
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfinement synthesis in porous molecule-based materials: a new opportunity for ultrafine nanostructures
Li-Ming
Cao†
,
Jia
Zhang†
,
Xue-Feng
Zhang
and
Chun-Ting
He
 *
*
Key Laboratory of Functional Small Molecules for Ministry of Education, College of Chemistry and Chemical Engineering, College of Life Science, Jiangxi Normal University, Nanchang 330022, China. E-mail: hct@jxnu.edu.cn
First published on 19th January 2022
Abstract
A balance between activity and stability is greatly challenging in designing efficient metal nanoparticles (MNPs) for heterogeneous catalysis. Generally, reducing the size of MNPs to the atomic scale can provide high atom utilization, abundant active sites, and special electronic/band structures, for vastly enhancing their catalytic activity. Nevertheless, due to the dramatically increased surface free energy, such ultrafine nanostructures often suffer from severe aggregation and/or structural degradation during synthesis and catalysis, greatly weakening their reactivities, selectivities and stabilities. Porous molecule-based materials (PMMs), mainly including metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and porous organic polymers (POPs) or cages (POCs), exhibit high specific surface areas, high porosity, and tunable molecular confined space, being promising carriers or precursors to construct ultrafine nanostructures. The confinement effects of their nano/sub-nanopores or specific binding sites can not only effectively limit the agglomeration and growth of MNPs during reduction or pyrolysis processes, but also stabilize the resultant ultrafine nanostructures and modulate their electronic structures and stereochemistry in catalysis. In this review, we highlight the latest advancements in the confinement synthesis in PMMs for constructing atomic-scale nanostructures, such as ultrafine MNPs, nanoclusters, and single atoms. Firstly, we illustrated the typical confinement methods for synthesis. Secondly, we discussed different confinement strategies, including PMM-confinement strategy and PMM-confinement pyrolysis strategy, for synthesizing ultrafine nanostructures. Finally, we put forward the challenges and new opportunities for further applications of confinement synthesis in PMMs.
1. Introduction
Catalysis occupies an extremely important status in industry and is essential for the synthesis of the vast majority of chemicals.1 The core of catalysis science is the construction of highly active catalysts, and its research has shown phenomenal growth.2 Metal nanoparticles (MNPs) are a very important class of nanocatalysts due to their abundant reactive surface atoms and special electronic configurations, which have been widely used in heterogeneous catalysis.2–6 The electronic and crystal structure on the surface of nanomaterials can undergone significant changes. The small size, surface, interface, and macro-quantum tunnelling and quantum size effects make nanomaterials have some excellent properties that traditional materials do not have, such as electromagnetic properties, corrosion resistance, mechanical properties, etc.7 The catalytic properties of MNPs are decisively influenced by their size. Reducing the size of MNPs to the atomic scale can not only vastly improve the specific surface areas, but also expose abundant catalytically active sites and surface dangling bonds, and/or even distort electronic/band structures.3 Particularly, ultrafine MNPs (typically < 5 nm) have been confirmed to have attractive catalytic activity and selectivity, as their surface atom ratio can reach up to over 50%, which could enormously change the surface atomic transport and coordination configurations and/or even change the electron spin conformations and the electron spectrum on the surface.8 Compared with bulk particles, their ultrafine nanostructures provide higher atom utilization, accessible catalytic sites, unique electronic properties, and specific surface/interface configurations, thus leading to superior catalytic reactivity.3 For example, bulk Au exhibits low reactivity for numerous catalytic reactions, yet exhibits efficient CO oxidation activity at sizes smaller than 5.0 nm.7 Bulk Pt shows low reactivity in catalytic hydrogenation, but ultrafine Pt NPs (<4.0 nm) demonstrate 100% conversion efficiency.9 Besides, other important factors determining the activity and selectivity of MNPs are their shape and dispersion. Many studies have shown that the different crystal faces exposed by MNPs exhibit different reactivity. For example, the catalytic rate of the Fe (111) surface surpasses those of its (100) and (110) surfaces by 16 and 418 times, respectively, in the catalytic synthesis of ammonia.3 For the deoxygenation of aromatic epoxides, the Cu (100) surface exhibits more than 90% conversion, while the Cu (111) surface is only 35%.10 However, due to the significant increase in surface free energy, ultrafine MNPs often encounter serious aggregation and poor stability during the synthesis as well as catalytic processes.4 In addition, structural degradation also leads to a precipitous decline in their reactivity and selectivity. Thus, the controllable synthesis of catalytically stable ultrafine MNPs remains a great challenge.Porous molecule-based materials (PMMs) mainly include metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and porous organic polymers (POPs) or cages (POCs), showing high specific surface areas and high porosity, and more importantly, highly tailorable nano/sub-nano-confined space.11–23 Thus, PMMs can be used as excellent carriers for confining and stabilizing MNPs.24–30 Their confinement effect of the nano/sub-nano-pores can not only effectively reduce the agglomeration problem of MNPs, but also can confine the growth of MNPs to obtain ultrafine MNPs.30 In addition, the confinement metal–organic/organic space can modulate the electronic configuration of MNPs and the host–guest chemistry near the reactive sites, boosting their inherent activity and selectivity.24 Moreover, PMMs can be used as extremely useful templates/precursors to derive a variety of ultrafine nanostructures, such as nanoclusters or single atom sites through their unique confinement effect.31 On one hand, PMMs contain a mass of internal nanocages/pores and abundant coordinating atoms, which can isolate metal precursors/metal ions, effectively reducing the diffusion and agglomeration of metals during the pyrolysis process; on the other hand, carbon layers in situ derived from PMMs can be electronic regulators and structural stabilizers for atomically dispersed MNPs.
In this review, we will emphasize the current developments in the confinement synthesis in PMMs for synthesizing atomic-scale nanostructures, e.g. ultrafine MNPs, nanoclusters, and single atoms. Firstly, we introduced typical confinement methods for the synthesis. Subsequently, we discussed the confinement strategy for fabricating ultrafine nanostructures, including the PMM-confinement strategy and PMM-confinement pyrolysis strategy. Finally, we put forward the challenges faced by PMM-confinement research.
2. Confinement methods for synthesis
In fact, it is still a major challenge to embed all MNPs in the nanocages/nanopores of PMMs. This challenge is attributed to the influence of the pore environment, the internal surface characteristics, and the kinetics of wetting and filling of PMMs, which lead to unavoidable loading of some MNPs with uneven size on the surface of PMMs. Generally, the strategies of introducing metal precursors into PMMs mainly include the impregnation method, double-solvent method, chemical vapor deposition method, in situ encapsulation method and so on.25 Subsequently, MNPs can be obtained through various reduction technologies of metal precursors, such as reduction by using NaBH4, hydrogen, ammonia borane, and hydrazine under different conditions.252.1. Impregnation method
Metal precursors can be uniformly inserted into PMMs via the impregnation method with the assistance of solvents. Specifically, the homogeneous solution formed by the metal precursors and the PMMs are thoroughly mixed.32–34 The capillary pressure existing in the system will cause self-infiltration of the solution into the internal spaces of the PMMs. Next, the metal precursors can be easily reduced in situ to MNPs without damaging the confinement frameworks. Theoretically, the internal voids of PMMs can absorb all kinds of metal precursors, and after reduction, uniformly dispersed ultrafine MNPs can be obtained. However, sometimes some metal precursors may be loaded (or diffused) outside the PMMs. Because there is no restriction of the nano-spaces, the metal precursors adsorbed on the surface can easily migrate and agglomerate during the reduction process, thus forming MNPs with a larger size and poor dispersion. Therefore, both the PMMs and metal precursors determine the dispersion and particle size of the prepared MNPs. For example, Huang et al. inserted Pd NPs into UiO-66 through the solution impregnation method.34 The activated UiO-66 powder was immersed in methylene chloride containing a certain concentration of palladium acetate. Pd NCs@UiO-66 was prepared by reducing Pd2+ under a H2/Ar flow at a low temperature. The average size of Pd NCs was less than 1.2 nm and they were uniformly dispersed in UiO-66. Dong et al. used a COF as a confinement framework to synthesize ultrafine Pd NPs (2–5 nm) by soaking the COF in a methanol solution of Pd(NO3)2, and subsequently, re-dispersing in water. Then, Pd@COF was constructed by adding NaBH4. In addition to crystalline porous materials, amorphous POPs are also suitable for introducing MNPs through this method. For example, Dong et al. constructed a POP (PC-POP) by the polymerization of cyanuric chloride and piperazine and introduced Pt4+ into the pores by immersing it in a methanol solution of chloroplatinic acid.35 After reduction with H2 at 250 °C, ultrasmall Pt NPs (2.96 nm) were successfully obtained.2.2. Double-solvent method
In the impregnation method, it is difficult to accurately manipulate the position of MNPs loaded into the internal pores of PMMs. As mentioned above, some metal precursors are probably adsorbed to the external surface of PMMs and easily form agglomerates during the reduction process. An effective alternative strategy is the double-solvent method (DSM), which can be effective in incorporating metal precursors into the voids of PMMs, so as to realize the precise encapsulation of MNPs in their pores without agglomerations.36–38 The different wettability of PMMs in different solvents is central to the DSM. Normally, hydrophobic organic solvents are employed to disperse hydrophilic PMMs, e.g. MOF powders. Then, homogeneous water solution with the dissolved metal precursor is added to the above suspension. Since the hydrophobic organic solvent and water are immiscible, the metal precursors in the system will form stable droplets, which are introduced into the confined channels by inherent capillary pressure.25 The ultrafine MNPs are well encapsulated inside the internal pores after reduction, and no large particles are scattered in the external layer of the matrix. For instance, Xu et al. proposed the DSM to synthesize MNPs within the MOFs' nanopores devoid of agglomerated MNPs on the crystal interface for the first time.36 For a typical step, activated MIL-101 powder was soaked in n-hexane, and a certain concentration of H2PtCl6·6H2O aqueous solution was added under vigorous stirring. After a solid–liquid separation by filtration and thermal reduction with H2, Pt@MIL-101 was prepared with ultrafine Pt NPs of ca. 1.8 nm, being obviously larger than the pore windows (1.2–1.6 nm) yet much smaller than the pore diameter (2.9–3.4 nm), indicating that the Pt NPs were well restricted to the isolated apertures of MIL-101 and meanwhile effectively avoided deposition or diffusion to the crystal surface. This method is not limited to the construction of ultrafine single metal NPs. Bimetallic or even multi-metallic alloy NPs can also be perfectly encapsulated inside MOFs by just changing the single metal aqueous solution to a multi-metal one.37 Since the DSM allows introducing metal ions into the specific pores as well as reducing them in situ in such confined spaces, the sizes of the nanoparticles are significantly restricted by the aperture sizes. Dong et al. selected two similar triazinyl-COFs called COF-Ph and COF-BPh with different aperture diameters of 2.3 and 3.9 nm, respectively, as confined templates to synthesize Pd NPs through the DSM.38 The sizes of the resultant Pd NPs were 0.79 and 0.81 nm, respectively, exhibiting strong correlation with that of the corresponding confined templates.2.3. Chemical vapor deposition method
Chemical vapor deposition (CVD) has been applied to construct MNP/PMM composite materials for a long time.25 This method usually selects organometallic compounds that are easy to sublime at high temperature or have high volatility as metal precursors. Generally, activated PMM powder and volatile metal precursors are packed in two separate bottles and placed together in a Schlenk tube that is kept under vacuum. Volatile organometallic compounds are converted to vapor at a suitable temperature. At this time, PMMs are exposed to metal precursor vapor, which will diffuse into their pores. The speed and extent of the vapor being sucked into the PMMs are determined by the steam pressure. After the final reduction treatment, MNPs can be introduced into PMMs. CVD is a convenient solvent-free process that can load large amounts of MNPs in PMMs. Fischer et al. utilized the CVD method to construct ultrafine Pd or Cu NPs (particle sizes of 1–2 nm) loaded inside MOF-5.39 In a sealed vacuum Schlenk tube, the MOF-5 powder was placed in the vapor of [(η5-C5H5)Cu(PMe3)], [(η5-C5H5)Pd(η3-C3H5)], or [(CH3)Au(PMe3)]. Kempe et al. fabricated highly distributed ultrafine bimetallic PdxNiy NPs (ca. 2–3 nm) embedded in MIL-101 by CVD with the assistance of dynamic vacuum.40 The activated MIL-101 powder and metal precursors of ([(η5-C5H5)Pd(η3-C3H5)] and [Ni(η5-C5H5)2]) were positioned in a divided three-lumen tube, and the system was maintained at 25 °C in a dynamic vacuum of 10−4 mbar for 3–8 h. The dynamic vacuum can greatly reduce the adsorption of metal precursors to the outer layer of MIL-101. The color of the MOF powder became darker after the metal precursor was adsorbed into the pores. PdxNiy@MIL-101 was finally obtained through a subsequent hydrogenolysis treatment. The CVD method can introduce a variety of metal clusters or even single atoms into specific porous structures or surfaces, but it brings about relatively higher requirements for the selection of metal precursors, and some volatile metal precursors are usually highly toxic, such as some metal carbonyl compounds.2.4. In situ encapsulation method
In situ encapsulation is considered to be a prospective method for embedding MNPs inside PMMs.25 Typically, metal precursors are added to the self-assembled system of PMMs. During the crystallization process of PMMs, the metal precursor molecules are encapsulated in the nanocages of PMMs. The frequently used metal precursor molecules mainly include metal acetylacetone complexes, metal carbonyl compounds, polyoxometalates (POMs) and so on. These metal precursors should have suitable molecular sizes, stable chemical structures, and good solubility in specific solvents. For example, Li et al. fabricated polyoxometalate-stabilized single atom site catalysts (SASCs) by the in situ encapsulation method.41 Platinum acetylacetonate Pt(acac)2 and H3PMo12O40 (PMo) were added in the crystallization process of HKUST-1 and the in situ encapsulation of Pt(acac)2 and PMo in its sub-nanopores (∼0.9 nm) was realized. Then, Pt1–PMo@HKUST-1 was constructed by reducing the metal precursor with 5% H2 for 1 h at 150 °C. Although this method is relatively simple and often requires only one step reaction to obtain metal precursor loaded PMMs, it is difficult for the metal precursors to be completely wrapped in the process of self-assembly of PMMs in the solution, which will result in the waste of a large amount of metal precursors. The more serious problem is that when using this method, it is usually difficult to accurately control the amount of metal precursor being encapsulated.3. PMM-confinement strategies to synthesize ultrafine nanostructures
Compared with other traditional confinement synthesis strategies (such as zeolite confinement or carbon confinement), PMM-confinement has the following prominent features. Firstly, PMMs can be designed and tailored to exhibit a wealth of versatile structures and morphologies. Secondly, PMMs create a molecular platform for atomic-level control of nano/sub-nano-confined space and functional groups, which can precisely regulate the size, dispersion and microenvironment of MNPs. Thirdly, PMMs are rich in highly ordered micropores (<2 nm), and their spatial confinement effect can realize the synthesis of highly dispersed ultrafine MNPs, nanoclusters or even single atom sites without using surfactants. In addition, PMMs also possess unique pore surfaces. On one hand, the coordination-confinement of functional groups can limit the nucleation and growth of MNPs. On the other hand, the functional groups can be used as structural stabilizers and electronic regulators for ultrafine MNPs. Both MOFs and COFs/POPs have their own benefits and drawbacks in confinement synthesis. The significant advantage of MOFs is that they are rich in atomically dispersed metal nodes, which can effectively regulate the electronic structure of MNPs or be synergistically involved in catalysis, thereby promoting the catalytic activity or selectivity of the composite systems. Nevertheless, the chemical stabilities of MOFs are relatively poor, especially in some strongly acidic or basic systems, which limit their applications under such harsh conditions. In contrast, COFs/POPs connected by robust covalent bonds usually have excellent chemical stabilities, showing a wide range of applications in confinement synthesis. The PMM confinement strategy can be divided into three types: space-confinement profiting from the unique nanocage/nanopore construction of PMMs, coordination-confinement through the functional groups on PMMs, decorating N, S, and O with lone pair electrons, and ion-confinement based on the charged PMM skeleton for the electrostatic interaction between anions and cations (Fig. 1). These three kinds of confinement effects can synergistically and significantly inhibit the shifting and clustering of MNPs, enabling the effective preparation of ultrafine nanostructures. In addition, profiting from the structural regularity and site uniformity of PMMs, MNPs can be accurately represented via X-ray diffraction, X-ray absorption and/or other spectrum technologies as the active centers of catalysis, which is helpful for the study of the catalytic mechanism.29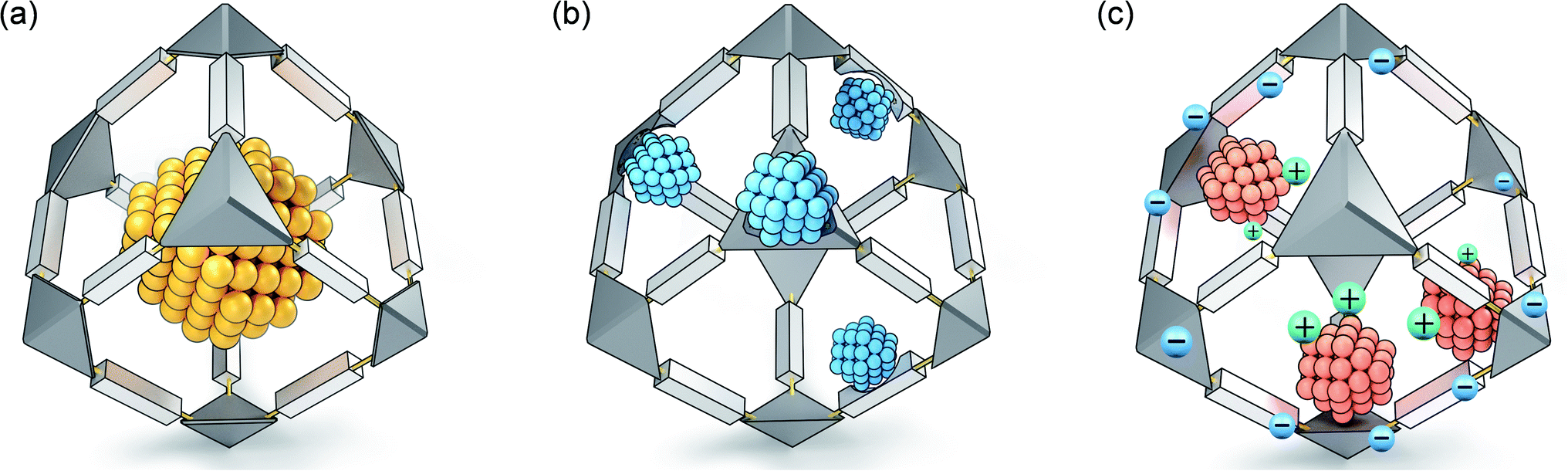 | ||
| Fig. 1 Three typical PMM-confinement strategies: (a) space-confinement, (b) coordination-confinement and (c) ion-confinement. | ||
3.1. Space-confinement
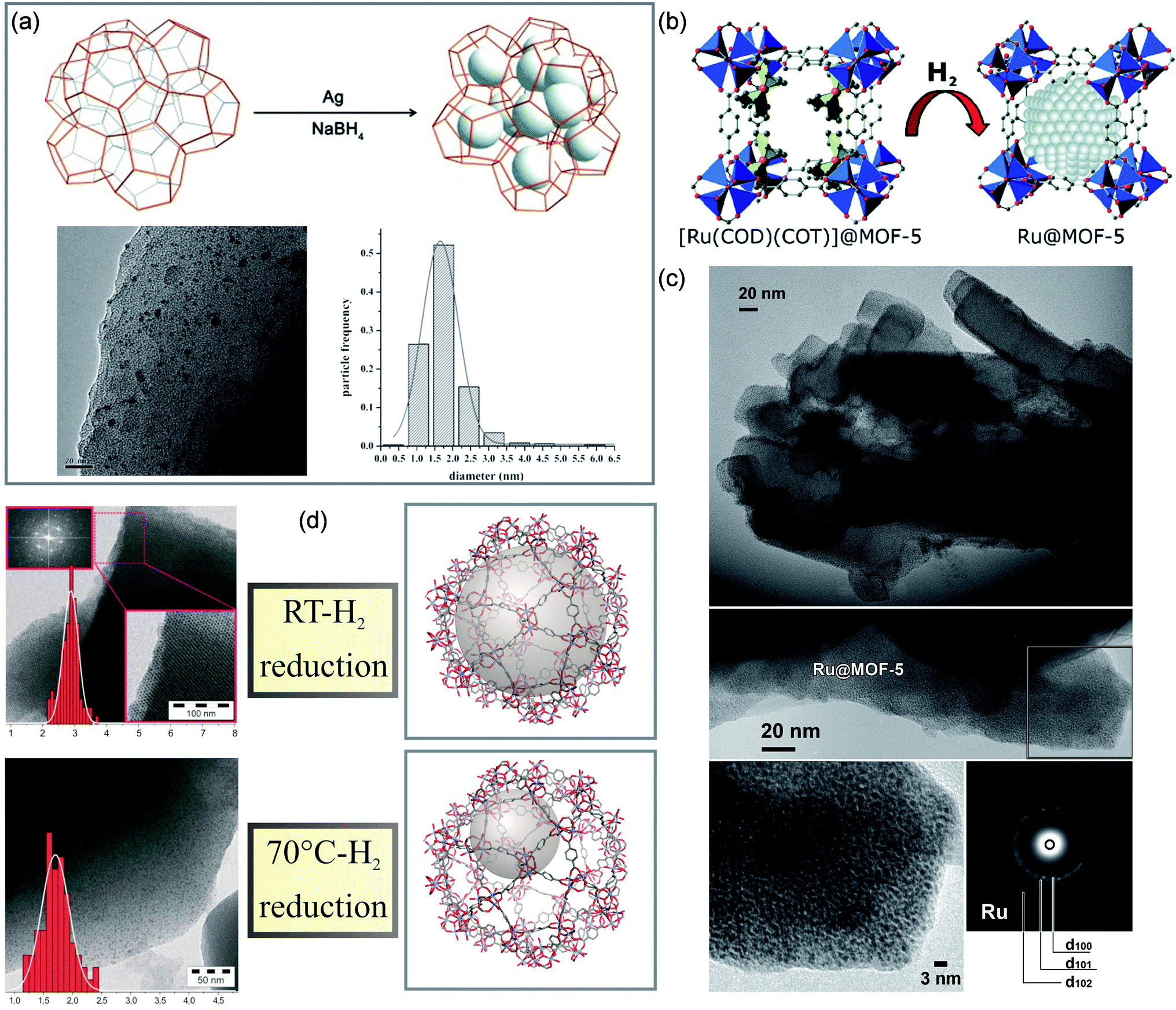 | ||
| Fig. 2 Cases of MOFs for space-confinement. Synthesis and TEM images of (a) Ag@MIL-101. Reproduced with permission.42 Copyright 2015, WILEY-VCH. (b and c) Ru@MOF-5. Reproduced with permission.43 Copyright 2008, American Chemical Society. (d) Pd@MIL-101. Reproduced with permission.44 Copyright 2011, WILEY-VCH. | ||
| MOF | Cavity size (nm) | Preparation method | Metal precursor | Reduction method | Particle size (nm) | Ref. |
|---|---|---|---|---|---|---|
| MIL-101 | 3.0–3.4 | Impregnation | Pd(NO3)2 | H2 | 2.5 ± 0.5 | 45 |
| Impregnation | Pd(NO3)2 | H2 | 2.6 ± 0.5 | 46 | ||
| Impregnation | Pd(NO3)2 | H2 | 1.9 ± 0.7 | 47 | ||
| Impregnation | Pd(NO3)2 | Microwave irradiation/N2H4·H2O | <3.0 | 48 | ||
| Impregnation | Pd(NO3)2 | H2 | 2.6 ± 0.5 | 49 | ||
| Impregnation | Pd(acac)2 | H2 | 2.0–3.0 | 50 | ||
| Impregnation | Pd(NO3)2 | H2 | 2.0–3.0 | 51 | ||
| Double-solvent | Pd(NO3)2 and CoCl2 | NH3BH3 | ∼2.5 | 52 | ||
| Double-solvent | H2PtCl6 | H2 | ∼2.5 | 53 | ||
| Chemical vapor deposition | Pd(C5H5)(C3H5) | H2 | 2.7 | 54 | ||
| Chemical vapor deposition | (η3-C3H5)Pd(η5-C5H5) and Ni(η5-C5H5)2 | H2 | ∼3.5 | 40 | ||
| Impregnation | AgNO3 | NaBH4 | 1.4 ± 0.4 | |||
| Al-MIL-100 | 2.5–3.0 | Impregnation | H2PtCl6 | H2 | 1.8 ± 0.4 | 55 |
| UiO-68 | 2.56 | Impregnation | Pd(NO3)2 | NaBH4 | <2.0 | 43 |
| MOF-5 | ∼1.5 | Chemical vapor deposition | Ru(cod)(cot) | H2 | 1.5–1.7 | 56 |
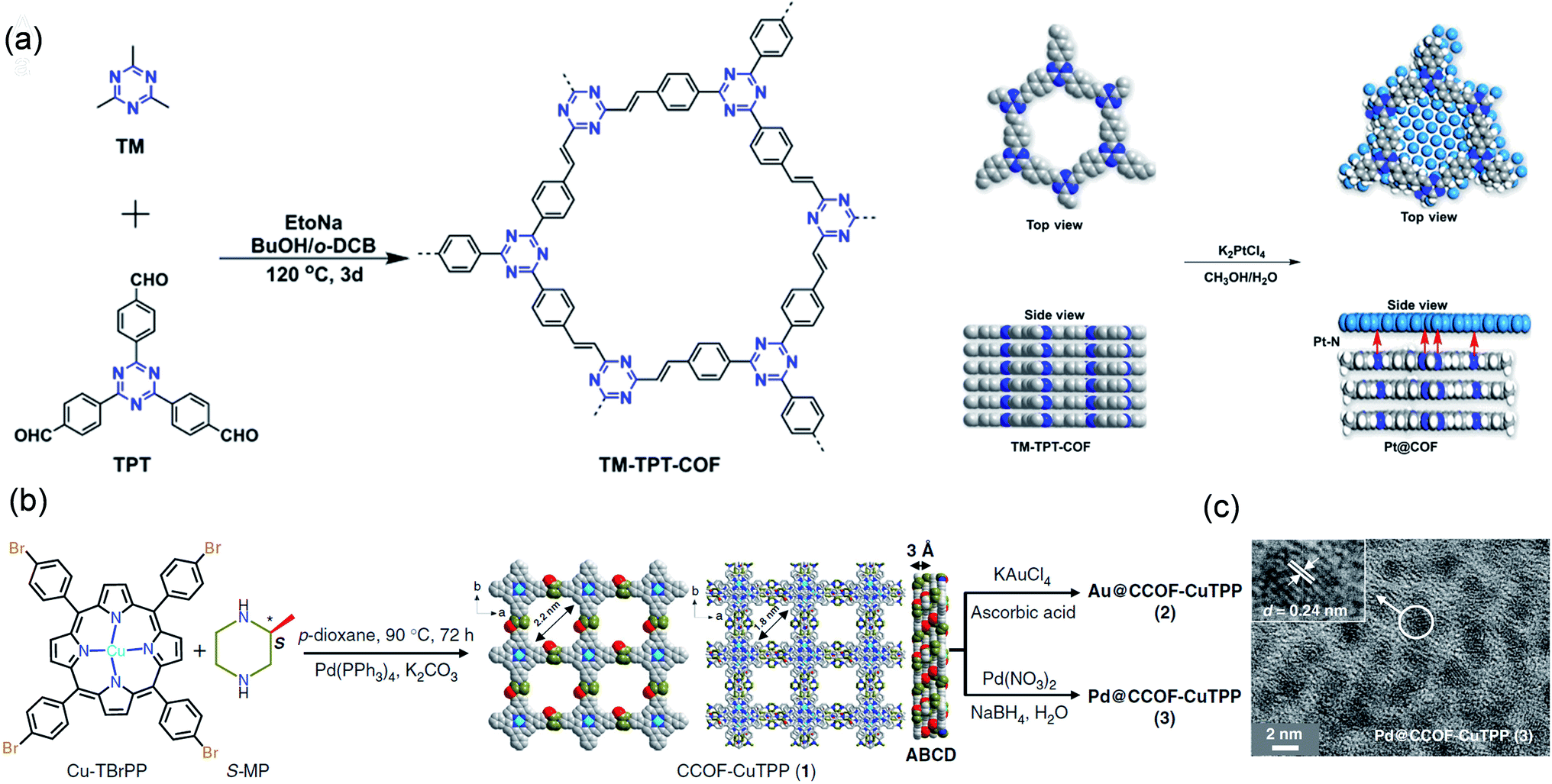
Jiang et al. constructed a TM-TPT-COF with a coordinated nitrogen-rich skeleton via using 2,4,6-trimethyl-1,3,5-trizaine (TM) and 1,3,5-tris-(4-formylphenyl)-triazine (TPT) as molecular building blocks (Fig. 3a).60 Its Brunauer–Emmett–Teller (BET) surface area was as high as 810 m2 g−1, and it possessed abundant nanopores, which could restrict the growth of Pt NPs, thereby preparing highly dispersed ultrafine Pt NPs (ca. 2.10 nm). The catalyst showed excellent oxygen reduction reaction (ORR) activity. In 0.1 M HClO4 solution, the catalytic onset potential and the half-wave potential (E1/2) were 1.05 and 0.89 V, respectively. Dong et al. synthesized COF-CuTPP through the polymerization of 2-methylpiperazine and copper tetrabromophenolphthalein (Cu-TBrPP) (Fig. 3b). Subsequently, Pd@CCOF-CuTPP was constructed by impregnation and reduction. COF-CuTPP was rich in nanopores, which could limit the growth of Pd NPs. Therefore, the as-prepared Pd NPs (2–5) nm were highly dispersed in COF-CuTPP (Fig. 3c).61 Pd@CCOF-CuTPP displayed a conversion efficiency of up to 98% in Henry and A3-coupling reactions.
Zhang et al. fabricated highly dispersed ultrafine Pd NPs through the space-confinement in a triazinyl-pentaerythritol POP (TP-POP) (Fig. 4a).71 TP-POP has a wealth of nanopores (pore size of ca. 3.0 nm) and excellent structural stability. Pd@TP-POP was prepared by the DSM. Firstly, TP-POP was immersed in H2O/CH2Cl2 solution containing Pd(OAc)2. Subsequently, Pd@TP-POP was constructed by reduction with NaBH4. These as-constructed Pd NPs exhibited a size range of 1.4–2.8 nm and were highly dispersed in the nanopores of TP-POP instead of appearing in the outer layer (Fig. 4b), indicating that the space-confinement of the nanopores plays a key role. The optimized Pd@TP-POP showed good catalytic properties in the reduction of nitro and aldehyde groups with high convertibility (>98%) and selectivity (100%). Xie et al. synthesized ultrafine Pd NPs using a series of microporous POPs (Fig. 4c).72 Pd(PPh3)4 was employed to facilitate the association of organic bromine monomer and 1,4-phenylenediboronic acid for constructing the POPs. In this process, Pd NPs were formed through the in situ transformation of Pd(PPh3)4 and were embedded in the pores of the POPs. Thanks to the restriction of a large number of nanopores in these POPs, Pd NPs (1.3 nm) presented high dispersion in the organic matrix (Fig. 4d). The ultrafine nanostructures endowed the catalysts with highly exposed catalytic sites, showing outstanding catalytic efficiency in the reduction of p-nitrophenol with a conversion rate of up to 99.9%.
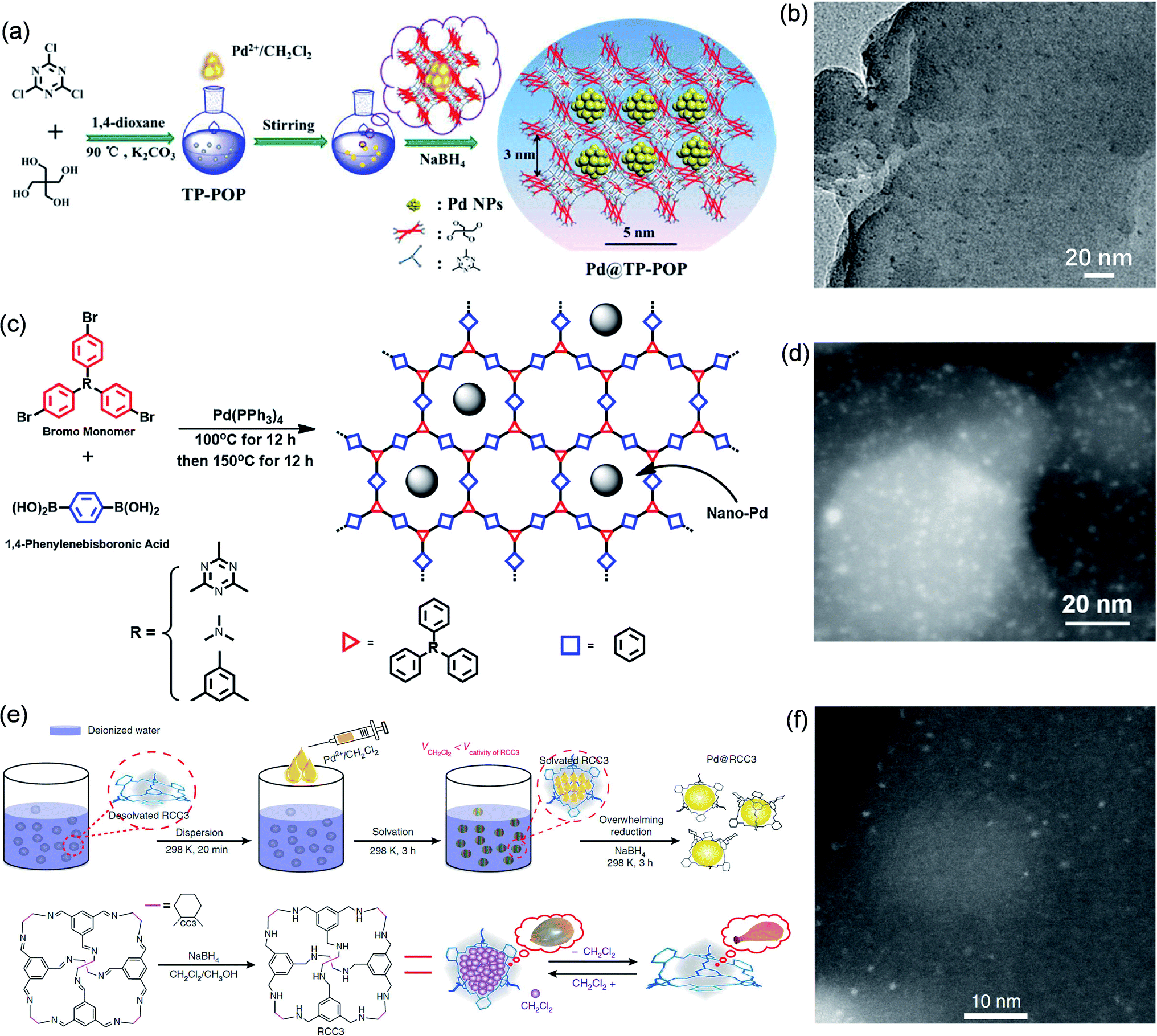 | ||
| Fig. 4 Cases of POPs or POCs for space-confinement. Construction schematic and TEM images of (a and b) Pd@TP-POP. Reproduced with permission.71 Copyright 2018, The Royal Society of Chemistry. (c and d) Pd@POP. Reproduced with permission.72 Copyright 2019, Elsevier. (e and f) Pd@RCC3. Reproduced with permission.73 Copyright 2018, Springer Nature. | ||
Xu et al. employed POCs as confinement templates for the synthesis of ultrafine Pd NPs (Fig. 4e).73 Firstly, the chiral imine cage CC3R was synthesized through the condensation reaction of the organic monomer 1,3,5-triformylbenzene and (R,R)-1,2-diaminocyclohexane. Next, the organic molecular cage (RCC3) was prepared by reducing the pre-fabricated CC3R. Since the cage of RCC3 is hydrophobic, they used the DSM to introduce Pd(OAc)2 into it. Due to the space confinement of the 0.7 nm organic cages, the prepared Pd nanoclusters encapsulated in the cage have an average diameter of just 0.72 nm (Fig. 4f). They found that the encapsulated Pd nanoclusters not only did not destroy the covalent structure of the cages, but also helped to increase the porosity of RCC3. Confining Pd nanoclusters in the cage cavity could largely reduce particle aggregation and enhance particle stability. Moreover, the Pd nanocluster core was covered by a discrete and soluble RCC3 shell, which exhibited excellent solubility and extremely high dispersibility in solvents, so that it had more accessible metal active centers in the liquid phase reaction. Therefore, Pd@RCC3 exhibited strengthened catalytic efficiency in a variety of reactions, including catalytic hydrogenation of nitroaromatics (up to 100% conversion), hydrogen production of ammonia borane (turnover frequency (TOF): 176 min−1), and reduction of organic dyes.
3.2. Coordination-confinement
 | ||
| Fig. 5 Cases of MOFs for coordination-confinement. Schematic illustration of constructing (a) Pd0/UiO-67 and (b) Pd@UiO-67 (C: black or grey, N: blue, O: red or green, Zr: cyan, Cl: purple, and Pd: orange; all the H atoms have been omitted for clarity). | ||
MOFs not only can be used as stabilizers for MNPs, but also provide a synergistic platform for their functional groups and MNPs to improve the catalytic performances. For instance, Pd NPs were anchored in the apertures of NH2-MIL-125 and MIL-125 through photo-assisted and ion-exchange digestion approaches (Pd-MIL-125 and Pd–NH2-MIL-125).76 The introduction of amino groups made the distribution of NPs more uniform, while reducing the size of Pd NPs from 5.5 to 3.1 nm. The Pd–NH2-MIL-125 catalyst exhibited better conversion efficiency than Pd-MIL-125 for hydrogen generation from formic acid, and the hydrogen production of Pd–NH2-MIL-125 and Pd-MIL-125 in the same time was 48.1 and 17.8 μmol, respectively, due to the introduction of amino groups rather than the size of Pd NPs. Being a proton remover, the basic amino groups facilitate the dissociation of the O–H bond by forming –+HNH2 species and benefit the β-hydride elimination from the reaction intermediate. Fei et al. successfully encapsulated ultrasmall Au NPs (1.8 ± 0.2 nm) into an NHC-modified MOF (Au-NC@UiO-68-NHC).7713C solid-state NMR spectroscopy confirmed that gold precursors can form strong chemical bonds with NHC moieties as nucleation sites, and finally formed ultrafine gold clusters inside the MOF pores through the coordination-confinement. As a control, an amine functionalized MOF was used to load Au NPs under identical conditions (UiO-68-NH2/Au), but Au NPs were observed to agglomerate at the outer surface of UiO-68-NH2 with a size of 5–20 nm, caused by the weak coordination between Au and amine groups. The covalent bonds between Au-NCs and NHCs enhanced their synergistic catalysis with MOFs and effectively promoted charge separation. The LSPR (local surface plasmon resonances) enhanced the light-catalytic CO2 conversion activity of Au-NC@UiO-68-NHC, with a CO yield of 57.6 μmol g−1 h−1, significantly superior to that of UiO-68-NH2/Au (14.3 μmol g−1 h−1).
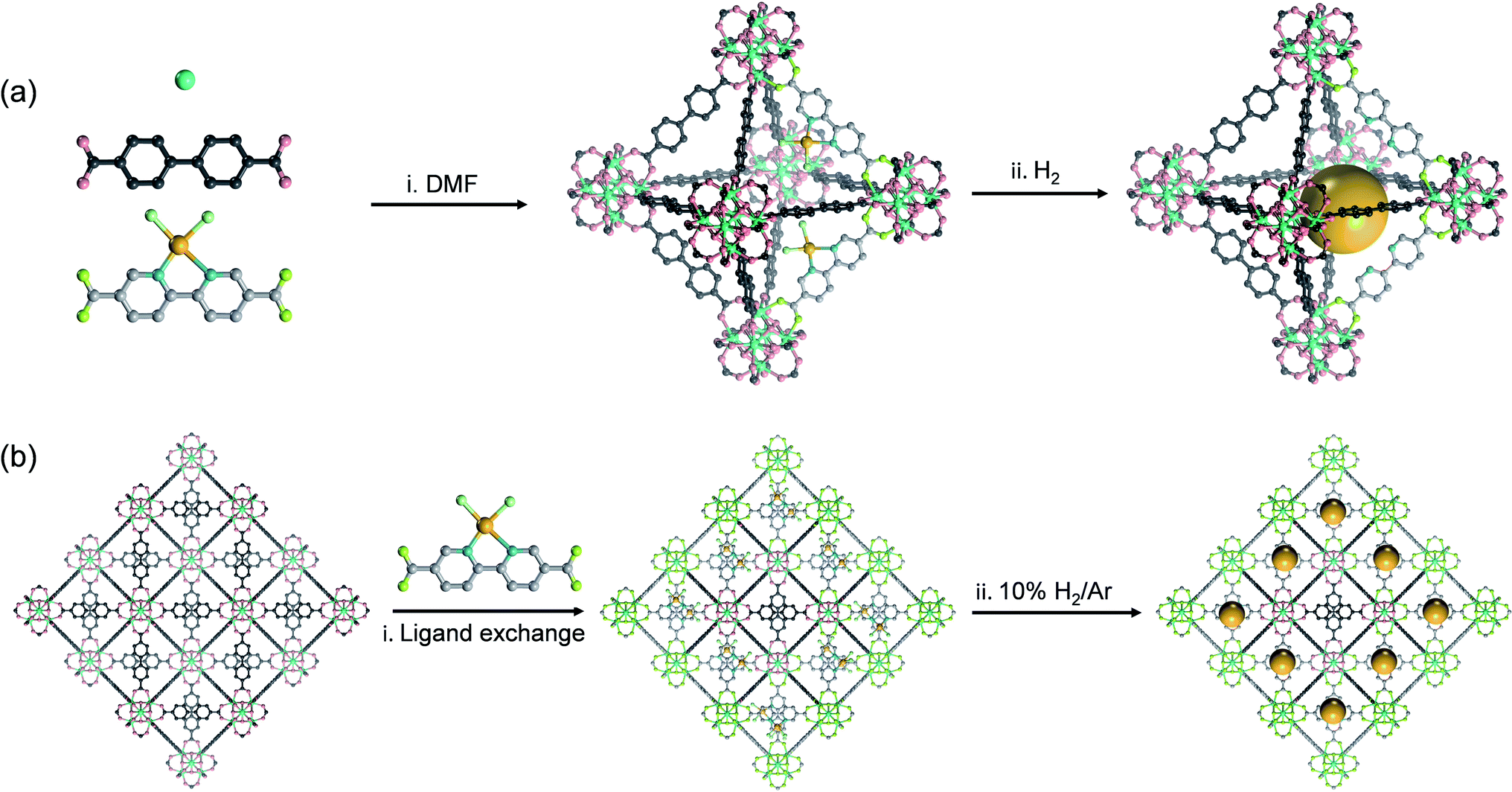
If the sizes of the MNPs are reduced to the extreme, single atoms (SAs) can be obtained, where the utilization efficiency of the atoms can reach 100 percent. Since the concept of SACs was put forward in 2011, this type of material has quickly become a hot research topic in the field of catalysis because of their exceptional atom utilization efficiency, controllable coordination environment and outstanding performance, which bridges the homogeneous and heterogeneous catalysis. However, SAs have higher surface energy and more easily migrate and agglomerate than MNPs, and thus the preparation of stable SACs remains a great challenge. MOFs can stabilize single metal sites through coordination-confinement, which provides an effective strategy to prepare a series of high-activity and reusable heterogeneous SAs@MOF catalysts. SAs@MOF catalysts can be divided into two types according to different single-atom coordination sites: metal node coordination and ligand coordination. Among them, metal nodes that usually serve as Lewis acid sites can create an electron-deficient environment for SAs and regulate their electronic structures. The coordination of SAs to the metal nodes in MOFs can be achieved through three strategies: solvent-assisted ligand incorporation, hydroxyl coordination, and metal ion exchange (Fig. 6). For instance, the Cr3(μ3-O) node in MIL-101(Cr) can be modified with amines and pyridyl molecules.78 Nguyen et al. modified MIL-101 with dopamine and subsequently interacted with vanadyl acetylacetonate to prepare a vanadium-anchored MOF (V(dop)-MIL-101), and the proportion of V/Cr was about 0.13 determined through ICP-OES (Fig. 6a). V(dop)-MIL-101 showed good conversion efficiency for the oxidation of organic sulfides and could be adjusted to specifically provide the sulfoxide product, and the value of TON to yield sulfoxide and sulfone was 73 and 13, respectively, in the 3rd cycle. NU-1000 was constructed from Zr6(μ3-O)4(μ3-OH)4(H2O)4(OH)4 (abbreviated to Zr6) and 1,3,5,8-(p-benzoate) pyrene. The hydroxyl groups enriched in the metal nodes could coordinate with bis(tert-butylimido)bis(dimethylamino)molybdenum(VI) (Mo(am)2(im)2) through thermal deposition, and after coming into contact with oxygen for full oxidation, molybdenum(VI) oxide was placed on the Zr6 node (denoted as Mo-SIM).79 Mo(VI) atoms were coordinated with oxygen-containing functional groups in Zr6, and ICP-OES analysis revealed 2.8 Mo per Zr6 (Fig. 6b). Mo-SIM achieved 93 ± 2% conversion in catalyzing cyclohexene epoxidation. As nodes of the Ti8-BDC MOF, Ti8(μ2-O)8(μ2-OH)4 can coordinate with Co2+ through ion exchange after Ti8-BDC was deprotonated with LiCH2SiMe3.80 ICP-MS analysis revealed the presence of 0.7 Co per Ti8 node in this single-site Co catalyst named Ti8-BDC-CoCl. TiIII2TiIV6-BDC-CoH was further prepared from Ti8-BDC-CoCl with NaBEt3H treatment, which has high catalytic activity for the hydrogenation of various heteroarenes (Fig. 6c). The yields of TiIII2TiIV6-BDC-CoH for the hydrogenation of pyridine, hindered pyridines (including 2,6-lutidine and 2,4,6-collidine), and quinoline derivatives (such as 3-methylquinoline and 2,6-dimethylquinoline) were all over 90%.
 | ||
| Fig. 6 The confinement strategies for SAs to the metal nodes in MOFs: (a) solvent-assisted ligand incorporation (H atoms have been omitted for clarity), (b) hydroxyl coordination and (c) metal ion exchange (C: black, N: blue, O: red, Cr: violet, Zr: cyan, Al: green, V: purple, Mo: magenta, Ti: olive, Cl: light pink, and H: white). | ||
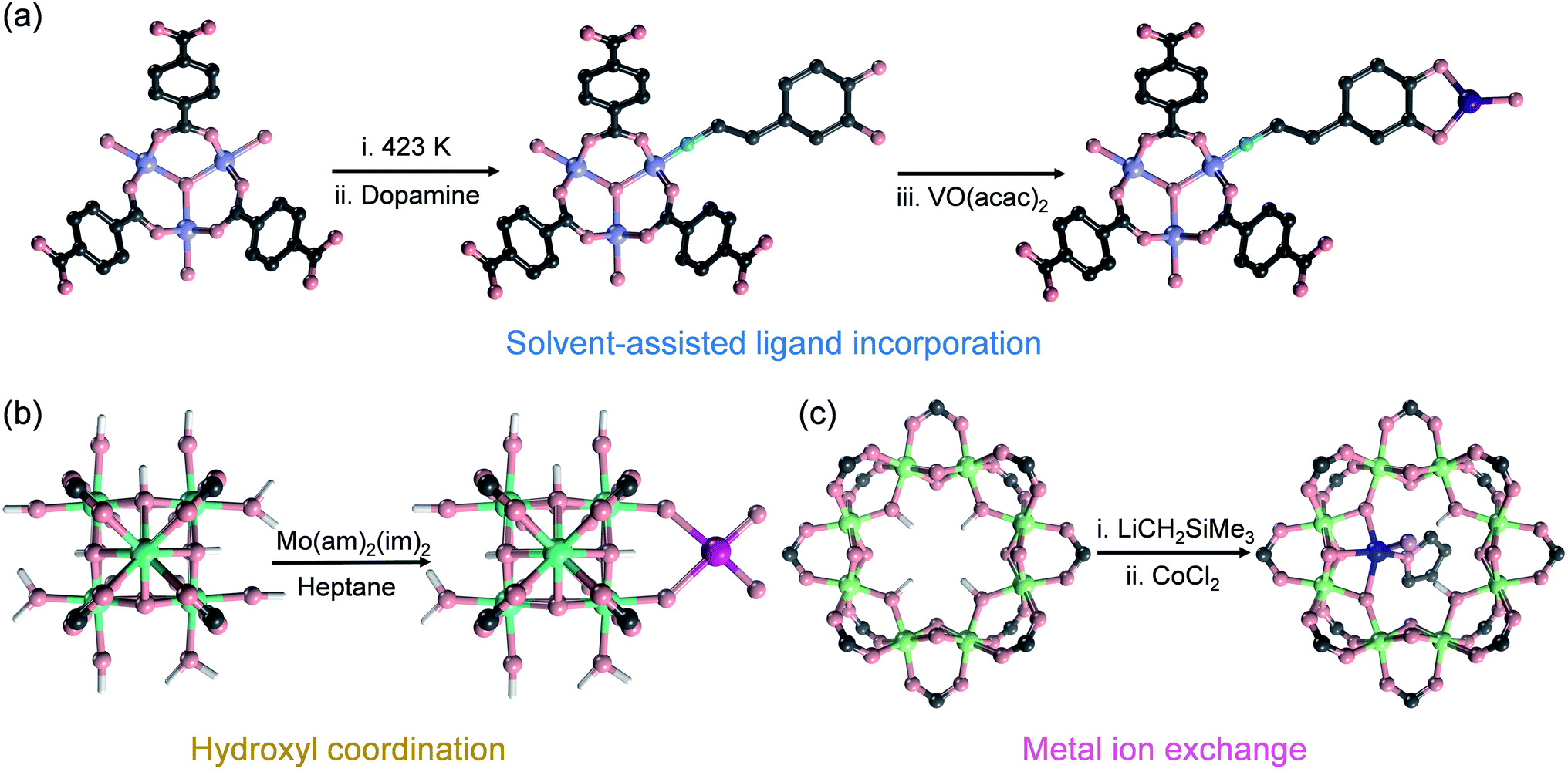
Salicylaldimine-, monophosphine-, β-diketimine-, and bipyridine-based ligands have a strong binding affinity to metals, which is beneficial for stabilizing single metal sites. At the same time, strategies such as post-synthesis modification, solvent-assisted linker exchange, and post-synthetic deprotection are conducive to the synthesis of such SAs@MOF catalysts (Fig. 7). Wang et al. reported that copper ions could be captured on the ligands of UiO-66-NH2 by –NH2, and Cu SAs were further anchored on MOFs (Cu SAs/UiO-66-NH2) after a photoinduced process (Fig. 7a).81 Cu SAs/UiO-66-NH2 can promote the reduction of carbon dioxide as an excellent photocatalyst, with the conversion rates of carbon dioxide to methanol and ethanol of 5.33 and 4.22 μmol h−1 g−1. Fontecave et al. exchanged the partial ligand of UiO-67 (bpydc, 2,2′-bipyridine-5,5′-dicarboxylic acid) with Cp*Rh(bpydc)Cl2 (Cp* = pentamethylcyclopentadiene) to generate a novel MOF loaded with rhodium SAs (Cp*Rh@UiO-67) (Fig. 7b).82 Cp*Rh@UiO-67 can be used as a photosensitive catalyst to convert CO2 to formate with a TOF of 7.5 h−1, and it could be recycled up to 6 times. Lin et al. modified NacNac (β-diketiminate) in Zr6O4(OH)4(TPDC-NH2)6, an amino-functionalized UiO-type MOF, by reaction with amino groups. They further utilized NacNac to coordinate with metal ions and then reduction with NaBHEt3 to exploit NacNac-M-MOF (M = Fe, Cu and Co) (Fig. 7c).83 NacNac-Fe-MOF, NacNac-Cu-MOF, and NacNac-Co-MOF can catalyze the hydrogenation of alkenes, amination of alkyl azides, and amination of cyclohexene, respectively. Wang et al. synthesized a Cu SAC by utilizing the coordination-confinement of N-heterocyclic carbene (NHC) encapsulated in UiO-67.84 Firstly, the NHC (1,3-dibenzyl-1H-imidazole-3-ium bromide, 2Bn·HBr) molecule was in situ encapsulated in UiO-67 (2Bn@UiO-67), thanks to the perfect size-matching between the UiO-67 cavity and 2Bn·HBr. Subsequently, a single-atom site Cu catalyst (2Bn-Cu@UiO-67) was constructed through the coordination of Cu and carbene C. 2Bn-Cu@UiO-67 exhibited excellent electrocatalytic performance for the conversion of CO2 to CH4 with a Faraday efficiency of 81%.
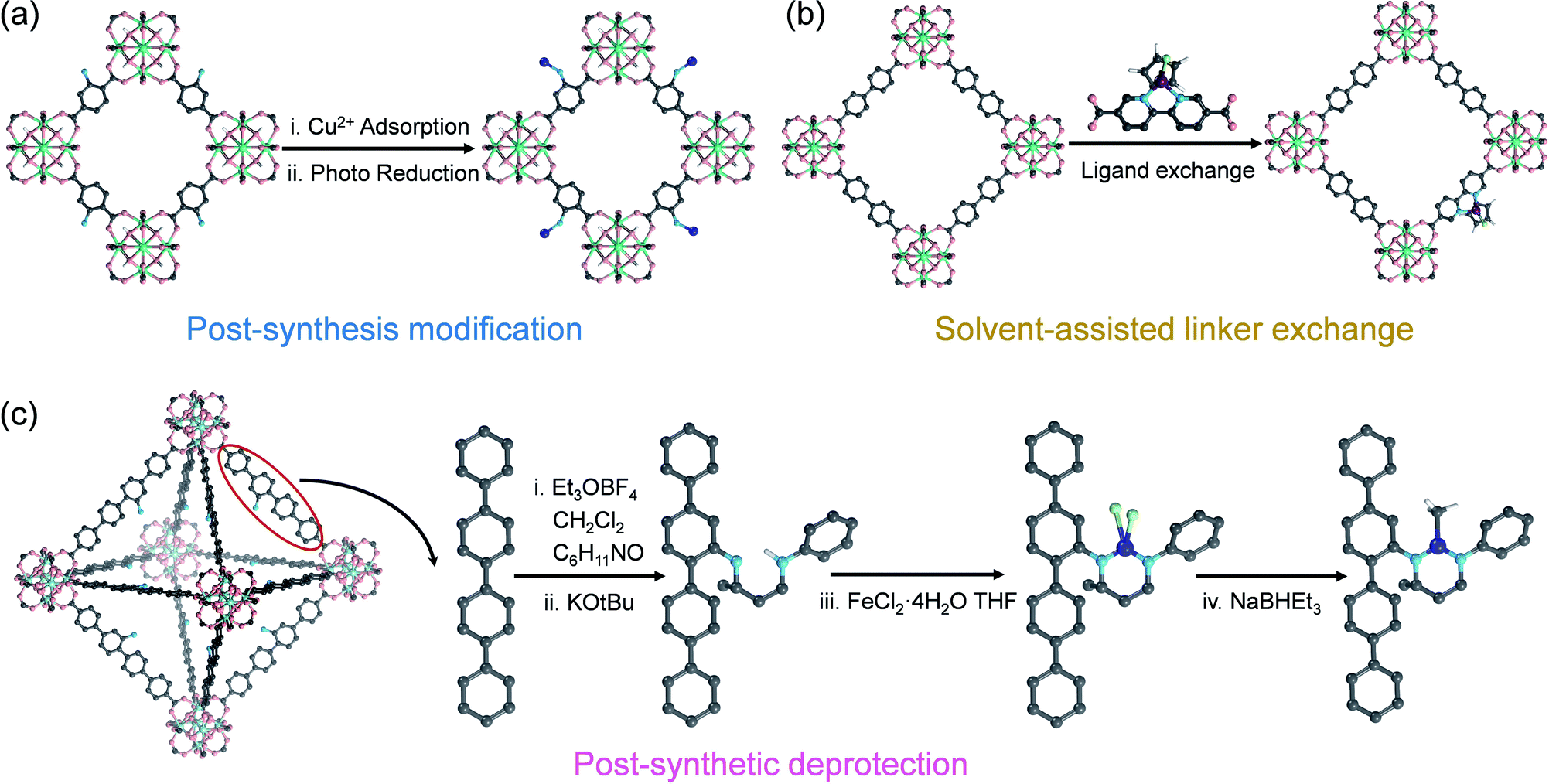 | ||
| Fig. 7 The confinement strategies for SAs to the ligands in MOFs: (a) post-synthesis modification, (b) solvent-assisted linker exchange and (c) post-synthetic deprotection (C: grey, N: blue, O: red, Zr: cyan, Cu: orange, Rh: wine, Cl: green, and Fe: dark blue; the H atoms on the frameworks have been omitted for clarity). | ||
Thiol/thioether groups have good coordination ability, can anchor a variety of metals, and are often modified to the side chains of COFs to confine the synthesis of MNPs and improve their catalytic activity and stability.93 Lu et al. successfully modified thiol chains in the COF channel using a post-modification synthesis strategy (Fig. 8a).94 Thanks to the strong binding force between –SH and Au, the thiol group in the pore could act as a nucleation site for Au NPs and limit the growth of Au nanoclusters. The as-prepared Au NPs with a very small diameter of 1.8 nm were uniformly distributed in Au@COF-S-SH. In contrast, the Au nanoclusters on COF-V (without thiol chain modification) tended to agglomerate with a larger size (Fig. 8b). Moreover, photoelectrochemical experiments and calculation of binding energy had shown that Au-S-COF bridging can improve the charge separation efficiency and keep Au nanoclusters highly dispersed under long-term light exposure. Therefore, Au@COF-S-SH exhibited enhanced performance and stability in the photodegradation of Rhodamine B. Zhang et al. constructed a thioether-containing COF (Thio-COF) via a bottom-up synthesis strategy, and then highly dispersed ultrafine Pt NPs (1.7 nm) and Pd NPs (1.78 nm) were synthesized by coordination-confinement from the thioether groups in the COF (Fig. 8c and d).95 Both PtNPs@Thio-COF and PdNPs@Thio-COF exhibited significant conversion efficiency (>95%) in reducing nitrophenol.
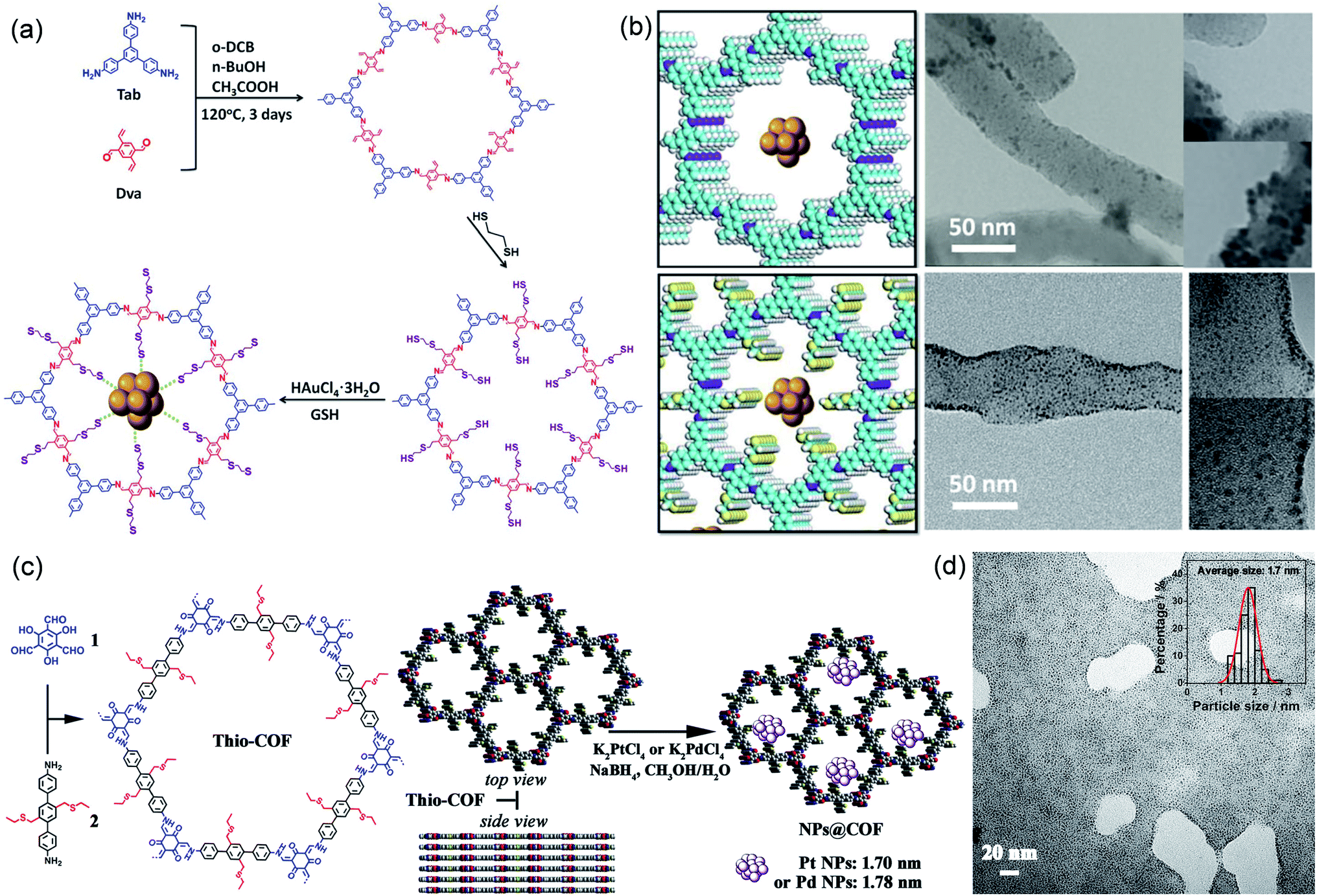 | ||
| Fig. 8 Cases of COFs for coordination-confinement. Construction schematic and TEM images of (a and b) Au@COF-S-SH. Reproduced with permission.94 Copyright 2020, WILEY-VCH. (c and d) Pt NPs@Thio-COF. Reproduced with permission.95 Copyright 2017, American Chemical Society. | ||
Nitrogen atoms can not only provide lone-pair electrons, but also have relatively small electronegativity and easily give electrons. Therefore, they have strong coordination ability and can coordinate with many kinds of metal ions. Amino groups and imines are also often used to anchor and limit the nucleation and growth of MNPs.96–102 For instance, Yue et al. adopted a COF possessing amine groups (COF-300) to confine the synthesis of ultrafine Pd NPs for benzyl alcohol oxidation.103 They prepared three different COFs via controlling the molar ratio of tetrakis(4-anilyl)methane (TAM) and terephthaldehyde (TPA) and studied the confinement effects of the three COFs on Pd NPs. With the optimized proportion of TAM and TPA, the prepared COF-300-1114 showed the best confinement effect. The size of the highly dispersed ultrafine Pd NPs was just 1.58 nm. The formation of ultrafine nanostructures was because when the organic monomer TAM increased, an excessive number of suspended amino groups could be introduced, which could act as nucleation sites for Pd and limited their migration and agglomeration during the reduction process. Pd/COF-300-1114 showed a good catalytic effect and stability for benzyl alcohol oxidation with a rather high conversion rate of 97% and selectivity as high as 99%. The high activity of Pd/COF-300-1114 was attributed to the existence of amino groups, which stabilized the Pd NPs and accelerated their activity through amino donor electrons.
Phosphine groups have high binding affinity to various metal ions and are widely used as organic ligands to prepare metal–organic compounds. The ordered phosphine groups introduced into COFs can not only anchor metal ions or MNPs, but also make the metal sites uniformly dispersed in the COF. For instance, Zhang et al. fabricated a phosphine-based COF (Phos-COF-1) for the first time and used its phosphine coordination site to control the synthesis of a series of highly dispersed ultrafine MNPs.104 Because triphenylphosphine (PPh3) has good binding affinity to various metal ions, the orderly distributed PPh3 functional groups in porous Phos-COF-1 can supply nucleation sites for MNPs. Attributed to the coordination and restriction of PPh3 in Phos-COF-1, the average sizes of the as-prepared Pd, Pt and Au were 1.62, 2.06, and 1.78 nm, respectively, and they were highly dispersed and evenly distributed in Phos-COF-1. MNPs@Phos-COF-1 showed good transformation efficiency in the reduction of 1-bromo-4-nitrobenzene and nitrophenol, coupling reaction, and tandem coupling and reduction of nitroiodobenzene. Moreover, MNPs@Phos-COF-1 was easy to recycle and can well maintain the nanostructure as well as high stability.
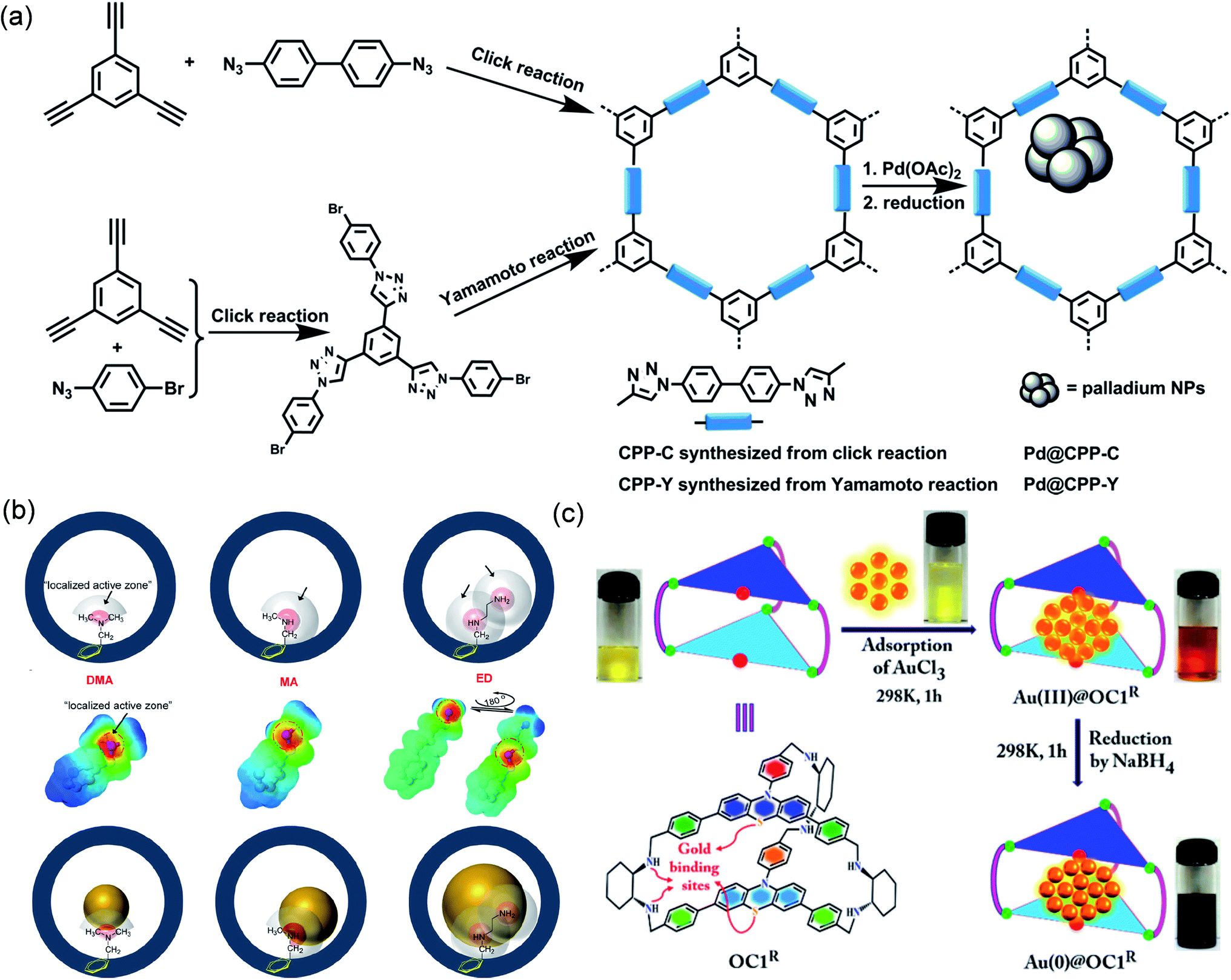 | ||
| Fig. 9 Cases of POPs or POCs for coordination-confinement. Schematic illustration of constructing (a) Pd@CPP-Y or Pd@CPP-C. Reproduced with permission.107 Copyright 2015, American Chemical Society. (b) Ag@OMP-DMA, Ag@OMP-MA, or Ag@OMP-ED. Reproduced with permission.114 Copyright 2019, American Chemical Society. (c) Au@OC1R. Reproduced with permission.119 Copyright 2017, American Chemical Society. | ||
The electron-rich amino group can coordinate with a variety of metal ions and is also often used for the confinement-synthesis of MNPs.108–113 Studies have found that the spatial site resistance and electron-giving capability of the amino substituents also affect the construction of MNPs. Wu et al. fabricated three different types of amino-functional ordered mesoporous polymers (OMP-DMA, OMP-MA, and OMP-ED), and explored their effects on the confinement-synthesis of Ag NPs (Fig. 9b).114 The aminos can be coordinated with Ag+, and their different types determine the activity range of their coordination. They called the enriched electron zones near N the “localized active zones (LAZs)”. The properties of LAZs depended on the substituents' site resistance and electron-giving capability. They found that modified electron-rich amines regulated the growth of Ag NPs by creating LAZs. As the extent of the LAZ surrounding N increased, excess Ag was confined to the area, which led to producing non-uniform larger MNPs. The DMA exhibited the smallest LAZ due to the presence of two –CH3 with the largest steric hindrance. Therefore, the average size of Ag NPs synthesized by OMP-DMA, OMP-MA and OMP-ED was 6, 9, and 11 nm, respectively. Moreover, they synthesized NH2-functionalized OMP (NOMP) through a bottom-up synthesis strategy. Because –NH2 had a smaller LAZ volume, the average size of Ag NPs in the as-synthesized Ag@NOMP was only 3–4 nm. Thus, the yield of Ag@NOMP in converting alkynes into alkynyl carboxylic acids reached more than 95%.
Upon introducing a variety of electron-rich coordination atoms into the organic cage, the coordination-confinement can cooperate with the space-confinement of the organic cage to limit the growth of MNPs.115–118 Mukherjee et al. reported the use of an organic cage OC1R to synthesize ultrafine Au NPs as a heterogeneous photocatalyst (Fig. 9c).119 Calculations showed that the cavity size of OC1R was between 1.67 and 1.82 nm. OC1R contained vicinal diamine clefts and cyclic thioether moieties, in which electron-rich N and S can coordinate with Au3+ to provide nucleation sites for Au NPs and stabilize metal active centers. The average particle size of the prepared ultrafine Au NPs was ca. 2.0 nm. 1H diffusion-ordered spectroscopy (DOSY) NMR revealed that free cages OC1R and Au@OC1R had similar diffusion coefficients, indicating that OC1R and Au@OC1R had comparable shapes and sizes, thus confirming that each Au NP was anchored in a single cage cavity instead of being combined by multiple cages. In addition, they found that the loading of Au in Au@OC1R was as high as 68 wt% without agglomeration. In contrast, when Au loaded by other methods reaches 20 wt%, significant agglomeration of MNPs will often occur. The formation of highly dispersed ultrafine nanostructures was attributed to not only the space-confinement of the ultrafine nanocage, but also the coordination-confinement of abundant N and S atoms. Au@OC1R can be used as a photocatalyst for the selective reduction of nitroaromatics to azo compounds, showing over 99% conversion and over 90% selectivity. Moreover, Au@OC1R exhibited excellent structural stability and reusability, and even though it was repeatedly used 5 times, there was no agglomeration of Au NPs.
3.3. Ion-confinement
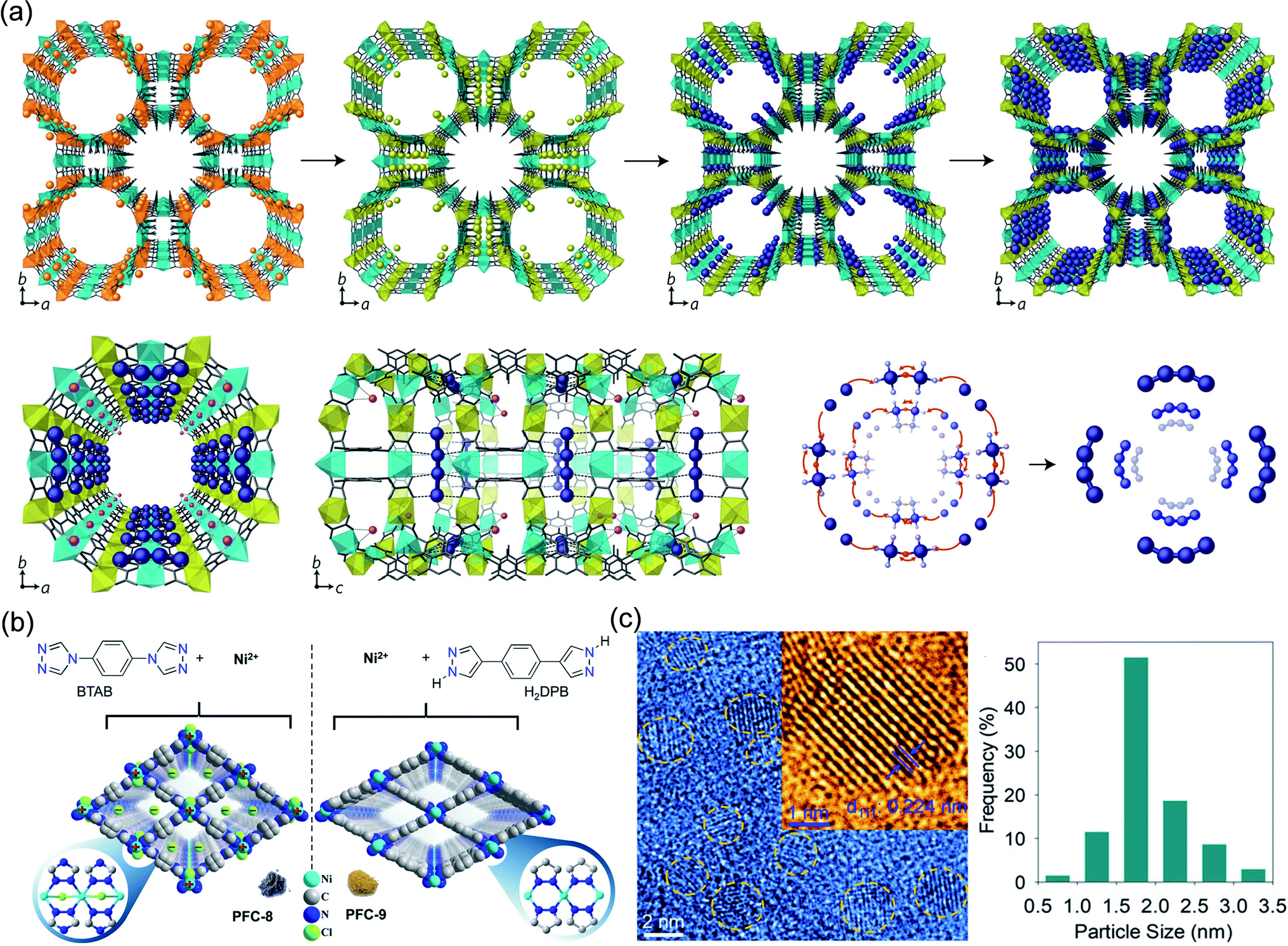 | ||
| Fig. 10 Cases of MOFs for ion-confinement. (a) Synthesis and X-ray crystal structure of Pd0/1+4-MOF. Reproduced with permission.120 Copyright 2017, Springer Nature. (b) Schematic representation of the structures of cationic PFC-8 and neutral PFC-9. (c) HRTEM images and Pd particle size distribution of Pd@Nano-PFC-8. Reproduced with permission.121 Copyright 2020, WILEY-VCH. | ||
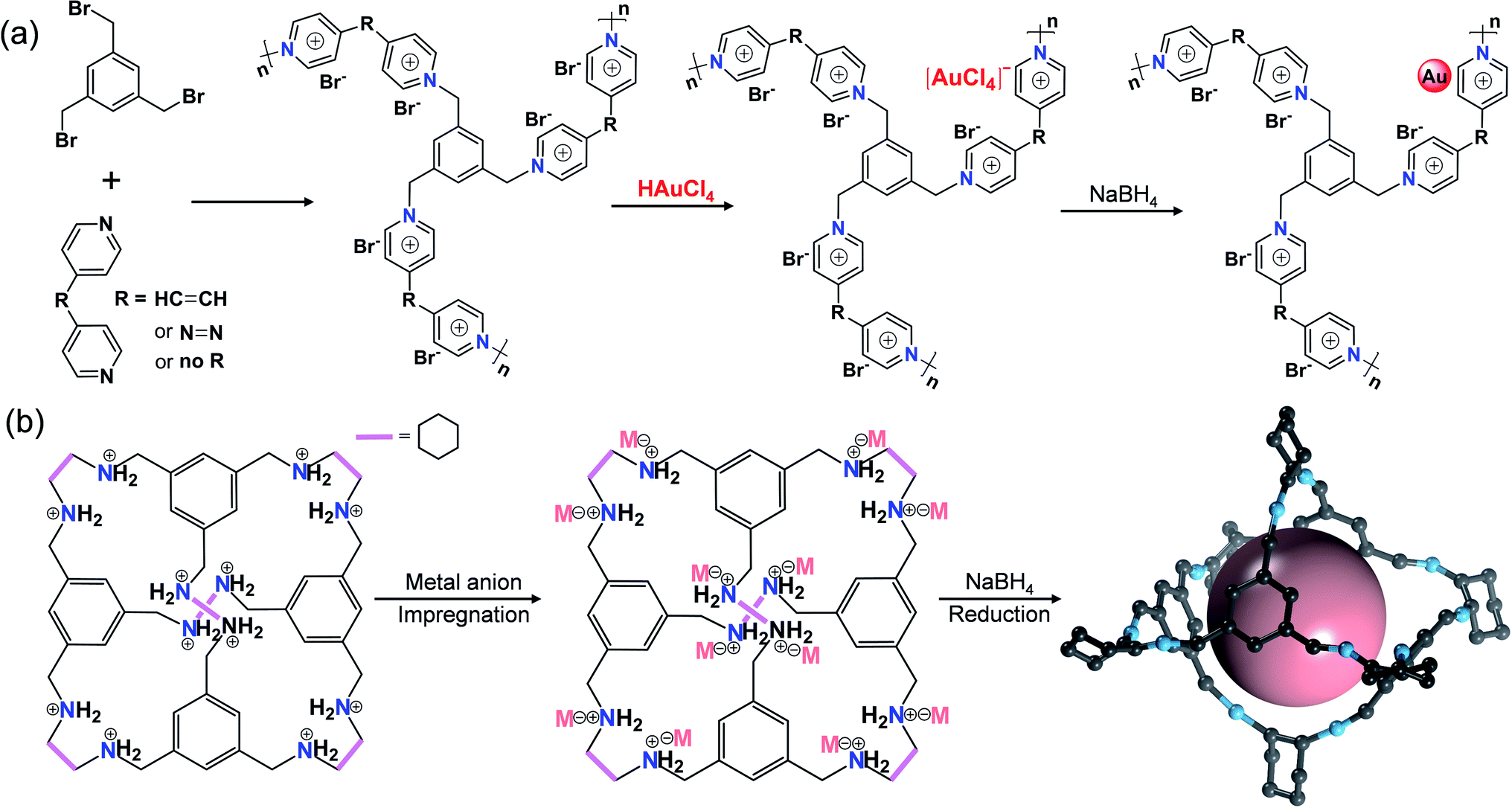 | ||
| Fig. 11 Cases of POPs or POCs for ion-confinement. Schematic illustration of constructing (a) Au@PION and (b) MCs@I-Cage-Cl. | ||
4. PMM-confinement pyrolysis to synthesize ultrafine nanostructures
Through the preparation of ultrafine nanostructures by pyrolysis of metal-contained precursors, it is easy to obtain thermodynamically stable crystal phases or to obtain phases that can only be formed at higher temperature, such as metal carbides, nitrides, etc. Commonly used synthesis strategies include direct metal precursor pyrolysis, mechanical mixed pyrolysis of metal ions and small organic molecules, high temperature vapor deposition, etc. However, through these methods it is often difficult to control the morphology and particle size of MNPs, and some methods encounter cumbersome operations or high costs. Therefore, the strategy of using PMMs as confinement templates has attracted attention in recent years due to their excellent adjustability and confinement effect.129–131 According to the structural features and confinement characteristics of PMMs, the confinement pyrolysis strategy can be divided into space-confinement pyrolysis (SCP) and coordination-confinement pyrolysis (CCP) (Fig. 12). In the PMM-confinement pyrolysis, an effective strategy to control the synthesis of ultrafine MNPs or SAs is to precisely regulate the ratios of PMM hosts to metal precursor guests. Besides, modulating the pyrolysis conditions (e.g. temperature, time, atmosphere, etc.) can also play important roles. Moreover, it is necessary to reveal the structures of ultrafine MNPs or SAs. Generally, the crystal structure, microscopic morphology, chemical composition, and bonding information of MNPs can be obtained by conventional powder X-ray diffraction (PXRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy and Fourier-transform infrared spectroscopy (FT-IR) techniques. X-ray absorption spectroscopy (XAS) and aberration-corrected scanning transmission electron microscopy (AC-STEM) can be helpful to identify single atom sites.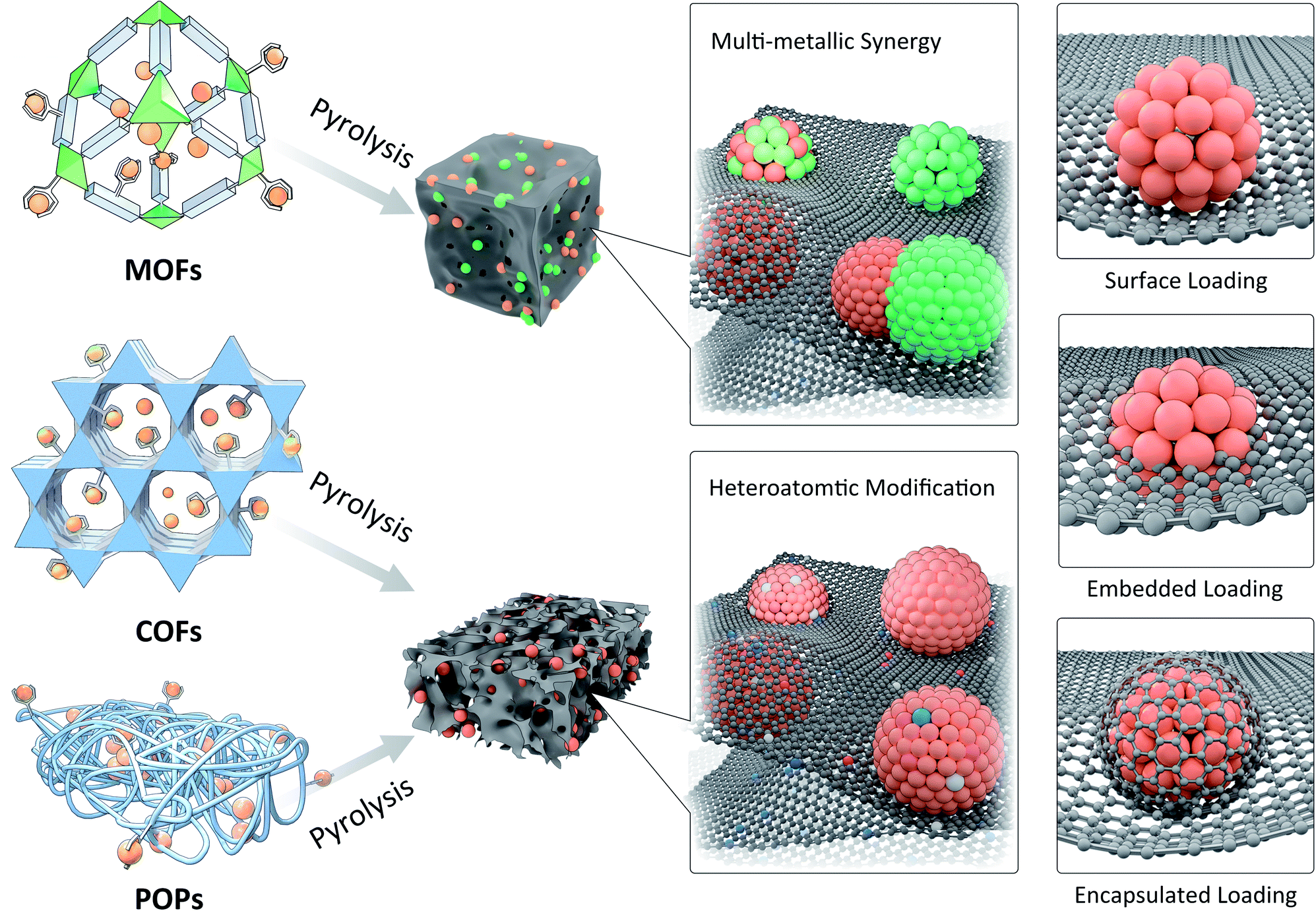 | ||
| Fig. 12 Confinement pyrolysis of MOFs, COFs, or POPs to synthesize ultrafine nanostructures with controllable compositions and atomic structures. | ||
4.1. Space-confinement pyrolysis
The SCP strategy is the use of nanocages/pores or bulky building blocks of PMMs to confine or isolate the metal precursor and then block its aggregation at high temperature.132 According to the different space types of PMMs, SCP can be divided into hybrid space-confinement pyrolysis (HSCP) and organic space-confinement pyrolysis (OSCP). Typically, MOFs are constructed from ligands and metal centers, which can provide organic–inorganic hybrid nano-space to confine guest molecules with suitable size based on host–guest chemistry. When the scale of the guest exactly matches the size of the nanocages and the pore windows of MOFs, each guest molecule is firmly confined in a separate nanocage, effectively avoiding their diffusion/agglomeration during the pyrolysis process. Besides, the other significant advantage of the HSCP is that additional metal elements from the hybrid frameworks can be purposefully introduced into the final materials. During the pyrolysis process, the metal atoms contained in MOFs can be doped into MNPs or form a heterostructure with them. On one hand, a large number of studies have confirmed that doping foreign metal atoms into the lattice of MNPs can significantly adjust the coordination and electronic structure of the active atoms, thereby adjusting their catalytic performance. On the other hand, heterostructures are beneficial to charge separation and interfacial charge transfer, provide more catalytic sites, and achieve synergistic catalytic effects, so that they exhibit unique properties in catalytic reactions. In comparison, COFs and POPs have abundant nanopores connected by organic monomers through covalent bonds and/or secondary bonds. The pre-designed nanopores in COFs or POPs can also isolate metal precursors and reduce their migration and agglomeration during the pyrolysis process, thereby forming ultrafine MNPs. Therefore, using the organic space in COFs or POPs to control the preparation of ultrafine MNPs is a typical OSCP strategy.133 The most significant advantage of this strategy is that it is particularly easy to introduce heteroatoms into the resultant materials. Generally, organic monomers are highly modifiable and can be decorated with various heteroatoms. Thus, these heteroatoms can be uniformly introduced into COFs or POPs through the polymerization reaction of organic monomers. During the pyrolysis process, these heteroatoms can be uniformly doped into the derived carbon supports or MNPs. For one thing, heteroatom-doped carbon can provide additional active sites, and for another, heteroatom doping into MNPs can also regulate the electronic structure of the metal centers, thereby regulating their catalytic activity. Furthermore, the carbon derived from PMMs can be both an electronic regulator and a structural stabilizer for atomically dispersed metal nanomaterials.The unique HSCP of MOFs can introduce additional metals to strengthen the catalytic properties of MNPs. Teng et al. constructed Pd–Cu NPs wrapped with porous N-doped carbon via such a strategy (Pd–Cu/PNC).138 A Cu–BTC (BTC = 1,3,5-benzenetricarboxylic acid) octahedral crystal with nano-scale cavities was selected as the host molecule. Palladium acetylacetone (Pd(acac)2) was selected as the guest molecule, which could enter the Cu–BTC nanocavities through immersion and capillary force. After pyrolysis, Pd–Cu/PNC was prepared (Cu NPs: ∼30 nm and Pd NPs: 2.0 nm). The formation of ultrafine Pd NPs was attributed to the space-confinement, which can reduce the migration and agglomeration of Pd precursors. Pd–Cu/PNC exhibited great denitrification, with a nitrate removal rate of about 97.1% and N2 selectivity of about 83%. In contrast, Cu/PNC without Pd exhibited rather poor denitrification, with a nitrate removal rate of only 62% and a N2 selectivity of only 28%. In addition, commercial Pd/C and porous carbon had worse effects on denitrification (<20%). These experimental results confirmed that Pd–Cu/PNC had high performance for denitrification on account of the cooperative catalysis of Pd nanoclusters and Cu NPs derived from the MOF metal nodes.
MAF-4 (also called ZIF-8) has an adjustable crystal morphology and a highly ordered porous structure. It is considered a promising precursor for the synthesis of well-defined nanocatalysts and is used as a good host molecule to control the synthesis of ultrafine MNPs, due to a relatively large cavity yet a small window.139 In addition, the carbon support derived from MAF-4 has a high specific surface area and porous characteristics. For instance, Li et al. adopted a host–guest strategy to prepare a series of single metal atoms (M1/CN) through HSCP (Fig. 13a).139 Taking the construction of Ir1/CN as an example, MAF-4 and Ir(acac)3 were selected as the host and guest molecules, respectively. Because the diameter of Ir(acac)3 (di = 9.8 Å) is exactly between the ZIF-8 aperture (dp = 3.4 Å) and the cavity diameter (dc = 11.6 Å), Ir(acac)3 molecules can be encaged in the cavities of MAF-4 without migrating out of the pores, realizing the effect of space-confinement, thereby avoiding the migration and accumulation of metal in pyrolysis. Extended X-ray absorption fine structure (EXAFS) illustrated that Ir was well dispersed on the CN substrate in the form of single atoms. Least-squares FT-EXAFS fitting of Ir1/CN confirmed that the Ir center was four-coordinated and the average bond length is 2 Å. The X-ray absorption near-edge structure (XANES) confirmed that the oxidation state of Ir was between 0 and +4. Ir1/CN exhibited excellent activity for electrocatalytic formic acid oxidation, with mass activity reaching 12.9 A mgIr−1. A wide range of SACs (Pd, Pt, Ru, Cu, Ni, Mo, Ga, and Mn) could be prepared using this HSCP strategy.
 | ||
| Fig. 13 Cases of space-confinement pyrolysis. (a) Schematic illustration of constructing M1/CN. Reproduced with permission.139 Copyright 2020, Springer Nature. Construction schematic and TEM images of (b and c) H2O-WOx@C. Reproduced with permission.141 Copyright 2019, American Chemical Society. and (d and e) WC@NPC. Reproduced with permission.142 Copyright 2017, American Chemical Society. | ||
Wang et al. constructed atomically dispersed uniform Ru3 clusters through the confinement of molecular cages in MAF-4.140 Ru3(CO)12 was selected as the guest molecule, because its diameter is about 8.0 Å, so it can be contained in a cage without being released. XANES, EXAFS, and wavelet transform (WT) confirmed the existence of Ru–N, Ru–Ru and Ru–C in Ru3/CN, and the atom-dispersed Ru3 clusters were stabilized by nitrogen in the carbon layer. Ru3/CN exhibited a good catalytic effect in the oxidation reaction of 2-aminobenzyl alcohol, with the conversion rate and chemoselectivity reaching 100%. The TOF value of Ru3/CN was superior to that of ultrafine Ru NPs (ca. 2.5 nm).
POMs have an adjustable molecular size, clear structure, and good solubility, making them ideal catalyst precursors. Niu et al. used molecular cages in MAF-4 to encapsulate Keggin-type K5BW12O40 (BW12), followed by pyrolysis and oxidation, to synthesize ultrafine H2O-WOx NPs (H2O-WOx@C) for the olefin epoxidation reaction (Fig. 13b).141 MAF-4 and BW12 were selected as the host and guest molecules due to the following reasons. Firstly, the size of a single BW12 anion is 0.9 nm, which is smaller than the cage size of MAF-4 (1.16 nm) and larger than the cage window size (0.34 nm), so it can be encapsulated in the cage without being leached from its window. Secondly, BW12 is very stable in the process of in situ packaging to form BW12@MAF-4. Thirdly, the K+ ions that compete with BW12 anions and can be located in adjacent molecular cages encapsulating BW12, which can ensure that BW12 was evenly distributed in MAF-4. Thanks to the space-confinement of the hybrid cages, the migration and agglomeration of metal precursors could be effectively reduced, thus preparing ultrafine W NPs. The average size of the highly dispersed ultrafine W NPs in the as-synthesized W@C was 2.28 nm (Fig. 13c). After oxidation, H2O-WOx still maintained the ultrafine nanoparticle structure with highly uniform dispersion. The final H2O-WOx@C showed high catalytic activity for the epoxidation of olefins with high conversion (98.6%) and selectivity (99%).
Compared with MAF-4, MAF-6 possesses a larger nanocage with a diameter of ca. 1.84 nm, a highly hydrophobic pore surface, a flexible small pore window (average diameter = 0.76 nm), and good thermal/chemical stability, which is also chosen as the host for even larger guest molecules (e.g. metal carbonyl compounds) to confine the construction of ultrafine MNPs.142
For example, we have reported the synthesis of ultrafine WC nanoclusters by using the HSCP method based on MAF-6 (Fig. 13d).142 The guest molecule W(CO)6 (molecular diameter of ca. 0.92 nm) could be encapsulated and isolated in the nanocavity of MAF-6 without leaching out of its window through low temperature vapor adsorption. Subsequently, WC@NPC was constructed by pyrolyzing the W(CO)6@MAF-6 precursor. The ultrafine WC NPs (ca. 2.0 nm) were highly dispersed and uniformly embedded in the carbon substrate (Fig. 13e). For comparison, a mechanically mixed precursor of W(CO)6/MAF-6 was constructed by adding W(CO)6 directly during the preparation of MAF-6, and in the same pyrolysis process, only W NPs could be obtained with obvious agglomeration that resulted in uneven sizes of 5–55 nm. The carbon protected WC nanoclusters showed excellent hydrogen evolution reaction activity and durability. In 0.5 M H2SO4, the overpotential for WC@NPC at 10 mA cm−2 was 51 mV with a small Tafel slope of 49 mV per decade. WC@NPC presented an extremely high exchange current density (2.4 mA cm−2), surpassing all tungsten/molybdenum-based electrocatalysts reported in advance. The different distribution of W(CO)6 molecules in MAF-6 and the confinement effect of the porous framework may determine the huge difference between the resultant MNPs, not just the particle sizes but even the crystalline phases.
Li et al. synthesized a metal isolated single-atom site (ISAS) catalyst (M-ISAS/p-CN) supported on porous nitrogen-doped carbon using the OSCP method.144 Firstly, Co(acac)2@PDA nanospheres were prepared by in situ encapsulation of cobalt acetylacetonate during the polymerization of dopamine monomers. Subsequently, Co-ISAS/p-CN was constructed via controlling the pyrolysis of the Co(acac)2@PDA precursor. This strategy had broad applicability and was suitable for the preparation of a variety of M-ISAS/p-CN (e.g. M = Cu, Co, Ni, Pd, Mn) catalysts, which still maintained the uniform nano-spherical appearance. Taking Co-ISAS/p-CN as an example, the EXAFS spectrum confirmed that there were no Co–Co chemical bonds, indicating that Co was dispersed in p-CN in the form of single-atom sites. In addition, a series of characterization techniques confirmed that the Co single atom existed in the form of coordination with four N atoms. Co-ISAS/p-CN showed excellent electrochemical ORR activity with a half-wave potential of 0.838 V.
Huang et al. also used the OSCP strategy for preparing ultrafine Mo2C NPs.145 Firstly, PMo12@PPy/CNT was constructed through polymerization, crosslinking and coating between CNTs, pyrrole, 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin (TCPP) and PMo12 molecules. In the process of pyrrole polymerizing around the outer layer of CNTs to form a polymer chain, the 4 carboxyl groups on the TCPP molecule and the N–H group on the PPy were cross-linked by 4 hydrogen bonds, resulting in nanopores with a diameter of ∼1.8 nm. The diameter of PMo12 is about 1.05 nm, so one nanopore can hold exactly one PMo12 molecule. Subsequently, size-controllable Mo2C NPs (Mo2C@NPC/CNT) coated with N and P co-doped carbon shells were prepared by pyrolyzing PMo12@PPy/CNT. The uniform embedding of Mo2C NPs in the carbon matrix during carbonization was due to the perfect confinement of these molecule-scale organic cages. The optimized Mo2C NPs were only 5.0 nm and were wrapped by 2–4 layers of ultrathin carbon and uniformly dispersed in Mo2C@NPC/CNT. Mo2C@NPC/CNT exhibited good electrochemical performance for the electrochemical sensing of acetaminophen (AC) and dopamine (DA) simultaneously, showing detection limits (S/N = 3) of 0.01 and 0.008 μM for DA and AC.
4.2. Coordination-confinement pyrolysis
The CCP strategy is the use of coordinated interactions between the coordinating atoms on PMMs and targeted metal ions to achieve confinement. Many PMMs are rich in coordination atoms (such as O, N, S, P, etc.), which can bind various metal ions to form monodisperse metal centers with appropriate distances, thus effectively reducing the diffusion and agglomeration of metals during the pyrolysis process.146–151 The coordination heteroatoms can be doped in situ on the carbon substrate formed by carbonization to modulate the electronic structures of MNPs or single atom sites and can also induce the metal precursors to form various functional metal compounds at high temperature, such as metal sulfides, metal phosphides, metal nitrides, etc., which are very difficult to make into ultrafine sizes by conventional methods.152–159Wu et al. reported an organic ligand CCP method to synthesize a Co single-atom catalyst and regulated the electrocatalytic CO2 reduction performance of the catalyst by adjusting the coordination number of a single Co site (Fig. 14a).160 Thanks to the coordination confinement effect of the N atoms on the ligands, the transport and accumulation of Co were significantly reduced to obtain a Co single-atom catalyst. The coordination number of Co (Co–N4, Co–N3, and Co–N2) could be regulated by the pyrolysis temperature, and the higher the temperature, the lower the coordination number. Co–N2 exhibited the best electrocatalytic CO2RR activity and selectivity, the CO Faraday efficiency was as high as 94%, and the TOF value was up to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h−1. Experiments and theoretical calculations confirmed that a lower coordination number was beneficial to the activation of CO2 to the
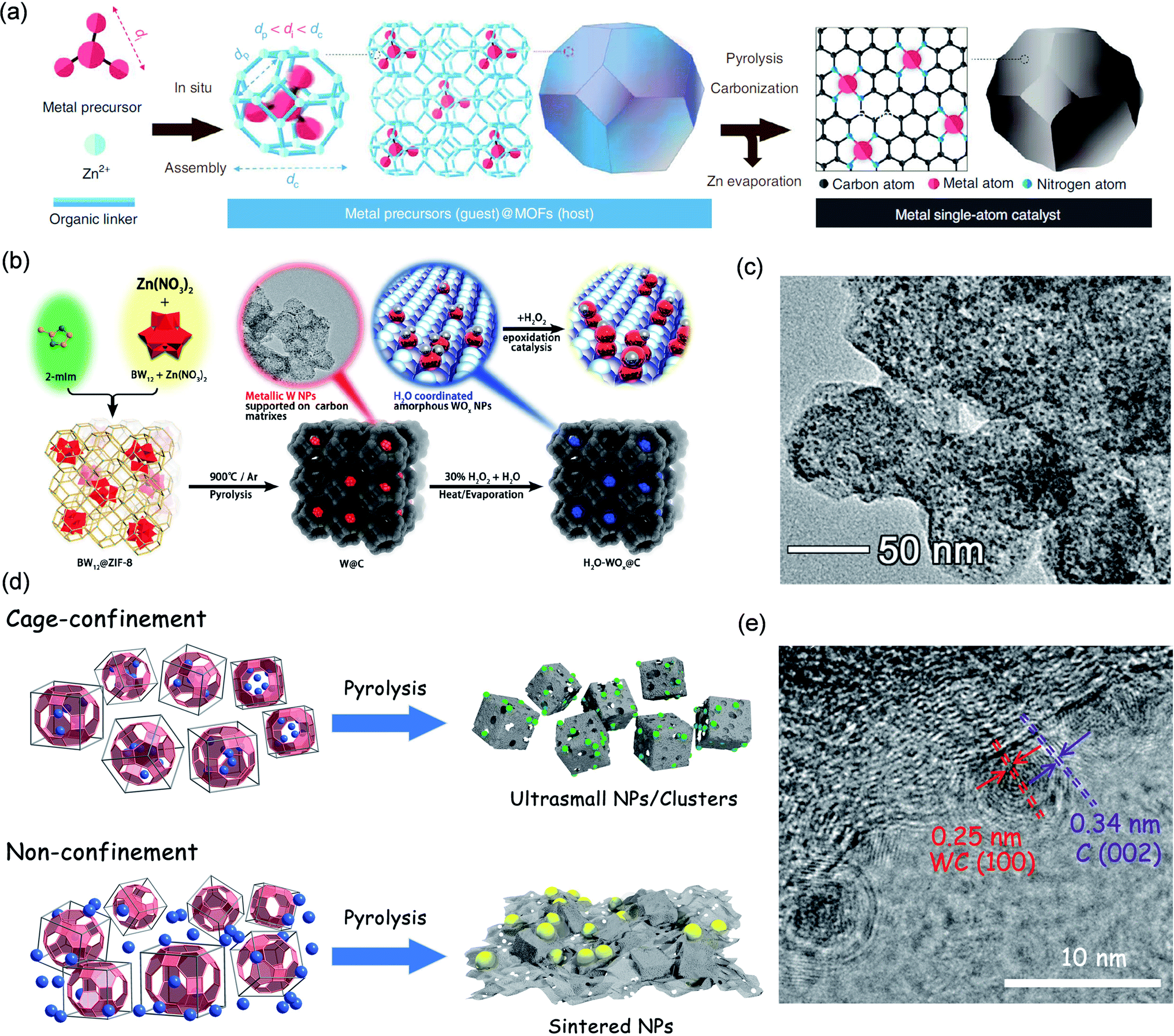
200 h−1. Experiments and theoretical calculations confirmed that a lower coordination number was beneficial to the activation of CO2 to the 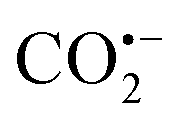 intermediate, which in turn promoted the CO2 reduction activity of the catalyst.
intermediate, which in turn promoted the CO2 reduction activity of the catalyst.
 | ||
| Fig. 14 Cases of MOF coordination-confinement pyrolysis. Schematic illustration of constructing (a) Co–N4, Co–N3, and Co–N2. Reproduced with permission.160 Copyright 2018, WILEY-VCH. (b) FeSA-N-C. Reproduced with permission.161 Copyright 2018, WILEY-VCH. (c) Ir@ZrO2@C. Reproduced with permission.162 Copyright 2019, Elsevier. | ||
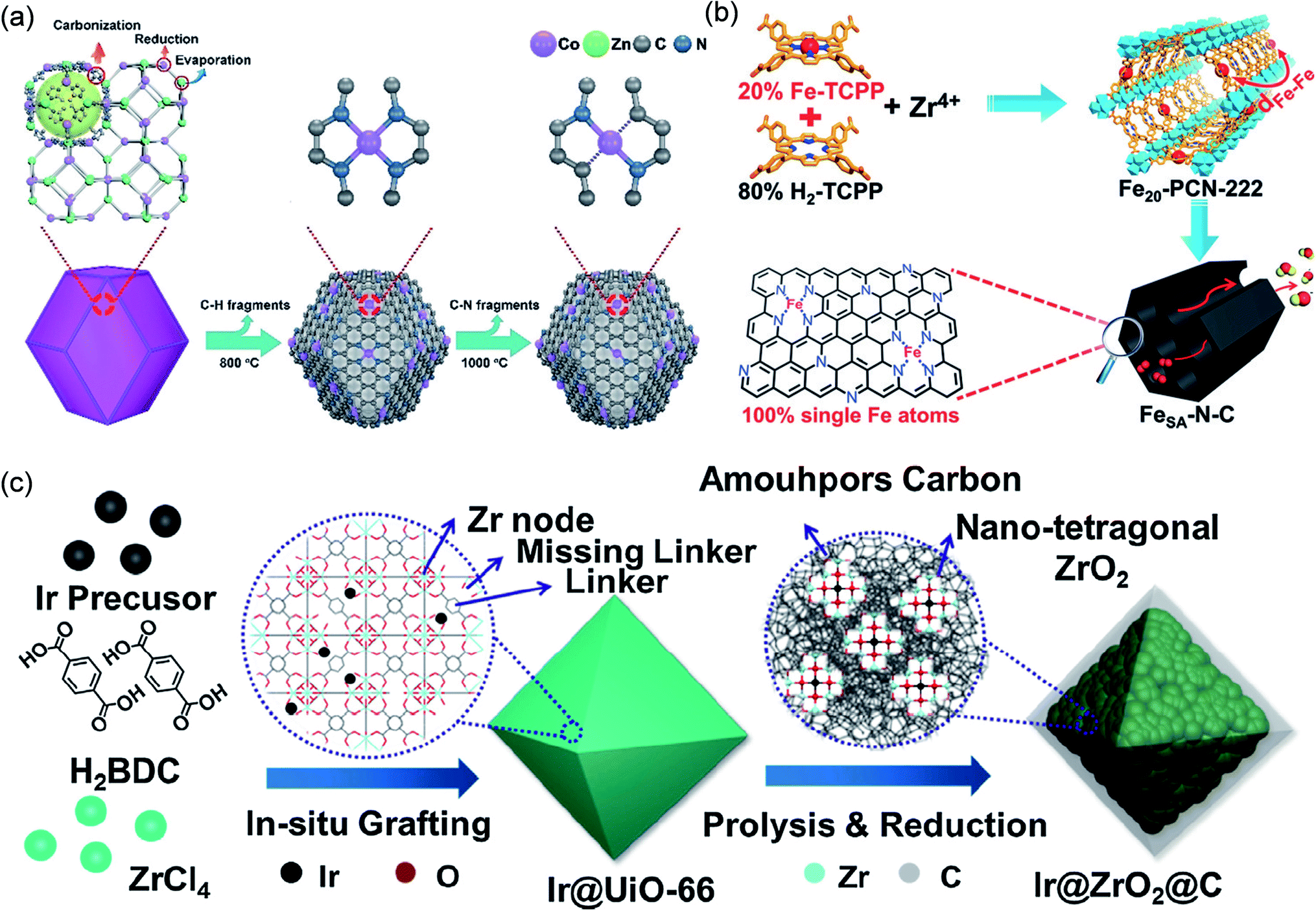
Jiang et al. used a porphyrinic MOF to coordinate Fe3+ for synthesizing Fe single atoms (Fig. 14b).161 Fex-PCN-222, being isomorphic to PCN-222, was constructed by the self-assembly of Fe-TCPP (TCPP = tetrakis-(4-carboxyphenyl)porphyrin), H2-TCPP and ZrOCl2·8H2O. By controlling the ratio of Fe-TCPP and H2-TCPP, the distance between Fe-TCPP units in adjacent Fex-PCN-222 could be optimized. The optimized Fe20-PCN-222 was converted into Fe single atoms (FeSA-N-C) implanted in porous nitrogen-doped carbon after pyrolysis and subsequent removal of ZrO2. EXAFS fitting confirmed that each Fe atom formed a four-coordinate structure with nitrogen. The formation of single-atom Fe was attributed to the coordination-confinement effect of four N, which could anchor Fe, and then directly retained in the final structure by high temperature pyrolysis. The ORR activity of FeSA-N-C (E1/2 = 0.891 V) was better than that of commercial Pt/C (E1/2 = 0.848 V).
In addition to organic ligand CCP, MOFs can also achieve metal node CCP. Zhang et al. used the confinement effect of metal nodes in UiO-66 to synthesize atomic-level Ir sites (Fig. 14c).162 The metal node Zr6 in UiO-66 is rich in hydroxyl groups, which can chelate metal ions. Firstly, Ir@UiO-66 was prepared by in situ grafting an Ir precursor during MOF synthesis. Subsequently, Ir@ZrO2@C was constructed by pyrolysis of Ir@UiO-66 to obtain Ir@ZrO2@C, which maintained well the octahedral morphology. A large number of ZrO2 NPs (ca. 3.0 nm) were formed in Ir@ZrO2@C, but no Ir NPs were found. In the PXRD pattern of Ir@ZrO2@C, there was only the diffraction peak of ZrO2 but no diffraction peak of Ir elementary substance, indicating the possible presence of monodisperse Ir. The formation of atomic-level Ir sites was attributed to the strong coordination of –OH to Ir on the Zr node in UiO-66, which made it difficult for Ir to migrate and aggregate during the pyrolysis process. Ir@ZrO2@C was a good acid-resistant catalyst, showing great conversion efficiency in the hydrogenation reaction of levulinic acid to c-valerolactone. The high performance was attributed to the enhanced synergy between the atomically dispersed Ir species and the ZrO2 carrier, as well as the protective effect of the in situ derived amorphous carbon.
COFs can also achieve organic CCP for synthesizing ultrafine MNPs.163–165 Li et al. used a COF-absorption-pyrolysis approach for preparing various isolated single atom site (ISAS) catalysts (Fig. 15a).166 Firstly, RT-COF-1 was constructed through the condensation reaction of organic monomers 1,3,5-tris(4-aminophenyl) benzene and 1,3,5-benzenetricarboxaldehyde. Then, the metal ions were uniformly anchored in RT-COF-1 using the coordination effect of the N atoms. Finally, the RT-COF-1 precursor with metal ions was pyrolyzed to construct ISAS catalysts (M-ISAS/CN, M = Fe, Co, Ni) embedded in a carbon substrate. M-ISAS/CN still maintained the morphology of RT-COF-1 precursor nanospheres, and no MNPs were found to be formed. Fe-ISAS/CN was taken as an example. FT-EXAFS revealed the existence of Fe–N/C bonds in Fe-ISAS/CN but no Fe–Fe bonds, indicating that Fe existed as a SA. Fe-ISAS/CN displayed good ORR activity with a kinetic current density as high as 5.47 mA cm−2 at 0.85 V. In addition, Fe-ISAS/CN exhibited good stability and methanol tolerance.
 | ||
| Fig. 15 Cases of COF or POP coordination-confinement pyrolysis. Schematic illustration of constructing (a) Fe-ISAS/CN and (b) Ru@C2N. (c) TEM image of Ru@C2N. Reproduced with permission.168 Copyright 2017, Springer Nature. | ||
Zhang et al. utilized a polymer-assisted pyrolysis strategy to construct a Co SAC and proved that the incorporated P can coordinate with Co to improve its ORR activity.167 The poly(HCCP–TA–BPS) (HCCP = hexachlorocyclotriphosphazene, TA = tannic acid, BPS = 4,4′-sulfonyldiphenol) nanospheres (PSTA) have a large number of catechol groups, which can chelate Co2+ ions to obtain PSTA-Co. Finally, PSTA-Co was pyrolyzed to prepare Co–N2P2 monoatomic sites uniformly dispersed in PSTA-Co-1000. Due to the chelation between the coordination atom and Co2+, the agglomeration of metals during the pyrolysis process could be reduced to prepare atomic-scale Co sites. EXAFS confirmed the existence of Co–N and Co–P in PSTA-Co-1000, but no Co–Co, indicating that a single Co atom was formed in the catalyst. EXAFS fitting revealed that the average coordination numbers of Co with N and P were 2.1 and 1.9, respectively. PSTA-Co-1000 exhibited good electrocatalytic ORR activity with a half-wave potential of 0.878 V. Compared with Co–N4, theoretical calculations showed that the Co–N2P2 active center could be more effectively combined with oxygen to drive the ORR, with faster ORR kinetics.
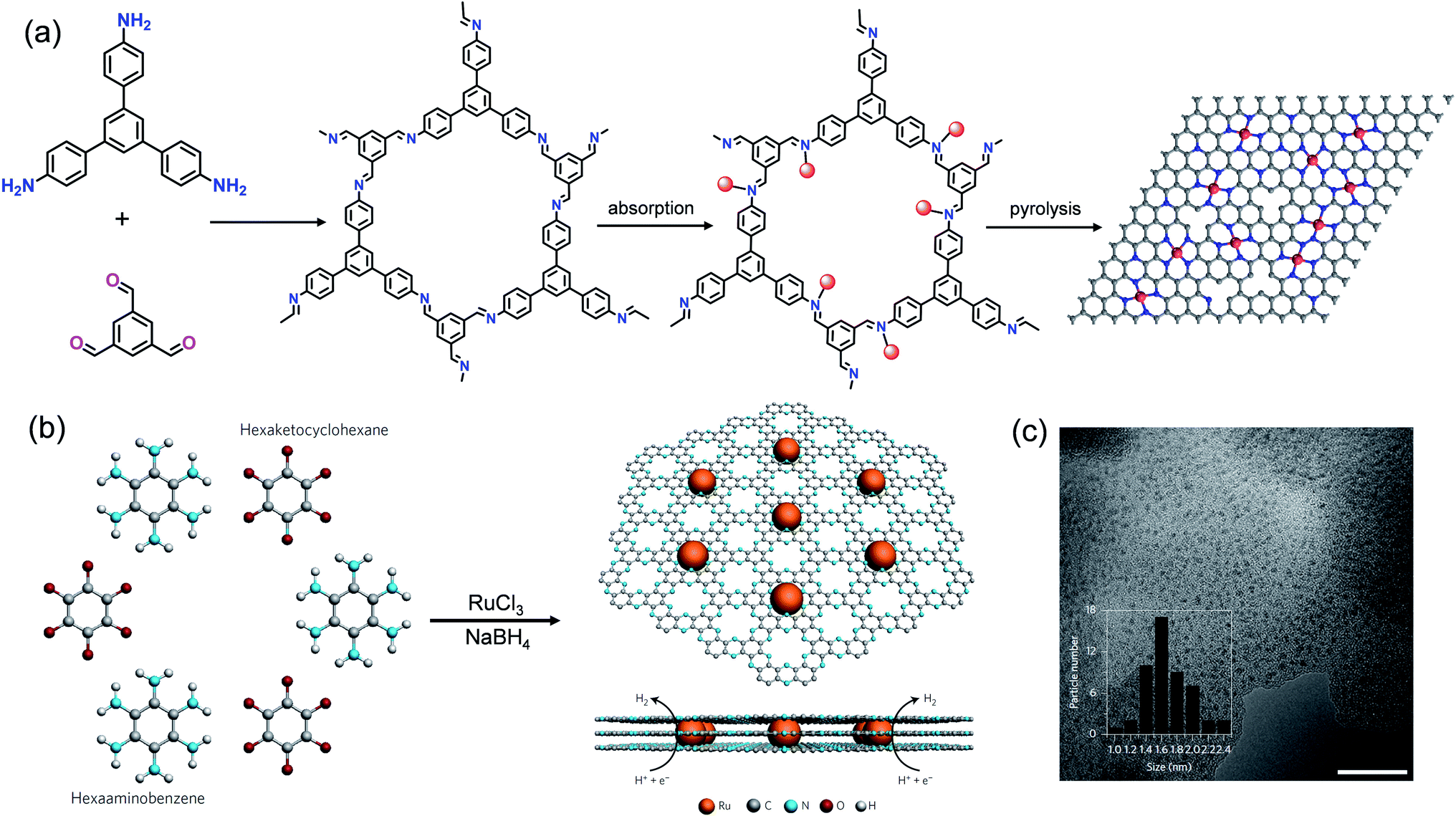
Baek et al. constructed a N-rich porous polymer (C2N) using the polycondensation reaction of organic monomers hexaketocyclohexane (HKH) and hexaaminobenzene (HAB) trihydrochloride, and synthesized Ru NPs (Ru@C2N) with the coordination-confinement of Ru3+ ions and subsequent pyrolysis (Fig. 15b).168 The BET surface area of Ru@C2N reached 400.1 m2 g−1. TEM showed that the ultrafine Ru NPs with a diameter of ca. 1.6 nm were uniformly distributed in the C2N supports (Fig. 15c). The formation of ultrafine nanostructures was attributed to the high density N sites in C2N, which could provide nucleation sites for Ru atoms and limit their migration and aggregation. The resultant Ru@C2N exhibited excellent HER activity and stability. The overpotentials for Ru@C2N at 10 mA cm−2 were only 17.0 (1.0 M KOH) and 13.5 mV (0.5 M H2SO4), surpassing commercial Pt/C and better than most reported catalysts.
5. Conclusions and prospects
This review article introduced the latest progress on the confinement synthesis in PMMs for synthesizing atomic-scale nanostructures. PMMs show superb advantages in constituent modulation and structural modification. The building blocks, organic functional groups, or metal nodes in PMMs can be accurately regulated at the atomic level. Therefore, it is possible to design and construct adjustable nano/sub-nano-confined spaces and modify various types of coordinated heteroatoms in PMMs. PMMs exhibit unique advantages of space-confinement, coordination-confinement, and ion-confinement, which can not only significantly reduce the agglomeration of MNPs, but also limit the growth of MNPs to obtain well-dispersed ultrafine MNPs. Additionally, PMMs can serve as desired templates or precursors to derive various ultrafine nanostructures benefiting from their special molecular construction and confinement effects. The abundant nanocages/pores and coordination atoms in PMMs can isolate and anchor metal precursors/metal ions to limit the atom diffusion and aggregation during the pyrolysis process. Moreover, PMMs can easily form hybrid carbon layers at high temperature, which not only regulate the electron structures of the atom-dispersed metal nanomaterials but also protect them from agglomeration or degradation in harsh chemical environments. By using these strategies, new multifunctional and highly active materials based on PMMs or their derivatives will appear in the future.Despite the tremendous successes, PMM-confinement research still faces some challenges. Firstly, most confinement strategies currently focus on the synthesis of ultrafine precious MNPs, like Au, Pt, Ru, etc. There are few reports on the construction of ultrafine non-noble MNPs by the PMM-confinement strategy, because non-precious MNPs present higher surface energy and more reactivity under oxidation and reduction conditions, tending to aggregate and become larger. Actually, easy-to-obtain and non-expensive MNPs have broader application prospects. Therefore, it is necessary to propose an effective PMM-confinement strategy to synthesize ultrafine yet stable non-noble MNPs. Secondly, the PMM-confinement strategy is mostly applied to the construction of metals or their alloys. The synthesis of ultrafine and stable metal phosphides, nitrides, and chalcogenides is rarely achieved, and these ultrafine nanostructures often show superior activity in heterocatalysis, especially the catalytic conversions of small energy molecules. Applying the PMM-confinement strategy to synthesize a richer variety of ultrafine nanostructures can make it more universal and expand its application fields. Thirdly, it is necessary to develop multi-confinement combination strategies, such as space-ion-confinement, space-coordination-confinement and so on, which may provide the possibility for the synthesis of more types of ultrafine nanostructures, such as multi-component ultrafine nanocrystals, single atom loaded nanoclusters, diatomic or triatomic site catalysts, etc. Moreover, it is also important to study the influence of different pore sizes and pore surface characteristics in PMMs, such as hydrophilicity and hydrophobicity, on the confinement synthesis of MNPs, as well as the influence of different functional groups or coordination on their morphologies and sizes, which would guide the rational design and construction of ideal PMMs for the confinement synthesis of specifically functional MNPs. Furthermore, the development of more widely sourced, lower cost PMM materials, such as biomass macromolecules, as domain-confinement templates is also desirable, which is conducive to the large-scale preparation of highly active ultrafine nanomaterials.
Author contributions
C.-T. H. conceived the concept and supervised the project. L.-M. Cao and J. Z. collected references, organized images and wrote the initial manuscript. X.-F. Z. modified the images and participated in discussions. All the authors discussed the results, revised and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21901088, 21901089, 32101750 and 22161021) and the Natural Science Foundation of Jiangxi Province (20212BAB203021 and 20202ZDB01004). C.-T. He acknowledges the support of Jiangxi Province (jxsq2018106041) and the “Young Elite Scientists Sponsorship Program” by CAST.Notes and references
- L. C. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- L. L. Zhang, M. X. Zhou, A. Q. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- Z. Li, S. F. Ji, Y. W. Liu, X. Cao, S. B. Tian, Y. J. Chen, Z. G. Niu and Y. D. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed.
- I. F. Teixeira, E. C. M. Barbosa, S. C. E. Tsang and P. H. C. Camargo, Chem. Soc. Rev., 2018, 47, 7783–7817 RSC.
- H. Tabassum, A. Mahmood, B. J. Zhu, Z. B. Liang, R. Q. Zhong, S. J. Guo and R. Q. Zou, Energy Environ. Sci., 2019, 12, 2924–2956 RSC.
- C. B. Gao, F. L. Lyu and Y. D. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- G. Palumbo, S. J. Thorpe and K. T. Aust, Nanocryst. Mater., 1990, 24, 1347–1350 CAS.
- J. N. Kuhn, W. Y. Huang, C. K. Tsung, Y. W. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026–14027 CrossRef CAS PubMed.
- B. Xiao, Z. Q. Niu, Y. G. Wang, W. Jia, J. Shang, L. Zhang, D. S. Wang, Y. Fu, J. Zeng, W. He, K. Wu, J. Li, J. L. Yang, L. Liu and Y. D. Li, J. Am. Chem. Soc., 2015, 137, 3791–3794 CrossRef CAS PubMed.
- M. L. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- M. T. Zhao, K. Yuan, Y. Wang, G. D. Li, J. Guo, L. Gu, W. P. Hu, H. J. Zhao and Z. Y. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- C. T. He, L. Jiang, Z. M. Ye, R. Krishna, Z. S. Zhong, P. Q. Liao, J. Q. Xu, G. F. Ouyang, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2015, 137, 7217–7223 CrossRef CAS PubMed.
- L. Y. Chen, R. Luque and Y. W. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC.
- M. T. Zhao, Y. Huang, Y. W. Peng, Z. Q. Huang, Q. L. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- D. P. Halter, R. A. Klein, M. A. Boreen, B. A. Trump, C. M. Brown and J. R. Long, Chem. Sci., 2020, 11, 6709–6716 RSC.
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396–9408 RSC.
- Z. W. Fu, X. Y. Wang, A. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. J. Liu, X. B. Li, A. Vogel, R. Clowes, M. Bilton, L. J. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. M. Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- X. Y. Guan, F. Q. Chen, Q. R. Fang and S. L. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- Z. F. Wang, S. N. Zhang, Y. Chen, Z. J. Zhang and S. Q. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- X. Kang, X. W. Wu, X. Han, C. Yuan, Y. Liu and Y. Cui, Chem. Sci., 2020, 11, 1494–1502 RSC.
- G. D. Li, S. L. Zhao, Y. Zhang and Z. Y. Tang, Adv. Mater., 2018, 30, 1800702 CrossRef PubMed.
- Q. H. Yang, Q. Xu and H. L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- R. Tao, X. R. Ma, X. L. Wei, Y. H. Jin, L. Qiu and W. Zhang, J. Mater. Chem. A, 2020, 8, 17360–17391 RSC.
- J. Y. Kim, H. Oh and H. R. Moon, Adv. Mater., 2019, 31, 1805293 CrossRef PubMed.
- N. Wang, Q. M. Sun and J. H. Yu, Adv. Mater., 2019, 31, 1803966 CrossRef PubMed.
- H. Y. Hu, S. L. Lu, T. Li, Y. Zhang, C. X. Guo, H. Zhu, Y. H. Jin, M. L. Du and W. Zhang, Nanoscale Adv., 2021, 3, 1865–1886 RSC.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- L. Y. Chen, H. F. Wang, C. X. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 RSC.
- P. Y. Ju, S. J. Wu, Q. Su, X. D. Li, Z. Q. Liu, G. H. Li and Q. L. Wu, J. Mater. Chem. A, 2019, 7, 2660–2666 RSC.
- H. C. Ma, J. L. Kan, G. J. Chen, C. X. Chen and Y. B. Dong, Chem. Mater., 2017, 29, 6518–6524 CrossRef CAS.
- X. L. Li, T. W. Goh, L. Li, C. X. Xiao, Z. Y. Guo, X. C. Zeng and W. Y. Huang, ACS Catal., 2016, 6, 3461–3468 CrossRef CAS.
- H. Zhao, G. Q. Yu, M. Yuan, J. Yang, D. Xu and Z. P. Dong, Nanoscale, 2018, 10, 21466–21474 RSC.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Ronnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- Q. L. Zhu, J. Li and Q. Xu, J. Am. Chem. Soc., 2013, 135, 10210–10213 CrossRef CAS PubMed.
- M. Y. Fan, W. D. Wang, Y. Y. Zhu, X. Sun, F. W. Zhang and Z. P. Dong, Appl. Catal., B, 2019, 257, 117942 Search PubMed.
- S. Hermes, M. K. Schroter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237–6241 CrossRef CAS PubMed.
- J. Hermannsdorfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kummel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed.
- Y. W. Liu, X. Wu, Z. Li, J. Zhang, S. X. Liu, S. J. Liu, L. Gu, L. R. Zheng, J. Li, D. S. Wang and Y. D. Li, Nat. Commun., 2021, 12, 4205 CrossRef CAS PubMed.
- X. H. Liu, J. G. Ma, Z. Niu, G. M. Yang and P. Cheng, Angew. Chem., Int. Ed., 2015, 54, 988–991 CrossRef CAS PubMed.
- F. Schroeder, D. Esken, M. Cokoja, M. W. E. van den Berg, O. I. Lebedev, G. van Tendeloo, B. Walaszek, G. Buntkowsky, H. H. Limbach, B. Chaudret and R. A. Fischer, J. Am. Chem. Soc., 2008, 130, 6119–6130 CrossRef CAS PubMed.
- J. Hermannsdofer and R. Kempe, Chem.–Eur. J., 2011, 17, 8071–8077 CrossRef PubMed.
- Y. Y. Pan, B. Z. Yuan, Y. W. Li and D. H. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- Y. B. Huang, Z. J. Lin and R. Cao, Chem.–Eur. J., 2011, 17, 12706–12712 CrossRef CAS PubMed.
- B. Z. Yuan, Y. Y. Pan, Y. W. Li, B. L. Yin and H. F. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed.
- M. S. El-Shall, V. Abdelsayed, A. E. R. S. Khder, H. M. A. Hassan, H. M. El-Kaderi and T. E. Reich, J. Mater. Chem., 2009, 19, 7625–7631 RSC.
- Y. B. Huang, T. Ma, P. Huang, D. S. Wu, Z. J. Lin and R. Cao, ChemCatChem, 2013, 5, 1877–1883 CrossRef CAS.
- X. M. Zhao, Y. Jin, F. M. Zhang, Y. J. Zhong and W. D. Zhu, Chem. Eng. J., 2014, 239, 33–41 CrossRef CAS.
- H. L. Liu, Y. W. Li, R. Luque and H. F. Jiang, Adv. Synth. Catal., 2011, 353, 3107–3113 CrossRef CAS.
- Y. Z. Chen, Q. Xu, S. H. Yu and H. L. Jiang, Small, 2015, 11, 71–76 CrossRef CAS.
- M. Yadav and Q. Xu, Chem. Commun., 2013, 49, 3327–3329 RSC.
- J. Hermannsdorfer, M. Friedrich and R. Kempe, Chem.–Eur. J., 2013, 19, 13652–13657 CrossRef.
- C. Zlotea, R. Campesi, F. Cuevas, E. Leroy, P. Dibandjo, C. Volkringer, T. Loiseau, G. Ferey and M. Latroche, J. Am. Chem. Soc., 2010, 132, 2991–2997 CrossRef CAS PubMed.
- Y. A. Li, S. Yang, Q. K. Liu, G. J. Chen, J. P. Ma and Y. B. Dong, Chem. Commun., 2016, 52, 6517–6520 RSC.
- Y. W. Liu, B. X. Wang, Q. Fu, W. Liu, Y. Wang, L. Gu, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2021, 60, 22522–22528 CrossRef CAS.
- X. W. Lan, C. Du, L. L. Cao, T. T. She, Y. M. Li and G. Y. Bai, ACS Appl. Mater. Interfaces, 2018, 10, 38953–38962 CrossRef CAS PubMed.
- R. Khatun, S. Biswas, I. H. Biswas, S. Riyajuddin, N. Haque, K. Ghosh and S. M. Islam, J. CO2 Util., 2020, 40, 101180 CrossRef CAS.
- L. P. Zhai, S. Yang, X. B. Yang, W. Y. Ye, J. Wang, W. H. Chen, Y. Guo, L. W. Mi, Z. J. Wu, C. Soutis, Q. Xu and Z. Jiang, Chem. Mater., 2020, 32, 9747–9752 CrossRef CAS.
- H. C. Ma, C. C. Zhao, G. J. Chen and Y. B. Dong, Nat. Commun., 2019, 10, 3368 CrossRef PubMed.
- Q. Zhuang, R. R. Gao, M. Y. Shi, X. P. Lin, A. M. Xie and W. Dong, ACS Appl. Nano Mater., 2021, 4, 3869–3876 CrossRef CAS.
- W. Gong, Q. Q. Wu, G. X. Jiang and G. J. Li, J. Mater. Chem. A, 2019, 7, 13449–13454 RSC.
- J. K. Sun, W. W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- Y. Zhang, Y. Xiong, J. Ge, R. Lin, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Chem. Commun., 2018, 54, 2796–2799 RSC.
- R. McCaffrey, H. Long, Y. H. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS PubMed.
- L. Qiu, R. McCaffrey, Y. H. Jin, Y. Gong, Y. M. Hu, H. L. Sun, W. Park and W. Zhang, Chem. Sci., 2018, 9, 676–680 RSC.
- B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed.
- G. J. Chen, W. L. Xin, J. S. Wang, J. Y. Cheng and Y. B. Dong, Chem. Commun., 2019, 55, 3586–3589 RSC.
- K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS PubMed.
- J. Yang, M. Yuan, D. Xu, H. Zhao, Y. Y. Zhu, M. Y. Fan, F. W. Zhang and Z. P. Dong, J. Mater. Chem. A, 2018, 6, 18242–18251 RSC.
- W. Zhao, Y. Z. Jiao, J. J. Li, L. P. Wu, A. Xie and W. Dong, J. Catal., 2019, 378, 42–50 CrossRef CAS.
- X. C. Yang, J. K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- L. Y. Chen, H. R. Chen, R. Luque and Y. W. Li, Chem. Sci., 2014, 5, 3708–3714 RSC.
- Z. M. Cui, T. Fan, L. Y. Chen, R. Q. Fang, C. M. Li and Y. W. Li, Sci. China: Chem., 2021, 64, 109–115 CrossRef CAS.
- M. Martis, K. Mori, K. Fujiwara, W. S. Ahn and H. Yamashita, J. Phys. Chem. C, 2013, 117, 22805–22810 CrossRef CAS.
- Y. L. Jiang, Y. Yu, X. Zhang, M. Weinert, X. L. Song, J. Ai, L. Han and H. H. Fei, Angew. Chem., Int. Ed., 2021, 60, 17388–17393 CrossRef CAS PubMed.
- H. G. T. Nguyen, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, Crystengcomm, 2012, 14, 4115–4118 RSC.
- H. Noh, Y. X. Cui, A. W. Peters, D. R. Pahls, M. A. Ortuno, N. A. Vermeulen, C. J. Cramer, L. Gagliardi, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2016, 138, 14720–14726 CrossRef CAS PubMed.
- P. F. Ji, Y. Song, T. Drake, S. S. Veroneau, Z. K. Lin, X. D. Pan and W. B. Lin, J. Am. Chem. Soc., 2018, 140, 433–440 CrossRef CAS PubMed.
- G. Wang, C. T. He, R. Huang, J. J. Mao, D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- M. B. Chambers, X. Wang, N. Elgrishi, C. H. Hendon, A. Walsh, J. Bonnefoy, J. Canivet, E. A. Quadrelli, D. Farrusseng, C. Mellot-Draznieks and M. Fontecave, ChemSusChem, 2015, 8, 603–608 CrossRef CAS PubMed.
- N. C. Thacker, Z. K. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. B. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed.
- S. Chen, W. H. Li, W. Jiang, J. Yang, J. Zhu, L. Wang, H. Oh, Z. Zhang, M. Chen, X. Sun, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202114450.
- J. H. Li, Z. W. Yu, Z. Gao, J. Q. Li, Y. Tao, Y. X. Xiao, W. H. Yin, Y. L. Fan, C. Jiang, L. J. Sun and F. Luo, Inorg. Chem., 2019, 58, 10829–10836 CrossRef CAS PubMed.
- K. Guo, X. L. Zhu, L. L. Peng, Y. H. Fu, R. Ma, X. Q. Lu, F. M. Zhang, W. D. Zhu and M. H. Fan, Chem. Eng. J., 2021, 405, 127011 CrossRef CAS.
- G. J. Chen, X. B. Li, C. C. Zhao, H. C. Ma, J. L. Kan, Y. B. Xin, C. X. Chen and Y. B. Dong, Inorg. Chem., 2018, 57, 2678–2685 CrossRef CAS PubMed.
- X. G. Li, C. L. Zhang, M. H. Luo, Q. L. Yao and Z. H. Lu, Inorg. Chem. Front., 2020, 7, 1298–1306 RSC.
- C. Zhang, M. Cui, J. J. Ren, Y. F. Xing, N. Li, H. Y. Zhao, P. Liu, X. P. Ji and M. Li, Chem. Eng. J., 2020, 401, 126025 CrossRef CAS.
- E. Park, J. Jack, Y. M. Hu, S. Wan, S. F. Huang, Y. H. Jin, P. C. Maness, S. Yazdi, Z. Y. Ren and W. Zhang, Nanoscale, 2020, 12, 2596–2602 RSC.
- R. Kamai, K. Kamiya, K. Hashimoto and S. Nakanishi, Angew. Chem., Int. Ed., 2016, 55, 13184–13188 CrossRef CAS PubMed.
- F. P. Lu, Y. Q. Li, Q. R. Shi, C. F. Zhao, S. H. Li and S. P. Pang, New J. Chem., 2020, 44, 15354–15361 RSC.
- Q. P. Zhang, Y. L. Sun, G. Cheng, Z. Wang, H. Ma, S. Y. Ding, B. Tan, J. H. Bu and C. Zhang, Chem. Eng. J., 2020, 391, 123471 CrossRef CAS.
- Y. Deng, Z. Zhang, P. Y. Du, X. M. Ning, Y. Wang, D. X. Zhang, J. Liu, S. T. Zhang and X. Q. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082–6089 CrossRef CAS PubMed.
- S. L. Lu, Y. M. Hu, S. Wan, R. McCaffrey, Y. H. Jin, H. W. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed.
- P. Pachfule, M. K. Panda, S. Kandambeth, S. M. Shivaprasad, D. D. Diaz and R. Banerjee, J. Mater. Chem. A, 2014, 2, 7944–7952 RSC.
- J. C. Wang, C. X. Liu, X. Kan, X. W. Wu, J. L. Kan and Y. B. Dong, Green Chem., 2020, 22, 1150–1155 RSC.
- J. Li, L. H. Zhang, X. T. Liu, N. Z. Shang, S. T. Gao, C. Feng, C. Wang and Z. Wang, New J. Chem., 2018, 42, 9684–9689 RSC.
- P. Pachfule, S. Kandambeth, D. D. Diaz and R. Banerjee, Chem. Commun., 2014, 50, 3169–3172 RSC.
- M. Y. Liu, X. Q. Wang, J. Liu, K. W. Wang, S. B. Jin and B. Tan, ACS Appl. Mater. Interfaces, 2020, 12, 12774–12782 CrossRef CAS PubMed.
- M. Y. Fan, W. D. Wang, X. Y. Wang, Y. Y. Zhu and Z. P. Dong, Ind. Eng. Chem. Res., 2020, 59, 12677–12685 CrossRef CAS.
- Q. Z. Sun, C. Y. Wu, Q. Y. Pan, B. J. Zhang, Y. M. Liu, X. Y. Lu, J. Sun, L. S. Sun and Y. J. Zhao, ChemNanoMat, 2021, 7, 95–99 CrossRef CAS.
- C. Y. Xu, J. Y. Lin, D. Yan, Z. Y. Guo, D. J. Austin, H. B. Zhan, A. Kent and Y. F. Yue, ACS Appl. Nano Mater., 2020, 3, 6416–6422 CrossRef CAS.
- R. Tao, X. R. Shen, Y. M. Hu, K. Kang, Y. Q. Zheng, S. C. Luo, S. Y. Yang, W. L. Li, S. L. Lu, Y. H. Jin, L. Qiu and W. Zhang, Small, 2020, 16, 1906005 CrossRef CAS PubMed.
- L. Y. Li, H. X. Zhao, J. Y. Wang and R. H. Wang, ACS Nano, 2014, 8, 5352–5364 CrossRef CAS PubMed.
- L. Y. Li, C. S. Zhou, H. X. Zhao and R. H. Wang, Nano Res., 2015, 8, 709–721 CrossRef CAS.
- L. Y. Li, H. X. Zhao and R. H. Wang, ACS Catal., 2015, 5, 948–955 CrossRef CAS.
- P. Bhanja, X. Liu and A. Modak, ChemistrySelect, 2017, 2, 7535–7543 CrossRef CAS.
- Z. F. Wang, C. B. Liu, Y. Huang, Y. C. Hu and B. Zhang, Chem. Commun., 2016, 52, 2960–2963 RSC.
- D. Xu, F. S. Wang, G. Q. Yu, H. Zhao, J. Yang, M. Yuan, X. Y. Zhang and Z. P. Dong, ChemCatChem, 2018, 10, 4569–4577 CrossRef CAS.
- X. Y. Yang, Y. J. He, L. Y. Li, J. N. Shen, J. H. Huang, L. Y. Li, Z. Y. Zhuang, J. H. Bi and Y. Yu, Chem.–Eur. J., 2020, 26, 1864–1870 CrossRef CAS PubMed.
- R. A. Molla, P. Bhanja, K. Ghosh, S. S. Islam, A. Bhaumik and S. M. Islam, ChemCatChem, 2017, 9, 1939–1946 CrossRef CAS.
- H. X. Fu, Z. H. Zhang, W. H. Fan, S. F. Wang, Y. Liu and M. H. Huang, J. Mater. Chem. A, 2019, 7, 15048–15053 RSC.
- W. Zhang, Y. Mei, X. Huang, P. Wu, H. H. Wu and M. Y. He, ACS Appl. Mater. Interfaces, 2019, 11, 44241–44248 CrossRef CAS PubMed.
- M. Nihei, H. Ida, T. Nibe, A. M. P. Moeljadi, Q. T. Trinh, H. Hirao, M. Ishizaki, M. Kurihara, T. Shiga and H. Oshio, J. Am. Chem. Soc., 2018, 140, 17753–17759 CrossRef CAS PubMed.
- X. X. Gou, T. Liu, Y. Y. Wang and Y. F. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS PubMed.
- Z. Z. Wang, C. B. Reddy, X. Zhou, J. J. Ibrahim and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 53141–53149 CrossRef CAS PubMed.
- N. N. Sun, C. M. Wang, H. L. Wang, L. Yang, P. Jin, W. Zhang and J. Z. Jiang, Angew. Chem., Int. Ed., 2019, 58, 18011–18016 CrossRef CAS PubMed.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- F. R. Fortea-Perez, M. Mon, J. Ferrando-Soria, M. Boronat, A. Leyva-Perez, A. Corma, J. M. Herrera, D. Osadchii, J. Gascon, D. Armentano and E. Pardo, Nat. Mater., 2017, 16, 760 CrossRef CAS PubMed.
- G. Huang, L. Yang, Q. Yin, Z. B. Fang, X. J. Hu, A. A. Zhang, J. Jiang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 4385–4390 CrossRef CAS PubMed.
- H. B. Fang, J. X. Chen, Y. L. Xiao and J. Y. Zhang, Appl. Catal., A, 2019, 585, 117186 CrossRef CAS.
- M. A. Ziaee, H. Zhong, C. Y. Cui and R. H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 10421–10428 CrossRef CAS.
- Y. Q. Su, X. J. Li, Y. X. Wang, H. Zhong and R. H. Wang, Dalton Trans., 2016, 45, 16896–16903 RSC.
- H. B. Fang, S. J. Sun, P. S. Liao, Y. Hu and J. Y. Zhang, J. Mater. Chem. A, 2018, 6, 2115–2121 RSC.
- H. X. Zhao, Y. X. Wang and R. H. Wang, Chem. Commun., 2014, 50, 10871–10874 RSC.
- P. F. Zhang, Z. A. Qiao, X. G. Jiang, G. M. Veith and S. Dai, Nano Lett., 2015, 15, 823–828 CrossRef CAS PubMed.
- S. Y. Zhang, Z. Kochovski, H. C. Lee, Y. Lu, H. M. Zhang, J. Zhang, J. K. Sun and J. Y. Yuan, Chem. Sci., 2019, 10, 1450–1456 RSC.
- Y. J. Chen, R. Gao, S. F. Ji, H. J. Li, K. Tang, P. Jiang, H. B. Hu, Z. D. Zhang, H. G. Hao, Q. Y. Qu, X. Liang, W. X. Chen, J. C. Dong, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- J. W. Wan, Z. H. Zhao, H. S. Shang, B. Peng, W. X. Chen, J. J. Pei, L. R. Zheng, J. C. Dong, R. Cao, R. Sarangi, Z. L. Jiang, D. N. Zhou, Z. B. Zhuang, J. T. Zhang, D. S. Wang and Y. D. Li, J. Am. Chem. Soc., 2020, 142, 8431–8439 CrossRef CAS PubMed.
- Q. Y. Qu, S. F. Ji, Y. J. Chen, D. S. Wang and Y. D. Li, Chem. Sci., 2021, 12, 4201–4215 RSC.
- X. L. Hu, G. Luo, Q. N. Zhao, D. Wu, T. X. Yang, J. Wen, R. H. Wang, C. H. Xu and N. Hu, J. Am. Chem. Soc., 2020, 142, 16776–16786 CrossRef CAS PubMed.
- D. Zhou, J. G. Yi, X. D. Zhao, J. Q. Yang, H. R. Lu and L. Z. Fan, Chem. Eng. J., 2021, 413, 127508 CrossRef CAS.
- Z. K. Kou, W. J. Zang, Y. Y. Ma, Z. H. Pan, S. C. Mu, X. R. Gao, B. S. Tang, M. Xiong, X. J. Zhao, A. K. Cheetham, L. R. Zheng and J. Wang, Nano Energy, 2020, 67, 104288 CrossRef CAS.
- H. B. Wu, B. Y. Xia, L. Yu, X. Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- Y. J. Chen, S. F. Ji, Y. G. Wang, J. C. Dong, W. X. Chen, Z. Li, R. A. Shen, L. R. Zheng, Z. B. Zhuang, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- Y. Wang, J. L. Li, W. X. Shi, Z. M. Zhang, S. Guo, R. Si, M. Liu, H. C. Zhou, S. Yao, C. H. An and T. B. Lu, Adv. Energy Mater., 2020, 10, 2002138 CrossRef CAS.
- T. H. Gu, W. Teng, N. Bai, Z. H. Chen, J. W. Fan, W. X. Zhang and D. Y. Zhao, J. Mater. Chem. A, 2020, 8, 9545–9553 RSC.
- Z. Li, Y. J. Chen, S. F. Ji, Y. Tang, W. X. Chen, A. Li, J. Zhao, Y. Xiong, Y. E. Wu, Y. Gong, T. Yao, W. Liu, L. R. Zheng, J. C. Dong, Y. Wang, Z. B. Zhuang, W. Xing, C. T. He, C. Peng, W. C. Cheong, Q. H. Li, M. L. Zhang, Z. Chen, N. H. Fu, X. Gao, W. Zhu, J. W. Wan, J. Zhang, L. Gu, S. Q. Wei, P. J. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. F. Duan, Y. Huang, X. M. Chen, D. S. Wang and Y. D. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed.
- S. F. Ji, Y. J. Chen, Q. Fu, Y. F. Chen, J. C. Dong, W. X. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. F. Duan, D. S. Wang, C. Draxl and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS PubMed.
- M. R. Zhang, V. Singh, X. F. Hu, X. Y. Ma, J. K. Lu, C. Zhang, J. P. Wang and J. Y. Niu, ACS Catal., 2019, 9, 7641–7650 CrossRef CAS.
- Y. T. Xu, X. F. Xiao, Z. M. Ye, S. L. Zhao, R. G. Shen, C. T. He, J. P. Zhang, Y. D. Li and X. M. Chen, J. Am. Chem. Soc., 2017, 139, 5285–5288 CrossRef CAS PubMed.
- D. Chakraborty, S. Nandi, R. Illathvalappil, D. Mullangi, R. Maity, S. K. Singh, S. Haldar, C. P. Vinod, S. Kurungot and R. Vaidhyanathan, ACS Omega, 2019, 4, 13465–13473 CrossRef CAS PubMed.
- A. J. Han, W. X. Chen, S. L. Zhang, M. L. Zhang, Y. H. Han, J. Zhang, S. F. Ji, L. R. Zheng, Y. Wang, L. Gu, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Adv. Mater., 2018, 30, 1706508 CrossRef PubMed.
- M. L. Wang, Y. Zhang, M. Z. Cui, Y. Lu, D. D. Peng, X. Cao, C. Wu, J. D. Zhou, Y. Feng, W. F. Liu, Z. F. Chen, X. G. Liu, T. Wang, P. Song and Y. Z. Huang, Biosens. Bioelectron., 2020, 165, 112373 CrossRef CAS PubMed.
- J. H. Zhang, W. Yang, M. Zhang, H. J. Wang, R. Si, D. C. Zhong and T. B. Lu, Nano Energy, 2021, 80, 105542 CrossRef CAS.
- K. X. Huang, J. H. Hua, G. G. Chang, Z. H. Li, G. Tian, M. J. Chen, J. X. Li, S. C. Ke, X. Y. Yang and B. L. Chen, Small, 2021, 17, 2002811 CrossRef CAS PubMed.
- T. He, S. M. Chen, B. Ni, Y. Gong, Z. Wu, L. Song, L. Gu, W. P. Hu and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 3493–3498 CrossRef CAS PubMed.
- X. Wang, W. X. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. X. Ju, J. C. Dong, L. R. Zheng, W. S. Yan, X. S. Zheng, Z. J. Li, X. Q. Wang, J. Yang, D. S. He, Y. Wang, Z. X. Deng, Y. E. Wu and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- X. F. Lu, L. Yu, J. T. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900699 CrossRef PubMed.
- Q. H. Li, W. X. Chen, H. Xiao, Y. Gong, Z. Li, L. R. Zheng, X. S. Zheng, W. S. Yan, W. C. Cheong, R. A. Shen, N. H. Fu, L. Gu, Z. B. Zhuang, C. Chen, D. S. Wang, Q. Peng, J. Li and Y. D. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- J. Zhao, R. X. Qin and R. Liu, Appl. Catal., B, 2019, 256, 117778 CrossRef.
- C. Wu, Z. F. Chen, M. M. Wang, X. Cao, Y. Zhang, P. Song, T. Y. Zhang, X. L. Ye, Y. Yang, W. H. Gu, J. D. Zhou and Y. Z. Huang, Small, 2020, 16, 2001686 CrossRef CAS PubMed.
- Z. F. Liang, D. W. Yang, P. Y. Tang, C. Q. Zhang, J. J. Biendicho, Y. Zhang, J. Llorca, X. Wang, J. S. Li, M. Heggen, J. David, R. E. Dunin-Borkowski, Y. T. Zhou, J. R. Morante, A. Cabot and J. Arbiol, Adv. Energy Mater., 2021, 11, 2003507 CrossRef CAS.
- P. Pachfule, X. C. Yang, Q. L. Zhu, N. Tsumori, T. Uchida and Q. Xu, J. Mater. Chem. A, 2017, 5, 4835–4841 RSC.
- J. Romero, D. Rodriguez-San-Miguel, A. Ribera, R. Mas-Balleste, T. F. Otero, I. Manet, F. Liscio, G. Abellan, F. Zamora and E. Coronado, J. Mater. Chem. A, 2017, 5, 4343–4351 RSC.
- J. T. Ren, L. Chen, D. D. Yang and Z. Y. Yuan, Appl. Catal., B, 2020, 263, 118352 CrossRef CAS.
- Z. P. Shi, K. Q. Nie, Z. J. Shao, B. X. Gao, H. L. Lin, H. B. Zhang, B. L. Liu, Y. X. Wang, Y. H. Zhang, X. H. Sun, X. M. Cao, P. Hu, Q. S. Gao and Y. Tang, Energy Environ. Sci., 2017, 10, 1262–1271 RSC.
- J. S. Li, Y. Wang, C. H. Liu, S. L. Li, Y. G. Wang, L. Z. Dong, Z. H. Dai, Y. F. Li and Y. Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- X. Q. Wang, Z. Chen, X. Y. Zhao, T. Yao, W. X. Chen, R. You, C. M. Zhao, G. Wu, J. Wang, W. X. Huang, J. L. Yang, X. Hong, S. Q. Wei, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- W. X. Cao, L. Lin, H. F. Qi, Q. He, Z. J. Wu, A. Q. Wang, W. H. Luo and T. Zhang, J. Catal., 2019, 373, 161–172 CrossRef CAS.
- F. T. Kong, X. H. Fan, X. Y. Zhang, L. Y. Wang, A. G. Kong and Y. K. Shan, Carbon, 2019, 149, 471–482 CrossRef CAS.
- Q. Zuo, P. P. Zhao, W. Luo and G. Z. Cheng, Nanoscale, 2016, 8, 14271–14277 RSC.
- L. Y. Chen, L. Zhang, Z. J. Chen, H. J. Liu, R. Luque and Y. W. Li, Chem. Sci., 2016, 7, 6015–6020 RSC.
- S. J. Wei, Y. Wang, W. X. Chen, Z. Li, W. C. Cheong, Q. H. Zhang, Y. Gong, L. Gu, C. Chen, D. S. Wang, Q. Peng and Y. D. Li, Chem. Sci., 2020, 11, 786–790 RSC.
- X. Wei, D. Zheng, M. Zhao, H. Z. Chen, X. Fan, B. Gao, L. Gu, Y. Guo, J. B. Qin, J. Wei, Y. L. Zhao and G. C. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14639–14646 CrossRef CAS PubMed.
- J. Mahmood, F. Li, S. M. Jung, M. S. Okyay, I. Ahmad, S. J. Kim, N. Park, H. Y. Jeong and J. B. Baek, Nat. Nanotechnol., 2017, 12, 441–446 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |