
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric synthesis of isochromanone derivatives via trapping carboxylic oxonium ylides and aldol cascade†
Jiawen
Lang
,
Siyuan
Wang
,
Changli
He
,
Xiaohua
Liu
 * and
Xiaoming
Feng
*
* and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: liuxh@scu.edu.cn; xmfeng@scu.edu.cn
First published on 27th December 2021
Abstract
An efficient asymmetric synthesis of isochromanone derivatives was realized through Z-selective-1,3-OH insertion/aldol cyclization reaction involving acyclic carboxylic oxonium ylides. The combination of achiral dirhodium salts and chiral N,N′-dioxide–metal complexes, along with the use of α-diazoketones instead of α-diazoesters, enables the cascade reaction efficiently. A variety of benzo-fused δ-lactones bearing vicinal quaternary stereocenters were obtained with good to excellent enantioselectivity, bypassing the competitive 1,1-OH insertion and racemic background aldol reaction.
Introduction
Transition-metal-catalyzed insertion of carbenes into heteroatom–hydrogen bonds (X–H, X = N, O, S, etc.)1 to form highly active onium ylides, undergoing either enantioselective insertion reaction2 or interception by electrophiles,3 has received considerable attention for effective construction of structurally complex molecules.4 To this end, enantioselective trapping of oxonium ylides initiated by the insertion of phenols and alcohols, one of the most common and important species, has been well studied.5 However, carboxylic oxonium ylides are largely undeveloped (Scheme 1a),6,8 and sporadic racemic cascade reactions by bypassing the competitive O–H insertion step (path I)7 have been disclosed, such as intermolecular trapping with imines (path II)6a and intramolecular aldol cascade to produce lactones (path III and IV).6c The enantioselective trapping of carboxylic oxonium ylides is rare with difficulties such as (1) low pKa values of protonated carboxylic acids in the zwitterionic intermediate leads to a rapid proton shift;8 (2) changing the electronics of the diazo-derived rhodium carbenoid to delay the proton shift and to facilitate additional cascade results in a strong racemic background reaction. Till recently, a representative capture of carboxylic oxonium ylides with imines for synthesis of chiral γ-butenolides was reported by Hu and coworkers, in which cycloprop-2-ene-1-carboxylic acids were employed as cyclic carboxylic oxonium ylide precursors.9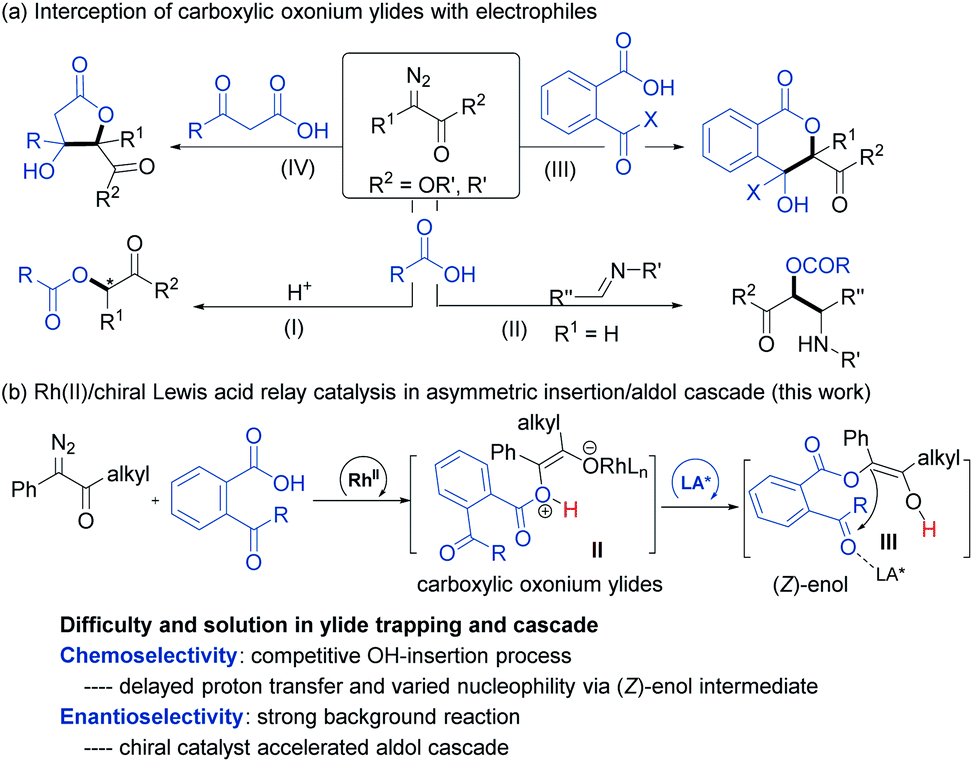 | ||
| Scheme 1 The reactions involving carboxylic oxonium ylides. | ||
In the course of our study related to the enantioselective O–H insertion reaction of carboxylic acid10 and dual metal complex relay catalysis,11 we are interested in trapping acyclic carboxylic oxonium ylides via an asymmetric cascade reaction to obtain chiral isochromanones with two vicinal quaternary centers. The δ-lactone moiety is attractive in food additives, pharmaceuticals and natural products.12 Nevertheless, to control the stereoselectivity of this cascade reaction, a chiral catalyst must engage in the final aldol reaction. The combination of dirhodium salt and the chiral Lewis acid catalyst would be a good choice. The chiral Lewis acid is able to do some multi-tasking under competition with the dirhodium catalyst: enhancing the electrophilicity of the ketone to facilitate the aldol addition to overtake the 1,1-OH insertion; directing the facial selectivity during the cyclization. On the other hand, we propose that the use of α-diazoketones instead of α-diazoesters would result in the formation of a transient (Z)-type enol intermediate for the aldol cascade (Scheme 1b), which has been proved in rhodium carbenoid-initiated Claisen rearrangement13 and others.14 Herein, we report the combination of achiral dirhodium salt and chiral N,N′-dioxide-Fe(III) or Sc(III) complex15 catalyzed cascade 1,3-OH insertion/asymmetric aldol cyclization reaction of ketoacids with α-diazoketones. The corresponding optically active benzo-fused δ-valerolactones were obtained in excellent diastereo- and enantioselectivity.
Results and discussion
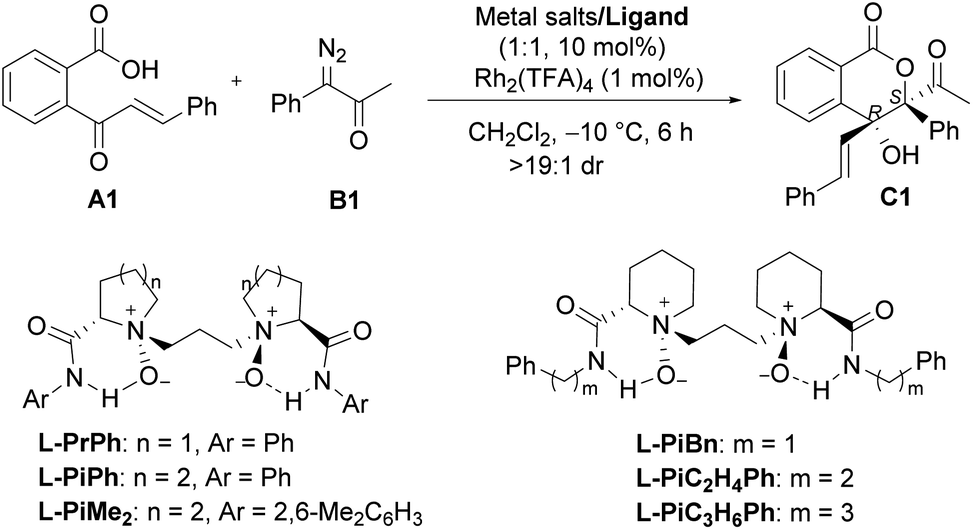
We initiated the investigation of the model reaction between 2-cinnamoylbenzoic acid (A1) and 1-diazo-1-phenylpropan-2-one (B1) in dichloromethane at −10 °C, using Rh2(TFA)4 and Sc(OTf)3 complexes of chiral N,N′-dioxide ligands as the catalytic system. The influence of chiral ligand structures on enantioselectivity and reactivity was evaluated (Table 1, entries 1–5). It was found that both the amino acid backbones and the amide subunits can have significant influence on the enantioselectivity (entries 1–3). L-Proline derivative and hindered amides were disadvantageous to the enantiocontrol. Phenylmethanamine-based L-PiBn and phenylethanamine-based L-PiC2H4Ph showed remarkably increased enantioselectivity (entries 4 and 5). Moreover, the screening of a series of metal salts combined with N,N′-dioxide L-PiC2H4Ph (entries 5–8) showed that Fe(OTf)3 could improve the yield to 84% with maintaining the ee value. It was interesting that Fe(OTf)2 yielded only a racemic product (entry 7). Noteworthily, an excess amount of Fe(OTf)3 benefited the reactivity, and a 91% yield with 97% ee was obtained for the lactone C1 even at an amount of 2 mol% catalyst loading (entries 9 and 10), meanwhile, only a trace amount of 1,1-OH insertion intermediate D1 was observed. In all cases, lactone was isolated as a single diastereoisomer (>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), and the absolute configuration of C1 was determined to be (3S,4R) by X-ray diffraction crystal analysis.16 Comparatively, other chiral ligands, such as BOX or PyBOX instead of N,N′-dioxide, or chiral [Rh2(S-DOSP)4] independently were employed, but low yields and ee values were observed for the product C1 (see the ESI for details†). The screening of other achiral rhodium salts gave similar ee values indicating that rhodium salts only took part in O–H insertion to generate oxonium ylides, implying the merit of dual catalysis. It was noteworthy that the addition of α-diazoketone performed in the one-pot and slow-addition procedure was unnecessary.
:1 dr), and the absolute configuration of C1 was determined to be (3S,4R) by X-ray diffraction crystal analysis.16 Comparatively, other chiral ligands, such as BOX or PyBOX instead of N,N′-dioxide, or chiral [Rh2(S-DOSP)4] independently were employed, but low yields and ee values were observed for the product C1 (see the ESI for details†). The screening of other achiral rhodium salts gave similar ee values indicating that rhodium salts only took part in O–H insertion to generate oxonium ylides, implying the merit of dual catalysis. It was noteworthy that the addition of α-diazoketone performed in the one-pot and slow-addition procedure was unnecessary.
|
|
||||
|---|---|---|---|---|
| Entry | Metal salts | Ligand | Yieldb (%) | eec (%) |
|
a Unless otherwise noted, all reactions were carried out with A1 (0.10 mmol), B1 (2.0 equiv.), metal salts/ligand (1:1, 10 mol%), and Rh2(TFA)4 (1 mol%), in CH2Cl2 (2.0 mL) at −10 °C for 6 h.
b Isolated yield.
c Determined by chiral HPLC analysis in a chiral stationary phase. All dr values were up to >19:1 detected by 1H NMR.
d Fe(OTf)3 (12 mol%).
e Fe(OTf)3/L-PiC2H4Ph (1.2:1, 2 mol%).
|
||||
| 1 | Sc(OTf)3 | L-PrPh | 34 | 6 |
| 2 | Sc(OTf)3 | L-PiPh | 40 | 66 |
| 3 | Sc(OTf)3 | L-PiMe2 | 25 | 8 |
| 4 | Sc(OTf)3 | L-PiBn | 66 | 94 |
| 5 | Sc(OTf)3 | L-PiC2H4Ph | 75 | 97 |
| 6 | Fe(OTf) 3 | L-PiC2H4Ph | 84 | 97 |
| 7 | Fe(OTf)2 | L-PiC2H4Ph | 67 | 0 |
| 8 | Co(OTf)2 | L-PiC2H4Ph | 57 | 80 |
| 9d | Fe(OTf)3 | L-PiC2H4Ph | 91 | 97 |
| 10 | Fe(OTf) 3 | L-PiC2H4Ph | 91 | 97 |
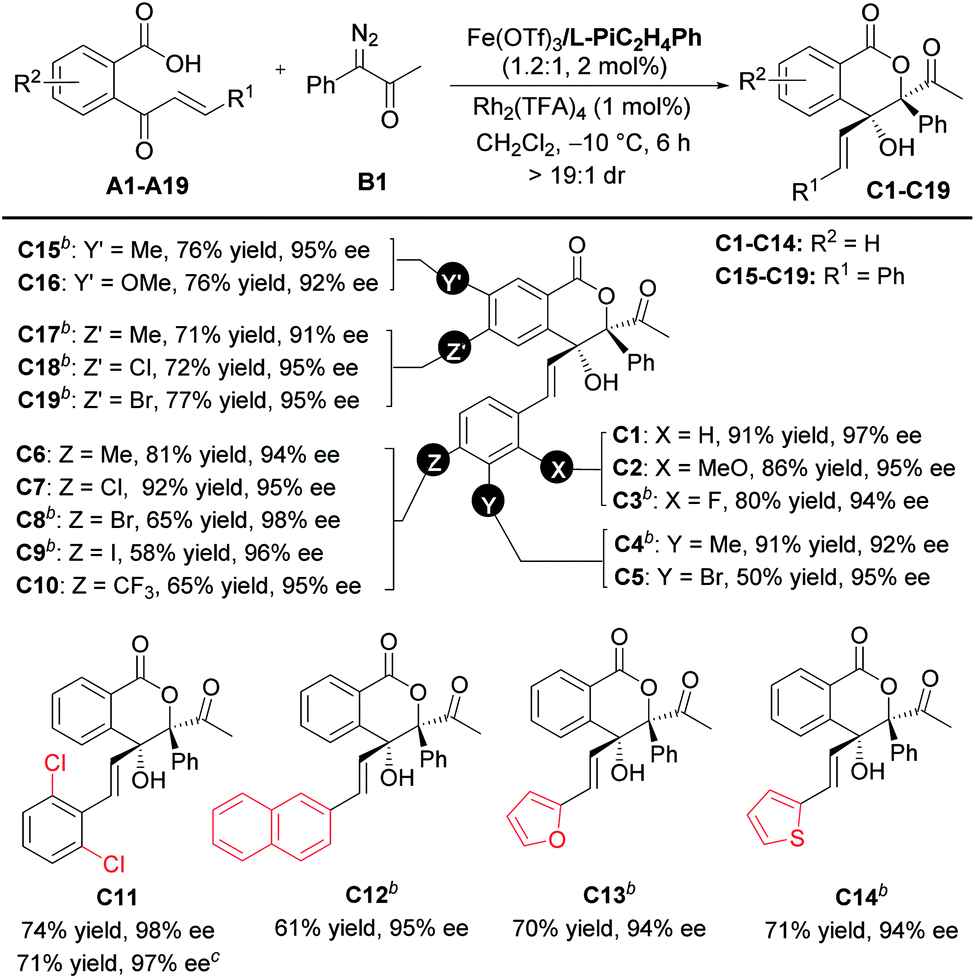
With the optimized reaction conditions in hand (Table 1, entry 10), the scope of cinnamoylbenzoic acid derivatives was examined first. As shown in Table 2, all the substrates containing electron-donating or electron-deficient substituents at the ortho-, meta-, or para-positions of the terminal-aryl group tolerated well, delivering C2–C10 in 50–92% yields with excellent enantioselectivities (92–98%). The 3-bromo substituted C5, 4-bromo-substituted C8 and 4-iodo-substituted C9 were obtained in lower yield than others, due to the increased amount of the interrupted 1,1-OH insertion products. The large-scale synthesis of the lactone C11 proceeded smoothly with good yield and excellent enantioselectivity (0.963 g, 71% yield, 97% ee). In addition, 2-naphthyl, 2-furanyl and 2-thiophenyl substituted cinnamoylbenzoic acids also performed well, delivering the corresponding products C12–C14 with good outcomes (61–71% yields, 94–95% ee). Furthermore, these cinnamoylbenzoic acid derivatives bearing electron-donating or electron-withdrawing groups at 4- or 5-positions of the phenyl ring were suitable to synthesize lactones C15–C19 in good yields with excellent ee values.
|
a Unless otherwise noted, all reactions were carried out with A (0.10 mmol), B1 (2.0 equiv.), Fe(OTf)3/L-PiC2H4Ph (1.2:1, 2 mol%), and Rh2(TFA)4 (1 mol%), in CH2Cl2 (2.0 mL) at −10 °C for 6 h. Isolated yields. The ee value was determined by HPLC analysis. All dr values were up to >19:1 detected by 1H NMR.
b Fe(OTf)3/L-PiC2H4Ph (1.2:1, 4 mol%).
c
A11 (3.0 mmol) and B1 (2.0 equiv.) in CH2Cl2 (60 mL) for 12 h.
|
|---|
|
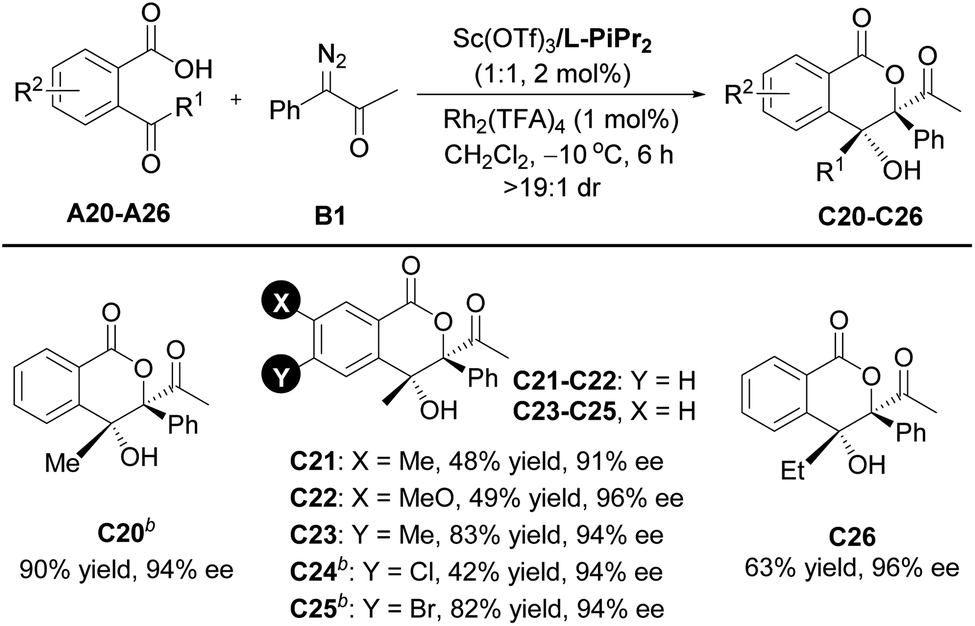
Encouraged by the above results, further efforts were made to expand the scope of ketoacids. A series of γ-ketoacids A20–A26 were explored (Table 3). By switching the catalyst to the Sc(OTf)3/L- PiPr2 complex, the cascade cyclization reaction with diazoketone B1 could efficiently construct the desired products C20–C26 in 42–90% yields with 91–96% ee values.
|
a Unless otherwise noted, all reactions were carried out with A (0.10 mmol), B1 (2.0 equiv.), Sc(OTf)3/L-PiPr2 (1:1, 2 mol%), and Rh2(TFA)4 (1 mol%), in CH2Cl2 (2.0 mL) at −10 °C for 6 h. Isolated yields and ee value was determined by HPLC analysis. All dr values were up to >19:1 detected by 1H NMR.
b Sc(OTf)3/L-PiPr2 (1:1, 4 mol%).
|
|---|
|
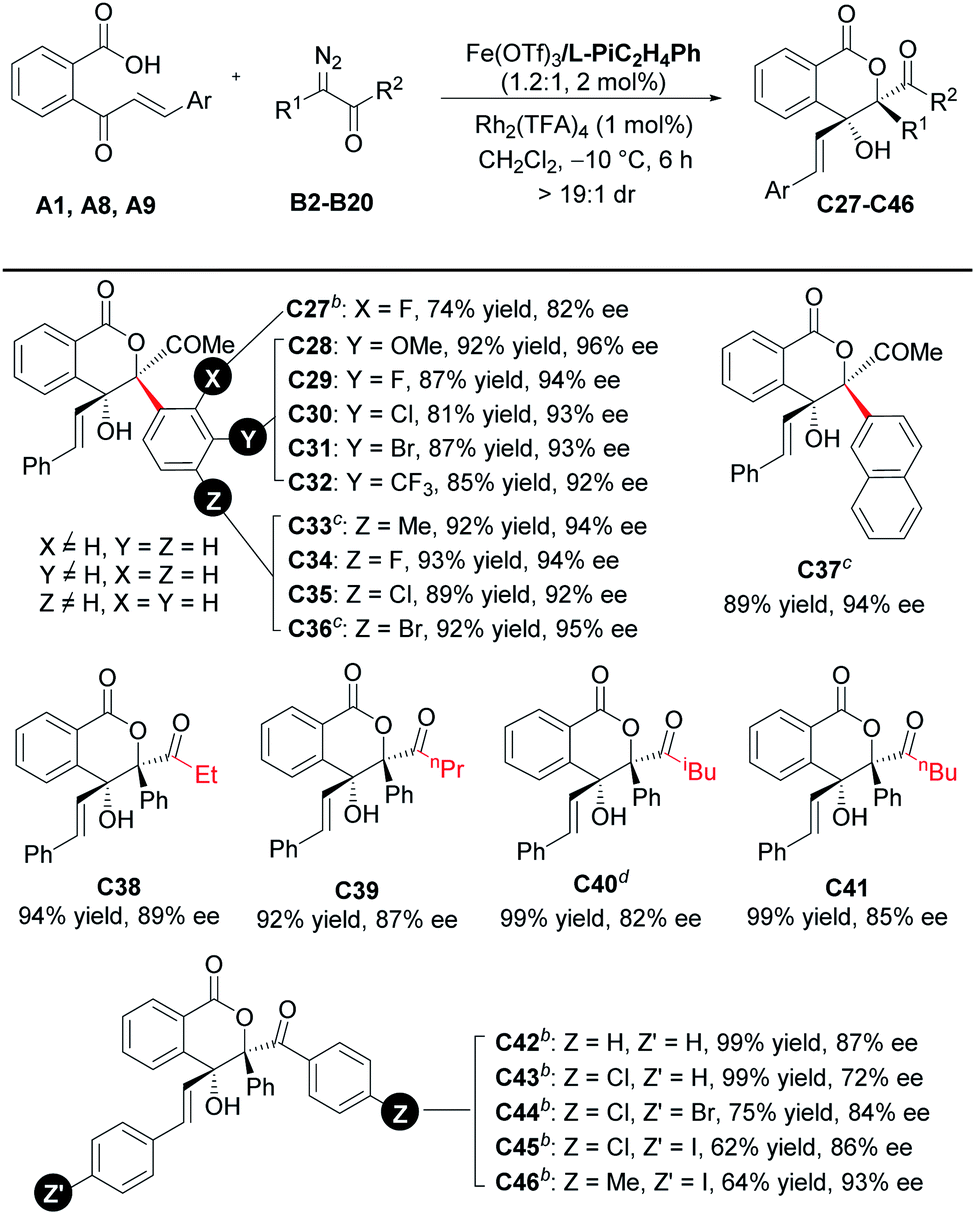
Subsequently, the scope with respect to the diazoketones was investigated (Table 4). All of the tested α-diazo-phenylpropanones (R2 = Me, R1, and B2–B11) reacted smoothly in 6 h to afford the corresponding chiral lactones C27–C36 in good to high yields with excellent enantioselectivities. The electronic properties of the substituents at the para- or meta-position on the phenyl ring of the diazoketones hardly affected the yields and enantioselectivities. 2-Naphthalenyl-bearing diazoketone performed the reaction well to generate C37 in 89% yield and 94% ee. We also studied the effect of the carbonyl substituent, and good enantioselectivities (72–93% ee) were obtained when R2 was either an alkyl group (C38–C41) or aryl group (C42–C46). It should be mentioned that the loading of the Lewis acid catalyst and subunits of the ligand needed to be adjusted in some cases. For instance, 3-phenylpropanamine-based ligand L-PiC3H6Ph was used instead to guarantee the good enantioselectivity. The electron-deficient substituent on 2-cinnamoylbenzoic acid could offset the loss of enantioselectivity caused by 4-chlorobenzoyl substitution (C44 and C45vs.C43).
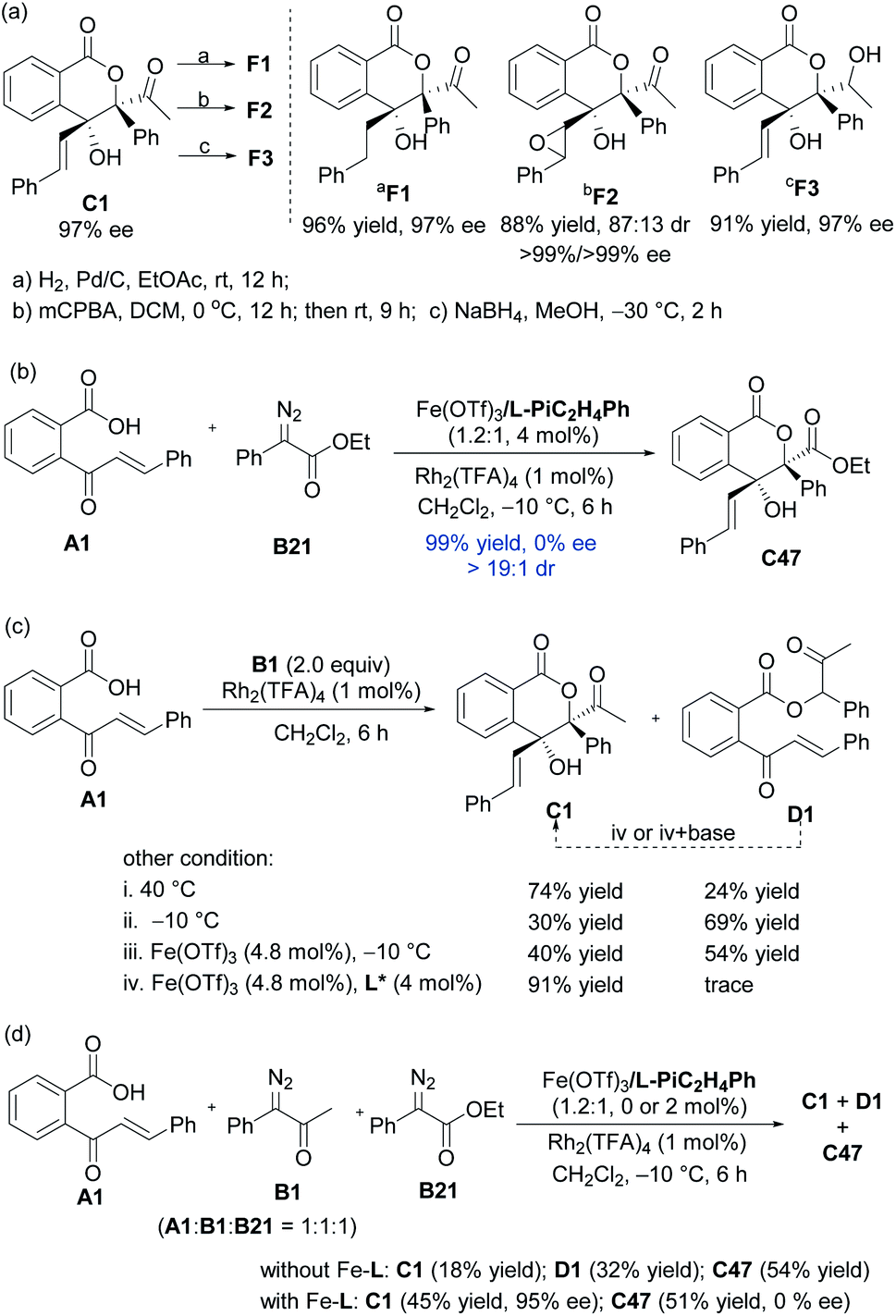
As shown in Scheme 2a, hydrogenation of C1 in the presence of Pd/C afforded derivative F1 in 96% yield with 97% ee. Upon addition of mCPBA, C1 could also be easily converted to an epoxide F2 (88% yield, 87:13 dr, 99% ee). Reduction of C1 by NaBH4 delivered alcohol F3 in high diastereo- and enantioselectivities (91% yield, 97% ee).16
 | ||
| Scheme 2 Transformation of the products and the control experiments. | ||
In order to understand the catalytic mechanism, we set out several control experiments (Scheme 2b–d). Intriguingly, when methyl 2-diazo-2-phenylacetate B21 was subjected to the standard catalytic system instead of α-diazoketone B1, the corresponding lactone product C47 was isolated in 99% yield as a racemate (Scheme 2b). Next, we probed into the role of each catalytic component and it could be seen from Scheme 2c that without the chiral Lewis acid, the 1,1-OH insertion intermediate D1 was coproduced in varying amounts even at low reaction temperature. Favoring a ligand-accelerated process, the 1,1-OH insertion product D1 was nearly bypassed. Nevertheless, the 1,1-OH insertion intermediate D1 produced none of lactone C1 when exposed to the standard reaction conditions, even in the presence of a base. This result suggested that the sequential transformations were not the case for the generation of C1.
The comparison experiment by mixing substrate A1, α-diazoketone B1 and α-diazoester B21 manifested that the rate of the cascade reaction of α-diazoesters exceeded α-diazoketones (Scheme 2d), and the generation of lactone from α-diazoesters occurred more readily than α-diazoketone via the intramolecular Mukaiyama aldol type reaction of metal ketene acetal with the Rh2(TFA)4 catalyst or spontaneously. The strong competition of the Rh2(TFA)4-involved racemic process might decrease the enantioselectivity in the reaction of α-diazoester B21 even at −60 °C (see the ESI for details†). Furthermore, the combination of other dirhodium salts and the chiral ferric catalyst led to complete conversion, containing racemic C47 and minor 1,1-OH insertion intermediates (see the ESI for details†). The prediction of the pKa value via Luo's molecular prediction platform17 showed that the enol intermediate of α-diazoester B21 is slight lower than the corresponding intermediates of α-diazoketones (see the ESI for details†). These results clearly indicate that stabilized ylide species affect the addition step in these cases.
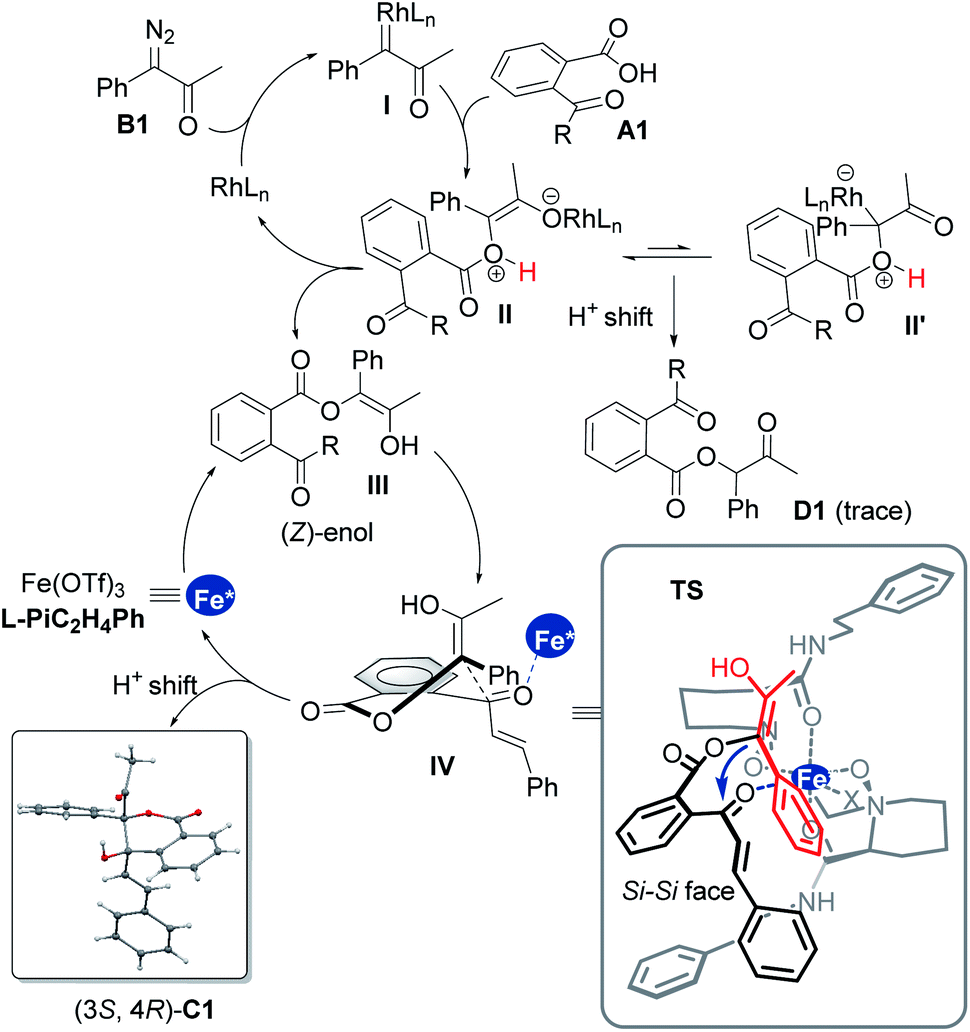
Based on these results and our previous studies,11,15 a possible catalytic cycle with a favorable transition-state model is illustrated in Scheme 3. Initially, nitrogen is extruded from the diazoketone compound to generate a Rh(II)-carbenoid intermediate I, engaging the carboxylic acid to form the rhodium-associated ylide species II′ and its isomer II. Bypassing tautomerization to afford the inertial 1,1-OH insertion byproduct D1, a (Z)-enol intermediate III is generated upon a 1,4-H shift to release the rhodium catalyst.11d,13,14 Furthermore, in the existence of chiral Lewis acid, which could coordinate with the carbonyl group of species III to advance the subsequent aldol reaction through the intermediacy of IV, where a half-chairlike transition state possesses axially disposed (Z)-enol substituents and vinyl substituents of ketones.
 | ||
| Scheme 3 Possible asymmetric catalytic model. | ||
Speeding the aldol cascade and transfer of the stereochemistry lie in the chiral ferric catalyst with external subunits of the ligand. The tetra-dentate coordination of L-PiC2H4Ph to ferric ions leads to an octahedral complex, preserving two similar sites at equatorial position for the coordination of the intermediates. As shown in TS, the two amide arms adpot reverse extension, leaving space to emplace both 2-acetylbenzoic acid A and α-diazoketone B. The Re-face of the ketone species bonds will be blocked by the below amide. Therefore, the favorable Si–Si facial approach produces the (3S,4R)-product C1 as the major isomer with excellent diastereo- and enantioselectivities after tautomerization and regeneration of the ferric catalyst.
Conclusions
In summary, a highly efficient asymmetric cascade O–H insertion/aldol cyclization of ketoacids with diazoketones was achieved with a Rh(II)/chiral N,N′-dioxide-Fe(III) or Sc(III) complex bimetallic relay catalytic system. Asymmetric intramolecular aldol addition of (Z)-enols which are generated from readily accessible ketoacids and α-keto Rh-carbenoids via acyclic carboxylic oxonium ylides was accomplished for the first time. A range of optically active lactone derivatives with two adjacent quaternary–quaternary stereocenters was afforded with good to excellent yields and stereoselectivities. A plausible catalytic cycle along with a working model was proposed to explain the formation of enantioenriched products according to the experimental evidence and single crystal data.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra and X-ray crystallographic data for C1 and F3 are available in the ESI.†Author contributions
J. W. L. performed the experiments and prepared the ESI† and paper. S. Y. W. and C. L. H. repeated some experiments. X. M. F. and X. H. L. supervised the project. X. M. F. and X. H. L. helped with modifying the paper and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 21890723, 21772127 and 21921002) for financial support.Notes and references
- For selected reviews, see: (a) T. Ye and M. A. Mckervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) P. Bulugahapitiya, Y. Landais, L. Parra-Rapado, D. Planchenault and V. Weber, J. Org. Chem., 1997, 62, 1630 CrossRef CAS; (c) Z. H. Zhang and J. B. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS; (d) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC; (e) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- For reviews, see: (a) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (b) Y.-Y. Ren, S.-F. Zhu and Q.-L. Zhou, Org. Biomol. Chem., 2018, 16, 3087 RSC. For selected examples, see: (c) H. Saito, T. Uchiyama, M. Miyake, M. Anada, S. Hashimoto, T. Takabatake and S. Miyairi, Heterocycles, 2010, 81, 1149 CrossRef CAS; (d) Y. Zhu, X. H. Liu, S. X. Dong, Y. H. Zhou, W. Li, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636 CrossRef CAS PubMed; (e) X.-L. Xie, S.-F. Zhu, J.-X. Guo, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 2978 CrossRef CAS PubMed; (f) K. Wang, P. Chen, D. Q. Ji, X. H. Zhang, G. Y. Xu and J. T. Sun, Angew. Chem., Int. Ed., 2018, 57, 12489 CrossRef CAS PubMed; (g) J. Yang, P. R. Ruan, W. Yang, X. M. Feng and X. H. Liu, Chem. Sci., 2019, 10, 10305 RSC; (h) H.-Q. Shen, H.-P. Xie, L. Sun and Y.-G. Zhou, Organometallics, 2019, 38, 3902 CrossRef CAS; (i) M.-L. Li, J.-H. Yu, Y.-H. Li, S.-F. Zhu and Q.-L. Zhou, Science, 2019, 366, 990 CrossRef CAS PubMed; (j) Y. Li, Y.-T. Zhao, T. Zhou, M.-Q. Chen, Y.-P. Li, M.-Y. Huang, Z.-C. Xu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2020, 142, 10557 CrossRef CAS PubMed; (k) W. Yang, M. P. Pu, X. B. Lin, M. Chen, Y. J. Song, X. H. Liu, Y.-D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 9648 CrossRef CAS PubMed.
- For reviews, see: (a) X. Guo and W. H. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed; (b) D. Zhang and W. H. Hu, Chem. Rec., 2017, 17, 739 CrossRef CAS PubMed.
- (a) D. M. Jaber, R. N. Burgin, M. Hepler, P. Zavalij and M. P. Doyle, Chem. Commun., 2011, 47, 7623 RSC; (b) N. J. Thumar, Q. H. Wei and W. H. Hu, Adv. Organomet. Chem., 2016, 66, 33 CrossRef.
- (a) Y. G. Zhu, C. W. Zhai, L. P. Yang and W. H. Hu, Chem. Commun., 2010, 46, 2865 RSC; (b) S. M. Nicolle, W. Lewis, C. J. Hayes and C. J. Moody, Angew. Chem., Int. Ed., 2015, 54, 8485 CrossRef CAS PubMed; (c) W. M. Yuan, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 8410 CrossRef CAS PubMed; (d) S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392 CrossRef CAS PubMed; (e) G. K. R. Alavala, F. Sajjad, T. D. Shi, Z. H. Kang, M. L. Ma, D. Xing and W. H. Hu, Chem. Commun., 2018, 54, 12650 RSC; (f) H. L. Wei, M. Bao, K. Y. Dong, L. H. Qiu, B. Wu, W. H. Hu and X. F. Xu, Angew. Chem., Int. Ed., 2018, 57, 17200 CrossRef CAS PubMed; (g) R. L. Sahani, M. D. Patil, S. B. Wagh and R.-S. Liu, Angew. Chem., Int. Ed., 2018, 57, 14878 CrossRef CAS PubMed; (h) Z. H. Kang, D. Zhang, J. Y. Shou and W. H. Hu, Org. Lett., 2018, 20, 983 CrossRef CAS PubMed; (i) X.-S. Liang, R.-D. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2019, 58, 13885 CrossRef CAS PubMed; (j) Z. H. Kang, Y. H. Wang, D. Zhang, R. B. Wu, X. F. Xu and W. H. Hu, J. Am. Chem. Soc., 2019, 141, 1473 CrossRef CAS PubMed; (k) A. G. K. Reddy, P. Niharika, S. Zhou, S.-K. Jia, T. D. Shi, X. F. Xu, Y. Qian and W. H. Hu, Org. Lett., 2020, 22, 2925 CrossRef PubMed; (l) K. M. Hong, S. L. Dong, X. X. Xu, Z. J. Zhang, T. D. Shi, H. X. Yuan, X. F. Xu and W. H. Hu, ACS Catal., 2021, 11, 6750 CrossRef CAS.
- (a) C. W. Zhai, D. Xing, Y. Qian, J. J. Ji, C. Q. Ma and W. H. Hu, Synlett, 2014, 25, 1745 CrossRef; (b) T.-S. Zhang, W.-J. Hao, N.-N. Wang, G. G. Li, D.-F. Jiang, S.-J. Tu and B. Jiang, Org. Lett., 2016, 18, 3078 CrossRef CAS PubMed; (c) N. P. Massaro, J. C. Stevens, A. Chatterji and I. Sharma, Org. Lett., 2018, 20, 7585 CrossRef CAS PubMed.
- For recent selected examples of O–H insertion reactions to carboxylic acids, see: (a) M. Kitamura, M. Kisanuki, R. Sakata and T. Okauchi, Chem. Lett., 2011, 40, 1129 CrossRef CAS; (b) Z. K. Wang, X. H. Bi, Y. J. Liang, P. Q. Liao and D. W. Dong, Chem. Commun., 2014, 50, 3976 RSC; (c) A. C. Hunter, K. Chinthapally and I. Sharma, Eur. J. Org. Chem., 2016, 13, 2260 CrossRef; (d) Y. L. Zhang, Y. Yao, L. He, Y. Liu and L. Shi, Adv. Synth. Catal., 2017, 359, 2754 CrossRef CAS; (e) I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC; (f) S. Harada, K. Tanikawa, H. Homma, C. Sakai, T. Ito and T. Nemoto, Chem.–Eur. J., 2019, 25, 12058 CrossRef CAS PubMed; (g) C. Empel, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2021, 23, 548 CrossRef CAS PubMed.
- (a) D. Zhang, X. Wang, M. C. Zhang and W. H. Hu, Org. Lett., 2020, 22, 5600 CrossRef CAS PubMed; (b) M. C. Zhang, T. Y. Zhang, D. Zhang and W. H. Hu, Adv. Synth. Catal., 2020, 362, 4662 CrossRef CAS.
- D. Zhang, X. Wang, M. C. Zhang, Z. H. Kang, G. L. Xiao, X. F. Xu and W. H. Hu, CCS Chem., 2020, 2, 432 CrossRef CAS.
- F. Tan, X. H. Liu, X. Y. Hao, Y. Tang, L. L. Lin and X. M. Feng, ACS Catal., 2016, 6, 6930 CrossRef CAS.
- For selected examples, see: (a) J. Li, L. L. Lin, B. W. Hu, X. J. Lian, G. Wang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2016, 55, 6075 CrossRef CAS PubMed; (b) T. Y. Huang, X. H. Liu, J. W. Lang, J. Xu, L. L. Lin and X. M. Feng, ACS Catal., 2017, 7, 5654 CrossRef CAS; (c) D. Zhang, L. L. Lin, J. Yang, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 12323 CrossRef CAS PubMed; (d) J. Yang, C. Q. Ke, D. Zhang, X. H. Liu and X. M. Feng, Org. Lett., 2018, 20, 4536 CrossRef CAS PubMed; (e) H. F. Zheng, Y. Wan, C. R. Xu, Q. Xiong, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327 CrossRef CAS PubMed; (f) C. R. Xu, K. X. Wang, D. W. Li, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 18438 CrossRef CAS PubMed; (g) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2020, 2, 1423 Search PubMed; (h) C. R. Xu, J. L. Qiao, S. X. Dong, Y. Q. Zhou, X. H. Liu and X. M. Feng, Chem. Sci., 2021, 12, 5458 RSC.
- (a) N. Matsumoto, T. Tsuchida, H. Nakamura, R. Sawa, Y. Takahashi, H. Naganawa, H. Iinuma, T. Sawa and T. Takeuchi, J. Antibiot., 1999, 52, 276 CrossRef CAS PubMed; (b) G. E. Croston, R. Olsson, E. A. Currier, E. S. Burstein, D. Weiner, N. Nash, D. Severance, S. G. Allenmark, L. Thunberg, J.-N. Ma, N. Mohell, B. O'dowd, M. R. Brann and U. Hacksell, J. Med. Chem., 2002, 45, 4950 CrossRef CAS PubMed; (c) F. Lehmann, E. A. Currier, R. Olsson, J.-N. Ma, E. S. Burstein, U. Hacksell and K. Luthman, Bioorg. Med. Chem., 2010, 18, 4844 CrossRef CAS PubMed; (d) V. Boucard, G. Broustal and J. M. Campagne, Eur. J. Org. Chem., 2007, 2, 225 CrossRef; (e) I. Shiina and K. Nakata, Medium-Sized Lactones, in Natural Lactones and Lactams, ed. T. Janecki, Wiley-VCH, Weinheim, 2013, pp. 193–227 Search PubMed; (f) E. Kim, S.-M. Lim, M.-S. Kim, S.-H. Yoo and Y. Kim, Nutrients, 2017, 9, 1049 CrossRef PubMed.
- (a) J. L. Wood, G. A. Moniz, D. A. Pflum, B. M. Stoltz, A. A. Holubec and H.-J. Dietrich, J. Am. Chem. Soc., 1999, 121, 1748 CrossRef CAS; (b) Y. S. Chen, S. X. Dong, X. Xu, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2018, 57, 16554 CrossRef CAS PubMed.
- Y. S. Chen, Y. Liu, Z. J. Li, S. X. Dong, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2020, 59, 8052 CrossRef CAS PubMed.
- For reviews of the metal/N,N′-dioxide complex, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791 CrossRef CAS.
- C1 (CCDC: 2004207) and F3 (CCDC: 2085693)..
- (a) Q. Yang, Y. Li, J.-D. Yang, Y. D. Liu, L. Zhang, S. Z. Luo and J.-P. Chen, Angew. Chem., Int. Ed., 2020, 59, 19282 CrossRef CAS PubMed; (b) http://isyn.luoszgroup.com/prediction .
Footnote |
| † Electronic supplementary information (ESI) available: 1H,13C{1H} and 19F{1H}NMR and HPLC spectra (PDF). X-ray crystallographic data for C1 and F3. C1 (CCDC: 2004207) and F3 (CCDC: 2085693). CCDC 2004207 and 2085693. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06025b |
| This journal is © The Royal Society of Chemistry 2022 |