectivities (mostly 99% ee), and the expected products were obtained along with modest to high yields when there was an EDG group on Ar.1 A fluorine-substituted phenylethyne-derived imine furnished the product with a diminished yield and excellent enantioselectivity. The heteroaryl substrate was also compatible with this protocol and afforded the desirable products with moderate stereoselectivities and yields, such as 4ka. The steric hindrance of the ester group in imines 1 was also investigated; when the COOEt group was changed to a COOMe or COOBn group, the corresponding α-amino acid derivatives were still obtained with excellent results. The absolute configuration of product 4cm (CCDC 2111690) was determined to be (2S,3S) by single-crystal X-ray diffraction analysis, and the structures of other products were assigned by analogy.24
| a All reactions were performed with 20 mol% of Cat.1, 0.1 mmol of 1, 0.12 mmol of 2, and 0.1 mmol i-Pr2EtN in 1 mL of EtOH at 0 °C for t h; isolated yields are provided. |
|---|
|
Having explored the reaction substrate scopes of this protocol, we turned our attention to the mechanism of this divergent synthesis strategy. First, under the optimal conditions to synthesize α-amino acid derivatives, the ethanolysis experiment of optically pure β-lactam 3ca was conducted (Scheme 3A and B). The ring opening product was not detected, and a diastereoisomer of 3ca was observed. These results elucidated that the α-amino acid derivatives do not come from the ethanolysis of β-lactams under the reaction conditions and supported the assumption that the epimerization of β-lactams contributed to lower diastereoselectivities under basic conditions,25 which was mentioned earlier in the article. A mixture solvent experiment was carried out under the optimal conditions of synthesizing β-lactams, and the only product was an α-amino acid derivative (Scheme 3B). We proposed that there would be two competitive routes in the process of regenerating the ITU catalyst. The oxygen atom in EtOH as a nucleophile prevails compared with the nitrogen atom in the substrate; therefore, the intramolecular esterification reaction occurs at a higher level than the intermolecular lactamization reaction. With this protocol, the chemical divergent synthesis of β-lactams and α-amino acid derivatives was truly directed by solvents.
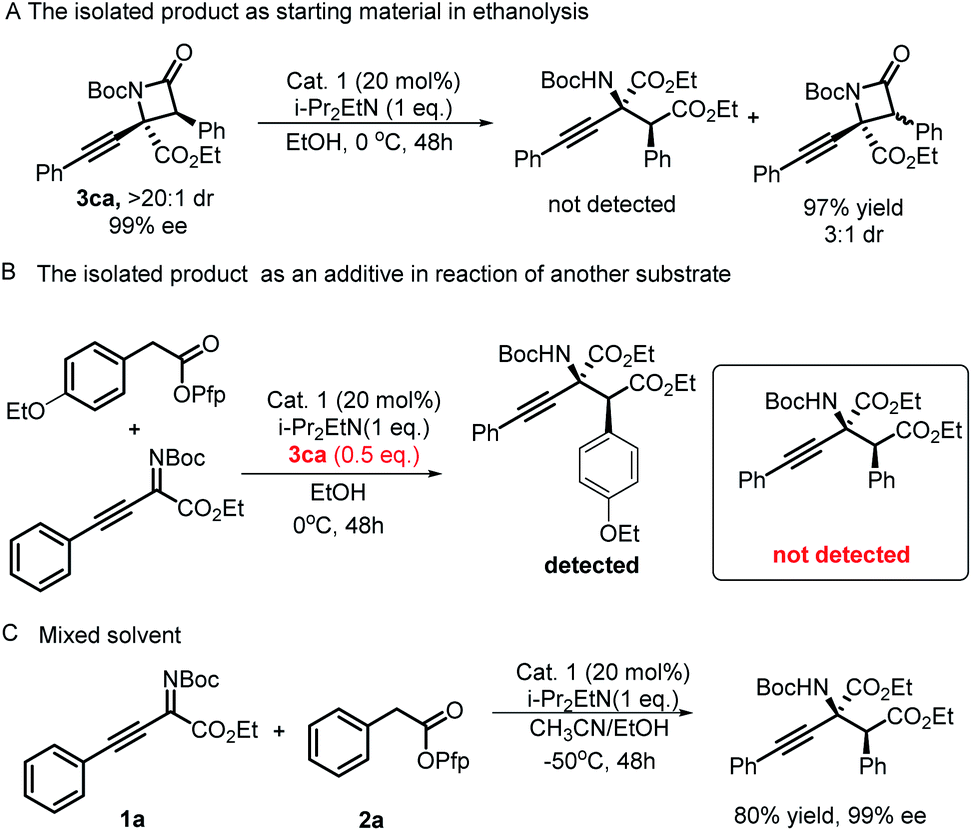 | ||
| Scheme 3 (A) The isolated product as starting material in ethanolysis. (B) The isolated product as an additive in reaction of another substrate. (C) Mixed solvent experiment. | ||
Based on these experimental results (nonlinear effect and deuterium experiments, see the ESI†) and other literature reports,12,14,16 a proposed mechanism is outlined in Fig. 2. Initially, an N-acylation occurs between BTM and the ester, which gives acyl ammonium ion pair I. Ammonium enolate II is generated from the process of deprotonation,16d,26 which is facilitated by i-Pr2NEt as an auxiliary base. Then, a stereoselective Mannich reaction of ammonium enolate II with imine occurs to form the key zwitterionic intermediate III. Subsequently, when acetonitrile is used as the solvent, lactamization will give β-lactam products and regenerate the catalyst BTM; otherwise, esterification gives α-amino acid derivatives and regenerates the catalyst BTM when ethanol is used as the solvent. The stereochemical outcome of the reaction is determined in the step of the Mannich reaction, where ammonium enolate II adopts a Z-conformation aided by the 1,5-O S interaction (chalcogen-bonding catalysis)27 between the enolate oxygen anion and the S atom of the catalyst, which provides a conformational lock. The phenyl group shields the Si face, and the imine approaches from the least hindered Re face and adopts the favored transition state28 with the help of π–π stacking interaction that comes from the phenyl group of the enolates and alkynyl group of the imines, which determines the configuration at C3 of the products. The stereochemical results are consistent with single X-ray crystal experiments.
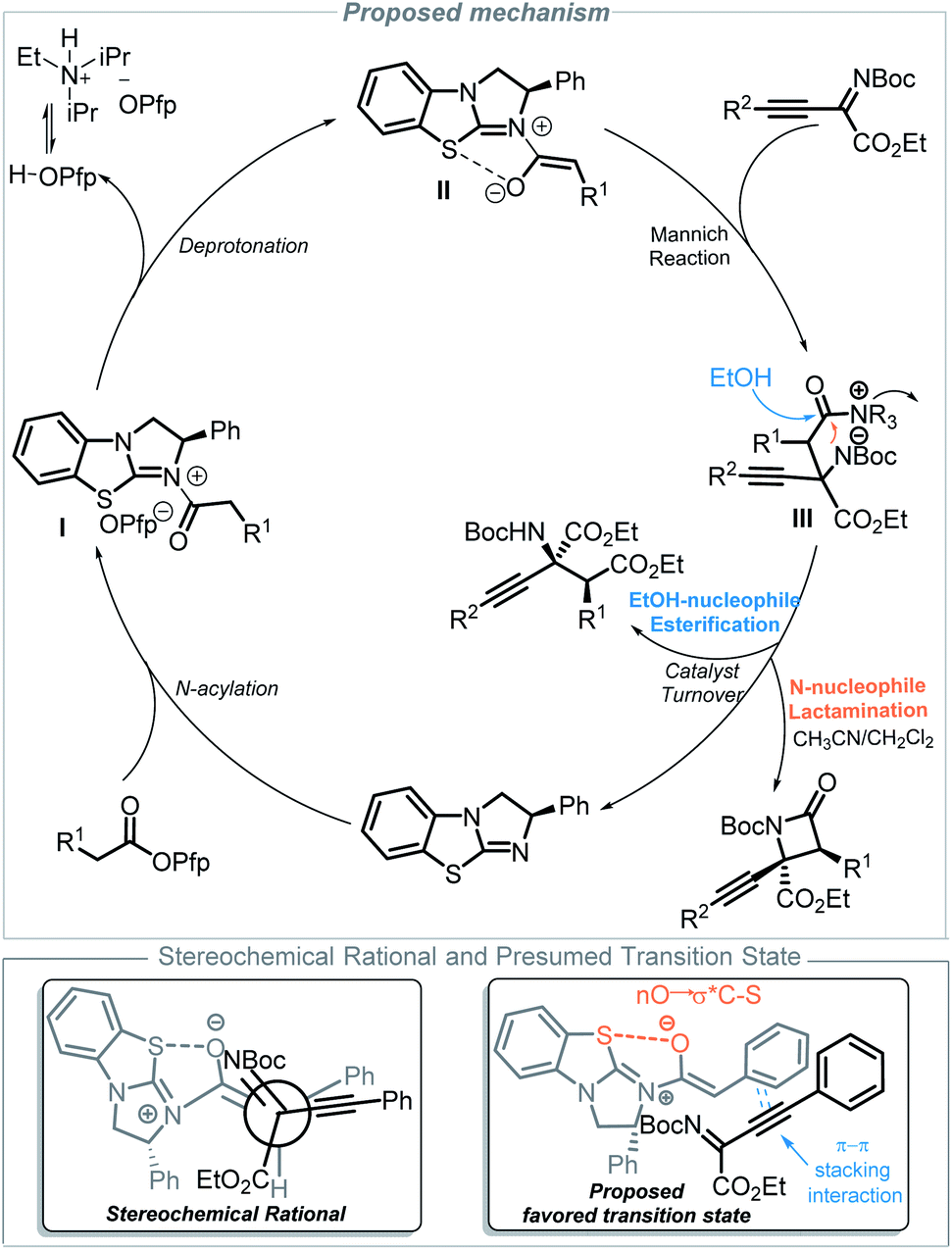 | ||
| Fig. 2 Proposed mechanism for the solvent directed divergent synthesis of β-lactam and α-amino acid derivatives. | ||
To prove the practicability of the protocol, some gram-scale reactions were conducted. Target products 3ca and 4ca were accessed with higher yields and similar stereoselectivities compared with the model reaction when the reaction scale was amplified 40-fold (Scheme 4A). In view of the potential value of alkynyl groups in synthetic chemistry, further transformation of the group was tested (Scheme 4B). It is worth emphasizing that a substituent-directed transformation of alkynyl groups in 3ca and 4ca was achieved. Under the conditions of using Ag/Au salts29 as catalysts in toluene, a process of intermolecular hydroxylation/isomerization mediated by Au occurred for 4ca and gave product 5 with 71% yield and 99% ee. Interestingly, an intramolecular process occurred for 3ca to give product 6 with a moderate yield but without a loss of enantioselectivity. Moreover, divergent hydrogenation of 4ca was achieved by using Pd/C or Pd/CaCO3 as the catalyst, which produced product 7 or 8, respectively, with good results.
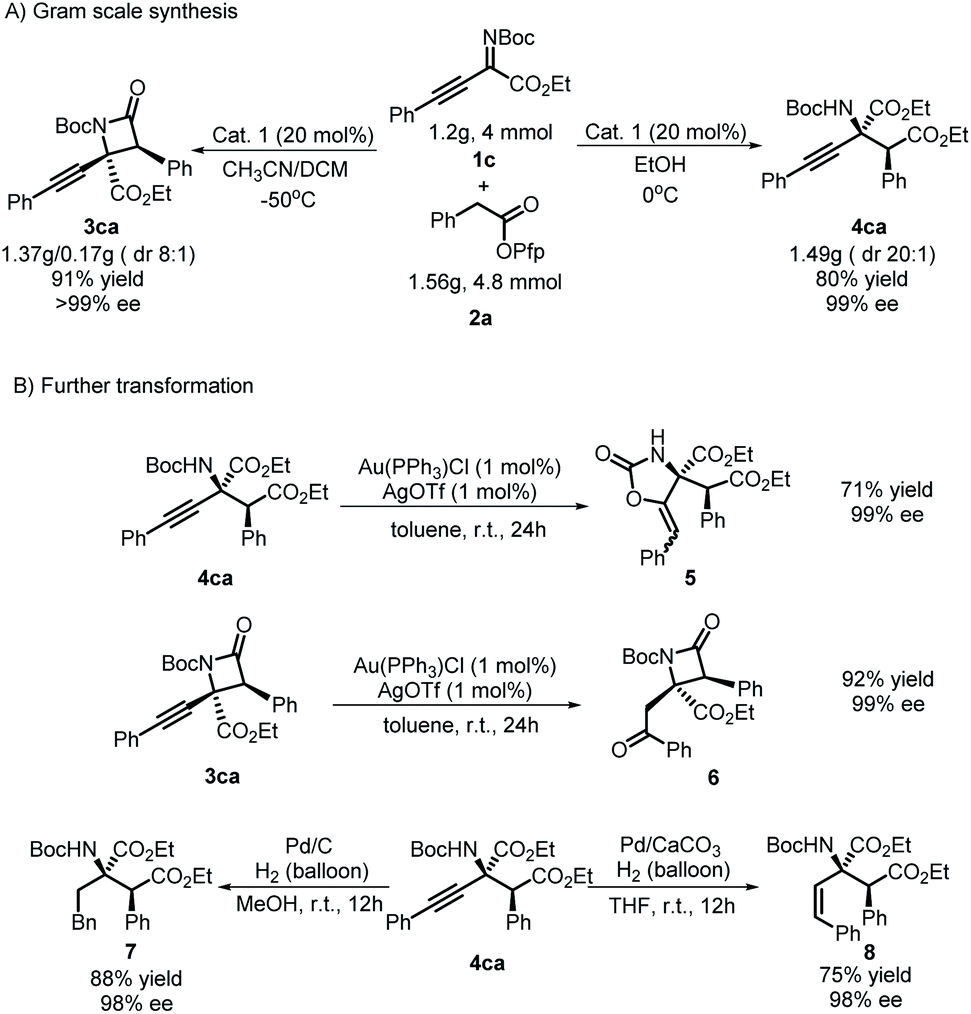 | ||
| Scheme 4 (A) Gram scale synthesis (B) divergent transformation. | ||
Conclusion
In conclusion, a highly stereoselective protocol for the chemically divergent synthesis of β-lactams and α-amino acid derivatives directed by solvents was successfully developed. In this protocol, not only was a quaternary carbon chiral center constructed, but two kinds of important molecules were also produced. Furthermore, substituent- and catalyst-directed divergent transformations of the alkynyl group were also achieved. It was found that the alkynyl group was the key to realizing this protocol, and noncovalent chalcogen-bonding catalysis and π–π stacking interactions played important roles in the process of steric control. This protocol provides a concise, green and effective synthetic method for β-lactams and α-amino acid derivatives. Further investigations of the divergent synthesis catalyzed by ITU are currently in progress in our laboratory.Author contributions
D.-S. J and K.-X. Y conducted all synthetic work. D.-S. J wrote the original manuscript. H. L. offered constructive suggestions and revised the manuscript. Z.-T. F offered constructive suggestions. G.-Q. X and Y.-C. L revised the manuscript. Y. C. G supervised and supported this project. P.-F. X. conceptualised and supervised the project, and provided critical feedback during the whole process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSFC (21632003 and 21871116), the key program of Gansu province (17ZD2GC011), and the “111” program from the MOE of P. R. China. We also gratefully acknowledge a Syngenta Postgraduate Fellowship to D.-S. Ji.Notes and references
- (a) R. S. Doerksen, C. C. Meyer and M. J. Krische, Angew. Chem., Int. Ed., 2019, 58, 14055 CrossRef CAS PubMed; (b) A. H. Hoveyda, Y. Zhou, Y. Shi, M. K. Brown, H. Wu and S. Torker, Angew. Chem., Int. Ed., 2020, 59, 21304 CrossRef CAS PubMed; (c) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed.
- (a) L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752 CrossRef CAS PubMed; (b) Y.-C. Lee, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2018, 57, 5212 CrossRef CAS PubMed.
- (a) J. Deng, Y. Ning, H. Tian and J. Gui, J. Am. Chem. Soc., 2020, 142, 4690 CrossRef CAS PubMed; (b) J. Zhou, D.-X. Tan and F.-S. Han, Angew. Chem., Int. Ed., 2020, 59, 18731 CrossRef CAS PubMed; (c) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432 CrossRef CAS PubMed.
- (a) Y. Wei and M. Shi, ACS Catal., 2016, 6, 2515 CrossRef CAS; (b) S. Jin, S.-J. Li, X. Ma, J. Su, H. Chen, Yu Lan and Q. Song, Angew. Chem., Int. Ed., 2021, 60, 881 CrossRef CAS PubMed; (c) Y. Xie, Y. Fang, Z. Huang, A. M. Tallon, C. W. Ende and J. M. Fox, Angew. Chem., Int. Ed., 2020, 59, 16967 CrossRef CAS PubMed; (d) P. Thilmany and G. Evano, Angew. Chem., Int. Ed., 2020, 59, 242 CrossRef CAS PubMed; (e) W. Luo, Z. Sun, E. H. N. Fernando, V. N. Nesterov, T. R. Cundari and H. Wang, Chem. Sci., 2020, 11, 9386 RSC.
- (a) J. F. Fisher and S. Mobashery, Chem. Rev., 2021, 121, 3412 CrossRef CAS PubMed; (b) D. T. Davies and M. Everett, Acc. Chem. Res., 2021, 54, 2055 CrossRef CAS PubMed; (c) C. C. Hanna, Y. O. Hermant, P. W. R. Harris and M. A. Brimble, Acc. Chem. Res., 2021, 54, 1878 CrossRef CAS PubMed.
- (a) S. Carosso, R. Liu, P. A. Miller, S. J. Hecker, T. Glinka and M. J. Miller, J. Med. Chem., 2017, 60, 8933 CrossRef CAS PubMed; (b) R. F. Abdulla and K. H. Fuhr, J. Med. Chem., 1975, 18, 625 CrossRef CAS PubMed; (c) P. W. Smith, F. Zuccotto, R. H. Bates, M. S. M. Martinez, K. D. Read, C. Peet and O. Epemolu, ACS Infect. Dis., 2018, 4, 1439 CrossRef CAS PubMed.
- (a) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807 CrossRef CAS PubMed; (b) O. V. Poorten, A. Knuhtsen, D. S. Pedersen, S. Ballet and D. Tourwé, J. Med. Chem., 2016, 59, 10865 CrossRef PubMed; (c) A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382 CrossRef CAS PubMed.
- (a) A. E. Taggi, A. M. Hafes and T. Lectka, Acc. Chem. Res., 2003, 36, 10 CrossRef CAS PubMed; (b) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437–4492 CrossRef CAS PubMed; (c) L.-L. Li, D. Ding, J. Song, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 7647 CrossRef CAS PubMed.
- S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985–3012 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2000, 33, 412–420 CrossRef CAS PubMed.
- (a) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; (b) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589–5610 CrossRef CAS; (c) S. R. Smith, J. Douglas, H. Prevet, P. Shapland, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1626 CrossRef CAS PubMed.
- (a) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214 RSC; (b) C. McLaughlin and A. D. Smith, Chem. - Eur. J., 2021, 27, 1533 CrossRef CAS PubMed.
- (a) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895 CrossRef CAS PubMed; (b) J. Song, Z.-J. Zhang, S.-S. Chen, T. Fan and L.-Z. Gong, J. Am. Chem. Soc., 2018, 140, 3177 CrossRef CAS PubMed.
- (a) J. N. Arokianathar, A. B. Frost, A. M. Z. Slawin, D. Stead and A. D. Smith, ACS Catal., 2018, 8, 1153 CrossRef CAS; (b) T. Fan, Z.-J. Zhang, Y.-C. Zhang and J. Song, Org. Lett., 2019, 21, 7897 CrossRef CAS PubMed.
- B. Kim, Y. Kim and S. Y. Lee, J. Am. Chem. Soc., 2021, 143, 73 CrossRef CAS PubMed.
- (a) F. Zhao, C. Shu, C. M. Young, C. C. Warren, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2021, 60, 11892 CrossRef CAS PubMed; (b) S. Zhang, M. D. Greenhalgh, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2020, 11, 3885 RSC; (c) H. Liu, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2020, 22, 1301 CrossRef CAS PubMed; (d) C. McLaughlin, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2019, 58, 15111 CrossRef CAS PubMed; (e) D.-J. B. Antunez, M. D. Greenhalgh, A. C. Brueckner, D. M. Walden, P. Elías-Rodríguez, P. Roberts, B. G. Young, T. H. West, A. M. Z. Slawin, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2019, 10, 6162 RSC.
- (a) K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214 CrossRef CAS PubMed; (b) L. H. Goetz, C. Yang and T. N. Snaddon, ACS Catal., 2018, 8, 10537 CrossRef PubMed; (c) C. M. Pearson, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2019, 58, 10521 CrossRef CAS PubMed.
- (a) L.-L. Li, D. Ding, J. Song, Z.-Y. Han and L.-Z. Gong, Angew. Chem., Int. Ed., 2019, 58, 7647 CrossRef CAS PubMed; (b) Y.-C. Zhang, R.-L. Geng, J. Song and L.-Z. Gong, Org. Lett., 2020, 22, 2261 CrossRef CAS PubMed; (c) J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212 CrossRef CAS PubMed.
- (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351 CrossRef CAS PubMed; (b) I. Shiina, K. Nakata, K. Ono, Y. Onda and M. Itagaki, J. Am. Chem. Soc., 2010, 132, 11629 CrossRef CAS PubMed; (c) S. Wang, C. Rodríguez-Escrich and M. A. Pericàs, Angew. Chem., Int. Ed., 2017, 56, 15068 CrossRef CAS PubMed; (d) J. Meng, W.-W. Ding and Z.-Y. Han, Org. Lett., 2019, 21, 9801 CrossRef CAS PubMed; (e) J.-Y. Ong, X. Q. Ng, S. Lu and Y. Zhao, Org. Lett., 2020, 22, 6447 CrossRef CAS PubMed; (f) D. Li, S. Wang, S. Ge, S. Dong and X. Feng, Org. Lett., 2020, 22, 5331 CrossRef CAS PubMed.
- (a) Y.-C. Luo, H.-H. Zhang, Y. Wang and P.-F. Xu, Acc. Chem. Res., 2010, 43, 1317 CrossRef CAS PubMed; (b) Y. Wang, H. Lu and P.-F. Xu, Acc. Chem. Res., 2015, 48, 1832 CrossRef CAS PubMed; (c) Y. Gu, Y. Wang, T.-Y. Yu, Y.-M. Liang and P.-F. Xu, Angew. Chem., Int. Ed., 2014, 53, 14128 CrossRef CAS PubMed; (d) Y. Wang, R.-G. Han, Y.-L. Zhao, S. Yang, P.-F. Xu and D. J. Dixon, Angew. Chem. Int. Ed., 2009, 48, 9834 CrossRef CAS PubMed; (e) Y. Wang and P.-F. Xu, Catalytic Cascade Reactions ed. P.-F. Xu and W. Wang, John Wiley & Sons: Canada, 2014; pp. 123–144 Search PubMed; (f) Y.-L. Zhao, Y. Wang, X.-Q. Hu and P.-F. Xu, Chem. Commun., 2013, 49, 7555–7557 RSC; (g) H. Lu, J.-L. Zhang, J.-Y. Liu, H.-Y. Li and P.-F. Xu, ACS Catal., 2017, 7, 7797–7802 CrossRef CAS; (h) H. Lu, J.-Y. Liu, C.-G. Li, J.-B. Lin, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2015, 51, 4473–4476 RSC; (i) L.-J. Zhang, Y. Wang, X.-Q. Hu and P.-F. Xu, Chem.–Asian J., 2016, 11, 834–838 CrossRef CAS PubMed; (j) J.-Y. Liu, J. Zhao, J.-L. Zhang and P.-F. Xu, Org. Lett., 2017, 19, 1846–1849 CrossRef CAS PubMed; (k) C.-G. Zhao, Z.-T. Feng, G.-Q. Xu, A. Gao, J.-W. Chen, Z.-Y. Wang and P.-F. Xu, Angew. Chem., Int. Ed., 2020, 59, 3058–3062 CrossRef CAS PubMed.
- We proposed that the electronegativity of the sp hybrid carbon atom in alkynyl was higher than that of the sp2 hybrid carbon atom, and therefore the cloud density of the CN bond in imine 1c is lower than that in 1a or 1b. Moreover, we speculated that the steric hindrance of the CN bond in imine 1c is less than that in 1a, the reason being that in imine 1c the phenyl group is away from the CN bond; furthermore, the phenyl group and the CN bond are in the same plane; in contrast, the phenyl group of imine 1a is perpendicular to the CN–CO face; therefore it is easy for C(1)-ammonium enolate to work with imine 1c.
- When imine 1b was used, only a trace product was detected.
- CCDC 2077824 (3cj) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. See the ESI† for details.
- CCDC 2111690 (4cm) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. See the ESI† for details.
- The proposed influences of pentafluorophenol or the correspnding pentafluorophenolate were also tested; the experiments showed that they don’t have obvious influences on epimerization under the optimal reaction conditions for the synthesis of β-lactams. For detailed experiments, see the ESI†.
- L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146 RSC.
- (a) C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. de la Houpliere, K. B. Ling, T. K. Smith, A. M. Z. Slawin, P. H. Willoughby, S. L. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705 CrossRef CAS PubMed; (b) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141(23), 9175 CrossRef CAS PubMed.
- For a discussion of favored and unfavored transition states as well as the rationale for diastereoselectivity see ESI† Part 6.
- (a) I. Lapuerta, S. Vera, M. Oiarbide and C. Palomo, Chem.–Eur. J., 2016, 22, 7229 CrossRef CAS PubMed; (b) E.-S. Lee, H.-S. Yeom, J.-H. Hwang and S. Shin, Eur. J. Org. Chem., 2007, 21, 3503 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2077824, 2111690, 2118205. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06127e |
| This journal is © The Royal Society of Chemistry 2022 |