
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOvercoming peri- and ortho-selectivity in C–H methylation of 1-naphthaldehydes by a tunable transient ligand strategy†
Yujian
Mao‡
,
Jing
Jiang‡
,
Dandan
Yuan‡
,
Xiuzhen
Chen
,
Yanan
Wang
,
Lihong
Hu
* and
Yinan
Zhang
 *
*
Jiangsu Key Laboratory for Functional Substances of Chinese Medicine, School of Pharmacy, Nanjing University of Chinese Medicine Nanjing, Jiangsu 210023, China. E-mail: yinanzhang@njucm.edu.cn; lhhu@njucm.edu.cn
First published on 6th January 2022
Abstract
Methyl groups widely exist in bioactive molecules, and site-specific methylation has become a valuable strategy for their structural functionalization. Aiming to introduce this smallest alkyl handle, a highly regioselective peri- and ortho-C–H methylation of 1-naphthaldehyde by using a transient ligand strategy has been developed. A series of methyl-substituted naphthalene frameworks have been prepared in moderate to excellent yields. Mechanistic studies demonstrate that peri-methylation is controlled by the higher electronic density of the peri-position of 1-naphthaldehyde as well as the formation of intermediary 5,6-fused bicyclic palladacycles, whereas experimental studies and theoretical calculations inferred that a 5-membered iridacycle at the ortho-position of 1-naphthaldehyde leads to energetically favorable ortho-methylation via an interconversion between the peri-iridacycle and ortho-iridacycle. Importantly, to demonstrate the synthetic utility of this method, we show that this strategy can serve as a platform for the synthesis of multi-substituted naphthalene-based bioactive molecules and natural products.
Introduction
The methylation strategy is of particular importance in accessing or optimizing bioactive molecules owing to the ubiquity of methyl groups in these pharmacological entities (Fig. 1A).1 The development of site-specific methylation has attracted significant attention in the rapid construction and late-stage functionalization of complex structures.2 Although a few regioselective methylation methods are well-developed for the transition metal (TM)-catalyzed ortho-C–H functionalization of benzene,3 the precise site-selectivity control on polyaromatic ring systems (e.g. naphthalene) remains largely absent,4 due to their possession of at least two proximal sites, the peri- and ortho-positions (Fig. 1B).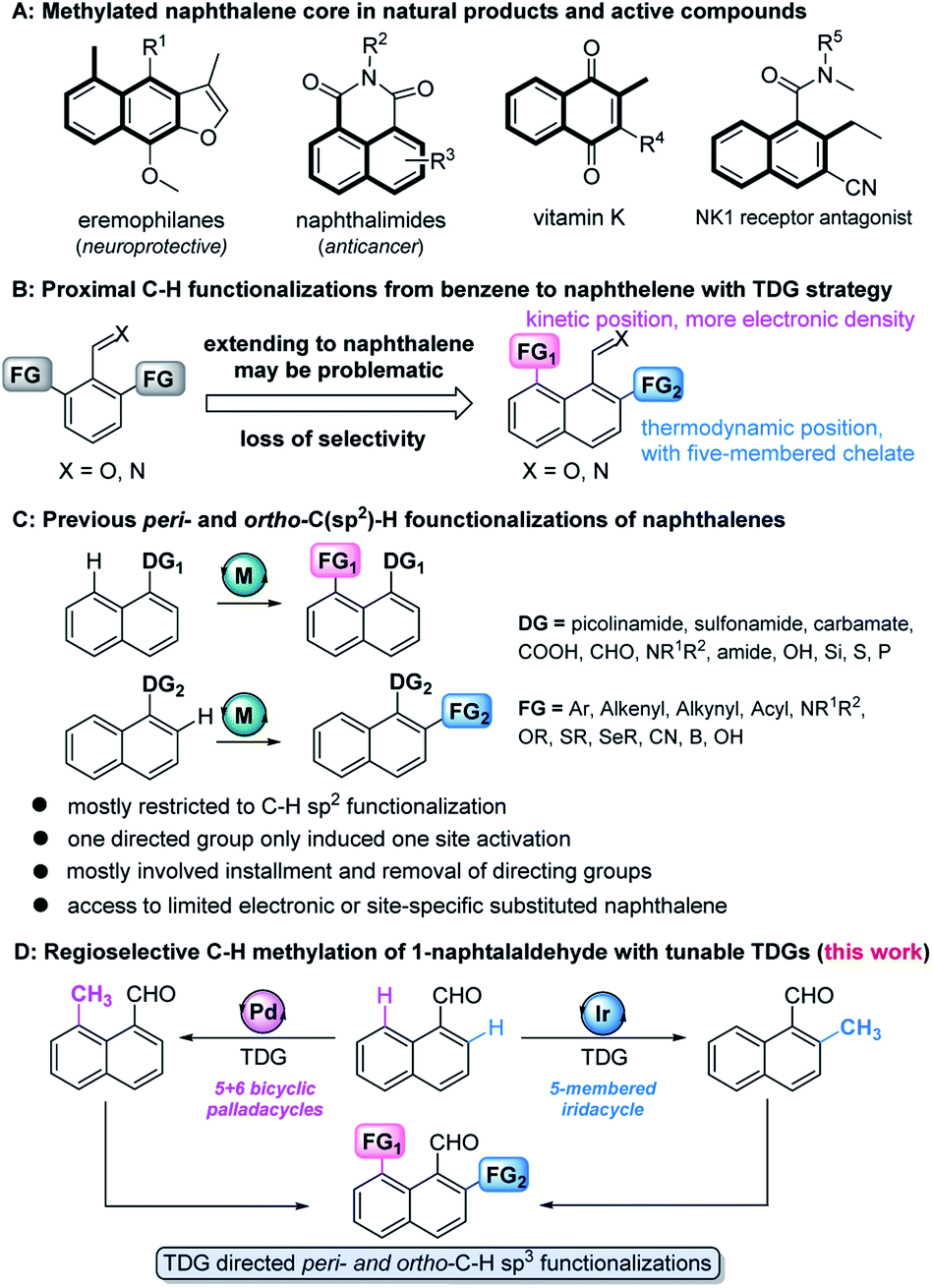 | ||
| Fig. 1 (A) Methyl groups in natural products and bioactive molecules. (B) Selectivity from benzene to naphthalene. (C) Peri- and ortho-C–H functionalizations of naphthalene. (D) Regioselective C–H methylation of 1-naphthaldehydes with tunable TDGs. | ||
With the help of immobilized directing groups, such as picolinamides,5 thioethers,6 carboxylic derivatives,7 amines,8 hydrosilyls,9 carbamates,10 pyridyls,11 phosphines,12 and sulfonamides,13advances in peri- and ortho-C–H functionalization have been made, but most of these ligands have been restricted to C–H sp2 functionalization (e.g. arylation, alkenylation, alkynylation, cyanation, etc.) and suffer from extra steps for installation and removal of directing groups (Fig. 1C). Although the transient directing group (TDG) strategy has been consequently devised to avoid using immobilized directing groups,14 TDG promoted C–H alkylation remains extremely challenging. The only example, demonstrated by the Sorensen group, showed that the in situ formation of Schiff bases with benzaldehydes and orthanilic acids promoted ortho-C–H methylation and fluorination.15 Our recent study has successfully extended the peri- and ortho-C–H oxygenation of polyaromatic rings.16 Inspired by these discoveries, we envisioned that the precise site-selectivity control of proximal C–H alkylation of the naphthalene ring might be achieved via the incorporation of TDGs (Fig. 1D). To the best of our knowledge, we herein wish to disclose the first regioselective peri- and ortho-C–H methylation of 1-naphthaldehyde via TDG catalysis. The study not only features the site-specific metal incorporation and C–H cleavage, but also details mechanistic driving forces by providing consistent experimental and computational evidence.
Results and discussion
Peri-C–H methylation
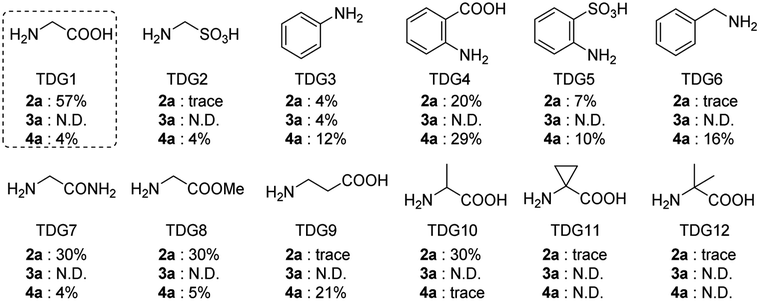
Initially, we began our investigation by conducting peri-C–H methylation of 1-naphthaldehyde. Interestingly, the desired peri-C–H methylated product was detected in low yield after a 36 h reaction that used 10 mol% of Pd(OAc)2 as the catalyst, 60 mol% of glycine (TDG1) as the TDG, 2.0 equivalents of AgOAc as the oxidant, 2.5 equivalents of potassium methyl trifluoroborate as the methylating reagent, and acetic acid (0.2 M) as the solvent (Table S1†). Striving to improve the reaction efficiency, we examined numerous oxidants and found that Cu(TFA)2-xH2O (ref. 17) performed better than other oxidants. Previously reported F+ oxidants failed to give the C–H methylation product at the peri-position.15 Next, screening of basic additives showed that two equivalents of CsOAc gave a better yield (35%). Surprisingly, the addition of H2O that facilitated the solubility of trifluoroborate salt improved the yield to 43%, and yet adopting soluble methylboronate MeBF3(Bu4N) and other methylating reagents inhibited the yield (Tables S1 and S2†). The screening of solvents, for example, hexafluoroisopropanol (HFIP) further promoted the reaction yield to 48%, while only traces of ortho-product 3a or bi-methylated product 4a were detected. Subsequent optimization of TDG ligands demonstrated that the presence of L,X-type TDGs is critical to C–H methylation, probably because the peri-selectivity relied on the formation of bicyclic palladacycles (Table 1). Using carbonyl as the X-branch of the ligand (TDG1, 7, 8) preferably gave the peri-product, whereas reactions involving the use of sulfonic acid as the X-branch (TDG2, 5) and monodentate ligands (TDG3, 6) were biased towards the bi-methylated product. Meanwhile, extended L,X-TDGs (TDG4, 5, 9) triggered a higher ratio of Pd insertion at the ortho-position, since the bicyclic palladacycle with a 5,6-fused ring has the most favorable ring strain.18 However, steric L,X-type TDGs (TDG10–12), which destabilized the conformation of bicyclic palladacycles, reduced the reactivity.|
|
|---|
a Experiments were performed under the following reaction conditions: 1a (0.3 mmol), CH3BF3K (1.05 mmol), Pd(OAc)2 (10 mol%), TDG (60 mol%), Cu(TFA)2-xH2O (0.6 mmol), CsOAc (0.6 mmol), HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH = 7:1 (1.5 mL), H2O (3.0 mmol), 90 °C, sealed tube, 36 h.
b Yields were determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. :AcOH = 7:1 (1.5 mL), H2O (3.0 mmol), 90 °C, sealed tube, 36 h.
b Yields were determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard.
|
|

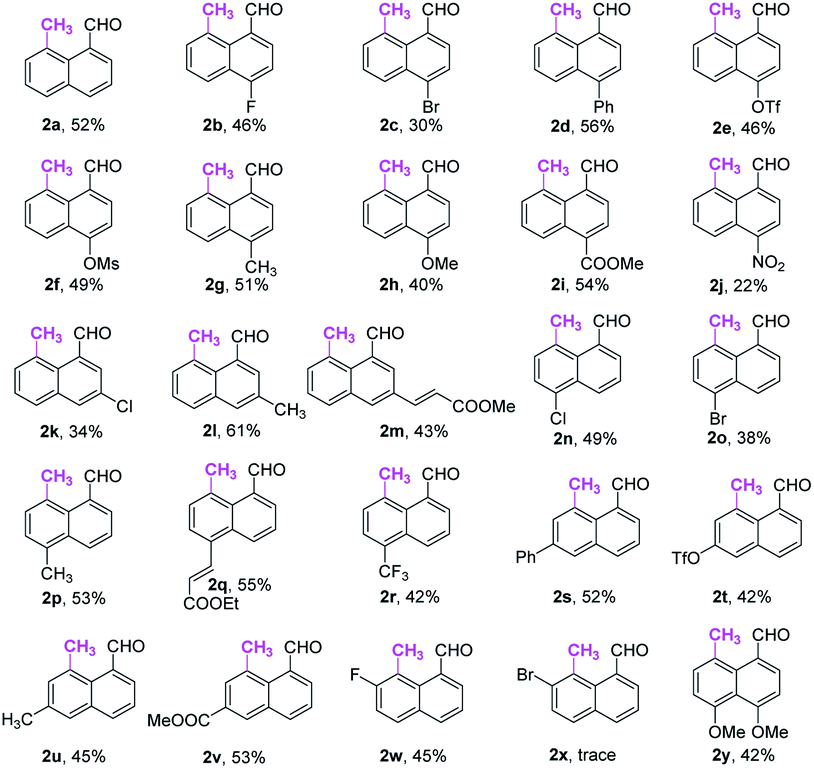
With the optimal reaction conditions in hand, we next explored the scope of peri-C–H methylation (Table 2). Various substituted 1-naphthaldehydes bearing electron-neutral, donating and withdrawing groups were smoothly methylated in uniformly moderate yields. Substrates with substituents in universal positions could provide the corresponding products. Gratifyingly, sulfonates (2e, 2f, 2t) and acrylates (2m, 2q) were amenable with the reaction system. Previously prohibited substrates, such as those with formidable withdrawing and donating groups (2j, 2y), also gave the desired products. The limitation of the method occurs only on the steric substitution at the 7-position (2x).

Ortho-C–H methylation
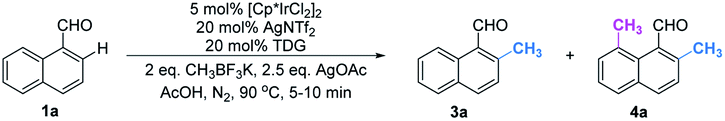
Although we have detected the existence of ortho-products (TDG3, Table 1) during the optimization of peri-C–H methylation, the yield with Pd catalyst is difficult to improve after futile efforts. The higher electron density at the peri-position allows the C–H activation to inevitably occur prior to the ortho-position. Inspired by previous studies,19 we hypothesized that iridium may override the unnecessary activation at the peri-position. To our delight, the ortho-C–H methylated product was found dominantly when using [Cp*IrCl2]2 and TDG3, although accompanied by a trace quantity of di-methylated 4a. Extensive screening of solvents, oxidants, additives and methylating reagents resulted in no yield enhancement (Tables S3 and S4†), while thorough investigation of the TDGs implicated that simple anilines were superior to the L,X-type ligands (Tables 3 and S3†). Finally, we identified that meta-EWG containing TDGs (TDG25, 28) were the optimal ligands for ortho-C–H methylation.|
|
|---|
| a Experiments were performed under the following reaction conditions: 1a (0.3 mmol), CH3BF3K (0.6 mmol), [Cp*IrCl2]2 (5 mol%), TDG (20 mol%), AgNTf2 (0.06 mmol), AgOAc (0.75 mmol), AcOH (1.5 mL), 90 °C, N2, sealed tube, 5–10 min. b Isolated yields. c Yields were determined by 1H NMR analysis of the crude reaction mixture using CH2Br2 as the internal standard. d TDG28 was used instead of TDG25. e TDG29 was used instead of TDG25, 30 min. |

|
With the optimized procedure in hand, we next explored the scope of ortho-C–H methylation. All the substituted 1-naphthaldehydes previously studied for peri-C–H methylation were revisited. It was found that not only all sorts of electron properties but also universal sites of substituted naphthaldehydes were well-tolerated to generate the expected ortho-products with excellent yields. Although substrates with steric hindrance and electron-donating substituents19a were found to inhibit the reactivity, 3-aminobenzonitrile (TDG28) dramatically promoted the reactivity of these steric (1k, 1l and 1m) and electron donating substrates (1h and 1y). When the di-CN TDG group was used (TDG29), the yield of 3y further increased to 71%, indicating that the electron-withdrawing TDG is favorable for ortho-C–H methylation. The poor convertion of peri-occupied substrates (2a and 1ab) inspired our further study of mechanism.
Synthetic applications to naphthalene-based derivatives and natural products
Based on the above studies, we devoted our efforts to demonstrating the synthetic utility of this protocol. As shown in Scheme 1, methylated products 2a and 3a can be obtained on gram scales with 48% and 81% yields respectively. Diverse di-substituted naphthalene derivatives were achieved via sequential C–H functionalization at the proximal positions of aldehydes with reactive groups, such as alkyl, aryl, halogen, sulfonamide, and alkoxy. Interestingly, 2,8-dimethyl-1-naphthaldehyde 4a could be obtained if the methyl group was first introduced at the 2-position (Table 3) rather than at the 8-position (Scheme 1). | ||
| Scheme 1 Synthesis of diverse naphthaldehydes via sequential C–H functionalizationsa. aReaction conditions: (a) 1a or 3a, CH3BF3K (3.5 eq.), Pd(OAc)2 (10 mol%), TDG (60 mol%), Cu(TFA)2-xH2O (2 eq.), CsOAc (2 eq.), HFIP:AcOH = 7:1, H2O (10 eq.), 90 °C, sealed tube, 36 h. (b) 1a, CH3BF3K (2 eq.), [Cp*IrCl2]2 (5 mol%), TDG (20 mol%), AgNTf2 (20 mol%), AgOAc (2.5 eq.), AcOH, 90 °C, N2, sealed tube, 4 h. (c) 2a, [Ru(p-cymene)Cl2]2 (5 mol%), AgSbF6 (20 mol%), 2-methyl-3-(trifluoromethyl)aniline (20 mol%), 4-chlorobenzoic acid (0.5 eq.), 1-ethyl-1H-pyrrole-2,5-dione (1.5 eq.), DCE:HFIP = 5:1, 60 °C, N2, sealed tube, 36 h. (d) 2a, Pd(OAc)2 (10 mol%), methyl 4-iodobenzoate (2 eq.), 2-amino-2-methylpropanoic acid (40 mol%), AgTFA (1 eq.), HFIP:TFA = 9:1, 110 °C, sealed tube, 24 h. (e) 2a, [CpIrCl2]2 (4 mol%), 3-trifluoromethylaniline (40 mol%), 4-methylbenzenesulfonyl azide (2 eq.), AgPF6 (24 mol%), DCE, 100 °C, N2, sealed tube, 36 h. (f) 2a or 3a, NCS (1.3 or 1.5 eq.), Pd(OAc)2 (10 mol%), anthranilic acid (30 mol%), AgTFA (10 mol%), TFA (10 eq.), DCE, 60 °C, sealed tube, 24 h. (g) 3a, Pd(OAc)2 (10 mol%), K2S2O8 (2 eq.), MeOH (20 eq.), 3-(trifluoromethyl)aniline (40 mol%), CH2Cl2, 60 °C, sealed tube, 36 h. (h) 3a, NBS (1.5 eq.), Pd(OAc)2 (10 mol%), 2-amino-4-nitrobenzoic acid (50 mol%), AgTFA (10 mol%), TfOH (50 mol%), DCE, 90 °C, sealed tube, 24 h. | ||
In our further attempt to demonstrate the synthetic utility, several bioactive compounds20 that bear alkyl-substituted naphthalene frameworks have been obtained efficiently (Scheme 2). For example, the preparation of nordehydrocacalohastine (11), an antimicrobial eremophilane sesquiterpene first isolated from Cacalia and Senecio plants,21 started from commercially available naphthol 5. Naphthaldehyde 7 was readily acquired by the Rieche reaction of compound 6 with high regioselectivity. After protection of the phenol group with α-bromoacetone, peri-methylation was successfully carried out to deliver the key intermediate 9 in a yield commensurate with the above standard conditions. Subsequent deformylation and cyclization accomplished the synthesis of the natural product in a total of 6 steps. Impressively, the employment of our methylation method saves 6 steps and improves the total yield 4.5-fold (11.7% vs. 2.6%) compared with the previously reported route.22 Meanwhile, gram methylation of 3-bromo-naphthaldehyde gave the ortho-methylated naphthaldehyde 13 in 80% yield. Next, treatment with the standard Bayer–Villiger reaction and oxidation smoothly produced the key precursor quinone 15, providing an alternative route to vitamin K.23 The final example was illustrated by the exchange of substitution on 4-fluoronaphthaldehyde. Peri-methylation and removal of the formyl group gave 16. Oxidation of the existing methyl group to another aldehyde 17 achieved ortho-methylation to form a rare 2-methyl-5-fluoronaphthaldehyde in 82% yield. These serial conversions highlighted the unique merit of our method in the preparation of naphthalene-based natural products and bioactive derivatives.
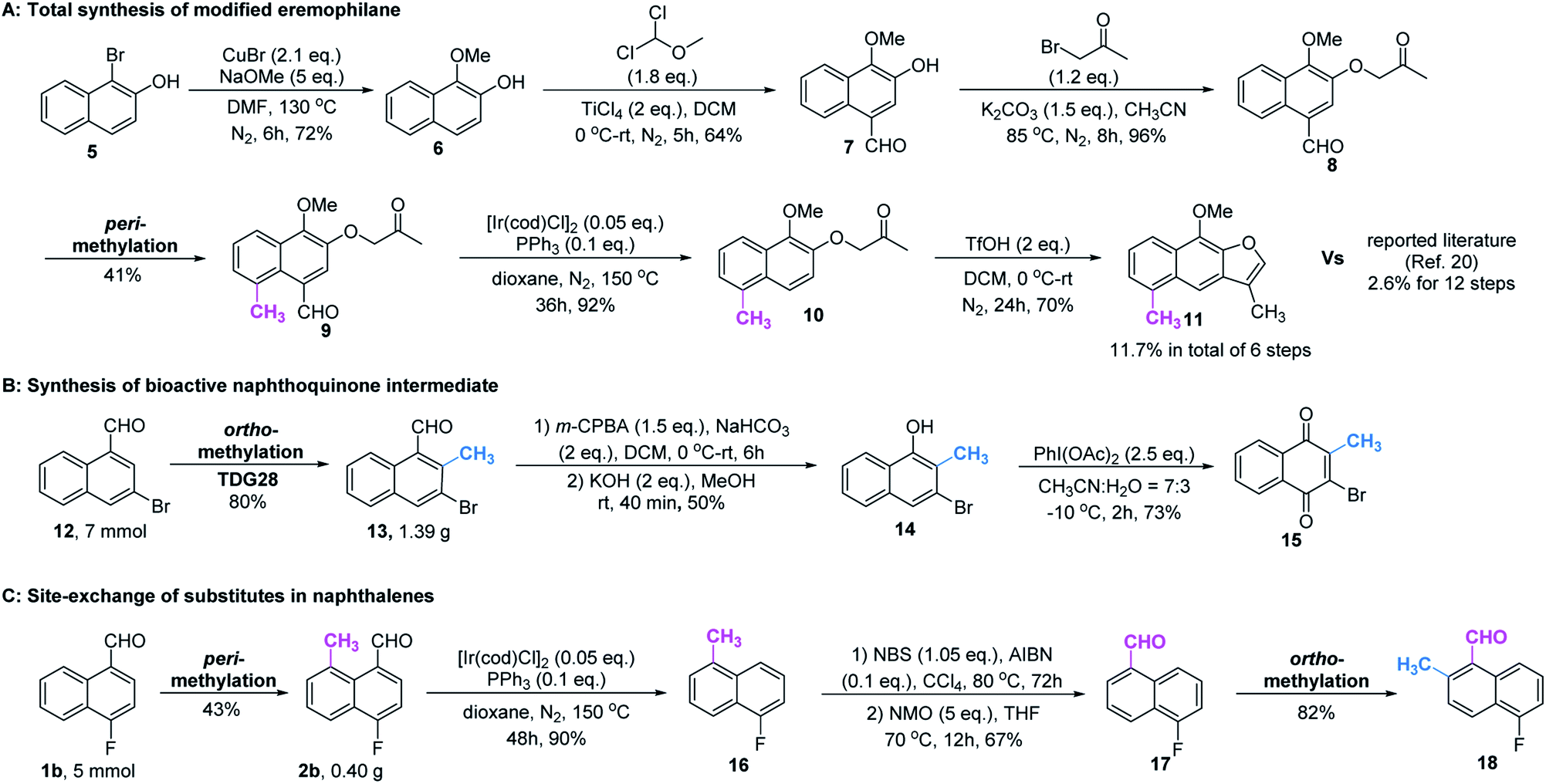 | ||
| Scheme 2 Synthetic transformation of regioselective peri- and ortho-C–H methylation. | ||
Mechanistic studies
To gain mechanistic insight into this reaction, a set of control experiments were carried out. In the absence of a catalyst or a TDG group, little product was detected under the standard conditions. The addition of a radical scavenger cannot quench the methylated products (Scheme 3, A1 and B1), indicating a non-radical process. Slightly reversible H/D exchange occurred at the peri-C–H position in the presence of Pd(OAc)2, while addition of glycine increased the ratio of the deuterated product. This observation verified the promotive effect of the TDG (Scheme 3, A2). Equivalent mixtures of EDG (1h) and EWG (1j) substrates under standard conditions of peri-methylation resulted in a large ratio of product 2h, illustrating that the electron-rich naphthaldehyde reacted faster than the electron-deficient one (Scheme 3, A3), while ortho-methylation had a much faster rate with electron-withdrawing substitutions (Scheme 3, B3). The stable palladacycle 19 was successfully captured by the pyridine ligand and could be further converted to the peri-methylated product 2a (Scheme 3, A4). Moreover, addition of product 2a was found to inhibit peri-methylation of 1a (Scheme S1†), which may explain the moderate yields of this conversion.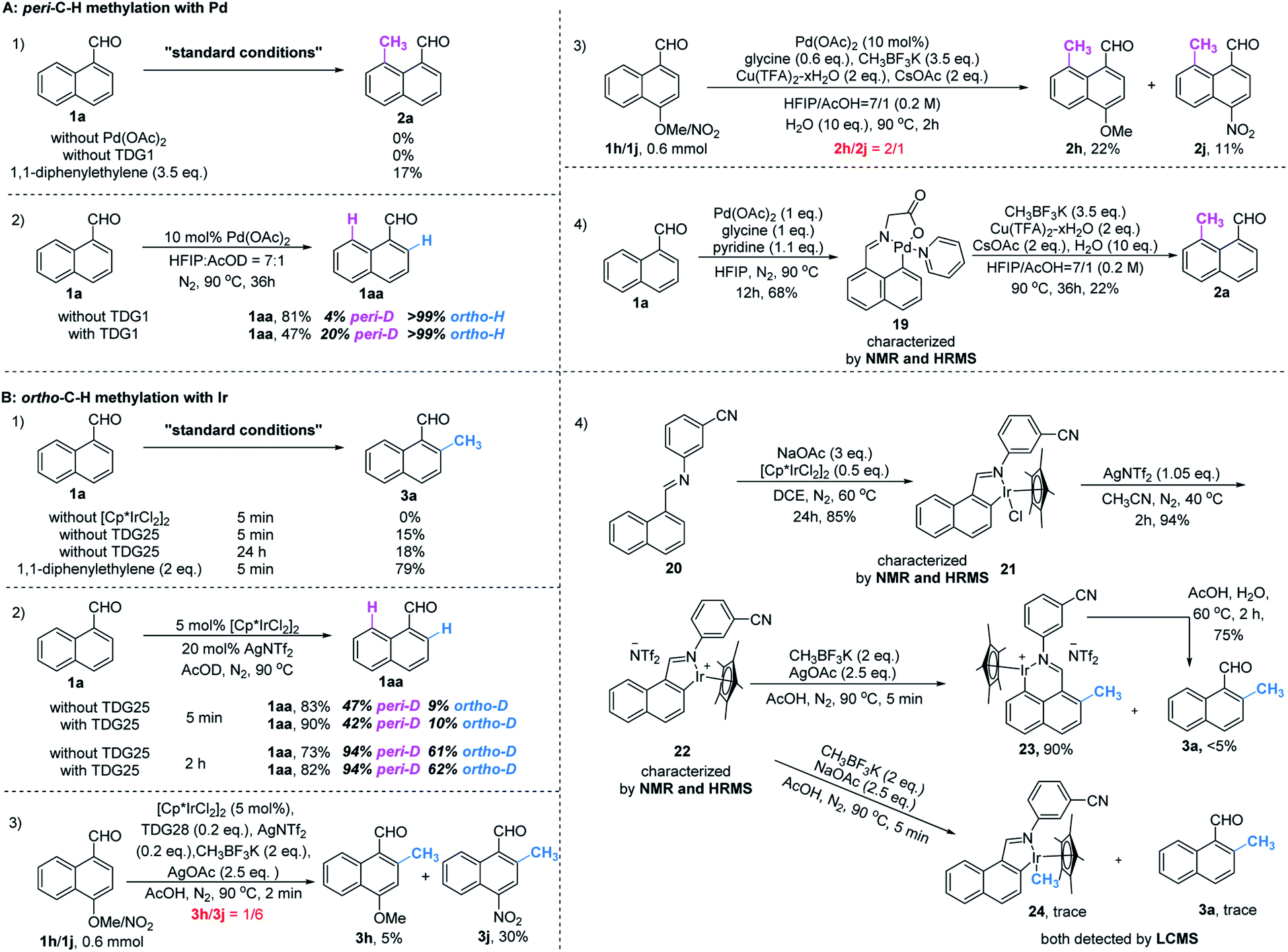 | ||
| Scheme 3 Mechanistic studies. | ||
However, treatment of 1a with [Cp*IrCl2]2 in AcOD delivered deuterated products at both peri- and ortho-positions. The higher D-ratio at the peri-position with standard and extended reaction times consistently showed that the peri-position is more likely to have the metal inserted due to its higher electronic density than the ortho-position,16 and even the introduction of TDG25 had no influence on this outcome (Scheme 3, B2). Although the rate of peri-metallation may be superior to the rate of methylation at the ortho-position in the C–H metal insertion step, it was found that the peri-iridacycle was readily hydrolysed, which also explains the higher yield of the deuterated product at this position. In contrast, resistance to ortho-hydrolysis resulted in the capture of iridacycle 21 and its NTf2 ligand exchanger 22 (Scheme 3, B5). Treatment with CH3BF3K produced iridacycle 24 and the desired ortho-methylated product 3a in the absence of an oxidant. However, in the presence of an oxidant, ortho-iridacycle 22 was converted to peri-iridacycle 23 when the ortho-position was occupied by the methyl group. And peri-iridacycle 23 was readily acid hydrolyzed to product 3a rather than successive methylation at the peri-position. Notably, the observations that ortho-iridacycle 22 was resistant to acid hydrolysis and peri-blocked substrates (3z and 4a, Table 3) diminished the ortho-productivity further corroborated the fact that the reactivity of and interconversion between these iridacycles together controlled the ortho-selectivity (Scheme 3, B4). Additionally, the poor reactivity of simple benzaldehyde in our method showed that the methylation method had unique properties in the selective activation of the proximal C–H bonds of naphthaldehyde (Scheme S2†). Finally, determination of the kinetic isotope effect (KIE) of the C–H cleavage was carried out (Scheme S3†). The intermolecular KIE of 1a and 1a-peri-D was found at 0.99 (kH/KD), which indicated that the cleavage of the peri-C–H bond is not involved in the rate-determining step. Similarly, the KIE of the ortho-C–H bond was studied at a low temperature to slow the conversion for measurement. The intermolecular KIE of 1a and 1a-ortho-D is 1.30 (kH/KD), which suggested that the cleavage of the ortho-C–H bond may not be involved in the rate-determining step.
Computational studies
Since the control experiment suggested that the peri-palladacycle directed the following methylation, the mechanism for the palladium-catalyzed peri-C–H activation process was thus investigated using M06-2X DFT calculations. As shown in Fig. 2A, the transition state of TS2 possessed a lower energy barrier (ΔG‡ = 11.9 kcal mol−1) at the peri-position, in comparison with the transition state TS1 guiding the ortho-C–H insertion (ΔG‡ = 16.5 kcal mol−1). At the same time, the lower energy level of peri-intermediate Int3 over Int2 also indicated that the higher electron density at the peri-position of 1-naphthaldehyde controlled the selectivity. In order to obtain details on the actual electron flow in the conversion, an intrinsic bond orbital (IBO) analysis was performed (Fig. 2B).24 In the TS2 structure, the electrons in the σ-orbital [aromatic C–H] interact with the π*-orbital [C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] of acetate to abstract a proton. The electrons of [σ(Pd–O)] become a lone pair [LP(Pd)] of the palladium complex, and at the same time, the electrons in the 4d-orbital of palladium [LP(Pd)] form a new palladium–carbon σ-bond [σ(Pd–C)] in the palladacycle intermediate. The bond recombination occurs via a concerted mechanism.25
O] of acetate to abstract a proton. The electrons of [σ(Pd–O)] become a lone pair [LP(Pd)] of the palladium complex, and at the same time, the electrons in the 4d-orbital of palladium [LP(Pd)] form a new palladium–carbon σ-bond [σ(Pd–C)] in the palladacycle intermediate. The bond recombination occurs via a concerted mechanism.25
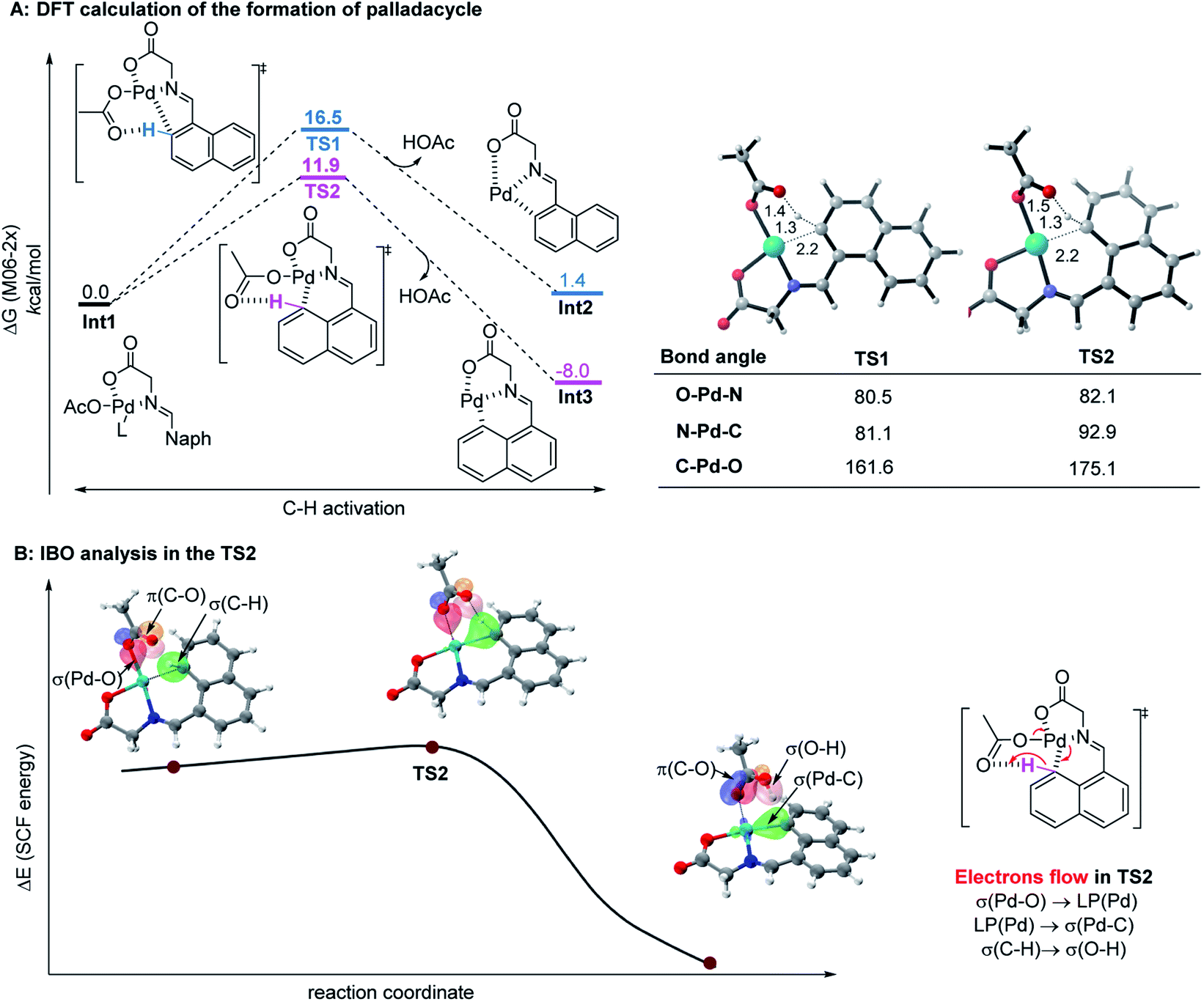 | ||
| Fig. 2 (A) The calculated reaction energy profile of palladacycle complex formation steps. (B) IBO analysis of the palladium-assisted peri-C–H activation of TS2. | ||
The mechanism of the ortho-C–H activation process with the iridium catalyst was also investigated following the experimental findings (Fig. 3A). As anticipated, starting from the relative zero point of the iridium-Schiff base complex, peri-metallation had a lower energy barrier of 3.5 kcal mol−1 than ortho-metallation. After C–H insertion, the peri-iridacycle was also found to be more stable than its ortho-counterpart 22. Subsequent transmetallation with CH3BF3K flipped over the Gibbs energy, in which ortho-product 24 was exothermic by 7.4 kcal mol−1, 11.1 kcal mol−1 higher than that of the process that yielded the peri-product. The addition of the Cl ligand to the intermediates Int5 and Int6 also reversed their stability, which was exactly validated by the isolation of iridacycle 21 rather than peri-21. In the intrinsic bond orbital (IBO) analysis, a similar pattern of electron flow to that of the palladacycle had been verified (Fig. 3B).
 | ||
| Fig. 3 (A) The calculated reaction energy profile of the formation of intermediate 24. (B) IBO analysis of the iridium-assisted ortho-C–H activation of TS3. (C) The calculated reaction energy profile of the formation of product 23. | ||
Previous findings prove that the energy barrier of reductive elimination is lowered at least by 15–30 kcal mol−1 by increasing the oxidation state of iridium from product-like Ir(III) to reactant-like Ir(V).26 We postulated that oxidatively enhanced reductive elimination was also dominant in our ortho-methylation after iridium was oxidized to hypervalent species by the silver oxidant. As shown in Fig. 3C, active ortho-Ir(V) species Int7 underwent reductive elimination and coupled the methyl group to the naphthalene ring releasing a large amount of energy (41.4 kcal mol−1), in which the energy barrier of ortho-methylation was 14.2 kcal mol−1 (2.7 kcal mol−1 lower than that of the unfavorable peri-methylation). Next, the iridacycle rebounded to the peri-position to produce intermediate 23 as experimentally observed, by continuing to release 4.9 kcal mol−1 of energy. Overall computational calculations indicated by the energy profile of transient states were in complete agreement with experimental investigations and supported the regioselectivity.
Collectively, we proposed a plausible catalytic cycle that is believed to be different from previously reported Pd(II)/(IV)15 and Ir(I)/(III)19a catalysis (Scheme 4). Starting from the formation of imine A or E, metal catalysts are incorporated into the activated C–H bond and afforded a 5,6-fused bicyclic or 6-membered ring at the peri-position. Insertion of CH3BF3K to the palladacycle induces subsequent reductive elimination to forge precursor D. For the iridacycle, hydrolysis and re-iridation promote the conversion of 6-membered ring F to 5-membered ring G, which is ready to perform transmetallation with a methylated reagent. After the oxidatively induced reductive elimination of iridacycle H,ortho-methylation brings about another peri-iridation to form intermediate I which is reactively inert to methylation. D and I further furnish the desired products 2a and 3a by in situ hydrolyzation, respectively. Concomitantly, the active catalysts are regenerated for the next catalytic cycle.
 | ||
| Scheme 4 Possible mechanistic pathway. | ||
Conclusions
We have developed a highly regioselective peri- and ortho-C–H methylation of 1-naphthaldehyde by using the transient ligand strategy. Mechanistically, capture of reaction intermediates and isotopic exchange experiments identified variable reactive metallacycles formed with different transient directing ligands in the metal-insertion and methylation steps. As suggested by DFT calculations and IBO analysis, although peri-metalation is energetically favorable compared to ortho-metalation in both palladium and iridium catalyses, transmetallation of CH3BF3K with iridium(III) readily takes place on the ortho-iridacycle rather than on the peri-iridacycle, which primarily explained the dichotomic selectivity on the two proximal positions of the naphthalene ring. We believe that this finding would greatly advance the future studies toward exquisite transformation control of polyaromatic rings. Moreover, the regioselectivity and broad utility of this method provide an unconventional tactic for the supply of multi-substituted naphthalene-based bioactive derivatives and natural products, thus building a stage for the use of dual metal/TDG catalysis approaches in the creation of molecules that are synthetically practicable and structurally attractive.Data availability
All the experimental data have been included in the ESI.†Author contributions
Y. M., J. J. and Y. Z. conceived and designed the project. Y. M., J. J. and D. Y. performed the experiments and collected the data. D. Y. and Y. M. contributed to the theoretical and experimental mechanistic investigation. X. C. and Y. W. contributed to the optimization of the transient directing strategy and the interpretation of the related results. Y. M., L. H. and Y. Z. prepared the manuscript with input from all contributing authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21877062, YZ), the “ShuangChuang” research team of Jiangsu Province (No. 20182036, YZ and LH), and the “Innovative and Entrepreneurial Talent” program (LH). We thank Prof. Donghui Wang for his constructive suggestion in the KIE study.Notes and references
- (a) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (b) H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- G. Yan, A. J. Borah, L. Wang and M. Yang, Adv. Synth. Catal., 2015, 357, 1333–1350 CrossRef CAS.
- (a) E. Gwilherm and T. Cédric, Angew. Chem., Int. Ed., 2019, 58, 7202–7236 CrossRef PubMed; (b) S. D. Friis, M. J. Johansson and L. Ackermann, Nat. Chem., 2020, 12, 511–519 CrossRef CAS PubMed.
- (a) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed; (b) S. Liang, M. Bolte and G. Manolikakes, Chem.–Eur. J., 2017, 23, 96–100 CrossRef CAS PubMed; (c) J. Li, Y. Wang, Y. Yu, R. Wu, J. Weng and G. Lu, ACS Catal., 2017, 7, 2661–2667 CrossRef CAS; (d) C. Jing, X. Chen, K. Sun, Y. Yang, T. Chen, Y. Liu, L. Qu, Y. Zhao and B. Yu, Org. Lett., 2019, 21, 486–489 CrossRef CAS PubMed; (e) M. Zhang, A. Luo, Y. Shi, R. Su, Y. Yang and J. You, ACS Catal., 2019, 9, 11802–11807 CrossRef CAS; (f) S. Prévost, ChemPlusChem, 2020, 85, 476–486 CrossRef; (g) B. Large and D. Prim, Synthesis, 2020, 52, 2600–2612 CrossRef CAS.
- Examples of picolinamide directed C8–H founctionalization of naphthalene, see: (a) L. Huang, Q. Li, C. Wang and C. Qi, J. Org. Chem., 2013, 78, 3030–3038 CrossRef CAS PubMed; (b) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS PubMed; (c) R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 11045–11052 CrossRef CAS PubMed; (d) L. Huang, X. Sun, Q. Li and C. Qi, J. Org. Chem., 2014, 79, 6720–6725 CrossRef CAS PubMed; (e) R. Shang, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 7660–7663 CrossRef CAS PubMed; (f) Z. Li, S. Sun, H. Qiao, F. Yang, Y. Zhu, J. Kang, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 4594–4597 CrossRef CAS PubMed; (g) Y. Xiong, Y. Yu, J. Weng and G. Lu, Org. Chem. Front., 2018, 5, 982–989 RSC; (h) S. Rej and N. Chatani, ACS Catal., 2018, 8, 6699–6706 CrossRef CAS; (i) T. Zhang, H. Zhu, F. Yang, Y. Wu and Y. Wu, Tetrahedron, 2019, 75, 1541–1547 CrossRef CAS; (j) X. Yu, F. Yang, Y. Wu and Y. Wu, Org. Lett., 2019, 21, 1726–1729 CrossRef CAS PubMed; (k) Y. Yao, Q. Lin, W. Yang, W. Yang, F. Gu, W. Guo and D. Yang, Chem.–Eur. J., 2020, 26, 5607–5610 CrossRef CAS PubMed; (l) C. Zu, T. Zhang, F. Yang, Y. Wu and Y. Wu, J. Org. Chem., 2020, 85, 12777–12784 CrossRef CAS PubMed.
- (a) M. Shigeno, Y. Nishii, T. Satoh and M. Miura, Asian J. Org. Chem., 2018, 7, 1334–1337 CrossRef CAS; (b) S. Moon, Y. Nishii and M. Miura, Org. Lett., 2018, 21, 233–236 CrossRef PubMed; (c) K. Yan, M. Liu, J. Wen, S. Wang, J. Li and H. Wang, Org. Lett., 2020, 22, 7825–7830 CrossRef CAS PubMed.
- (a) J. Karthikeyan and N. Yoshikai, Org. Lett., 2014, 16, 4224–4227 CrossRef CAS PubMed; (b) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS; (c) A. Biafora, T. Krause, D. Hackenberger, F. Belitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752–14755 CrossRef CAS PubMed; (d) S. Li, G. Deng, F. Yin, C. Li and H. Gong, Org. Chem. Front., 2017, 4, 417–420 RSC; (e) G. Tan, Q. You and J. You, ACS Catal., 2018, 8, 8709–8714 CrossRef CAS; (f) J. Garrec, M. Cordier, G. Frison and S. Prévost, Chem.–Eur. J., 2019, 25, 14441–14446 CrossRef CAS PubMed.
- (a) R. Shi, L. Lu, H. Xie, J. Yan, T. Xu, H. Zhang, X. Qi, Y. Lan and A. Lei, Chem. Commun., 2016, 52, 13307–13310 RSC; (b) S. Rej and N. Chatani, Chem.–Eur. J., 2020, 26, 11093–11098 CrossRef CAS PubMed.
- B. Su and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 10163–10167 CrossRef CAS PubMed.
- X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4830–4833 CrossRef CAS PubMed.
- (a) M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem.–Eur. J., 2014, 20, 2459–2462 CrossRef CAS PubMed; (b) M. Kondrashov, S. Raman and O. F. Wendt, Chem. Commun., 2015, 51, 911–913 RSC.
- X. Luo, J. Yuan, C. Yue, Z. Zhang, J. Chen, G. Yu and C. Che, Org. Lett., 2018, 20, 1810–1814 CrossRef CAS PubMed.
- (a) Y. Nishinaka, T. Satoh, M. Miura, H. Morisaka, M. Nomura, H. Matsui and C. Yamaguchi, Bull. Chem. Soc. Jpn., 2001, 74, 1727–1735 CrossRef CAS; (b) X. Li, X. Gong, M. Zhao, G. Song, J. Deng and X. Li, Org. Lett., 2011, 13, 5808–5811 CrossRef CAS PubMed.
- Examples of TDG strategy, see: (a) Y. Wu, Y. Chen, T. Liu, M. D. Eastgate and J. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (b) F. L. Zhang, K. Hong, T. J. Li, H. Park and J. Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS PubMed; (c) X. Liu, H. Park, J. Hu, Y. Hu, Q. Zhang, B. Wang, B. Sun, K. Yeung, F. Zhang and J. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed; (d) X. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 6280–6283 CrossRef CAS PubMed; (e) Y. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS PubMed; (f) H. Park, P. Verma, K. Hong and J. Yu, Nat. Chem., 2018, 10, 755–762 CrossRef CAS PubMed; (g) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC; (h) F. Li, Y. Zhou, H. Yang, Z. Wang, Q. Yu and F. Zhang, Org. Lett., 2019, 21, 3692–3695 CrossRef CAS PubMed; (i) Y. Chen, Y. Wu, Z. Wang, J. X. Qiao and J. Yu, ACS Catal., 2020, 10, 5657–5662 CrossRef CAS PubMed; (j) M. Ding, W. Hua, M. Liu and F. Zhang, Org. Lett., 2020, 22, 7419–7423 CrossRef CAS PubMed; (k) G. Li, Q. Liu, L. Vasamsetty, W. Guo and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 3475–3479 CrossRef CAS PubMed; (l) N. Goswami, T. Bhattacharya and D. Maiti, Nat. Rev. Chem., 2021, 5, 646–659 CrossRef CAS; (m) S. Bag, S. Jana, S. Pradhan, S. Bhowmick, N. Goswami, S. K. Sinha and D. Maiti, Nat. Commun., 2021, 12, 1393–1400 CrossRef CAS PubMed; (n) N. Thrimurtulu, A. Dey, K. Pal, D. Maiti and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 1441–1446 CrossRef CAS.
- J. Jiang, D. Yuan, C. Ma, W. Song, Y. Lin, L. Hu and Y. Zhang, Org. Lett., 2021, 23, 279–284 CrossRef CAS PubMed.
- X. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS PubMed.
- (a) X. Chen, J. Li, X. Hao, C. E. Goodhue and J. Yu, J. Am. Chem. Soc., 2006, 128, 78–79 CrossRef CAS PubMed; (b) X. Chen, C. E. Goodhue and J. Yu, J. Am. Chem. Soc., 2006, 128, 12634–12635 CrossRef CAS PubMed.
- G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J. Yu, Nat. Chem., 2019, 11, 571–577 CrossRef CAS PubMed.
- (a) X. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951–8956 RSC; (b) A. Woźniak, J. Tan, Q. Nguyen, A. Madron Du Vigné, V. Smal, Y. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed.
- (a) E. A. Doisy, S. B. Binkleya and S. A. Thayer, Chem. Rev., 1941, 28, 477–517 CrossRef CAS; (b) J. S. Albert, C. Ohnmacht, P. R. Bernstein, W. L. Rumsey, D. Aharony, Y. Alelyunas, D. J. Russell, W. Potts, S. A. Sherwood, L. Shen, R. F. Dedinas, W. E. Palmer and K. Russell, J. Med. Chem., 2004, 47, 519–529 CrossRef CAS PubMed; (c) D. Heckmann, A. Meyer, B. Laufer, G. Zahn, R. Stragies and H. Kessler, ChemBioChem, 2008, 9, 1397–1407 CrossRef CAS PubMed; (d) P. Kasaplar, Ö. Yılmazer and A. Çağır, Bioorg. Med. Chem., 2009, 17, 311–318 CrossRef CAS PubMed; (e) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef PubMed; (f) T. Liu, H. Wu, J. Zhang, W. Deng and L. Yang, J. Nat. Prod., 2020, 83, 185–193 CrossRef CAS PubMed.
- P. Torres, R. Chinchilla, M. C. Asensi and M. Grande, Phytochemistry, 1989, 28, 3093–3095 CrossRef CAS.
- Y. Hirai, M. Doe, T. Kinoshita and Y. Morimoto, Chem. Lett., 2004, 33, 136–137 CrossRef CAS.
- M. Braasch-Turi and D. C. Crans, Molecules, 2020, 25, 4477–4513 CrossRef CAS PubMed.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- K. Shin, Y. Park, M. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, computational details, and copies of 1H and 13C NMR spectra for all compounds featured in this manuscript. See DOI: 10.1039/d1sc05899a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |