
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorine-induced diastereodivergence discovered in an equally rare enantioselective syn-aza-Henry reaction†
Jade A.
Bing
 ,
Nathan D.
Schley
and
Jeffrey N.
Johnston
*
,
Nathan D.
Schley
and
Jeffrey N.
Johnston
*
Department of Chemistry, Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, Tennessee 37235-1822, USA. E-mail: jeffrey.n.johnston@vanderbilt.edu
First published on 14th February 2022
Abstract
Attention to the aza-Henry reaction, particularly over the past two decades, has resulted in a wide range of effective catalysts for the enantio- and diastereoselective versions, driven by the versatility of the β-amino nitroalkane products as precursors to secondary amines and vic-diamines. Despite this broad effort, syn-diastereoselective variants are exceedingly rare. We have discovered a subset of α-fluoro nitroalkane additions that are characterized by an unusual crossover in diastereoselection, often delivering the products with high selectivities. We report here a rigorous comparative analysis of non-fluorinated and α-fluoro nitroalkanes in their additions to azomethines. Both homogeneous and heterogeneous catalysis were applied to probe the possibility that this phenomenon might be more widely operative in the enantioselective additions of fluorine-substituted carbon nucleophiles. A complete correlation within four categories is described that uncovered a clear trend, while revealing a dramatic and distinct reversal of diastereoselection that would normally go undetected.
Introduction
The addition of nitroalkanes to imine azomethines is often referred to as the aza-Henry or nitro-Mannich reaction (Scheme 1, eqn (1)). It has been successfully developed into a leading method for the stereocontrolled synthesis of vic-diamines1 owing to the ease of subsequent nitro reduction (Scheme 1, eqn (2)).2 The discovery of a wide array of Lewis acid and metal-free chiral catalysts that accelerate the aza-Henry reaction has greatly expanded the amine products readily accessed, in particular diamine and secondary amine precursors to small molecules with relevance to therapeutic development.3,4 Despite these advances, nearly all aza-Henry reactions promoted by chiral catalysts favor the anti-diastereomer.5,6 Simultaneous observation of high syn-diastereoselection and high enantioselection (up to 98% ee) has been achieved only once7 with α-alkyl or α-aryl nitromethane derivatives, and twice8,9 with α-nitro ester derivatives.10 The absence of general syn-selective aza-Henry reactions reflects a gap in our understanding of the factors responsible for selectivity, while diminishing the reaction's otherwise broad utility as a source of diamines for therapeutic development and asymmetric synthesis.1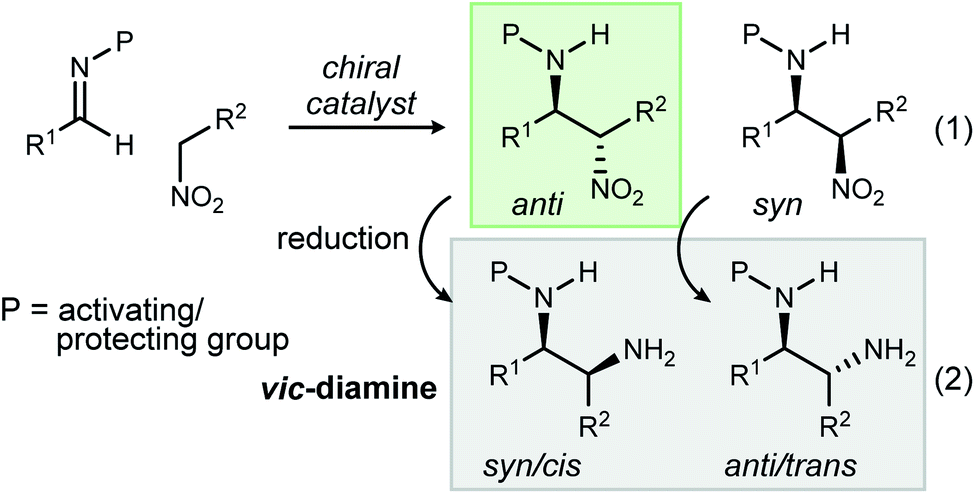 | ||
| Scheme 1 The aza-Henry reaction as a source of enantioenriched β-amino nitroalkanes (eqn (1)) and vic-diamines (eqn (2)). | ||
We have discovered an unusual reversal of diastereoselectivity, favoring the syn-aza-Henry product, that arises within a subset of α-fluoro nitroalkane pronucleophiles. A notable feature is that the reversal is mediated, but not determined by the catalyst, leading to the discovery of an example of fluorine-based diastereodivergence.11 This behavior is outlined by a methodical investigation of this substrate control using aryl and aliphatic aldimines, combined with aryl and aliphatic α-fluoro nitromethane derivatives. An underlying hierarchy has been uncovered in these additions whereby fluorine reverses the inherent anti-selectivity of nitroalkane additions in cases not including aryl nitromethanes. These nitronates, bearing geminal aryl and fluorine, remain unaffected when changing from hydrogen to fluorine at nitronate carbon, suggesting a dominant directing effect provided by the aromatic ring. As a result of our observations, a classification system (Fig. 1, Types I–IV) is proposed by which to organize these behaviors, one that might be adaptable to future studies.
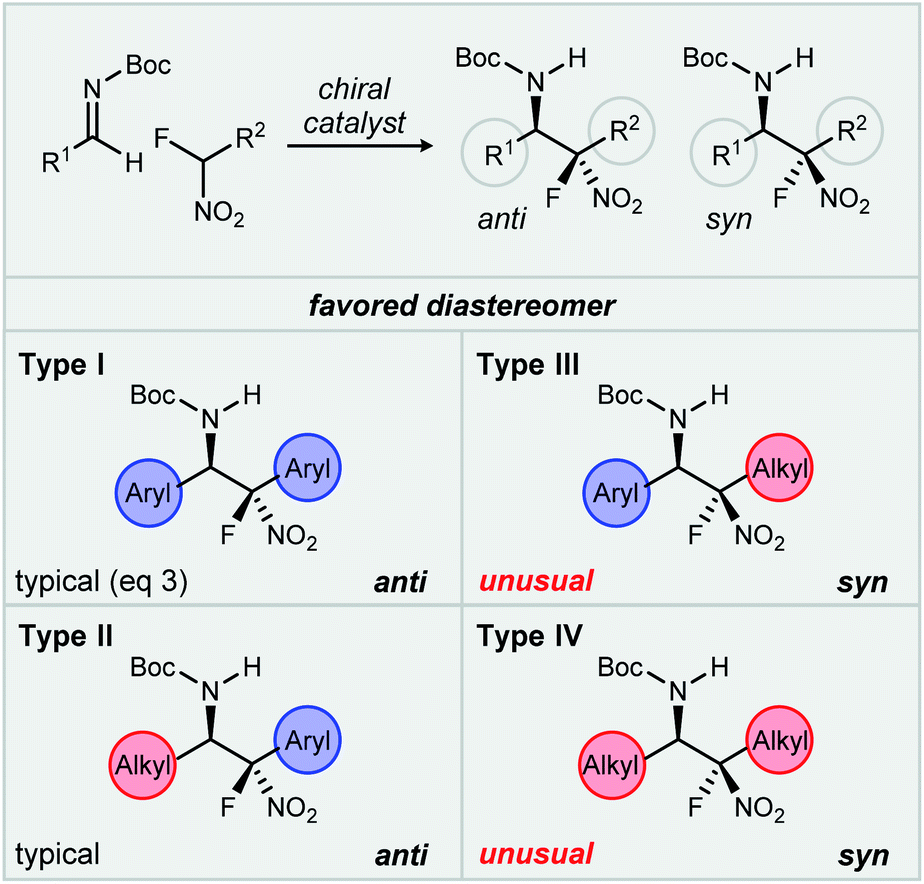 | ||
| Fig. 1 Proposed classification system for catalyzed enantioselective aza-Henry reactions studied (this work), highlighting the substituent-dependence of syn-selectivity. | ||
Results
We recently reported the catalyzed addition of α-fluoro nitroalkanes to N-Boc aldimines using chiral bis(amidine) [BAM] catalyst 1 (Scheme 2, eqn (3)).12 The inclusion of fluorine in the nitroalkane pronucleophile attenuated the overall rate relative to its nonfluorinated counterpart, but otherwise provided for the synthesis of β-fluoro amino stilbenes (after reduction).13–16 The relative configuration of the β-amino nitroalkanes (Fig. 1, Type I)17 was assigned by X-ray diffractometry, establishing that the fluorine's effect on rate blunted but did not reverse anti-selectivity in this case. Other cases exhibited similarly high selectivity, encouraging a tentative assignment of configuration by analogy.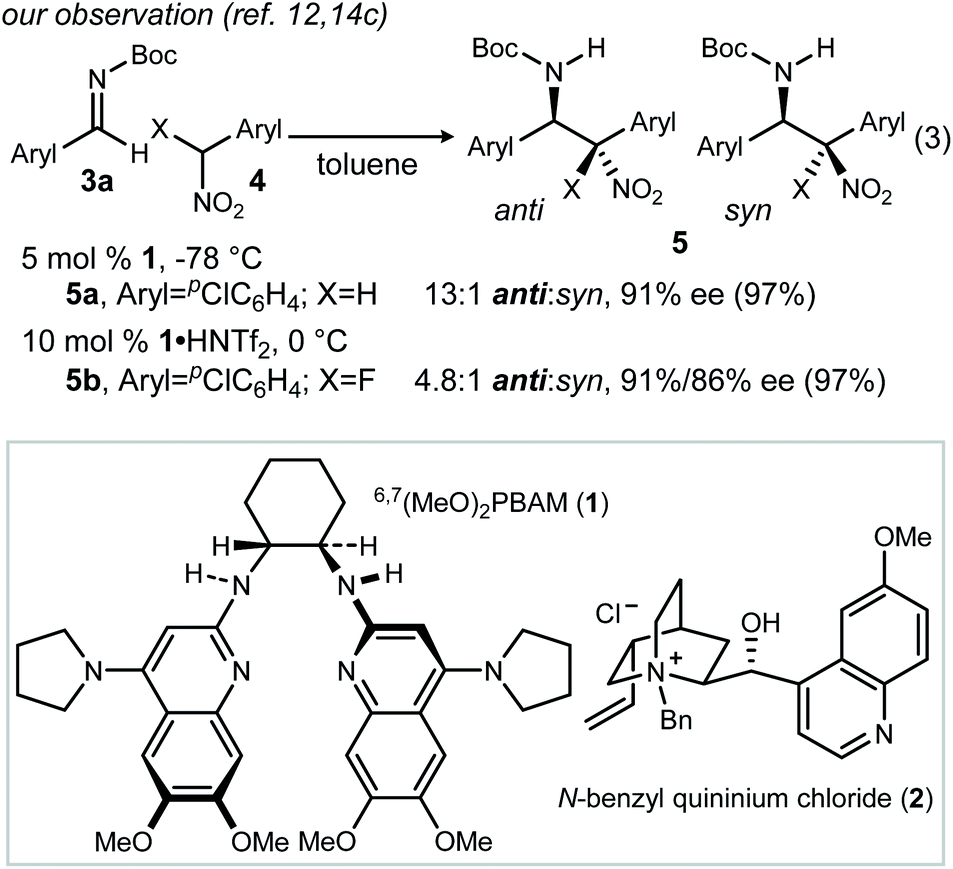 | ||
| Scheme 2 The aza-Henry reaction: typical anti-selective reactions. | ||
During our pursuit of applications requiring Type IV additions, we continued to observe high diastereo- and enantioselectivity. These studies provided an additional opportunity to rigorously determine absolute and relative stereochemistry by X-ray diffraction. In doing so, we made the surprising discovery that selectivity favored the syn-β-amino-α-fluoro nitroalkane. This led us to launch a comprehensive study of each category of aza-Henry, with the objective to identify the control element(s) leading to syn-selectivity, and ultimately an understanding of this phenomenon of diastereodivergence.18
Standard conditions were established using 3b, phenyl nitromethane (4a), and 1 as the catalyst (Table 1, entry 1). As previously reported, the product (6a) is produced with high anti-selectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and 78% ee. The use of heterogeneous catalysis was examined, specifically phase transfer catalysis (PTC) with N-benzyl quininium chloride (2, 12 mol%) and cesium hydroxide. When this protocol was applied to the α-amido sulfone precursor to N-Boc imine, the addition product was formed with high diastereoselectivity, but low enantioselection for the major (anti) diastereomer (Table 1, entry 2: 6a, 15:1 dr, 39% ee). Catalysts 1 (homogeneous) and 2 (heterogeneous) not only exhibited similar behavior with phenyl nitromethane, but also α-fluoro phenyl nitromethane (4b) (Table 1, entries 3–4). This suggested a robust correlation between nitronate-azomethine orientation during activation by catalyst, and relative insensitivity of this stereochemistry-determining arrangement to the presence of fluorine at a reacting carbon. It should be noted, however, that fluorine notably slowed the aza-Henry additions relative to those of non-fluorinated aryl nitromethanes. This is a phenomenon already noted for BAM catalyst 1 (including the effects of ligand protonation), consistent with a combination of rate-limiting nitroalkane deprotonation, and the higher effective pKa exhibited by α-fluoronitroalkanes.19 In summary, homogeneous catalyst 1 is superior to heterogeneous catalyst 2 in Type I additions, but both favor anti-diastereoselectivity, and the same absolute configuration for the major and minor diastereomers.
:1 dr) and 78% ee. The use of heterogeneous catalysis was examined, specifically phase transfer catalysis (PTC) with N-benzyl quininium chloride (2, 12 mol%) and cesium hydroxide. When this protocol was applied to the α-amido sulfone precursor to N-Boc imine, the addition product was formed with high diastereoselectivity, but low enantioselection for the major (anti) diastereomer (Table 1, entry 2: 6a, 15:1 dr, 39% ee). Catalysts 1 (homogeneous) and 2 (heterogeneous) not only exhibited similar behavior with phenyl nitromethane, but also α-fluoro phenyl nitromethane (4b) (Table 1, entries 3–4). This suggested a robust correlation between nitronate-azomethine orientation during activation by catalyst, and relative insensitivity of this stereochemistry-determining arrangement to the presence of fluorine at a reacting carbon. It should be noted, however, that fluorine notably slowed the aza-Henry additions relative to those of non-fluorinated aryl nitromethanes. This is a phenomenon already noted for BAM catalyst 1 (including the effects of ligand protonation), consistent with a combination of rate-limiting nitroalkane deprotonation, and the higher effective pKa exhibited by α-fluoronitroalkanes.19 In summary, homogeneous catalyst 1 is superior to heterogeneous catalyst 2 in Type I additions, but both favor anti-diastereoselectivity, and the same absolute configuration for the major and minor diastereomers.
|
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Type | R1 | Conditions | R2 | H/F | 4 → 6 | Temp. (°C) | Catalyst |
anti:synb |
eeb (%) | Yieldc (%) | ||
| 3/3′ | anti | syn | NMR | Isol | |||||||||
|
a Conditions: (A) the α-amido sulfone 3′ is treated with Cs2CO3 in toluene to form imine 3. After filtration, this solution is used directly in the aza-Henry reaction which is carried out in toluene (0.1 M) using the nitro- or fluoronitroalkane (1.2 equiv.) and 6,7(MeO)2PBAM (1) or 6,7(MeO)2PBAM·HNTf2 (1·HNTf2, 10 mol%) for 24 hours. (B) Using 3 (neat, preformed from 3′), the aza-Henry reaction is run in dry toluene (0.1 M) under argon using the nitro- or fluoronitroalkane (1.2 equiv.) with 1 or 1·HNTf2. (C) Reaction run in dry toluene (0.1 M) under argon using the nitro- or fluoronitroalkane (4.5 equiv.), N-benzylquininium chloride (2, 12 mol%), and CsOH·H2O (1.3 equiv.) for 48–72 hours. See ESI for complete details.
b Diastereomeric ratios measured by 1H NMR analysis of the crude reaction mixture. Enantiomeric excess determined by HPLC using a chiral stationary phase.
c Yields over 2 steps (from α-amido sulfone). NMR: yield measured using an internal standard when 6 is present at high apparent purity in crude reaction mixture. Isol: isolated yield obtained when impurities are evident alongside 6 in crude reaction mixture. Selected cases analyzed using both methods for comparison.
d Data from ref. 12.
e 20 mol% catalyst. Using 10 mol% catalyst provides 6d with 5.8:1 dr, 87/>99% ee, and 43% yield.
|
|||||||||||||
| 1 | I | Ph (a) | B | Ph | H | 4a/6a | −55 | 1 | >20:1 |
78 | 99 | 79 | — |
| 2 | I | Ph (a) | C | Ph | H | 4a/6a | −50 | 2 | 15:1 |
39 | 99 | 30 | — |
| 3d | I | Ph (a) | B | Ph | F | 4b/6b | 0 | 1·HNTf2 | 3.5:1 |
94 | 84 | — | 88 |
| 4 | I | Ph (a) | C | Ph | F | 4b/6b | −55 | 2 | 4.2:1 |
52 | 31 | 60 | — |
| 5 | II | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2 (b) CHCH2CH2 (b) |
A | Ph | H | 4a/6c | −55 | 1 | >20:1 |
60 | 99 | 35 | 35 |
| 6 | II | CH2CHCH2CH2 (b) |
C | Ph | H | 4a/6c | −35 | 2 | 11:1 |
89 | 99 | 33 | 31 |
| 7e | II | CH2CHCH2CH2 (b) |
A | Ph | F | 4b/6d | −20 | 1·HNTf2 | 5.2:1 |
83 | 99 | — | 53 |
| 8 | II | CH2CHCH2CH2 (b) |
C | Ph | F | 4b/6d | −35 | 2 | 2.7:1 |
91 | 93 | — | 43 |
| 9 | III | p ClC6H4 (c) | B | PhCH2CH2 | H | 4c/6e | −20 | 1·HNTf2 | 20:1 |
87 | 51 | — | 49 |
| 10 | III | p ClC6H4 (c) | C | PhCH2CH2 | H | 4c/6e | −55 | 2 | 3:1 |
33 | 7 | 86 | — |
| 11d | III | p ClC6H4 (c) | B | PhCH2CH2 | F | 4d/6f | 25 | 1·HNTf2 | 1:5.0 |
99 | 93 | — | 85 |
| 12 | III | p ClC6H4 (c) | C | PhCH2CH2 | F | 4d/6f | −35 | 2 | 1:2.5 |
24 | 60 | 89 | 79 |
| 13 | IV | t BuCH2 (d) | A | PhCH2CH2 | H | 4c/6g | −55 | 1 | 1:1 |
20 | 11 | — | 21 |
| 14 | IV | t BuCH2 (d) | C | PhCH2CH2 | H | 4c/6g | −55 | 2 | >20:1 |
99 | — | 94 | 90 |
| 15 | IV | t BuCH2 (d) | A | PhCH2CH2 | F | 4d/6h | 0 | 1·HNTf2 | 1:2.4 |
81 | 80 | 47 | — |
| 16 | IV | t BuCH2 (d) | C | PhCH2CH2 | F | 4d/6h | −35 | 2 | 1:7.2 |
76 | 91 | — | 84 |
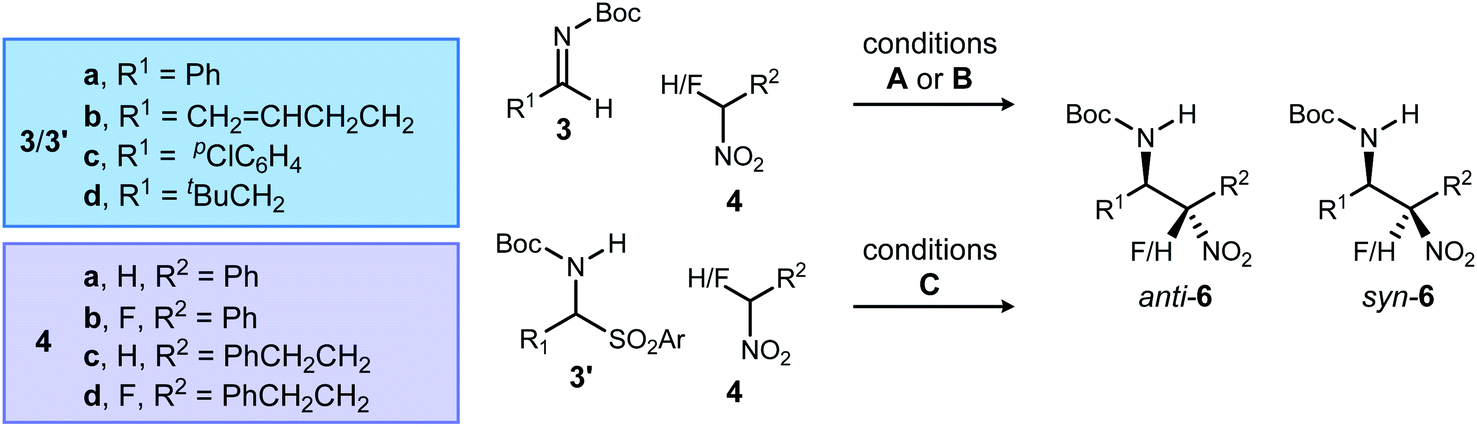
We next investigated an alkyl aldimine electrophile (Table 1, entries 5–8) with the same pair of nitroalkanes (4a–b), observing varying levels of selectivity. Catalysts 1 and 2 favored the same relative and absolute configurations for the products of these reactions. The level of diastereoselection during formation of 6c was highest using BAM catalysis (Table 1, entry 5 vs. entry 6), but enantioselection was higher using phase transfer catalyst (PTC) 2 (89% ee vs. 60% ee). Extending this examination to α-fluoronitroalkane 4b conversion to 6d, both catalysts 1 and 2 featured anti-selective additions for α-fluoro aryl nitromethane additions to aliphatic aldimines. For example, α-fluoro phenyl nitromethane provided the β-amino-α-fluoro-nitroalkane in 5.2:1 anti:syn, 83% ee (Table 1, entry 7). Temperature in this and later examples was most often selected to optimize conversion and yield for specific cases, while seeking the lowest functional temperature to favor higher selectivity. Also of note, our desire for rigorous assignment of the major stereoisomer for the non-fluorinated adduct went unfulfilled, since crystalline anti-6c formed only thin, feathery needles. As a result, the assignment was made by analogy to a similar case examined by Duan.26 Absolute and relative stereochemistry for the major isomer of the fluorinated substrate was assigned by X-ray diffraction (anti-6d, Fig. 2).
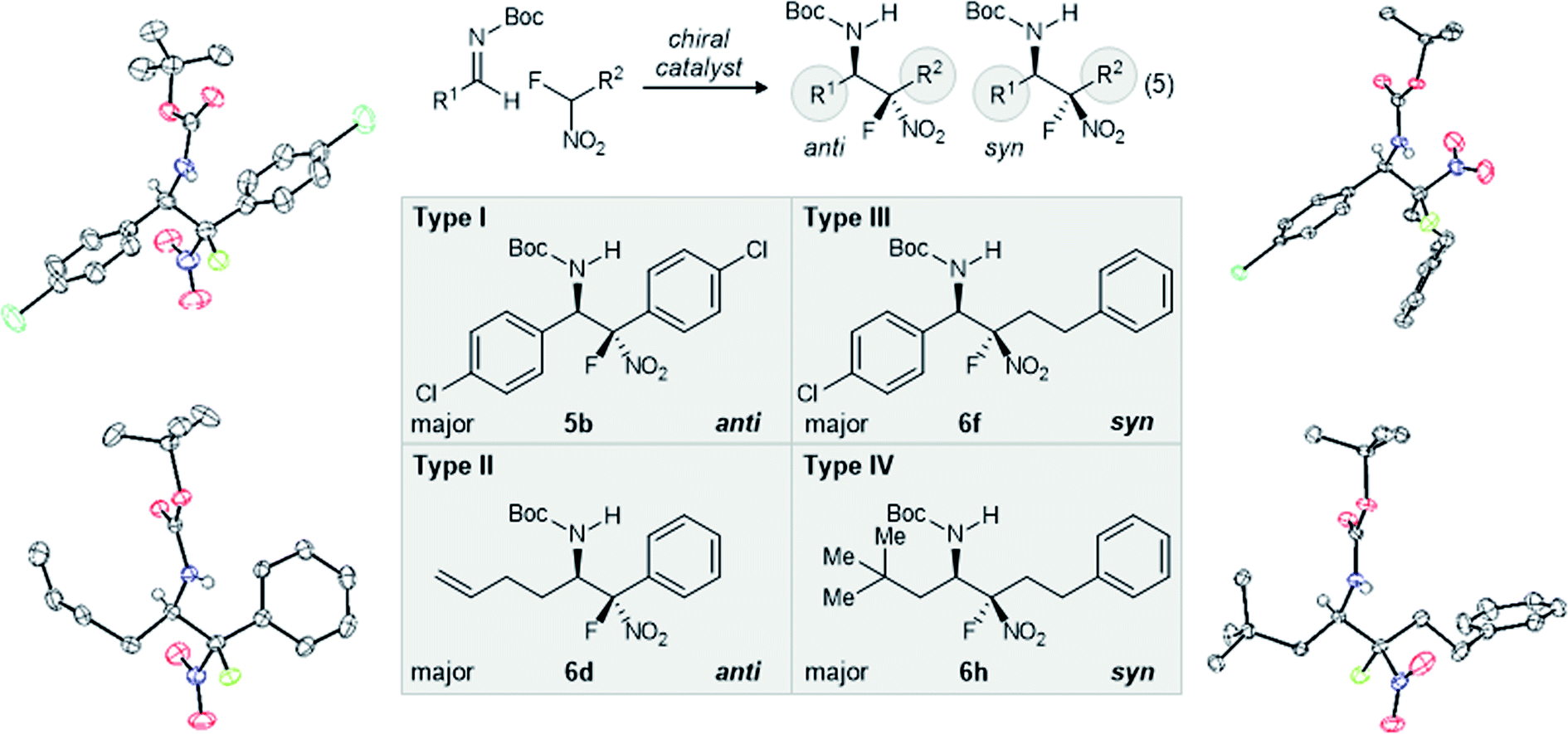 | ||
| Fig. 2 X-Ray analysisa for major (relative and absolute) stereoisomer formed in experiments detailed by Table 1, and categorization of aza-Henry reactions by the type (aryl vs. alkyl) of the azomethine electrophile substituent, and the nitromethane substitutent(s). a ORTEPs shown at 50% probability. | ||
Within Type III additions, non-fluorinated alkyl-substituted nitronates provided addition products with aryl aldimines with good selectivity (20:1 dr, 87% ee) when using 1·HNTf2 (Table 1, entry 9). Dixon20 and Palomo21a have independently assigned anti-selectivity to this type of addition. In the formation of 6e from 4c, catalyst 2 was not competitive, providing low diastereoselection (3:1) and enantioselection (33% ee, Table 1, entry 10) relative to BAM catalysis. Despite the low selectivity, both catalysts favor the same stereoisomer. In Type III cases involving α-fluoro aliphatic nitroalkane additions (4d) and aromatic aldimine electrophiles, good diastereo- and enantioselectivity (5.0:1 dr, 99% ee: Table 1, entry 11) was observed. This behavior followed the trends outlined for all of the additions described to this point. Fortunately, we did not rely on analogy for stereochemical assignment since it would have predicted conservation of anti-selectivity based on all Type I/II additions, and non-fluorinated Type III additions. We instead sought rigorous stereochemical assignment by X-ray diffraction. Single crystals of the major diastereomer for 6f revealed its syn-relationship (Fig. 2). Comparison of homo- and heterogeneous catalysis confirmed again that both methods favored the same relative and absolute stereochemistry in the product, albeit with low dr and ee for 2 (Table 1, entry 12).
For Type IV additions, we selected 3-phenyl-1-nitropropane for evaluation in both hetero- and homogeneous catalyst protocols, delivering the product of addition to 3d with high selectivity only when using 2 (Table 1, entries 13–14). A strength of phase transfer catalysis is the pairing of rates for imine formation and consumption, a feature that is particularly impactful when the imine can tautomerize to the N-acyl enamide. In this comparison, catalyst 1 provided 6g in low yield and with minimal stereoselection. However, PTC 2 delivered 6g with exceptional selectivity (>20:1 dr, 99% ee) and yield (Table 1, entry 14). Rigorous assignment of anti-selectivity for this reaction was made by X-ray diffraction analysis of the major product, anti-6g.
Our examination of the corresponding Type IV additions of α-fluoronitroalkane using either homogeneous BAM catalysis (1·HNTf2) or heterogeneous catalysis (2) revealed similar selectivity trends again, with the former providing product with low diastereoselection (2.4:1) and moderate enantioselection (81% ee) at 0 °C. Catalyst 2 led to the same major diastereomer with high selectivity (7.2:1), and moderate enantioselection (76% ee) at −35 °C (Table 1, entry 16). In this case, both the major and minor diastereomers of 6h formed good quality single crystals that allowed the major to be assigned as syn-6h and the minor to be assigned as anti-6h. This also allowed us to further confirm that the stereochemistry at the azomethine carbon is conserved. Once again, the addition of alkyl-substituted α-fluoro nitronates displayed a reversal of diastereoselection, favoring syn-selectivity. Notably, this behavior stands in contrast to the anti-selectivity observed with non-fluorinated alkyl-substituted nitronates 4c.
Discussion
An important aspect of this study is the dedicated effort to rigorously assign relative and absolute configuration for each example while placing it in the context of the same efforts by others. In Type I (Ar/Ar) additions, aryl nitromethanes undergo diastereo- and enantioselective addition to N-Boc benzaldimines using bis(amidine) [BAM] catalysis, providing anti-addition products with high yield and selectivity (Scheme 2, eqn (3), 5a). In addition to assignment of the favored stereoisomer by X-ray diffraction (R13-Br-4-MeOC6H3, R2C6H5), a key analogue (R1R24-ClC6H4) was converted to the p53/MDM2 inhibitor Nutlin-3a,14b and its potency recapitulated in vitro.14,22 Advances for Type I additions have been made by others (Fig. 3). Ooi developed a chiral ammonium betaine (C1) that further improves the selectivity of this addition, providing the Type I product (Ph/Ph) in 97% ee. Ooi's investigation of aryl nitromethane, and α-vinyl-α-aryl-nitromethane addition to imines exhibited highly conserved anti-selectivity.23,24 Kozlowski identified cinchonidinium acetate (C3·HOAc) as an effective homogeneous catalyst for Type I product (Ph/tBuC6H4: 97:3 anti:syn, 70% ee).25 Finally, Duan used a urea/tetralkyl ammonium bifunctional catalyst (C4) to provide 6a (Ph/Ph) in 99:1 anti:syn and 99% ee.26,27
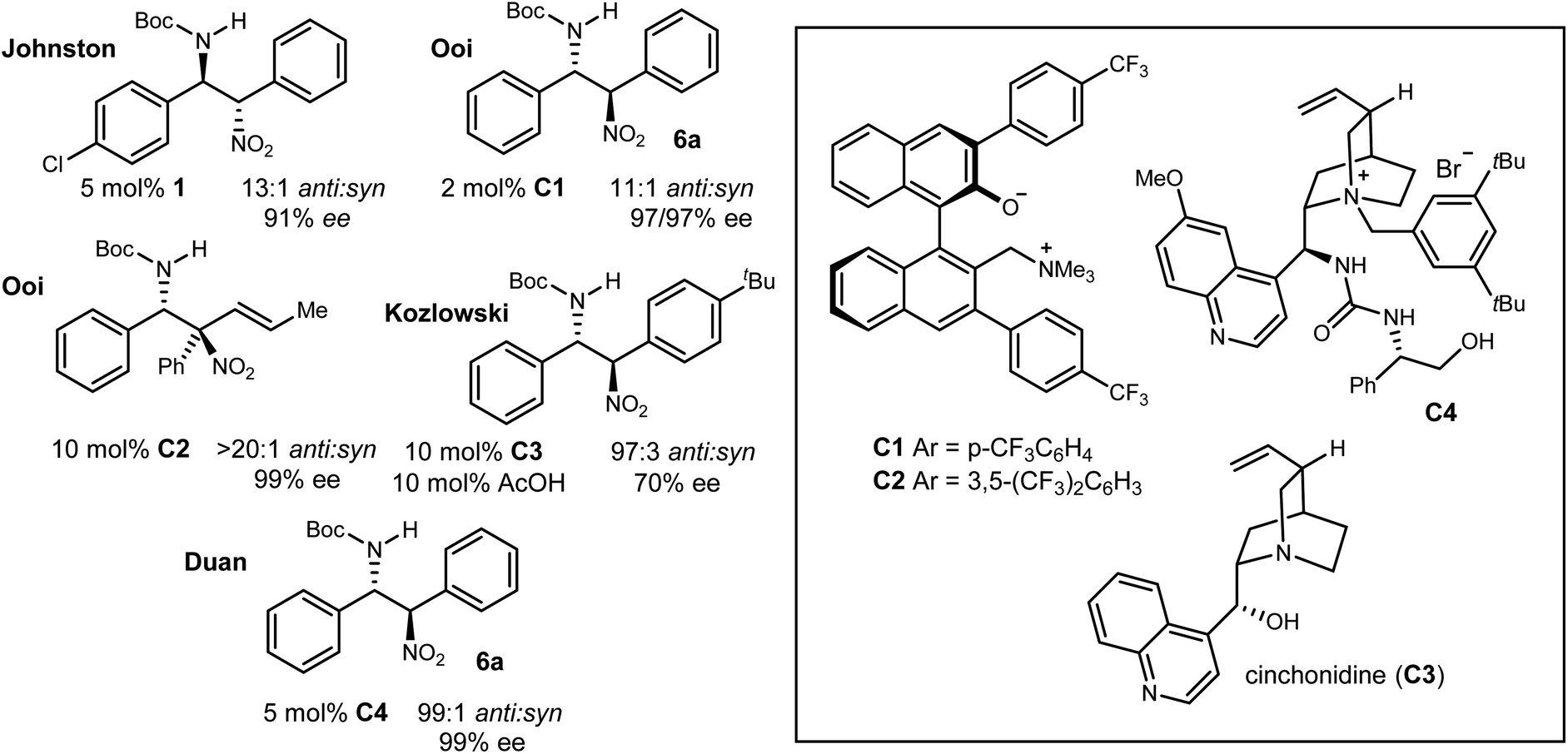 | ||
| Fig. 3 The aza-Henry reaction of aryl nitromethanes to aryl aldimines: anti-selectivity across diverse catalysts. | ||
In Type II (Alkyl/Ar) additions, the reaction of aryl nitronates with alkyl aldimines have been established as anti-selective. Aliphatic N-Boc aldimines are best addressed by phase transfer-catalysis since the formation of N-Boc enamide by tautomerization is minimized.21,28 Duan extended the catalyst effective for Type I additions to a single example involving an aliphatic aldimine, which provided the product in 91% yield (PhCH2CH2/Ph: 94:6 dr, 97% ee). The stereochemical assignment of this example was made by analogy to the associated Type I cases, specifically 6a.
Our assignment of product configuration (vide supra) confirmed that Type II additions exhibit robust anti-selectivity regardless of fluorine substitution of the intermediate aryl nitronate. Importantly, these results also establish that this behavior is independent of catalyst type (homo- vs. heterogeneous).
Type III additions are undoubtedly the most studied cases, thereby providing a greater pool of catalysts for comparison. Among these, the catalysts 1·HNTf2 and 2 have been shown to be highly effective. However, the high degree of consistency exhibited by Type I–II additions using non-fluorinated and fluorinated nitroalkanes 4 did not accurately forecast the behaviors to be uncovered by Type III additions.
Limited literature precedent exists for the favored stereoisomer in additions of alkyl-substituted nitronates to aliphatic aldimines (Type IV). This includes the early examples (2005) by Palomo citing anti-selectivity when drawing analogy to Type III additions. This Type IV selectivity was more rigorously established in 2008 using X-ray diffraction.21b Shibasaki's singular syn-selective Type IV additions featured a Cu(II)/Sm(III) protocol for nitroethane addition to an aliphatic aldimine (65% yield, 20:1 dr, 80% ee), and this product was converted to nemonapride, an antipsychotic agent.7 In other cases, Shibasaki's bimetallic catalyst provided the typical anti-selectivity with good enantioselection.29 Anderson rigorously assigned silyl nitronate addition products within the Type III class for which a copper(II)–bis(oxazoline) system provided high dr/ee. Analogy was then used to assign those belonging to the Type IV class, which included two cases: the product from cyclohexanal N-PMP imine (88% yield, >15:1 dr, 87% ee) and hexanal imine (88% yield, 1:1 dr, 86% ee).30 Finally, Duan used a phase transfer catalyst equipped with hydrogen bonding ability to report a single example: the addition of nitroethane to the cyclohexyl carboxaldehyde imine, with up to 99:1 dr, 97% ee. There is minimal literature related to the effect of fluorine on the diastereo- and enantioselective addition of nitroalkanes bearing alternative activating groups.31–34
To summarize, across Types I–IV, anti-diastereoselection is observed when using non-fluorinated nitroalkanes. This is observed regardless of catalyst (1 or 2), reaffirming reports with PTC,21 BAM,35 and a wide range of other catalysts4,36–38 with alkyl-substituted nitronates, and PTC24–26 or BAM14 catalysts applied to aryl-substituted nitronates. Departing from this behavior39 for enantioselective aza-Henry reactions, Shibasaki's report is a standout. It is the most successful syn-diastereo- and enantioselective reaction to-date: using a mixed Cu(II)/Sm(III) complex, excellent syn-diastereoselection was achieved, with good enantioselection (43–83% ee). Aliphatic aldimines were less successful, reaching as high as 81% ee, and the scope was also limited to three linear aldimines.7 The syn-selectivity in that work is attributed to reagent control by formation of a samarium(III) nitronate and copper(II)-activated Boc-imine supported simultaneously by the same chiral ligand.
Two basic stereochemistry-determining arrangements of azomethine and nitronate are summarized in Fig. 4: pre-anti (7 or 9) and pre-syn (8 or 10). Regardless of catalyst deployed in experiments here, the azomethine Si face is favored. This is evident from the configuration at the aminomethyl carbon for both diastereomers, indicating that the azomethine-catalyst binding is relatively conserved (this was further confirmed in this work by X-ray analysis, see ESI† for details). Therefore, each Newman projection in Fig. 4 illustrates nitronate approach to the azomethine Si face. A second guiding principle is the bifunctional activation of azomethine and nitronate which favors a synclinal arrangement of azomethine nitrogen and nitronate-NO2 units. This feature has been supported by Dudding's analysis,40,41 it is consistent with the bifunctional character of BAM catalysis, and it is also invoked elsewhere.7,21b Type I–II aza-Henry reactions are anti-selective and insensitive to the presence of fluorine (R1H vs. F). The arrangement in Fig. 4A provides for the first two factors, and placement of the aromatic ring of the nitronate between the smaller aldimine-hydrogen and Ar/Alkyl substituents (i.e. R2) of the azomethine predicts anti-selectivity on purely steric grounds. The cases of alkyl-substituted nitronates are more nuanced (Fig. 4B), with the non-fluorinated nitronate (R1H) favoring anti-selectivity for the same reasons as listed above.
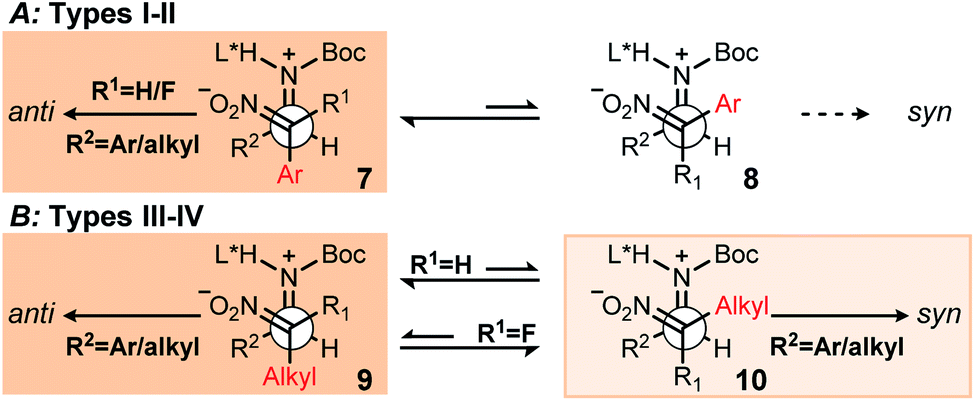 | ||
| Fig. 4 Working models (i.e., L = 1) to identify dominant effects present during C–C bond formation as a function of nitronate substituent combination (aryl/alkyl, H/F). | ||
Fluorine-substituted nitronates favor syn-selectivity (Fig. 4B) when geminally-substituted with an alkyl substituent, but not aryl. Although fluorine is highly electronegative and a weak hydrogen bond acceptor, the consistent azomethine face-selectivity suggests that the electrostatically-driven hydrogen bonding between ligand and azomethine is not dramatically affected by the presence of fluorine in the nitroalkane.42 Contemporary models advancing the gauche effect for fluorine43 align with anti-selectivity, wherein iminium and fluorine are gauche (Fig. 4) to allow σCC → 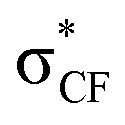 σCF* overlap involving the aldimine R2–carbon bond.44 While this reasoning is consistent with Type I–II (R1F) additions, it should also apply to Type III–IV additions (R1F), but these lead to syn-selectivity instead.
σCF* overlap involving the aldimine R2–carbon bond.44 While this reasoning is consistent with Type I–II (R1F) additions, it should also apply to Type III–IV additions (R1F), but these lead to syn-selectivity instead.
The origin of diastereodivergence may emanate from a secondary interaction between the iminium and the nitronate substituent in an anti relationship in the transition state, favoring Ar > F > alkyl. We speculated that the extensive literature describing the Diels–Alder reaction might harbor fluorine-based diastereodivergence, particularly in cases where directing groups compete in a thermal reaction. Indeed, the competition between phenyl and fluorine for endo-positioning in Diels–Alder reactions favors slightly (2–3:1) endo-fluorine with an electron-rich benzoisofuran and α-fluoro styrene.45 Moreover, (E)-β-fluoro styrene is also slightly endo-fluorine selective, while (Z)-β-fluoro styrene produces only endo-product (both Ph and F are positioned endo).46 The substituent effect of fluorine (vs. hydrogen) is perhaps most pronounced in enantioselective catalysis with α-fluoro enones and esters. In these cases, fluorine overrides ketone and ester carbonyl for endo-positioning by 3:1.47 Computational methods have been applied to thermal and Lewis acid-promoted Diels–Alder cycloadditions of cyclopentadiene with enones, leading to the conclusion that destabilization of the endo-pathway for 3-fluorobutenone is primarily responsible for its endo-fluorine (exo-CO) selectivity.48 Overall, these substituent effects follow a hierarchy F > Ph/carbonyl > H consistent across cyclopentadiene and diphenyl isobenzofuran.
Unlike the thermal Diels–Alder reactions where fluorine effected diastereodivergence when competitive with both phenyl (aryl) and carbonyl geminal substituents, the hierarchy here is Ar > F > alkyl. Of course, the transition states for nitronate additions to imines with catalysis are quite different, and the role of fluorine would be expected to be more pronounced than an alkyl group in the transition state. The unusual behavior lies in the contrasting effects when phenyl nitromethane derivatives are involved. We speculate that in these cases the electronic nature of an aryl ring prevails, perhaps through a secondary interaction between anti-aryl or anti-F and imine π* orbitals: πAr → π*-imine > nF → π*-imine > σC-alkyl → π*-imine.
Conclusion
Despite extensive investigation of the aza-Henry reaction using a broad range of chiral catalysts, highly enantio- and syn-selective variations are rare.3 Moreover, with one exception, the only known examples require an α-nitro ester.8–10 Only one syn-selective aza-Henry of a nitroalkane without an additional activating group has been reported, with enantioselection as high as 98%.7 A thorough study of catalyzed nitronate additions to N-Boc imines derived from aliphatic and aromatic aldehydes revealed exceptions to the otherwise common finding of anti-selectivity that is broadly observed in enantioselective, catalyzed aza-Henry reactions. This selectivity is particularly characteristic of terminal nitroalkanes lacking activating groups, and we replicated and extended this trend, using X-ray diffraction to rigorously assign product stereochemistry when needed. The exceptions were found among fluorine-substituted nitronates, but only those with an aliphatic substituent. In cases where an aromatic ring substituent was geminal to the C–F bond, anti-selectivity prevailed. The fluorine effect, and its compartmentalization into a subset of nitronates is unprecedented. We speculate that a hierarchy of directing effects is responsible for the selectivity, with the nitro group's position conserved among Type I–IV, but a phenyl ring overriding the fluorine's additional effect in Type I–II additions. Remarkable aspects of this discovery include the turnover in diastereoselection to syn-selectivity simultaneous with high enantioselection, in some cases setting it apart from prior highs established by others.7 That these trends are relatively independent of catalysis method (hetero- vs. homogeneous) when using α-fluoronitroalkane pronucleophiles suggests that the behavior may be more generally observed. Examples of diastereodivergence associated with substrate control, using a single catalyst, are increasing.49–51 As solutions to the stereocontrolled aza-Henry reaction increase, so will their impact on concise preparations of small molecules in drug development, and innovative entry to peptides based solely on catalytic, enantioselective methods.52Data availability
All experimental and characterization data in this article are available in the ESI.† Crystallographic data for compounds anti-6d, syn-6f, anti-6g, anti-6h, and syn-6h have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC 2118052–2118056.Author contributions
J. B. and J. J. conceived the project, J. B. completed the experimental work, N. S. provided X-ray crystallography support. All authors wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. 1445197/1937963 (J. A. B.). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. J. A. B. was supported in part by a VU Provost's Fellowship. We are grateful to the National Institute of General Medical Sciences (NIH GM 084333) for financial support. The Indiana University Mass Spectrometry Facility acknowledges support from the NSF (CHE1726633).References
- D. Lucet, T. Le Gall and C. Mioskowski, The chemistry of vicinal diamines, Angew. Chem., Int. Ed., 1998, 37, 2580 CrossRef CAS PubMed.
- For reviews on the aza-Henry reaction see: (a) M. F. P. Ana, Recent Advances on the Organocatalytic Asymmetric Aza-Henry Reaction, Curr. Organocatal., 2016, 3, 222 CrossRef; (b) E. Marqués-López, P. Merino, T. Tejero and R. P. Herrera, Catalytic Enantioselective Aza-Henry Reactions, Eur. J. Org. Chem., 2009, 2401 CrossRef; (c) B. Westermann, Asymmetric Catalytic Aza-Henry Reactions Leading to 1,2-Diamines and 1,2-Diaminocarboxylic Acids, Angew. Chem., Int. Ed., 2003, 42, 151 CrossRef CAS PubMed.
- A. Y. Sukhorukov, A. A. Sukhanova and S. G. Zlotin, Stereoselective reactions of nitro compounds in the synthesis of natural compound analogs and active pharmaceutical ingredients, Tetrahedron, 2016, 72, 6191 CrossRef CAS.
- A. Noble and J. C. Anderson, Nitro-Mannich Reaction, Chem. Rev., 2013, 113, 2887 CrossRef CAS PubMed.
- The use of relative stereochemical terms (anti, syn) here conforms to the most common convention: the aza-Henry product is drawn in zigzag form using the carbon backbone, and anti characterizes this drawing when the nitrogens of amine and nitro reside on opposite sides.
- Ketimine electrophiles in enantioselective aza-Henry reactions are far less common, but syn-selective variants are known: G. Hong, D. Mao, X. Zhu, S. Wu and L. Wang, Metal free access to amide compounds via peroxide-mediated N/N double bond cleavage of azobenzenes, Org. Chem. Front., 2015, 2, 985 RSC.
- (a) S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, syn-Selective catalytic asymmetric nitro-Mannich reactions using a heterobimetallic Cu-Sm-Schiff base complex, J. Am. Chem. Soc., 2007, 129, 4900 CrossRef CAS PubMed; (b) S. Handa, V. Gnanadesikan, S. Matsunaga and M. Shibasaki, Heterobimetallic Transition Metal/Rare Earth Metal Bifunctional Catalysis: A Cu/Sm/Schiff Base Complex for syn-Selective Catalytic Asymmetric Nitro-Mannich Reaction, J. Am. Chem. Soc., 2010, 132, 4925 CrossRef CAS PubMed.
- A. Singh and J. N. Johnston, A Diastereo- and Enantioselective Synthesis of a-Substituted syn-α,β-Diamino Acids, J. Am. Chem. Soc., 2008, 130, 5866 CrossRef CAS PubMed.
- D. Uraguchi, K. Koshimoto and T. Ooi, Chiral Ammonium Betaines: A Bifunctional Organic Base Catalyst for Asymmetric Mannich-Type Reaction of a-Nitrocarboxylates, J. Am. Chem. Soc., 2008, 130, 10878 CrossRef CAS PubMed.
- α-Substituted (alkyl or aryl) α-nitroesters are conventionally drawn with the ester in the plane, leading to these summary comments, and interpretations of behavior should consider this. In the current context, therefore, this statement might summarize that syn-selective additions of α-nitroesters are common, while there are currently only two anti-selective exceptions.
- H. G. Adolph and M. J. Kamlet, Fluoronitroaliphatics. I. The Effect of α Fluorine on the Acidities of Substituted Nitromethanes, J. Am. Chem. Soc., 1966, 88, 4761 CrossRef CAS.
- B. A. Vara and J. N. Johnston, Enantioselective Synthesis of β-Fluoro Amines Via β-Amino α-Fluoro Nitroalkanes and a Traceless Activating Group Strategy, J. Am. Chem. Soc., 2016, 138, 13794 CrossRef CAS PubMed.
- Some stilbene β-fluoroamines can be prepared by enantioselective phase transfer catalysis of aziridine ring-opening. Leading reference: G. Pupo, A. C. Vicini, D. M. H. Ascough, F. Ibba, K. E. Christensen, A. L. Thompson, J. M. Brown, R. S. Paton and V. Gouverneur, Hydrogen Bonding Phase-Transfer Catalysis with Potassium Fluoride: Enantioselective Synthesis of β-Fluoroamines, J. Am. Chem. Soc., 2019, 141, 2878 CrossRef CAS PubMed.
- (a) B. A. Vara, A. Mayasundari, J. C. Tellis, M. W. Danneman, V. Arredondo, T. A. Davis, J. Min, K. Finch, R. K. Guy and J. N. Johnston, Organocatalytic, Diastereo- and Enantioselective Synthesis of Nonsymmetric cis-Stilbene Diamines: A Platform for the Preparation of Single-Enantiomer cis-Imidazolines for Protein–Protein Inhibition, J. Org. Chem., 2014, 79, 6913 CrossRef CAS PubMed; (b) T. A. Davis, A. E. Vilgelm, A. Richmond and J. N. Johnston, Preparation of (−)-Nutlin-3 Using Enantioselective Organocatalysis at Decagram Scale, J. Org. Chem., 2013, 78, 10605 CrossRef CAS PubMed; (c) T. A. Davis and J. N. Johnston, Catalytic, enantioselective synthesis of stilbene cis-diamines: a concise preparation of (−)-nutlin-3, a potent p53/MDM2 inhibitor, Chem. Sci., 2011, 2, 1076 RSC; (d) S. V. Tsukanov, M. D. Johnson, S. A. May, S. P. Kolis, M. H. Yates and J. N. Johnston, Continuous Platform to Generate Nitroalkanes on-Demand (In Situ) Using Peracetic Acid-Mediated Oxidation in a PFA Pipes-in-Series Reactor, Org. Process Res. Dev., 2018, 22, 971 CrossRef CAS PubMed; (e) S. V. Tsukanov, M. D. Johnson, S. A. May, M. Rosemeyer, M. A. Watkins, S. P. Kolis, M. H. Yates and J. N. Johnston, Development of an Intermittent-Flow Enantioselective Aza-Henry Reaction Using an Arylnitromethane and Homogeneous Brønsted Acid–Base Catalyst with Recycle, Org. Process Res. Dev., 2016, 20, 215 CrossRef CAS PubMed.
- The effect of added Brønsted acid to ligand 1 when using non-fluorinated aryl nitromethanes was negligible, owing to the acidity of the nitroalkanes (i.e. the nitroalkane serves as an acid modifier). The activity/selectivity increase of added acid (HNTf2) returned when using less acidic (higher pKa: ref. 12) α-fluoro aryl nitromethanes. The specific effect can be acid-dependent (c.f. ref. 14c (HOTf) and Table 1, entry 1: 1·HNTf2 provided the product with 42% ee).
- For a general review of fluorine effects on fluoroalkylation reactions, see: C. Ni and J. Hu, The unique fluorine effects in organic reactions: recent facts and insights into fluoroalkylations, Chem. Soc. Rev., 2016, 45, 5441 RSC.
- The Type framework is based on the hypothesis that the relative stereoselectivity may be influenced by the nature of the reactive functionality’s substituents, organized into four categories, Types I–IV (Fig. 1), determined by the N-Boc aldimine and nitroalkane substituents (aryl vs. alkyl). For example, a Type I aza-Henry involves an aryl nitromethane addition to aryl aldimine, while a Type III addition involves a nitroalkane addition to an aryl aldimine.
- In total, three cases (7h–j in ref. 12) must be reassigned as syn-diastereomers. See ESI† for complete experimental data.
- (a) F. G. Bordwell and J. E. Bartmess, Taft Equation as Applied to Equilibrium Acidities of Nitroalkanes, G(CH2)NO2, J. Org. Chem., 1978, 43, 3101 CrossRef CAS; (b) ref. 11; (c) V. I. Slovetskii, L. V. Okhobystina, A. A. Feinzil'berg, A. I. Ivanov, L. J. Birynkova and S. S. Novikov, Spectrophotometric determination of the ionization constant of fluorodinitromethane, Izv. Akad. Nauk SSSR, Ser. Khim., 1965, 2063 CAS.
- A Type III addition product was assigned by analogy to a Type IV product: K. M. Johnson, M. S. Rattley, F. Sladojevich, D. M. Barber, M. G. Nunez, A. M. Goldys and D. J. Dixon, A New Family of Cinchona-Derived Bifunctional Asymmetric Phase-Transfer Catalysts: Application to the Enantio- and Diastereoselective Nitro-Mannich Reaction of Amidosulfones, Org. Lett., 2012, 14, 2492 CrossRef CAS PubMed.
- (a) A Type III product assigned by optical rotation: C. Palomo, M. Oiarbide, A. Laso and R. Lopez, Catalytic Enantioselective Aza-Henry Reaction with Broad Substrate Scope, J. Am. Chem. Soc., 2005, 127, 17622 CrossRef CAS PubMed; (b) E. Gomez-Bengoa, A. Linden, R. Lopez, I. Mugica-Mendiola, M. Oiarbide and C. Palomo, Asymmetric Aza-Henry Reaction under Phase Transfer Catalysis: An Experimental and Theoretical Study, J. Am. Chem. Soc., 2008, 130, 7955 CrossRef CAS PubMed.
- For use of this material in vivo, see: (a) A. E. Vilgelm, J. S. Pawlikowski, Y. Liu, O. E. Hawkins, T. A. Davis, J. Smith, K. P. Weller, L. W. Horton, C. M. McClain, G. D. Ayers, D. C. Turner, D. C. Essaka, C. F. Stewart, J. A. Sosman, M. C. Kelley, J. A. Ecsedy, J. N. Johnston and A. Richmond, Mdm2 and Aurora Kinase A Inhibitors Synergize to Block Melanoma Growth by Driving Apoptosis and Immune Clearance of Tumor Cells, Cancer Res., 2015, 75, 181 CrossRef CAS PubMed; (b) A. E. Vilgelm, N. Saleh, R. Shattuck-Brandt, K. Riemenschneider, L. Slesur, S.-C. Chen, C. A. Johnson, J. Yang, A. Blevins, C. Yan, D. B. Johnson, R. N. Al-Rohil, E. Halilovic, R. M. Kauffmann, M. Kelley, G. D. Ayers and A. Richmond, MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21, Sci. Transl. Med., 2019, 11, eaav7171 CrossRef PubMed.
- K. Oyaizu, D. Uraguchi and T. Ooi, Vinylogy in nitronates: utilization of [α]-aryl conjugated nitroolefins as a nucleophile for a highly stereoselective aza-Henry reaction, Chem. Commun., 2015, 51, 4437 RSC.
- D. Uraguchi, K. Oyaizu, H. Noguchi and T. Ooi, Chiral Ammonium Betaine-Catalyzed Highly Stereoselective Aza-Henry Reaction of α-Aryl Nitromethanes with Aromatic N-Boc Imines, Chem.–Asian J., 2015, 10, 334 CrossRef CAS PubMed.
- R. R. Walvoord and M. C. Kozlowski, Cinchonidinium acetate as a convenient catalyst for the asymmetric synthesis of cis-stilbenediamines, Tetrahedron Lett., 2015, 56, 3070 CrossRef CAS PubMed.
- N. Lu, R. Li, Z. Wei, J. Cao, D. Liang, Y. Lin and H. Duan, Enantio- and Diastereoselective Nitro-Mannich Reaction of α-Aryl Nitromethanes with Amidosulfones Catalyzed by Phase-Transfer Catalysts, J. Org. Chem., 2017, 82, 4668 CrossRef CAS PubMed.
- An immobilized thiourea provided low selectivity (up to 91:9 anti:syn, 42% ee): R. Pedrosa, J. M. Andrés, D. P. Ávila, M. Ceballos and R. Pindado, Chiral ureas and thioureas supported on polystyrene for enantioselective aza-Henry reactions under solvent-free conditions, Green Chem., 2015, 17, 2217 RSC.
- F. Fini, V. Sgarzani, D. Pettersen, R. P. Herrera, L. Bernardi and A. Ricci, Phase-transfer-catalyzed asymmetric aza-Henry reaction using N-carbamoyl imines generated in situ from α-amido sulfones, Angew. Chem., Int. Ed., 2005, 44, 7975 CrossRef CAS PubMed.
- T. Nitabaru, N. Kumagai and M. Shibasaki, Catalytic Asymmetric Nitro-Mannich Reactions with a Yb/K Heterobimetallic Catalyst, Molecules, 2010, 15, 1280 CrossRef CAS PubMed.
- J. C. Anderson, G. P. Howell, R. M. Lawrence and C. S. Wilson, An Asymmetric Nitro-Mannich Reaction Applicable to Alkyl, Aryl, and Heterocyclic Imines, J. Org. Chem., 2005, 70, 5665 CrossRef CAS PubMed.
- Y. Pan, Y. Zhao, T. Ma, Y. Yang, H. Liu, Z. Jiang and C.-H. Tan, Enantioselective Synthesis of α-Fluorinated β-Amino Acid Derivatives by an Asymmetric Mannich Reaction and Selective Deacylation/Decarboxylation Reactions, Chem.–Eur. J., 2010, 16, 779 CrossRef CAS PubMed.
- Enantioselective α-fluoro-α-sulfonyl nitroalkanes additions to isatin-derived ketimines catalyzed by cinchonine are also known: M. Urban, M. Franc, M. Hofmanová, I. Císařová and J. Veselý, The enantioselective addition of 1-fluoro-1-nitro(phenylsulfonyl)methane to isatin-derived ketimines, Org. Biomol. Chem., 2017, 15, 9071 RSC.
- For enantioselective Michael additions of α-fluoro nitroester to enones, see: H.-F. Cui, P. Li, X.-W. Wang, Z. Chai, Y.-Q. Yang, Y.-P. Cai, S.-Z. Zhu and G. Zhao, Highly enantioselective synthesis of α-fluoro-α-nitro esters via organocatalyzed asymmetric Michael addition, Tetrahedron, 2011, 67, 312 CrossRef CAS.
- R. Blaszczyk, A. Gajda, S. Zawadzki, E. Czubacka and T. Gajda, N-Carbamate α-aminoalkyl-p-tolylsulfones-convenient substrates in the nitro-Mannich synthesis of secondary N-carbamate protected syn-2-amino-1-nitroalkanephosphonates, Tetrahedron, 2010, 66, 9840 CrossRef CAS.
- (a) B. M. Nugent, R. A. Yoder and J. N. Johnston, Chiral proton catalysis: a catalytic enantioselective direct aza-Henry reaction, J. Am. Chem. Soc., 2004, 126, 3418 CrossRef CAS PubMed; (b) A. Singh, R. A. Yoder, B. Shen and J. N. Johnston, Chiral Proton Catalysis: Enantioselective Brønsted Acid Catalyzed Additions of Nitroacetic Acid Derivatives as Glycine Equivalents, J. Am. Chem. Soc., 2007, 129, 3466 CrossRef CAS PubMed; (c) J. C. Wilt, M. Pink and J. N. Johnston, A Diastereo- and Enantioselective Synthesis of α-Substituted anti-α,β-Diaminophosphonic Acid Derivatives, Chem. Commun., 2008, 4177 RSC; (d) T. A. Davis, J. C. Wilt and J. N. Johnston, Bifunctional Asymmetric Catalysis: Amplification of Bronsted Basicity Can Orthogonally Increase the Reactivity of a Chiral Bronsted Acid, J. Am. Chem. Soc., 2010, 132, 2880 CrossRef CAS PubMed; (e) D. J. Sprague, A. Singh and J. N. Johnston, Diastereo- and enantioselective additions of [α]-nitro esters to imines for anti-[α],[β]-diamino acid synthesis with [α]-alkyl-substitution, Chem. Sci., 2018, 9, 2336 RSC.
- G. K. Friestad and A. K. Mathies, Recent developments in asymmetric catalytic addition to C=N bonds, Tetrahedron, 2007, 63, 2541 CrossRef CAS.
- T. Akiyama, J. Itoh and K. Fuchibe, Recent progress in chiral Bronsted acid catalysis, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS.
- T. Vilaivan, W. Bhanthumnavin and Y. Sritana-Anant, Recent advances in catalytic asymmetric addition to imines and related C=N systems, Curr. Org. Chem., 2005, 9, 1315 CrossRef CAS.
- syn-Selectivity in aza-Henry additions was also observed in cases where an achiral catalyst and/or racemic product is formed, and in others where a solvent effect was noted: (a) D. Kundu, R. K. Debnath, A. Majee and A. Hajra, Zwitterionic-type molten salt-catalyzed syn-selective aza-Henry reaction: solvent-free one-pot synthesis of β-nitroamines, Tetrahedron Lett., 2009, 50, 6998 CrossRef CAS; (b) J. C. Anderson, G. J. Stepney, M. R. Mills, L. R. Horsfall, A. J. Blake and W. Lewis, Enantioselective Conjugate Addition Nitro-Mannich Reactions: Solvent Controlled Synthesis of Acyclic anti- and syn-β-Nitroamines with Three Contiguous Stereocenters, J. Org. Chem., 2011, 76, 1961 CrossRef CAS PubMed; (c) X. Wang, Y.-F. Chen, L.-F. Niu and P.-F. Xu, Diastereo- and Enantioselective Aza-MBH-Type Reaction of Nitroalkenes to N-Tosylimines Catalyzed by Bifunctional Organocatalysts, Org. Lett., 2009, 11, 3310 CrossRef CAS PubMed.
- L. Belding, S. M. Taimoory and T. Dudding, Mirroring Enzymes: The Role of Hydrogen Bonding in an Asymmetric Organocatalyzed Aza-Henry Reaction—A DFT Study, ACS Catal., 2015, 5, 343 CrossRef CAS.
- S. M. Taimoory and T. Dudding, An Evolving Insight into Chiral H-Bond Catalyzed Aza-Henry Reactions: A Cooperative Role for Noncovalent Attractive Interactions Unveiled by Density Functional Theory, J. Org. Chem., 2016, 81, 3286 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches and J. F. Paquin, Organic Fluorine as a Hydrogen-Bond Acceptor: Recent Examples and Applications, Synthesis, 2015, 47, 306 CAS.
- (a) C. R. S. Briggs, M. J. Allen, D. O'Hagan, D. J. Tozer, A. M. Z. Slawin, A. E. Goeta and J. A. K. Howard, The observation of a large gauche preference when 2-fluoroethylamine and 2-fluoroethanol become protonated, Org. Biomol. Chem., 2004, 2, 732 RSC; (b) E.-M. Tanzer, L. E. Zimmer, W. B. Schweizer and R. Gilmour, Fluorinated Organocatalysts for the Enantioselective Epoxidation of Enals: Molecular Preorganisation by the Fluorine-Iminium Ion Gauche Effect, Chem.–Eur. J., 2012, 18, 11334 CrossRef CAS PubMed . For reviews see: D. O'Hagan, Understanding organofluorine chemistry. An introduction to the C–F bond, Chem. Soc. Rev., 2008, 37, 308 Search PubMed; (c) L. Hunter, The C–F bond as a conformational tool in organic and biological chemistry, Beilstein J. Org. Chem., 2010, 6, 38 Search PubMed.
- G. K. S. Prakash, F. Wang, N. Shao, T. Mathew, G. Rasul, R. Haiges, T. Stewart and G. A. Olah, A Persistent α-Fluorocarbanion and Its Analogues: Preparation, Characterization, and Computational Study, Angew. Chem., Int. Ed., 2009, 48, 5358 CrossRef CAS PubMed.
- T. Ernet and G. Haufe, Diels-Alder reactions of vinyl fluorides with 1,3-diphenylisobenzofuran, Tetrahedron Lett., 1996, 37, 7251 CrossRef CAS.
- G. Landelle, M.-O. Turcotte-Savard, L. Angers and J.-F. Paquin, Stereoselective Synthesis of Both Stereoisomers of β-Fluorostyrene Derivatives from a Common Intermediate, Org. Lett., 2011, 13, 1568 CrossRef CAS PubMed.
- K. Shibatomi, K. Futatsugi, F. Kobayashi, S. Iwasa and H. Yamamoto, Stereoselective Construction of Halogenated Quaternary Stereogenic Centers Via Catalytic Asymmetric Diels–Alder Reaction, J. Am. Chem. Soc., 2010, 132, 5625 CrossRef CAS PubMed.
- L. Merzoud, A. Saal, R. Moussaoui, O. Ouamerali, C. Morell and H. Chermette, Fluorine substituent effect on the stereochemistry of catalyzed and non-catalyzed Diels–Alder reactions. The case of R-butenone with cyclopentadiene: a computational assessment of the mechanism, Phys. Chem. Chem. Phys., 2018, 20, 16102 RSC.
- J. Hernández-Toribio, R. Gómez Arrayás and J. C. Carretero, Substrate-Controlled Diastereoselectivity Switch in Catalytic Asymmetric Direct Mannich Reaction of Glycine Derivatives with Imines: From anti- to syn-α,β-Diamino Acids, Chem.–Eur. J., 2010, 16, 1153 CrossRef PubMed.
- L. Lin and X. Feng, Catalytic Strategies for Diastereodivergent Synthesis, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed.
- R. Ding, Z. A. De los Santos and C. Wolf, Catalytic Asymmetric Mannich Reaction of α-Fluoronitriles with Ketimines: Enantioselective and Diastereodivergent Construction of Vicinal Tetrasubstituted Stereocenters, ACS Catal., 2019, 9, 2169 CrossRef CAS PubMed.
- (a) B. Shen, D. M. Makley and J. N. Johnston, Umpolung reactivity in amide and peptide synthesis, Nature, 2010, 465, 1027 CrossRef CAS PubMed; (b) J. P. Shackleford, B. Shen and J. N. Johnston, Discovery of competing anaerobic and aerobic pathways in umpolung amide synthesis allows for site-selective amide 18O-labeling, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 44 CrossRef CAS PubMed; (c) K. E. Schwieter, B. Shen, J. P. Shackleford, M. W. Leighty and J. N. Johnston, Umpolung Amide Synthesis Using Substoichiometric N-Iodosuccinimide (NIS) and Oxygen as a Terminal Oxidant, Org. Lett., 2014, 16, 4714 CrossRef CAS PubMed; (d) K. E. Schwieter and J. N. Johnston, Enantioselective synthesis of D-α-amino amides from aliphatic aldehydes, Chem. Sci., 2015, 6, 2590 RSC; (e) K. E. Schwieter and J. N. Johnston, Enantioselective Addition of Bromonitromethane to Aliphatic N-Boc Aldimines Using a Homogeneous Bifunctional Chiral Organocatalyst, ACS Catal., 2015, 5, 6559 CrossRef CAS PubMed; (f) K. E. Schwieter and J. N. Johnston, A one-pot amidation of primary nitroalkanes, Chem. Commun., 2016, 52, 152 RSC; (g) V. T. Lim, S. V. Tsukanov, A. B. Stephens and J. N. Johnston, Enantioselective Synthesis of α-Bromonitroalkanes for Umpolung Amide Synthesis: Preparation of tert-Butyl((1R)-1-(4-(benzyloxy)phenyl)-2-bromo-2-nitroethyl)carbamate, Org. Synth., 2016, 93, 88 CrossRef CAS PubMed; (h) M. T. Knowe, S. V. Tsukanov and J. N. Johnston, Preparation of Benzyl((R)-2-(4-(benzyloxy)phenyl)-2-((tert-butoxycarbonyl)amino)acetyl)-D-phenylalaninate Using Umpolung Amide Synthesis, Org. Synth., 2017, 94, 388 CrossRef CAS; (i) M. S. Crocker, H. Foy, K. Tokumaru, T. Dudding, M. Pink and J. N. Johnston, Direct Observation and Analysis of the Halo-Amino-Nitro Alkane Functional Group, Chem, 2019, 5, 1248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details (PDF); NMR and HPLC trace data (PDF); data for anti-6d, syn-6f, anti-6g, anti-6h, and syn-6h (CIF). CCDC 2118052–2118056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05910f |
| This journal is © The Royal Society of Chemistry 2022 |