
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh strength, epoxy cross-linked high sulfur content polymers from one-step reactive compatibilization inverse vulcanization†
Sangwoo
Park‡§
a,
Minju
Chung‡¶
 a,
Alexandros
Lamprou
b,
Karsten
Seidel
c,
Sanghoon
Song
d,
Christian
Schade
e,
Jeewoo
Lim
*d and
Kookheon
Char
*a
a,
Alexandros
Lamprou
b,
Karsten
Seidel
c,
Sanghoon
Song
d,
Christian
Schade
e,
Jeewoo
Lim
*d and
Kookheon
Char
*a
aSchool of Chemical and Biological Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: khchar@snu.ac.kr
bFunctional Polymers Global Research, Innovation Campus Asia Pacific, BASF, 200137 Shanghai, China
cMaterial Physics, Analytics & Formulation Research, BASF SE, 67056 Ludwigshafen, Germany
dDepartment of Chemistry, Kyung Hee University, Seoul 02447, Republic of Korea. E-mail: jeewoo@khu.ac.kr
eFunctional Polymers Global Research, BASF SE, 67056 Ludwigshafen, Germany
First published on 15th December 2021
Abstract
Inverse vulcanization provides a simple, solvent-free method for the preparation of high sulfur content polymers using elemental sulfur, a byproduct of refining processes, as feedstock. Despite the successful demonstration of sulfur polymers from inverse vulcanization in optical, electrochemical, and self-healing applications, the mechanical properties of these materials have remained limited. We herein report a one-step inverse vulcanization using allyl glycidyl ether, a heterobifunctional comonomer. The copolymerization, which proceeds via reactive compatibilization, gives an epoxy cross-linked sulfur polymer in a single step, as demonstrated through isothermal kinetic experiments and solid-state 13C NMR spectroscopy. The resulting high sulfur content (≥50 wt%) polymers exhibited tensile strength at break in the range of 10–60 MPa (70–50 wt% sulfur), which represents an unprecedentedly high strength for high sulfur content polymers from vulcanization. The resulting high sulfur content copolymer also exhibited extraordinary shape memory behavior along with shape reprogrammability attributed to facile polysulfide bond rearrangement.
Introduction
Sulfur possesses a variety of interesting properties, ranging from high molar refraction to high theoretical capacity in cathodes of Li–S batteries. Furthermore, sulfur, in its elemental form, is environmentally benign, and due to its production as a byproduct of petroleum refining, inexpensive and abundant. Such characteristics of sulfur, along with the poor physical properties of sulfur in its elemental form, have imparted considerable importance to novel methods for the utilization of sulfur for the preparation of advanced materials, especially for optical and energy storage applications.While numerous compounding methods for achieving physical blends of sulfur with carbon-based materials,1,2 inorganic nanoparticles,3 and construction materials (e.g. sulfur cement/concrete)4,5 have been reported, chemical methods of sulfur utilization, such as the direct use of elemental sulfur in polymerization reactions, had been scarce, largely due to the low stability of high sulfur content polymers. Recently, bulk free-radical copolymerization of molten sulfur with 1,3-diisopropenylbenzene (DIB) has been reported.6 The method, dubbed “inverse vulcanization” allowed for the formation of polysulfide polymers with unprecedentedly high sulfur contents (50–90 wt%) which were much less prone to depolymerization through cyclooctasulfur elimination.7,8 Since the first report of inverse vulcanization in 2013, a large number of multifunctional comonomers, such as divinylbenzene,9,10 diethynyl benzene,11 myrcene,12 limonene,13 dicyclopentadiene,12 and diallyl disulfide14 have been used in inverse vulcanization.
The polymers from inverse vulcanization display attractive properties such as high (n > 1.80) refractive index,7,15,16 high energy density in Li–S batteries,6,17–19 and self-healing.8,20 Despite this, their relatively poor mechanical properties have limited further expansion of the scope of applications. Recent studies show that the strength of inverse vulcanization polymers are much lower than those of conventional polymers, with an upper limit of tensile strength at break (σmax) of lower than 10 MPa.8,20–22 Employing a mixture of crosslinkers21,23 have allowed for some control over the shear modulus of inverse vulcanization products, but the approach has yet to provide higher strength materials. The crosslinkers used in inverse vulcanizations thus far were limited to homobifunctional molecules with unsaturated carbon–carbon bonds, where addition reaction of sulfur radicals (R–S·) is the only available chemistry (Scheme 1),6,10,11,14,21,23 and introducing new types of chemistry for crosslink formation has recently emerged as a compelling strategy for the enhancement of mechanical properties of inverse vulcanized polymers. In 2020, Hasell and coworkers reported a two-step process involving a reaction of sulfur with sorbitan oleate (Span 80), a non-ionic surfactant composed of a polar headgroup containing three hydroxyl groups and a hydrophobic tail composed of an oleyl group.24 The reaction of sulfur and Span 80 gave a prepolymer, which was subsequently treated with a diisocyanate to form urethane crosslinks. The resulting polymers showed shape-memory behavior with σmax as high as 20.56 MPa for the products with the highest degree of crosslinking, representing a significant improvement in strength compared to previously reported inverse vulcanization products. Further work by the group introduced similar polymers with improved tensile elongation and toughness.25 Recently, Pyun and coworkers synthesized segmented block copolymer sulfur polyurethanes showing both high tensile strength (13–24 MPa) and ductility (348% strain at break), using inverse vulcanization products as prepolymers.26
 | ||
| Scheme 1 Overall scheme for inverse vulcanization process illustrating comparison of adopted reaction chemistry and suitable monomers of previous reports and this work. | ||
Based on our recent finding that inducing radical propagation of divinylbenzene (DVB) during inverse vulcanization leads to products with enhanced thermal properties,9 we envisioned that heterobifunctional comonomers, which could simultaneously undergo addition reactions with sulfur radicals and cross-linking involving a different mechanism, may lead to inverse vulcanization polymers with unprecedented strength. We report herein an inverse vulcanization using a single comonomer, allyl glycidyl ether (AGE), to give high sulfur content (>50 wt% S) polymers having σmax of over 60 MPa in a single step (Scheme 1). Although AGE is immiscible with sulfur even at elevated temperatures, the bulk polymerization of sulfur and AGE leads to reactive compatibilization, yielding a homogeneous polymer over a wide range of sulfur contents. Isothermal kinetic studies showed two different types of reactions occurring simultaneously, an observation attributed to the concurrent radical polymerization of sulfur and epoxide crosslinking. The resulting polymer exhibited stable macroscopic shape memory properties with excellent shape recovery and reprogrammability, properties which are attributed to the presence of both strong and weak linkages within the polymer network. High contents of polysulfide groups within the polymer matrix allowed for facile reprogramming and reprocessing of the shape memory polymer.
Results and discussion
Inverse vulcanization of elemental sulfur and AGE
Inverse vulcanization of elemental sulfur and AGE (50 wt%) was carried out at 150 °C. The initially biphasic reaction mixture gradually became monophasic over three hours and subsequently underwent vitrification to yield a rubbery solid (Fig. 1a). Substituting AGE for allyl butyl ether (ABE) in an otherwise identical polymerization also gave a monophasic mixture. The mixture, however, did not become viscous, and the reaction mixture remained a monophasic liquid after 5 hours at 150 °C. The observation suggested that, while the reaction between molten sulfur and AGE led to polymerization, the reaction between sulfur and ABE did not. The reaction between elemental sulfur and butyl glycidyl ether (BGE), an analog lacking the olefin functional group, was conducted under identical conditions to probe the role of the epoxy group. Heating the mixture of elemental sulfur and BGE at 150 °C for 5 hours did not result in any apparent reaction, with the reaction mixture remaining biphasic. When 1,1-dimethyl allyl glycidyl ether (DAGE) was used, the reaction mixture turned monophasic after 15 hours, but did not vitrify even after 36 hours at 150 °C. The results indicated that the presence of both an allylic hydrogen and a glycidyl group was crucial, with the glycidyl group associated with the vitrification of the reaction product.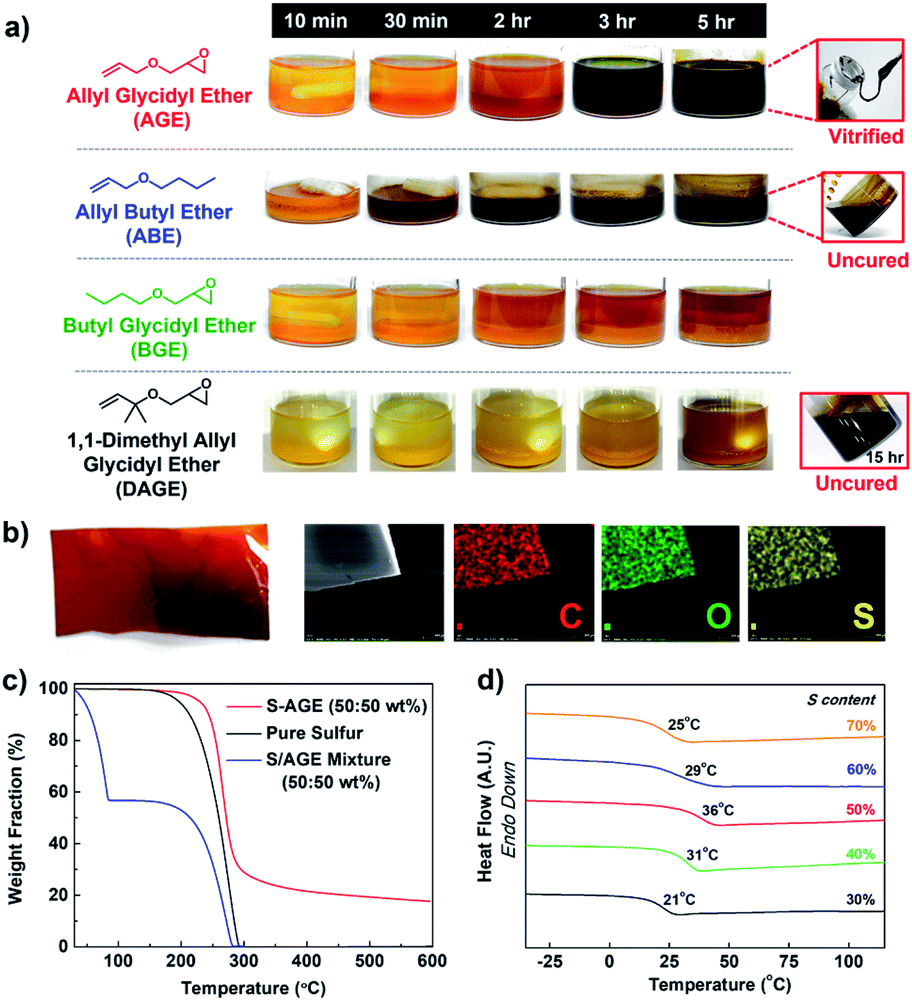 | ||
| Fig. 1 Inverse vulcanization of elemental sulfur and AGE. (a) Photographs of reaction between equal masses of elemental sulfur and allyl glycidyl ether (AGE), butyl glicidyl ether (BGE), allyl butyl ether (ABE), and 1,1-dimethyl allyl glycidyl ether (DAGE). Casted free-standing film of poly(S-r-AGE) and the corresponding SEM-EDS elemental mapping (carbon, oxygen and sulfur) images are shown in (b). (c) TGA thermogram of the poly(S-r-AGE), elemental sulfur, and a mixture of sulfur and AGE, and (d) glass transition temperatures, obtained from differential scanning calorimetry, of poly(S-r-AGE) with varying sulfur contents. | ||
The vitrified product of the reaction of sulfur and AGE (poly(S-r-AGE), S–AGE) was insoluble in organic solvents, similarly to previously reported thermosets from the copolymerization of molten sulfur, limiting the number of available analytical methods for its characterisation.12,16 The composition of S–AGE was probed using scanning electron microscopy (SEM). SEM-EDS analysis of free-standing films of S–AGE indicated a uniform distribution of carbon, oxygen, and sulfur atoms, suggesting that S–AGE is a homogeneous polymeric material (Fig. 1b). Thermogravimetric analysis (TGA) of S–AGE showed typical one-step mass loss of random copolymers with higher thermal onset temperature of decomposition compared to that of elemental sulfur (Fig. 1c). A simple mixture of sulfur and AGE displayed a much lower onset temperature, attributed to the evaporation of AGE, followed by the loss of elemental sulfur. Powder X-ray diffraction (PXRD) profile of S–AGE prepared from 50 wt% sulfur did not feature peaks associated with elemental sulfur, suggesting the absence of unreacted sulfur (Fig. S1†). Elemental analysis results showed carbon, hydrogen, and sulfur composition nearly identical to theoretical values (Table S1†). The formation of a homogeneous polymeric product from an initially biphasic mixture suggested that the polymerization proceeded via reactive compatibilization with initially formed polymers possibly acting as compatibilizers.
Differential scanning calorimetry (DSC) of S–AGE revealed a gradual increase in the glass transition temperatures (Tg) with increasing AGE content up to 50 wt% (Fig. 1d). This ascending trend of Tg along with increasing comonomer content is similar to the trends observed in the bulk copolymerization of elemental sulfur with homobifunctional comonomers such as divinylbenzene10 and diisopropenylbenzene,6 suggesting that AGE is serving as a cross-linker for sulfur polymerization despite having only one double bond per molecule. Increase in AGE content beyond 50 wt% resulted in decreasing Tg, presumably due to the plasticizing effect of dangling or unreacted AGE molecules present in the polymer matrix.27 When AGE content was increased to 80 wt%, the resulting reaction mixture did not vitrify and remained a non-viscous liquid, and Tg could not be measured.
Mechanistic studies
Isothermal kinetic studies and time-dependent NMR studies on the soluble fraction of the reaction mixture were conducted to elucidate the mechanism of the polymerization reaction between sulfur and AGE (Fig. 2). The DSC thermogram of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (weight ratio) mixture of elemental sulfur and AGE exhibited two discernable exothermic events, the termination of which occurred earlier with increasing temperature (Fig. 2a). DSC thermograms obtained from the reaction of ABE, an analog of AGE without the epoxide group, featured only a single exotherm, corresponding to the first of the two exothermic events in the reaction between elemental sulfur and AGE (Fig. 2b). No apparent thermal events were observed when BGE, an analog without the olefin, was used instead. Based on the results, the first and second exothermic events were attributed to the reaction involving the allyl group and the glycidyl group, respectively. Furthermore, the reaction of the allyl group apparently sets the stage for the subsequent reaction of the glycidyl group. Time-dependent 1H NMR spectra were collected to monitor the consumption of each functional group during the reaction at 140 °C (Fig. 2c). The intensity of resonances from allylic hydrogens (5.0–5.2 ppm and 5.8–6.0 ppm) rapidly diminished within the first 10 hours of the reaction, while the consumption of the epoxide groups started later.
:1 (weight ratio) mixture of elemental sulfur and AGE exhibited two discernable exothermic events, the termination of which occurred earlier with increasing temperature (Fig. 2a). DSC thermograms obtained from the reaction of ABE, an analog of AGE without the epoxide group, featured only a single exotherm, corresponding to the first of the two exothermic events in the reaction between elemental sulfur and AGE (Fig. 2b). No apparent thermal events were observed when BGE, an analog without the olefin, was used instead. Based on the results, the first and second exothermic events were attributed to the reaction involving the allyl group and the glycidyl group, respectively. Furthermore, the reaction of the allyl group apparently sets the stage for the subsequent reaction of the glycidyl group. Time-dependent 1H NMR spectra were collected to monitor the consumption of each functional group during the reaction at 140 °C (Fig. 2c). The intensity of resonances from allylic hydrogens (5.0–5.2 ppm and 5.8–6.0 ppm) rapidly diminished within the first 10 hours of the reaction, while the consumption of the epoxide groups started later.
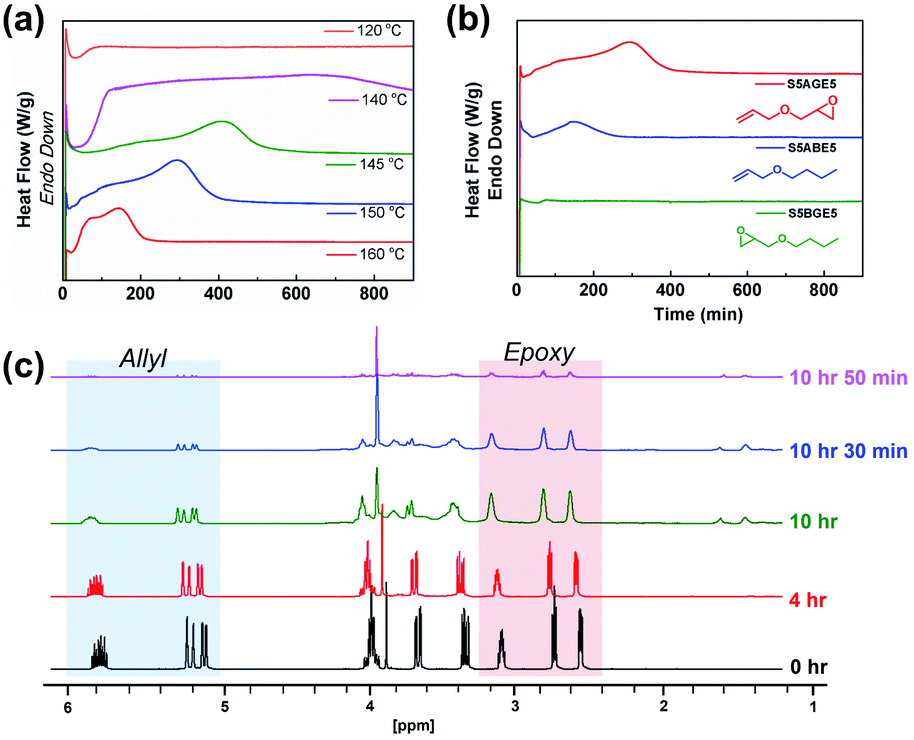 | ||
| Fig. 2 Isothermal DSC and time-dependent NMR studies. Isothermal DSC thermogram of (a) elemental sulfur–AGE mixture (1:1 weight ratio) under various temperatures and (b) mixtures of elemental sulfur with AGE, ABE, and BGE conducted at 150 °C. The 1H NMR spectrum of sulfur–AGE mixtures upon reaction at 140 °C is shown in (c). | ||
Inverse vulcanization involves the reactions of sulfur radicals generated through thermal homolytic ring-opening of cyclooctasulfur.6 While the radicals from the ring-opening of sulfur readily add to olefins, hydrogen atom abstraction that results in the formation of nucleophilic radicals is also a viable reaction pathway, given that sulfur radicals are electrophilic. Since allyl radicals are nucleophilic,28 abstraction of allylic hydrogen of AGE by sulfur is favorable. The reaction would produce polysulfanes in situ, which, like thiol, is able to attack the epoxide ring of the glycidyl group. A similar sequence of reactions had been proposed in a mixture of 2-allylphenol and bisphenol-A diglycidyl ether, which was cured at 190–210 °C with a small (∼7 wt%) amount of elemental sulfur.29 Based on the results and the reactivity of sulfur radicals, a possible mechanism of the polymerization of sulfur and AGE is proposed, where sulfur radicals participate both in addition reactions and hydrogen abstraction reactions with the allyl group of AGE. Compatibilization of sulfur and AGE is attributed to the formation of a polymer from the radical addition reaction of sulfur radicals to the allyl double bonds. The polysulfane resulting from the hydrogen abstraction reaction subsequently reacts with the epoxide, resulting in a vitrified network polymer (Scheme 2).
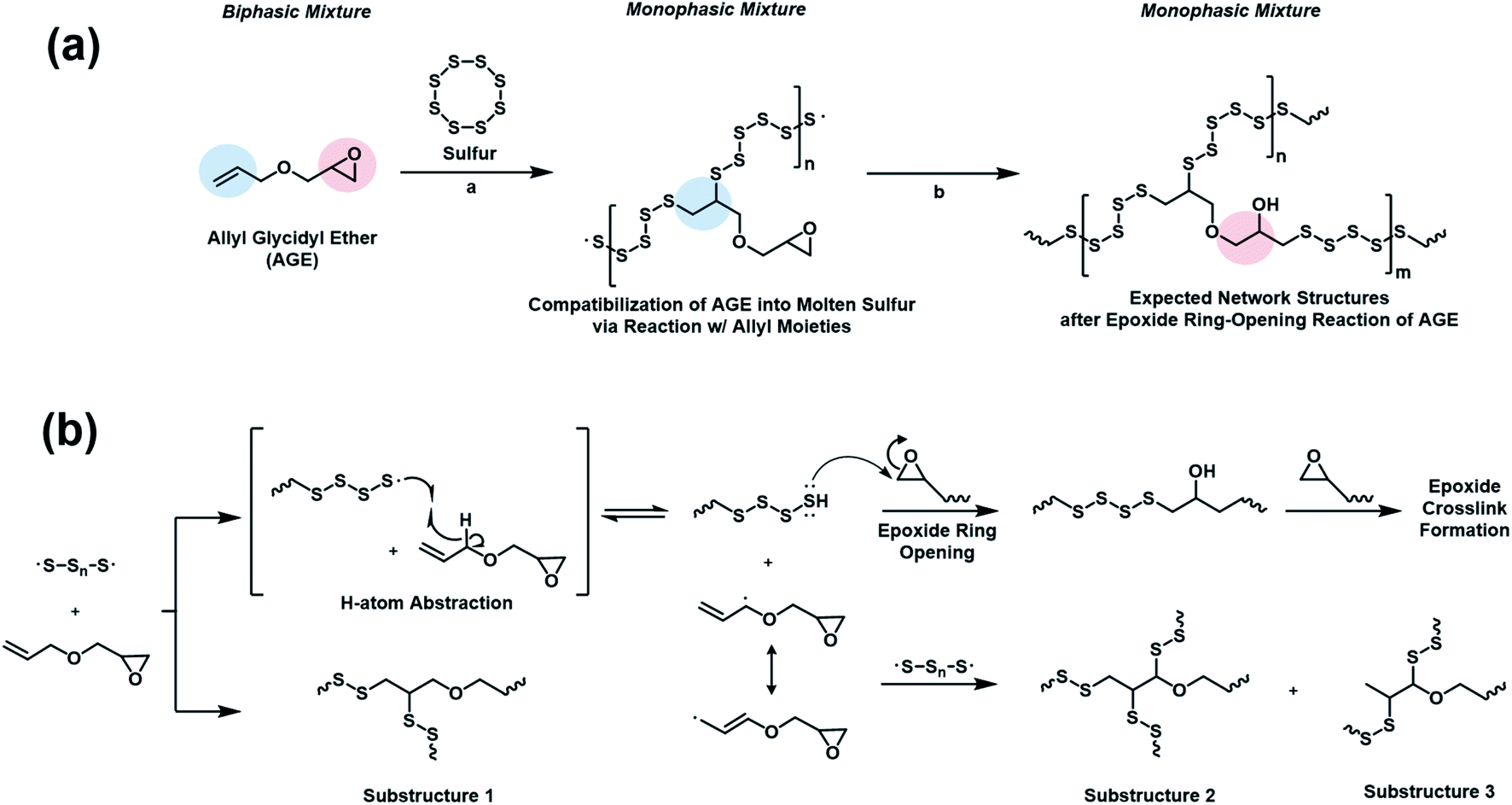 | ||
| Scheme 2 Proposed mechanism of sulfur–AGE copolymerization showing (a) addition of elemental sulfur to the allyl group followed by (b) polysulfane-initiated epoxide ring-opening. | ||
Probing the bond connectivity of S–AGE
To further elucidate the bond connectivity within the S–AGE matrix, Fourier-transform infrared (FTIR) spectroscopy and solid-state 13C NMR analysis were conducted (Fig. 3). The presence of O–H stretching bands at ∼3440 cm−1 in the FTIR spectrum of S–AGE indicated the presence of hydroxyl groups from the ring-opening of epoxides (Fig. 3a). The FTIR spectrum of neat AGE exhibited absorption bands at 3000–2850 cm−1 (2998 cm−1, 2922 cm−1, 2858 cm−1) and at 3100–3000 cm−1 (3081 cm−1, 3057 cm−1), which correspond to sp3 and sp2 C–H stretching, respectively. The FTIR spectrum of S–AGE, however, contains absorption bands exclusively at the former range of 3000–2850 cm−1 (2916 cm−1, 2857 cm−1). The disappearance sp2 C–H stretching bands, along with the intense C–H wag at 990 cm−1, indicates complete consumption of the vinyl groups of AGE during the polymerization.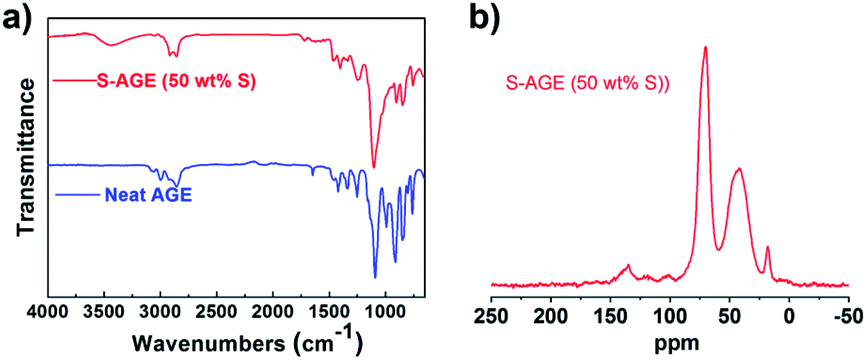 | ||
| Fig. 3 Probing the bond connectivity of S–AGE. (a) FTIR spectra and (b) semi-quantitative 13C solid-state NMR spectrum from 1H–13C cross-polarisation of S–AGE. | ||
The 1H–13C cross-polarization (CP) spectrum (Fig. 3b) exhibits rather broad signals, which suggests a lack of crystallinity. Signal buildup under CP (Fig. S2†), as well as additional direct excitation experiments with short recycle delays (Fig. S3†), point to a very low local molecular mobility and a rather uniform proton density. This suggests a high degree of cross-linking. Also, the absence of significant differences between the CP and the fully relaxed direct polarization spectra confirms that the CP spectrum can be considered quantitative to a good approximation (Fig. S4†). The absence of resonances around 130 ppm also indicates the nearly complete consumption of vinyl double bonds. The resonances at 48.1 ppm and 41.6 ppm could be attributed to C–S connectivity, i.e. R2C–S and RC–S, respectively. The intense resonances at 70 ppm and 73.8 ppm, within the same broad peak, could be attributed to ring-opened epoxides, i.e. R2C–OH and R(OH)C–O, respectively, consistent with Scheme 2a. A spinning sideband from that resonance is clearly visible around 138 ppm. Because of the relatively broad resonances, the possibility of remaining epoxide rings cannot be fully excluded. The resonance around 18 ppm could be due to terminal methyl groups formed according to the substructure 3 (Scheme 2b).
Mechanical properties of S–AGE
With the results thus far suggesting a highly cross-linked structure for S–AGE, its tensile strength, along with the potential for its application as a shape memory polymer (SMP) was evaluated. The tensile strength was measured using a universal testing machine with a crosshead speed of 10 mm min−1 (Fig. 4). The strength of S–AGE varied over a wide range depending on sulfur content, with 30 wt% AGE content leading to soft and ductile polymer showing σmax of 13.55 MPa at the strain of 16.7%, values similar to those from other inverse vulcanization polymers. A dramatic enhancement in Young's modulus was observed as AGE content was increased to 50 wt%. At 50 wt% AGE, a high tensile strength was observed, with σmax of 60.44 MPa. This value is by far the highest value, to the best of our knowledge, from an inverse vulcanization product. The high strength was attributed to the high degree of crosslinking afforded by the high functional group density of the comonomer and crosslinker, AGE. The drastic change of physical properties as the AGE content was changed from 30 to 50 wt% is attributed to the significant enhancement of epoxide crosslink formation with increasing epoxide functional group concentration within the polymerization mixture. However, increasing the degree of crosslinking results in decreased ductility and the sulfur/AGE ratio should be tuned to obtain the optimal strength and ductility for each target application. Further investigations into the morphology of S–AGE in relation to AGE content is currently under investigation.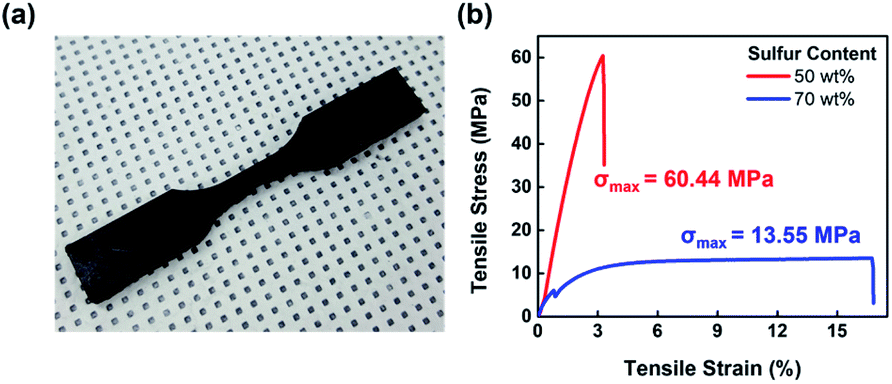 | ||
| Fig. 4 Tensile strength measurement. (a) Digital photograph of the dogbone sample of S–AGE with 50 wt% sulfur and (b) stress–strain curve of S–AGE with sulfur contents of 50 and 70 wt%. | ||
SMPs constitute a class of stimuli-responsive polymers that revert to a certain shape (permanent shape) from a temporarily deformed shape upon exposure to appropriate stimulus such as heat.30,31 Interestingly, we found that free-standing films of S–AGE exhibit thermally induced dual-shape memory behavior with Tg as the transition temperature (Ttrans). The dual-shape memory effect was quantitatively studied through dynamic mechanical analysis (DMA) under single-cantilever bending mode, using S–AGE with 50 wt% sulfur (Fig. 5a).
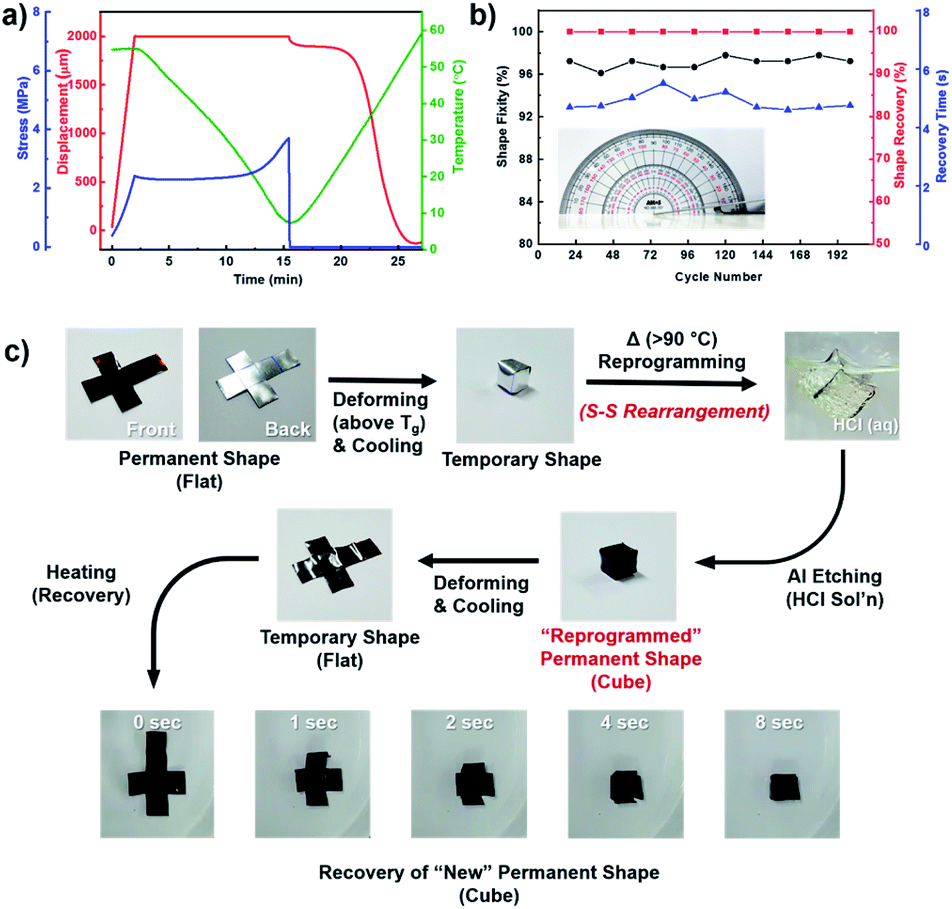 | ||
| Fig. 5 Evalution and demonstration of shape-memory behavior. (a) Quantitative analysis of dual-shape memory cycle process, (b) shape-memory performances under repetitive bending cycles, and (c) demonstration of dual-shape memory behavior and polysulfide bonds rearrangement-induced shape reconfigurability of S–AGE. Polymerization allows for the formation of highly cross-linked polymer, which, despite being a thermoset, can be thermally reprocessed due to the presence of large quantities of dynamic S–S bonds. The S–AGE displayed excellent shape-memory properties, and we believe that our studies would open a new class of polysulfide materials that could be applied in various fields requiring simple and well-characterized stimuli-responsive material which could be readily prepared in large scales (Video S2.† Spontaneous folding of S–AGE (50 wt% sulfur) film into a cube). | ||
The dual-shape memory cycle consisted of (1) gradual deformation of the film to its maximum displacement (εm) under 55 °C (∼20 °C higher than Ttrans) followed by cooling the film to 5 °C to fix the temporary shape, followed by (2) removal of the deforming force and subsequent heating of the sample to 60 °C to induce shape recovery. The displacement, stress, and temperature profiles were recorded with respect to time. The shape fixity (Rf), which is an important performance factor of SMPs, was calculated as the ratio of εu (unloading strain) to εm (maximum strain) (εu/εm). S–AGE film was found to exhibit excellent shape fixity of 95%. Also, the strain at the end of recovery (εr) approached zero, which means the copolymer showed a nearly complete (100%) shape recovery ratio (Rr = (εm − εr)/εm) as seen in the displacement curve. In addition to quantitative DMA, a manual bending cycle test was also conducted using a film having dimensions of 90 mm × 12 mm and 1 mm thickness to test the durability of S–AGE as an SMP (Fig. 5b and Video S1.† Spontaneous bending of 90 mm × 12 mm and 1 mm thick S–AGE (50 wt% sulfur)). The S–AGE film maintained excellent shape fixity (Rf > 95%) and recovery ratio (Rr ∼ 100%) over 200 cycles of repetitive bending to 180° followed by full recovery, demonstrating a reliable shape memory performance of poly(S-r-AGE).
Since sulfur–sulfur bonds are often utilized in dynamic covalent bond chemistry32,33 and applied in self-healing and other thermoresponsive polymers,34–36 we envisioned that the sulfur-rich S–AGE could readily undergo reprogramming into a new permanent shape through facile S–S bond rearrangement. To demonstrate the reprogrammability of S–AGE SMP, S–AGE film was fashioned into a cube layout (Fig. 5c, flat). Then, the film was folded into a cube and annealed in a convection oven held at 90 °C for 30 minutes to induce thermal S–S bond rearrangements to fix a new permanent shape (Fig. 5c, cube). It should be noted that aluminum foil was used as a substrate to prevent unintended shape recovery during annealing. The aluminum foil was then removed by submerging the cube in aqueous HCl. High chemical resistance, typical of high sulfur-content polymers, allowed for a clean removal of the substrate without affecting physicochemical properties of S–AGE. Shape memory cycle test using the cube gave excellent shape memory behavior, with the new permanent shape being recovered from the flat layout within 8 seconds (Fig. 5c). These results demonstrate the merit of using S–AGE as SMP, since the permanent shape can be easily tuned with simple heat treatment.
Another advantage of the high sulfur content in S–AGE SMP manifested in the reprocessability of the material (Fig. S5†). Hot-pressing ground powder of S–AGE at 150 °C gave a monolithic specimen which exhibited nearly identical shape memory performance to cast films. Despite the high crosslink density of S–AGE with 50 wt% AGE which renders the material with high tensile strength, the presence of dynamic S–S bonds allows the material to be readily recycled.
Lastly, it should be noted that, although many different classes of shape memory polymers (SMP) have been carefully designed and developed, preparation of such SMPs are often complicated and involve multiple components polymerized via multistep procedures. Many SMPs are made of phase-segregated multi-block copolymer structures consisting of hard segments as physical cross-links and soft segments as thermoresponsive domains for shape-memory actuation.37–39 The S–AGE described herein is prepared from a one-step, solvent-free bulk polymerization, which, together with its unprecedentedly high strength, excellent performance as a SMP, reprogrammability, and reprocessability, render the new material worthy of further investigations and applications.
Conclusions
In summary, we synthesized novel high sulfur content (up to 70 wt%) copolymers through inverse vulcanization involving elemental sulfur and a heterobifunctional comonomer, AGE. The unique mechanism involved in the polymerization allows for the formation of highly cross-linked polymers, with the highest tensile strength among inverse vulcanization polymers reported to date, in a single polymerization step. The product, S–AGE, despite being a thermoset, can be thermally reprocessed due to the presence of large quantities of dynamic S–S bonds. The S–AGE displayed excellent shape-memory properties, and we believe that our studies would open a new avenue towards a class of scalable, high-strength inverse vulcanization materials that could be applied in various fields requiring simple and well-characterized stimuli-responsive polymers.Data availability
The experimental data supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization, S. P., M. C., J. L., and K. C.; methodology, S. P., M. C., and J. L.; investigation, S. P., M. C., A. L., K. S., and S. S.; writing – original draft, S. P., M. C., A. L., K. S., and J. L.; writing – review & editing, S. P., M. C., A. L., K. S., S. S., C. S., J. L., and K. C.; funding acquisition, C. S., J. L., and K. C.; project supervision, J. L. and K. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. C. acknowledges the financial support from NRF of Korea for the National Creative Research Initiative Center for Intelligent Hybrids (Grant No. 2010-0018290) and the World Class University Program through the NRF of Korea (Grant No. R31-10013). J. L. would like to acknowledge the financial support from NRF of Korea (Grant No. NRF-2019R1C1C1011191). K. C. and J. L. both acknowledge the Network for Asian Open Research (NAO) program by BASF. The Institute of Engineering Research at Seoul National University provided research facilities for this work.Notes and references
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- W. J. Chung, A. G. Simmonds, J. J. Griebel, E. T. Kim, H. S. Suh, I.-B. Shim, R. S. Glass, D. A. Loy, P. Theato, Y.-E. Sung, K. Char and J. Pyun, Angew. Chem., Int. Ed., 2011, 50, 11409–11412 CrossRef CAS PubMed.
- M. M. Vlahovic, S. P. Martinovic, T. D. Boljanac, P. B. Jovanic and T. D. Volkov-Husovic, Constr. Build. Mater., 2011, 25, 3926–3934 CrossRef.
- A.-M. O. Mohamed and M. M. El Gamal, Sulfur Concrete for the Construction Industry: A Sustainable Development Approach, J. Ross Pub., Ft. Lauderdale, FL, 2010 Search PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y. E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K. J. Kim, J. Van Der Laan, N. A. Nguyen, E. L. Dereniak, M. E. MacKay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- S. Park, D. Lee, H. Cho, J. Lim and K. Char, ACS Macro Lett., 2019, 8, 1670–1675 CrossRef CAS.
- I. Gomez, D. Mecerreyes, J. A. Blazquez, O. Leonet, H. Ben Youcef, C. Li, J. L. Gómez-Cámer, O. Bundarchuk and L. Rodriguez-Martinez, J. Power Sources, 2016, 329, 72–78 CrossRef CAS.
- Z. Sun, M. Xiao, S. Wang, D. Han, S. Song, G. Chen and Y. Meng, J. Mater. Chem. A, 2014, 2, 9280–9286 RSC.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- I. Gomez, O. Leonet, J. A. Blazquez and D. Mecerreyes, ChemSusChem, 2016, 9, 3419–3425 CrossRef CAS PubMed.
- L. E. Anderson, T. S. Kleine, Y. Zhang, D. D. Phan, S. Namnabat, E. A. LaVilla, K. M. Konopka, L. Ruiz Diaz, M. S. Manchester, J. Schwiegerling, R. S. Glass, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2017, 6, 500–504 CrossRef CAS.
- T. S. Kleine, N. A. Nguyen, L. E. Anderson, S. Namnabat, E. A. Lavilla, S. A. Showghi, P. T. Dirlam, C. B. Arrington, M. S. Manchester, J. Schwiegerling, R. S. Glass, K. Char, R. A. Norwood, M. E. Mackay and J. Pyun, ACS Macro Lett., 2016, 5, 1152–1156 CrossRef CAS.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y. E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. Mackay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- H. Kang, H. Kim and M. J. Park, Adv. Energy Mater., 2018, 8, 1802423 CrossRef.
- Y. Xin, H. Peng, J. Xu and J. Zhang, Adv. Funct. Mater., 2019, 29, 1808989 CrossRef.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem.–Eur. J., 2019, 25, 10433–10440 CrossRef CAS PubMed.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- B. Zhang, S. Petcher and T. Hasell, Chem. Commun., 2019, 55, 10681–10684 RSC.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- P. Yan, W. Zhao, S. J. Tonkin, J. M. Chalker, T. L. Schiller and T. Hasell, ChemRxiv, 2021, this content is a preprint and has not been peer-reviewed.
- K. S. Kang, A. Phan, C. Olikagu, T. Lee, D. A. Loy, M. Kwon, H. jong Paik, S. J. Hong, J. Bang, W. O. Parker, M. Sciarra, A. R. de Angelis and J. Pyun, Angew. Chem., Int. Ed., 2021, 60, 22900–22907 CrossRef CAS PubMed.
- Z. Zhang and S. Fang, Electrochim. Acta, 2000, 45, 2131–2138 CrossRef CAS.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2720–2724 CrossRef PubMed.
- Q. Lian, Y. Li, T. Yang, K. Li, Y. Xu, L. Liu, J. Zhao, J. Zhang and J. Cheng, J. Mater. Sci., 2016, 51, 7887–7898 CrossRef CAS.
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 49–50, 3–33 CrossRef CAS.
- D. Habault, H. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- Q. Guan, S. J. Picken, S. S. Sheiko and T. J. Dingemans, Macromolecules, 2017, 50, 3903–3910 CrossRef CAS.
- J. R. Kumpfer and S. J. Rowan, J. Am. Chem. Soc., 2011, 133, 12866–12874 CrossRef CAS PubMed.
- C. L. Xu, J. B. Zeng, Q. Y. Zhu and Y. Z. Wang, Ind. Eng. Chem. Res., 2013, 52, 13669–13676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures and videos. See DOI: 10.1039/d1sc05896g |
| ‡ These authors contributed equally. |
| § Current address: Battery R&D Center, Samsung SDI, 130 Samsung-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do 16678, Republic of Korea. |
| ¶ Current address: Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2022 |