
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIterative addition of carbon nucleophiles to N,N-dialkyl carboxamides for synthesis of α-tertiary amines†
Jiahua
Chen
,
Jun Wei
Lim
,
Derek Yiren
Ong
and
Shunsuke
Chiba
 *
*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: shunsuke@ntu.edu.sg
First published on 26th November 2021
Abstract
A protocol for the synthesis of α-tertiary amines was developed by iterative addition of carbon nucleophiles to N,N-dialkyl carboxamides. Nucleophilic 1,2-addition of organolithium reagents to carboxamides forms anionic tetrahedral carbinolamine (hemiaminal) intermediates, which are subsequently treated with bromotrimethylsilane (Me3SiBr) followed by organomagnesium (Grignard) reagents, organolithium reagents or tetrabutylammonium cyanide, affording α-tertiary amines. Employment of (trimethylsilyl)methylmagnesium bromide as the 2nd nucleophile allowed for aza-Peterson olefination of the resulting α-tertiary (trimethylsilyl)methylamines with acidic work-up, resulting in the formation of 1,1-diarylethylenes.
Introduction
α,α,α-Trisubstituted amines (α-tertiary amines) are found as core structural motifs in various biologically active natural alkaloids1 and are identified as a key functional unit for drug discovery programs.2 Although the sterically hindered nature of α-tertiary amines has rendered their synthetic approaches more challenging, significant advancements of their synthesis have recently been made.3–6 Among them, geminal difunctionalization on carbonyl oxygen of readily accessible and bench-stable carboxamides via deoxygenative installation of two carbon–carbon bonds has been considered as one of the most practical routes to α-tertiary amines.7,8 However, this approach generally necessitates pre-conversion of carboxamides to methylthioiminium salts via thiocarboxamides (Scheme 1A)9 or trifluoromethanesulfonyloxyiminium salts (Scheme 1B),10 prior to adding two fold organomagnesium or organolithium reagents. There are several exceptions that enable the direct use of carboxamides without their preactivation for the geminal difunctionalization. For example, dimethylation of carboxamides with methylmagnesium bromide (MeMgBr) could be performed by the stoichiometric use of oxophilic Lewis acids such as TiCl4 and ZrCl4, whereas carbanion reagents other than MeMgBr were not examined (Scheme 1C).11 The use of N-alkoxyamides allowed for iterative installation of two carbon nucleophiles, where the 2nd carbon nucleophile should be added in the presence of acid additives (Scheme 1D).12 A sequential transformation of potassium acyltrifluoroborates has recently been developed for the synthesis of α-tertiary amines via the geminal difunctionalization on carbonyl oxygen via (i) conversion of acyltrifluoroborates to acyltrifluoroborate-iminiums by treatment with N,N-dialkylamines; (ii) addition of Grignard reagents (the 1st carbanion) to form α-aminoalkyltrifluoroborates; (iii) oxidation with bis(pyridine)iodonium(I) tetrafluoroborate (py2IBF4) to iminium ions followed by addition of Grignard reagents (the 2nd carbanion) (Scheme 1E).13 Nonetheless, synthesis of α-tertiary amines via the iterative installation of two different carbon substituents to carboxamides without their prefunctionalization remains an unmet challenge. | ||
| Scheme 1 Prior arts on the synthesis of α-tertiary amines via geminal difunctionalization of carbonyl groups. | ||
To develop a more concise and straightforward route to α-tertiary amines from carboxamides, we wondered if anionic tetrahedral carbinolamine (hemiaminal) intermediates formed upon addition of the 1st carbanion reagents14 could successively incorporate the 2nd carbanion reagents via deoxygenation. Building on the recent precedents on the successful functionalization of O-silylated hemiaminal intermediates, which are generated via transition-metal catalyzed hydrosilylation15 or controlled hydride reduction followed by silylation16 with carbon nucleophiles such as Grignard reagents to form α-secondary amines (Scheme 2A), we surmised that the synthesis of α-tertiary amines from carboxamides might be realized by the silylation of anionic hemiaminal intermediates upon addition of the 1st carbanion nucleophiles and the ensuing engagement of the 2nd carbanion reagents (Scheme 2B). The essential keys to enable this proposed process are (i) a proper choice of the 1st carbon nucleophiles for the smooth construction of anionic hemiaminal intermediates with prevention of their decomposition before the engagement of the 2nd nucleophiles (phase I); (ii) an efficient O-silylation of sterically hindered anionic hemiaminal intermediates (phase II) followed by the 2nd C–C bond formation (phase III).17 We describe our findings herein.
 | ||
| Scheme 2 Geminal difunctionalization of carbonyl groups via O-silylated hemiaminal intermediates. | ||
Results and discussion
We embarked on our investigation for the geminal functionalization of N,N-dimethyl-2-naphthamide (1) via phenylation and methylation (Table 1). We observed that addition of pre-prepared and titrated phenyllithium (PhLi) proceeded smoothly and subsequent treatment with chlorotrimethylsilane (Me3SiCl) and methylmagnesium bromide (MeMgBr) at 60 °C afforded desired α-tertiary amine 2 in 68% yield along with the formation of α-tertiary alcohol 3 in 25% yield (entry 1). We found that the use of bromotrimethylsilane (Me3SiBr) could improve the yield of α-tertiary amine 2 to 89% (84% isolated) yield (entry 2). This protocol was found to be scalable up to 15 mmol scale without detrimental impact on the yield of 2 (entry 3). The use of PhLi prepared in situ via halogen–lithium exchange using bromobenzene and tert-butyllithium afforded amine 2 in 70% yield (entry 4). Despite slightly diminished efficiency, this approach circumvents pre-preparation of organolithium reagents (vide infra). However, the reaction with phenylmagnesium bromide (PhMgBr) as the 1st carbanion did not afford desired amine 2 at all (entry 5). In this case, formation of alcohol 3 was observed in 13% yield along with 2-naphthyl phenyl ketone (3′) in 52% yield, indicating insufficient O-silylation of the corresponding anionic carbinol amine intermediate generated from 2-naphthamide 1 and PhMgBr.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 (mmol) | Ph-[M] | Temp [°C] | Me3SiX | Yieldc [%] | |
| 2 | 3 | |||||
| a 1H NMR yields based on the internal standard. The isolated yield is in parentheses. b PhLi was pre-prepared from PhBr and Li in Et2O and titrated before use. c PhLi was generated in situ from PhBr (0.75 mmol) and tert-BuLi (1.5 mmol, 1.32 M in pentane) in THF, and directly used for the reaction with 1. d PhMgBr was pre-prepared from PhBr and Mg in THF and titrated before use. e 2-Naphthyl phenyl ketone (3′) was formed in 52% yield. | ||||||
| 1 | 0.5 | PhLib | −78 to 0 | Me3SiCl | 68 | 25 |
| 2 | 0.5 | PhLib | −78 to 0 | Me3SiBr | 89 (84) | 8 |
| 3 | 15 | PhLib | −78 to 0 | Me3SiBr | 90 (86) | 10 |
| 4 | 0.5 | PhLic | −78 to 0 | Me3SiBr | 70 | 5 |
| 5 | 0.5 | PhMgBrd | 0 to 60 | Me3SiBr | 0 | 13e |

The proposed mechanism of the geminal difunctionalization of 2-naphthamide 1 is depicted in Scheme 3. Addition of PhLi to the amide carbonyl group forms anionic hemiaminal intermediate A, which could be detected as a stable form even at ambient temperature by 1H and 13C NMR analyses (see the ESI†). O-Silylation of A with halotrimethylsilane (Me3SiX) resulted in the formation of B and ensuing addition of MeMgBr results in the formation of amine 2via iminium ion intermediate C. On the other hand, electrophilic activation of the amino group of A by Me3SiX collapses A into ketone 3′, which is trapped with MeMgBr to provide alcohol 3. We speculated that the use of more electrophilic Me3SiBr renders the reaction course more selective toward the formation of iminium Cvia efficient O-silylation of A over that of ketone Dvia N-silylation.
 | ||
| Scheme 3 Proposed reaction pathways. | ||
We next investigated the scope of the reaction with respect to the carbanion reagents using 2-naphthamide 1 (Scheme 4). As for the organolithium reagents as the 1st carbanion, the method was found to be compatible with the use of both electron-rich and electron-deficient aryllithium as well as 2-thienyllithium, 2-furyllithium, 5-indolyllithium and 2-pyridyllithium, providing the corresponding amines 4–9 in good to moderate yields (Scheme 4A). The protocol is amenable to use lithium phenylacetylide to form propargylamine 10. As for alkyllithium reagents, the present protocol could employ methyllithium and butyllithium, efficiently providing the corresponding α-tertiary amines 11 and 12, while the use of isopropyllithium resulted in moderate efficiency for the formation of amine 13 (31% yield). We then examined the compatibility of the 2nd carbanion reagents using PhLi or MeLi as the 1st carbanion reagent (Scheme 4B). With PhLi as the 1st carbanion, the method was amenable to engage allyl and benzyl Grignard reagents (for 14 and 15). Installation of acetylenic moieties could also be implemented with good efficiency using ethynylmagnesium bromide and lithium phenylacetylide, respectively (for 16 and 17). We found that the use of tetrabutylammonium cyanide enables the downstream Strecker reaction to form α-cyano amine 18 in good yield. With MeLi as the 1st carbanion, similarly, the protocol was compatible to incorporate allyl, benzyl, and alkynyl motifs in the corresponding α-tertiary amines 19–21.18
 | ||
| Scheme 4 Scope with respect to the nucleophiles. aThe reactions conditions: 2-naphthamide 1 (0.5 mmol), 1st organolithium reagent (1.5 equiv.) at −78 to 0 °C for 1 h, followed by Me3SiBr (2 equiv.) and MeMgBr (2.5 equiv.) at 0 °C, then stirring at 60 °C for 2 h. Isolated yields were recorded. bAryllithium was prepared by the treatment of the corresponding aryl bromide (0.75 mmol) with t-BuLi (1.5 mmol) in THF at −78 °C. c2-Furanyl- or 2-thienyllithium was prepared by the treatment of furan or thiophene (0.75 mmol) with BuLi (0.75 mmol) in THF at −78 to 0 °C. dThe commercially available organolithium reagents were used after titration. eLithium phenylacetylide (3 equiv.) at 60 °C for 2 h, followed by Me3SiBr (2 equiv.) and MeMgBr (3 equiv.) at 0 °C, then stirring at 60 °C for 2 h. fMeMgBr (3 equiv.) was used. gThe reaction conditions: carboxamide 1 (0.5 mmol), PhLi or MeLi (1.5 equiv.) at −78 to 0 °C for 1 h, followed by Me3SiBr (2 equiv.) and organomagnesium reagents (2.5 equiv.) as the 2nd nucleophile (2.5 equiv.) at 0 °C, then stirring at 60 °C for 2 h. Isolated yields were recorded. hLithium phenylacetylide was used as the 2nd nucleophile. iBu4NCN was used as the 2nd nucleophile. | ||
Substituent compatibility of amide nitrogen was investigated next (Scheme 5). This method allowed for the synthesis of α-tertiary amines starting from N-benzyl-N-methylamide (for 22) and those based on azetidine, pyrrolidine, piperidine, morpholine and azepane (for 23–27).
 | ||
| Scheme 5 Substituent compatibility of amide nitrogen. aThe reactions conditions: carboxamides (0.5 mmol), PhLi (1.5 equiv.) at −78 to 0 °C for 1 h, followed by Me3SiBr (2 equiv.) and MeMgBr (2.5 equiv.) at 0 °C, then stirring at 60 °C for 2 h. Isolated yields were recorded. bAfter addition of Me3SiBr at 0 °C, the mixture was stirred at room temperature for 0.5 h before the mixture was treated with MeMgBr at 0 °C and heated at 60 °C for 2 h. cAfter addition of Me3SiBr at 0 °C, the mixture was stirred at 60 °C for 1 h before the mixture was treated with MeMgBr at 0 °C and heated at 60 °C for 2 h. | ||
We also examined the compatibility of various carboxamides, using a combination of PhLi and MeMgBr for the geminal difunctionalization (Scheme 6). As for aromatic amides, the process tolerated substituents with different electronic properties such as a biphenyl moiety (for 28), electron-donating groups (for 29–31) and electron-withdrawing groups including a trifluoromethyl group (for 32) and halogen atoms (for 33–35). Carboxamides based on electron-rich benzofuran (for 36) as well as electron-deficient 6-membered rings including pyridine, quinoline, and isoquinoline moieties (for 37–39) were also compatible. The ability for concise synthesis of 1,1-disubstituted tetrahydroisoquinoline 40 (ref. 19) from the corresponding lactam is also one of the advantageous features of the method. The present protocol also allowed for the geminal difunctionalization of aliphatic amides. The reactions of α-primary and α-secondary alkyl carboxamides having enolizable α-protons proceeded to afford the corresponding amines 41–44 in good to moderate yields. However, the reaction of sterically congested 1-adamantyl carboxamide 45 resulted in the formation of alcohol 47 as the major product.
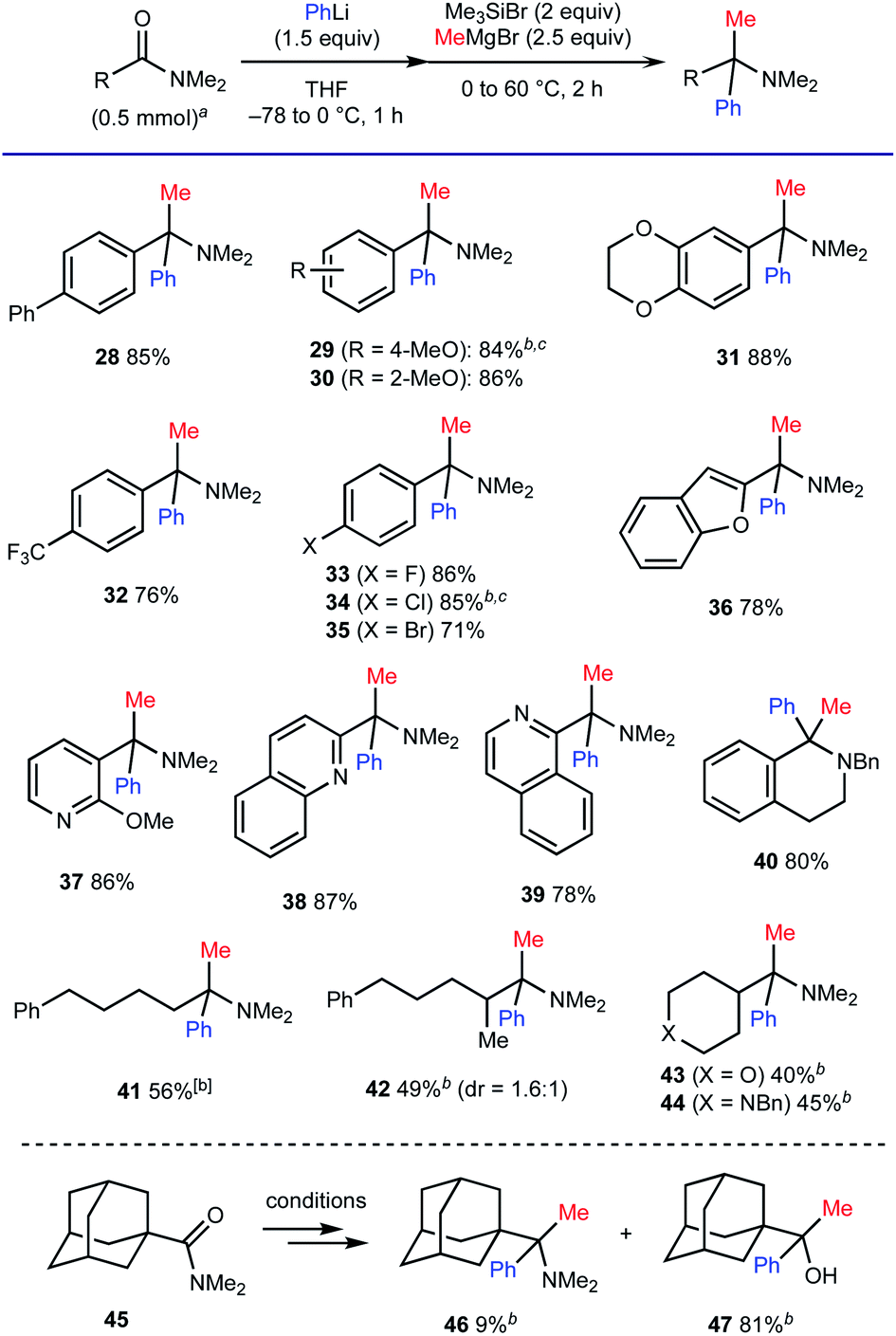 | ||
| Scheme 6 Scope with respect to the carboxamides. aThe reactions conditions: carboxamides (0.5 mmol), PhLi (1.5 equiv.) at −78 to 0 °C for 1 h, followed by Me3SiBr (2 equiv.) and MeMgBr (2.5 equiv.) at 0 °C, then stirring at 60 °C for 2 h. Isolated yields were recorded. b3 equiv. of MeMgBr was used. cAfter addition of MeMgBr, the reaction mixture was stirred at 60 °C for 3 h. Bn = benzyl. | ||
When (trimethylsilyl)methylmagnesium chloride was used as the 2nd carbanion reagent upon treatment of 2-naphthamide 1 with PhLi, we observed that the resulting α-tertiary amine 48 was further converted into 1,1-diarylethylene 49 upon the aqueous acid work-up via elimination of silylamine (Scheme 7A). This process could be regarded as phenylative aza-Peterson olefination20 of carboxamide 1 and we found that this protocol is applicable for the facile construction of unsymmetrical 1,1-diarylethylenes 50–55 (Scheme 7B).
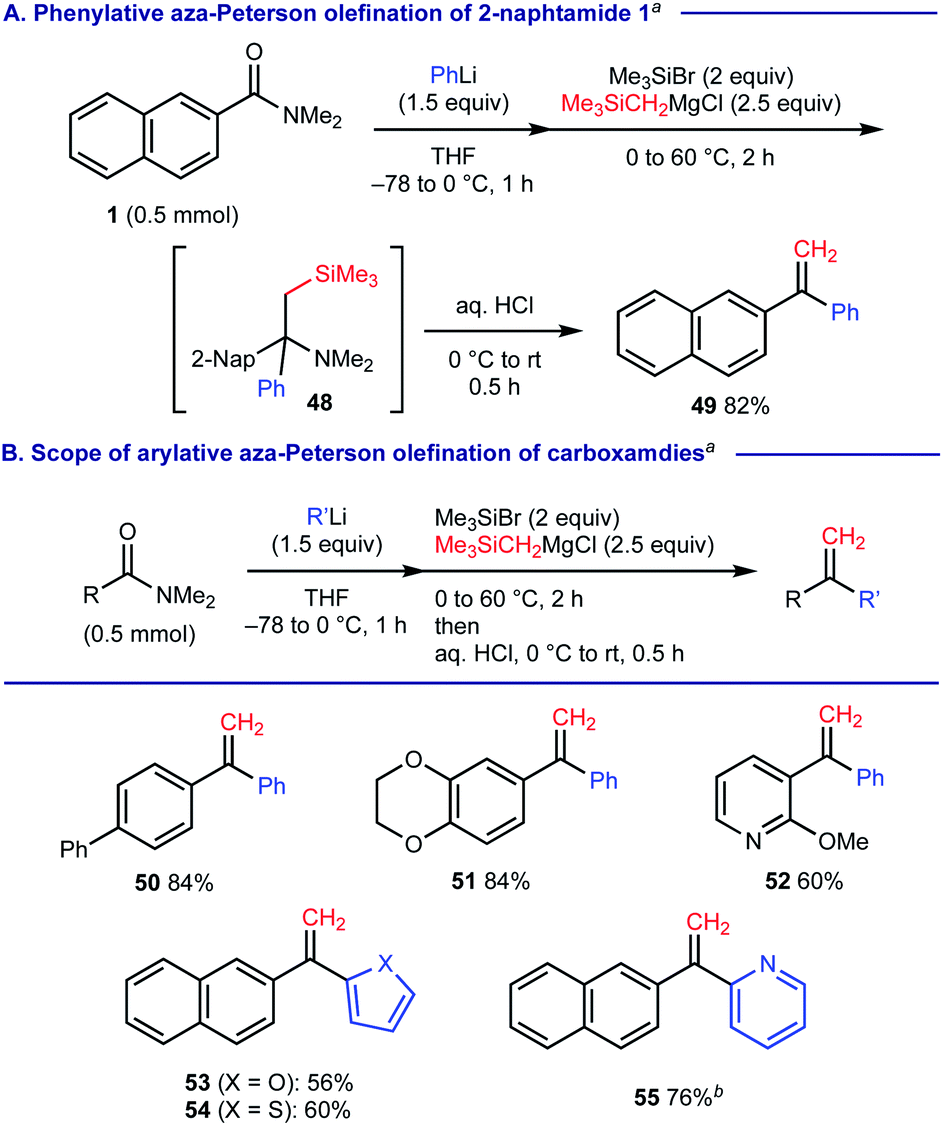 | ||
| Scheme 7 Arylative aza-Peterson olefination of carboxamides. aThe reactions conditions: carboxamides (0.5 mmol), PhLi (1.5 equiv.) at −78 to 0 °C for 1 h, followed by Me3SiBr (2 equiv.) and Me3SiCH2MgCl (2.5 equiv.) at 0 °C, then stirring at 60 °C for 2 h; water (1 mL) at 0 °C, and then 3 M HCl aqueous solution (5 mL), stirring at room temperature for 0.5 h. Isolated yields were recorded. bAfter addition of 6 M HCl aqueous solution, the mixture was stirred at 60 °C for 2 h. | ||
Conclusions
In summary, we have developed a transition-metal free protocol for the synthesis of α-tertiary amines by iterative addition of carbon nucleophiles to readily available and bench stable N,N-dialkyl carboxamides. Given the broad scope and operationally simple protocol of the method, we view it to be adaptable in various synthetic endeavours.Data availability
All synthetic procedures, characterization data, spectroscopic data, supplementary schemes, figures and tables, and detailed crystallographic information are provided in the ESI.†Author contributions
S. C. conceived the project and designed the studies. J. C., J. W. L. and D. Y. O. carried out the experiments. All the authors discussed the results and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Nanyang Technological University (NTU) and the Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2019-T2-1-089) (for S. C.).Notes and references
- A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491–522 RSC.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed; (b) M. S. M. Pearson-Long, F. Boeda and P. Bertus, Adv. Synth. Catal., 2017, 359, 179–201 CrossRef CAS; (c) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (d) J. Clayden, M. Donnard, J. Lefranc and D. J. Tetlow, Chem. Commun., 2011, 47, 4624–4639 RSC.
- For addition of carbon nucleophiles to N-substituted ketimines, see: (a) T. Kano, Y. Aota and K. Maruoka, Angew. Chem., Int. Ed., 2017, 56, 16293–16296 CrossRef CAS; (b) L. Yin, Y. Otsuka, H. Takada, S. Mouri, R. Yazaki, N. Kumagai and M. Shibasaki, Org. Lett., 2013, 15, 698–701 CrossRef CAS; (c) C. J. Pierce, M. Nguyen and C. H. Larsen, Angew. Chem., Int. Ed., 2012, 51, 12289–12292 CrossRef CAS PubMed; (d) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS; (e) R. Wada, T. Shibuguchi, S. Makino, K. Oisaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7687–7691 CrossRef CAS; (f) D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1999, 121, 268–269 CrossRef CAS.
- For C–H amination, see: (a) K. Kiyokawa, T. Watanabe, L. Fra, T. Kojima and S. Minakata, J. Org. Chem., 2017, 82, 11711–11720 CrossRef CAS PubMed; (b) J. L. Roizen, D. N. Zalatan and J. Du Bois, Angew. Chem., Int. Ed., 2013, 52, 11343–11346 CrossRef CAS; (c) Q. Michaudel, D. Thevenet and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 2547–2550 CrossRef CAS PubMed.
- For C–H alkylation/arylation of amines via photoredox catalysis, see: (a) M. C. Nicastri, D. Lehnherr, Y.-h. Lam, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 987–998 CrossRef CAS; (b) D. Lehnherr, Y.-h. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478 CrossRef CAS PubMed; (c) A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS; (d) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed., 2019, 58, 4002–4006 CrossRef CAS; (e) D. Vasu, A. L. Fuentes de Arriba, J. A. Leitch, A. de Gombert and D. J. Dixon, Chem. Sci., 2019, 10, 3401–3407 RSC; (f) J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS; (g) J. Rong, P. H. Seeberger and K. Gilmore, Org. Lett., 2018, 20, 4081–4085 CrossRef CAS.
- D. Seebach, Angew. Chem., Int. Ed., 2011, 50, 96–101 CrossRef CAS.
- For selected reviews on functionalization of carboxamides, see: (a) D. Y. Ong, J.-h. Chen and S. Chiba, Bull. Chem. Soc. Jpn., 2020, 93, 1339–1349 CrossRef CAS; (b) D. Kaiser, A. Bauer, M. Lemmerer and N. Maulide, Chem. Soc. Rev., 2018, 47, 7899–7925 RSC; (c) A. Chardon, E. Morisset, J. Rouden and J. Blanchet, Synthesis, 2018, 50, 984–997 CrossRef CAS; (d) T. Sato, M. Yoritate, H. Tajima and N. Chida, Org. Biomol. Chem., 2018, 16, 3864–3875 RSC; (e) A. Volkov, F. Tinnis, T. Slagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC; (f) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS PubMed; (g) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697–3736 CrossRef CAS.
- (a) T. Murai and Y. Mutoh, Chem. Lett., 2012, 41, 2–8 CrossRef CAS; (b) A. Agosti, S. Britto and P. Renaud, Org. Lett., 2008, 10, 1417–1420 CrossRef CAS PubMed; (c) T. Murai, R. Toshio and Y. Mutoh, Tetrahedron, 2006, 62, 6312–6320 CrossRef CAS; (d) T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968–5969 CrossRef CAS PubMed.
- (a) H. Chen, Y.-H. Huang, J.-L. Ye and P.-Q. Huang, J. Org. Chem., 2019, 84, 9270–9281 CrossRef CAS PubMed; (b) K.-J. Xiao, J.-M. Luo, X.-E. Xia, Y. Wang and P.-Q. Huang, Chem.–Eur. J., 2013, 19, 13075–13086 CrossRef CAS PubMed; (c) H.-H. Huo, X.-E. Xia, H.-K. Zhang and P.-Q. Huang, J. Org. Chem., 2013, 78, 455–465 CrossRef CAS PubMed; (d) G. Bélanger, G. O'Brien and R. Larouche-Gauthier, Org. Lett., 2011, 13, 4268–4271 CrossRef; (e) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS.
- S. M. Denton and A. Wood, Synlett, 1999, 55–56 CrossRef CAS.
- (a) Y. Yanagita, H. Nakamura, K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Chem.–Eur. J., 2013, 19, 678–684 CrossRef CAS; (b) G. Vincent, R. Guillot and C. Kouklovsky, Angew. Chem., Int. Ed., 2011, 50, 1350–1353 CrossRef CAS.
- (a) M. K. Jackl, A. Schuhmacher, T. Shiro and J. W. Bode, Org. Lett., 2018, 20, 4044–4047 CrossRef CAS; (b) T. Shiro, A. Schuhmacher, M. K. Jackl and J. W. Bode, Chem. Sci., 2018, 9, 5191–5196 RSC.
- (a) M. Adler, S. Adler and G. Boche, J. Phys. Org. Chem., 2005, 16, 193–209 CrossRef; (b) S. Ghinato, D. Territo, A. Maranzana, V. Capriati, M. Blangetti and C. Prandi, Chem.–Eur. J., 2021, 27, 2868–2874 CrossRef CAS PubMed; (c) J. de Jong, D. Heijnen, H. Helbert and B. L. Feringa, Chem. Commun., 2019, 55, 2908–2911 RSC; (d) C. Liu, M. Achtenhagen and M. Szostak, Org. Lett., 2016, 18, 2375–2378 CrossRef CAS PubMed; (e) M. Adler, M. Marsch, N. S. Nudelman and G. Boche, Angew. Chem., Int. Ed., 1999, 38, 1261–1263 CrossRef CAS; (f) G. A. Olah, G. K. S. Prakash and M. Arvanaghi, Synthesis, 1984, 228–230 CrossRef CAS.
- For selected reports, see: (a) D. Matheau-Raven, P. Gabriel, J. A. Leitch, Y. A. Almehmadi, K. Yamazaki and D. J. Dixon, ACS Catal., 2020, 10, 8880–8897 CrossRef CAS; (b) P. Gabriel, A. W. Gregory and D. J. Dixon, Org. Lett., 2019, 21, 6658–6662 CrossRef CAS PubMed; (c) Y. Takahashi, T. Sato and N. Chida, Chem. Lett., 2019, 48, 1138–1141 CrossRef CAS; (d) L.-G. Xie and D. J. Dixon, Nat. Commun., 2018, 9, 2841 CrossRef; (e) P. Trillo, T. Slagbrand and H. Adolfsson, Angew. Chem., Int. Ed., 2018, 57, 12347–12351 CrossRef CAS PubMed; (f) W. Ou, F. Han, X.-N. Hu, H. Chen and P.-Q. Huang, Angew. Chem., Int. Ed., 2018, 57, 11354–11358 CrossRef CAS PubMed; (g) Á. L. Fuentes de Arriba, E. Lenci, M. Sonawane, O. Formery and D. J. Dixon, Angew. Chem., Int. Ed., 2017, 56, 3655–3659 CrossRef; (h) L.-G. Xie and D. J. Dixon, Chem. Sci., 2017, 8, 7492–7497 RSC; (i) P.-Q. Huang, W. Ou and F. Han, Chem. Commun., 2016, 52, 11967–11970 RSC; (j) M. Nakajima, T. Sato and N. Chida, Org. Lett., 2015, 17, 1696–1699 CrossRef CAS PubMed.
- D. Y. Ong, D. Fan, D. J. Dixon and S. Chiba, Angew. Chem., Int. Ed., 2020, 59, 11903–11907 CrossRef CAS PubMed.
- The synthesis of α-secondary amines via two-fold addition of Grignard reagents to N,N-dialkylformamides in the presence of titanium(IV) isopropoxide [Ti(O-iPr)4] and chlorotrimethylsilane (Me3SiCl) was developed by de Meijere. However, this method is only applicable to the conversion of formamides and thus not suitable for the synthesis of α-tertiary amines, see: O. Tomashenko, V. Sokolov, A. Tomashevskiy, H. A. Buchholz, U. Welz-Biermann, V. Chaplinski and A. de Meijere, Eur. J. Org. Chem., 2008, 5107–5111 CrossRef CAS.
- The method was not capable for installation of a phenyl group using PhMgBr as the 2nd carbanion reagent (see the ESI†).
- (a) X. Li and I. Coldham, J. Am. Chem. Soc., 2014, 136, 5551–5554 CrossRef CAS PubMed; (b) M. Ludwig, C. E. Hoesl, G. Höfner and K. T. Wanner, Eur. J. Med. Chem., 2006, 41, 1003–1010 CrossRef CAS PubMed; (c) M. Ohkubo, A. Kuno, K. Katsuta, Y. Ueda, K. Shirakawa, H. Nakanishi, I. Nakanishi, T. Kinoshita and H. Takasugi, Chem. Pharm. Bull., 1996, 44, 95–102 CrossRef CAS PubMed.
- (a) M. Das and D. F. O'Shea, Org. Lett., 2016, 18, 336–339 CrossRef CAS PubMed; (b) M. Das, A. Manvar, M. Jacolot, M. Blangetti, R. C. Jones and D. F. O'Shea, Chem.–Eur. J., 2015, 21, 8737–8740 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including procedures, syntheses and characterization of new compounds; 1H and 13C NMR spectra. CCDC 2105303. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05876b |
| This journal is © The Royal Society of Chemistry 2022 |