
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An organophotocatalytic late-stage N–CH3 oxidation of trialkylamines to N-formamides with O2 in continuous flow†
Mark John P.
Mandigma
 a,
Jonas
Žurauskas
a,
Callum I.
MacGregor
b,
Lee J.
Edwards
b,
Ahmed
Shahin
ac,
Ludwig
d'Heureuse
a,
Philip
Yip
d,
David J. S.
Birch
d,
Thomas
Gruber
a,
Jörg
Heilmann
a,
Matthew P.
John
b and
Joshua P.
Barham
*a
a,
Jonas
Žurauskas
a,
Callum I.
MacGregor
b,
Lee J.
Edwards
b,
Ahmed
Shahin
ac,
Ludwig
d'Heureuse
a,
Philip
Yip
d,
David J. S.
Birch
d,
Thomas
Gruber
a,
Jörg
Heilmann
a,
Matthew P.
John
b and
Joshua P.
Barham
*a
aFakultät für Chemie und Pharmazie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: Joshua-Philip.Barham@chemie.uni-regensburg.de
bGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, UK
cChemistry Department, Faculty of Science, Benha University, 13518 Benha, Egypt
dDepartment of Physics, SUPA, University of Strathclyde, 107 Rottenrow East, Glasgow, G4 0NG, UK
First published on 28th December 2021
Abstract
We report an organophotocatalytic, N–CH3-selective oxidation of trialkylamines in continuous flow. Based on the 9,10-dicyanoanthracene (DCA) core, a new catalyst (DCAS) was designed with solubilizing groups for flow processing. This allowed O2 to be harnessed as a sustainable oxidant for late-stage photocatalytic N–CH3 oxidations of complex natural products and active pharmaceutical ingredients bearing functional groups not tolerated by previous methods. The organophotocatalytic gas–liquid flow process affords cleaner reactions than in batch mode, in short residence times of 13.5 min and productivities of up to 0.65 g per day. Spectroscopic and computational mechanistic studies showed that catalyst derivatization not only enhanced solubility of the new catalyst compared to poorly-soluble DCA, but profoundly diverted the photocatalytic mechanism from singlet electron transfer (SET) reductive quenching with amines toward energy transfer (EnT) with O2.
Introduction
A quintessential theme in medicinal chemistry is probing structure activity relationships. While strategies such as de novo and diversity-oriented synthesis are powerful tools to achieve this task, late-stage functionalization (LSF) has gained traction over the past decade as it offers a quicker route to access libraries of complex bioactive molecules from a defined core structure.1,2 Among the myriad of methods that are applied in LSF, C–H functionalization is undeniably an attractive and potent addition to a synthetic chemist's arsenal, given the ubiquity of C–H bonds in molecules.1–3 This umbrella term stretches over traditional transition metal catalysis to alkali and base-metal catalysis to organocatalysis and photocatalytically-enabled transformations. Recent examples demonstrate the value of C–H functionalization of simple and complex amides through ionic4 or radical5 mechanisms. Trialkylamines are especially important targets since they are well represented in the alkaloids, a family of potent bioactive molecules that has shaped the natural sciences.6,7 N–CH3 groups are attractive loci for C–H functionalization in pharmaceutical research, since incremental structural variations carry substantial pharmacological effects (Fig. 1), for example in bioactivities of opiates.8 However, C–H bonds α to N are relatively inert. Access to derivatives was historically carried out stepwise, leveraging the nucleophilicity of the N atom, usually requiring initial demethylations of trialkylamine N–CH3 groups to free N–H secondary amines for subsequent transformations.9 That is until the renaissance of single electron transfer (SET) redox methods, partly driven by photoredox catalysis, which have revolutionized practices in organic synthesis.10 This allowed direct C–H functionalizations α to N, of benzylic amines with nucleophiles11 and a few examples of aliphatic amines with electrophiles.12 A direct and highly N–CH3-selective LSF of trialkylamines was achieved using stoichiometric quantities of an SET-generated hydrogen atom transfer agent (DABCO˙+).13 Powerful catalytic or photocatalytic LSF strategies for complex trialkylamines have emerged, but remain scarce.14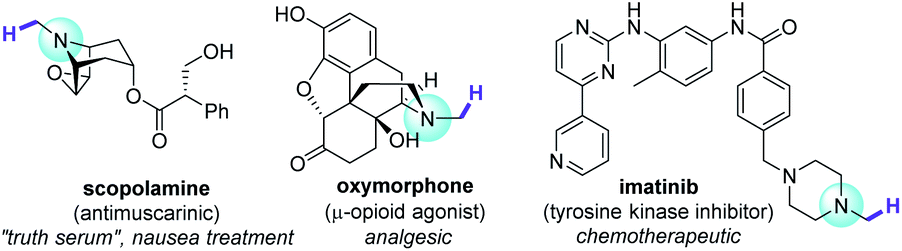 | ||
| Fig. 1 Bioactive trialkylamines and target sites for N–CH3 C–H functionalization. | ||
Direct C–H oxidation of a trialkylamine's N–CH3 group to an N-formyl group is a worthy endeavour as N-formamide products (and mechanisms to access them) are relevant to oxidative metabolite research,15 are natural products,6,16 and serve as synthetic handles17 for further modifications including Barbier-type amidations,18 C–C cross-couplings,19 amino-carbonylations of alkenes or alkynes,20 and couplings with phenols21 or amines22 (affording carbamates or ureas, respectively). N-Formamides are classically accessed from trialkylamines by toxic Ru(IV) or Os(IV) oxidants (Fig. 2A).23–27
 | ||
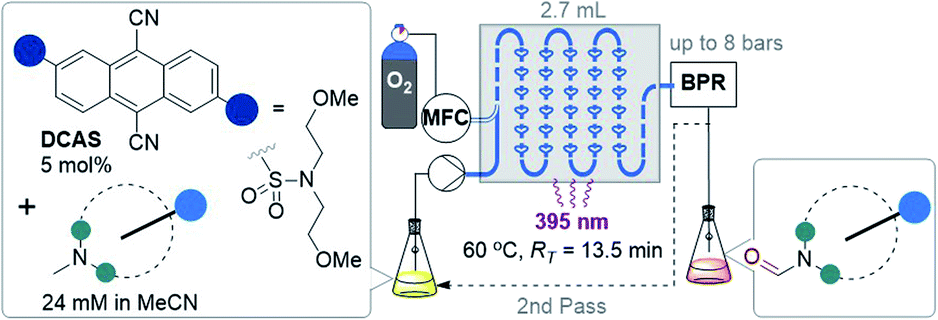
| Fig. 2 Strategies for selective N–CH3 to N-formyl oxidations. PC = photocatalyst, MFC = mass flow controller (O2), BPR = back pressure regulator, RT = retention time. | ||
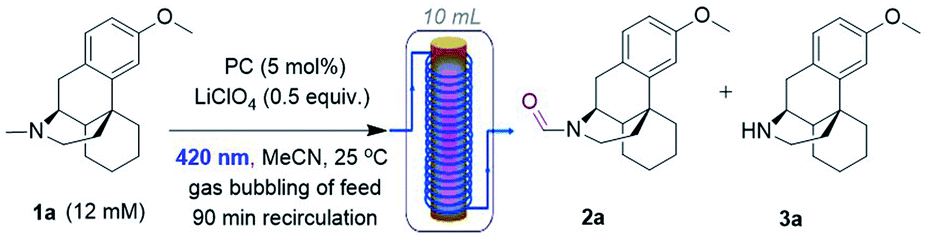
Recently, Yamaguchi and co-workers circumvented this via an elegant Cu(II)/Cu(I) and nitroxyl radical catalyst system.28 Song and co-workers reported a transition metal (T.M.)-free deconstructive formylation reaction.29 The main drawbacks of such previous methods are (i) the incompatibility of redox-sensitive functionalities common to complex pharmaceuticals, hence limiting their application to relatively simple amines, and (ii) the expense of reagents (hindered nitroxyl radical catalysts28b and excess difluorocarbene reagents) which are economically impractical for scale-up. We contemplated an alternative strategy using an organophotocatalyst, in order to avoid toxic, precious, unsustainable T.M.-based photocatalysts.30 Simultaneously, and inspired by numerous reports of gas–liquid photocatalytic flow processes,31 we envisaged that continuous flow would allow us to efficiently and safely handle O2 as a simple, abundant, sustainable terminal oxidant. The rapid uptake of continuous flow reactors in the synthesis of fine materials and pharmaceuticals is worth noting, as is their innovative marriage with visible light irradiation which drastically enhanced the efficiency, sustainability and safety of photochemical processes.32 Previous organophotocatalytic activations of trialkylamines using O2 were reported, but those instead targeted (i) N-demethylations of opiates and tropanoids,33 (ii) endocyclic C–H cyanations α to N34 or (iii) oxidations of benzylic amines.35 We were particularly drawn to 9,10-dicyanoanthracene (DCA) as used by Santamaria and co-workers (Fig. 2B). Using DCA as a potent photooxidizing catalyst (E1/2[1DCA*/DCA˙−] = +1.99 V vs. SCE),30 air (O2) as terminal oxidant, and an LiClO4 additive, they reported variable amounts of N-formyl products (2) in batch, however N-demethylation (nor-amines (3)) competed or dominated reactions.36,37 Herein, we report a late-stage organophotocatalytic oxidation of N–CH3 groups that selectively delivers N-formyl compounds (2). Our method leverages mild conditions and continuous flow processing to handle O2 safely as a terminal oxidant (Fig. 2C). Key to the aforementioned achievements was the design of a novel dicyanoanthracene catalyst that not only enhanced solubility for flow processing, but switched the excited state mechanism from single electron transfer with amines toward energy transfer with O2.
Results and discussion
Photocatalyst and process design
At the onset, our attempts to use DCA using modified reaction conditions of Santamaria in batch (and in flow) were severely obstructed by its poor solubility in MeCN (i.e., turbidity and sedimentation were observed). The suspended, undissolved photocatalyst was detrimental to photochemistry due to hindering light penetration of the reaction. Furthermore, in continuous flow this often led to flow channel blockages and longer reaction times (for details, see ESI† file). Thus, design of a catalyst with enhanced solubility was required (Fig. 3).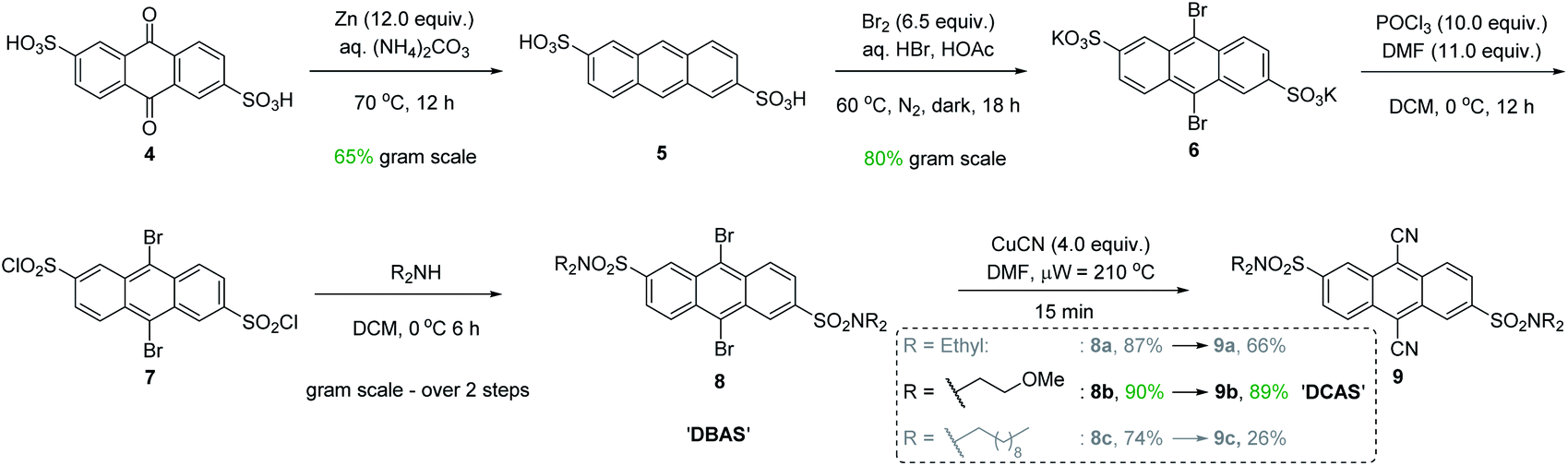 | ||
| Fig. 3 Chromatography-free gram scale synthesis of DCAS photocatalyst. | ||
Intuitively, introduction of polar substituents improves solubility of compounds in polar aprotic solvents. Nitro- and sulfonic acid- groups are good choices for polyaromatic compounds as the synthetic process to access them is straightforward. Glöcklhofer and co-workers reported the synthesis of a dinitro derivative of DCA with improved solubility.38 On the other hand, sulfonic acids carry the advantage of further derivatization via their sulfonyl chlorides. Inspired by intermediates reported in the synthesis of a water-soluble DCA analogue,39 we began our catalyst synthesis (Fig. 3). Anthraquinone-2,6-disulfonic acid 4, commercially supplied or easily synthesized from cheap anthraquinone,40 was reduced by activated Zn in aq. (NH4)2CO3 to afford anthracene-2,6-disulfonic acid 5 in good (65%) yield after acidic workup and recrystallisation from aq. KCl. Electrophilic bromination of the central ring of 5 gave 6 in high (80%) yield. At this stage, our synthesis deviated from the literature cyanation which digested the crude product (containing CuCN) in conc. HNO3 and liberated toxic HCN gas. However, both Rosenmund von-Braun and Pd-catalysed cyanations failed to cyanate 6 due to its poor solubility in organic solvents. Tohnai and co-workers had reported that the derivatization of anthraquinone disulfonic acids (ADS) as their organic ammonium salts (i.e., n-heptyl and n-pentyl) prevented π-stacking interactions of ADS as observed by crystallography.41,42 Instead of ammonium salts which would hinder characterization and reaction workup, we achieved this covalently with sulfonamides.
Therefore, 6 was derivatized to increase its solubility in polar aprotic organic solvents and to increase prospects for successful cyanation. Chlorination of 6 with POCl3 and subsequent trapping of 7 with secondary amines of various chain lengths gave 9,10-dibromoanthracene-2,6-disulfonamides (DBAS) 8a, 8b and 8c in 87, 90 and 74% yields, respectively. Pleasingly, Rosenmund von-Braun cyanations under microwave-assisted (15 min) or thermal (see ESI†) heating afforded 9,10-dicyanoanthracene-2,6-disulfonamides 9a, 9b (DCAS) and 9c as ‘brilliant yellow’ solids in 66%, 89% and 26% yields, respectively. We note that our entire synthesis to 9 is carried out on gram scale, with straightforward purification via recrystallisation instead of chromatography. Photocatalyst 9b (henceforth coined ‘DCAS’) was progressed to evaluation in reactions since it: (i) displayed the highest solubility in MeCN (1.900 ± 0.100 mg mL−1vs. 0.340 ± 0.006 mg mL−1 for DCA) consistent with its calculated physical property values (which showed that it was the least lipophilic and had the highest topological polar surface area, see ESI†),43 and (ii) was obtained in the highest overall yield (42% over 5 steps).
Studies using a homogeneous liquid flow photoreactor
Next, DCAS was tested under some initial photocatalytic flow conditions (Table 1) in a commercial tubular coil continuous flow photoreactor (Vapourtec Ltd R-series/UV-150). Using 1a (12 mM) as our substrate and 5 mol% of DCAS at rt, a maximum yield of 25% for 2a (with 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of 2a:3a selectivity) was obtained under recycling conditions (90 min) no matter whether dry air, O2, or (1:1) N2/O2 were used (entry 2). The absence of catalyst (entry 2) or O2 led to no reaction. We found out that in the absence of LiClO4, single pass conditions gave a similar yield (25%) and with much improved selectivity for 2a (entry 4, 3a was not detected). When the temperature was increased to 40 °C, the yield improved to 40% (entry 5). Under similar conditions but employing DCA as catalyst afforded 2a in 15% yield, confirming superiority of DCAS under flow conditions. A batch reaction mimicking Santamaria and co-workers' condition (entry 8) afforded a complex reaction mixture (see ESI†). Our previously reported batch anaerobic conditions for SET oxidation of N-alkyl tetrahydroisoquinolines with [Ru(bpy)3]2+ photocatalysis11b in batch (entry 7) gave no reaction, and when the more potent photooxidizing SET catalyst [Ru(bpz)3]2+ was used only traces of 3a were observed. As such and due to cost of the catalysts, we did not examine these any further in flow.
:1 of 2a:3a selectivity) was obtained under recycling conditions (90 min) no matter whether dry air, O2, or (1:1) N2/O2 were used (entry 2). The absence of catalyst (entry 2) or O2 led to no reaction. We found out that in the absence of LiClO4, single pass conditions gave a similar yield (25%) and with much improved selectivity for 2a (entry 4, 3a was not detected). When the temperature was increased to 40 °C, the yield improved to 40% (entry 5). Under similar conditions but employing DCA as catalyst afforded 2a in 15% yield, confirming superiority of DCAS under flow conditions. A batch reaction mimicking Santamaria and co-workers' condition (entry 8) afforded a complex reaction mixture (see ESI†). Our previously reported batch anaerobic conditions for SET oxidation of N-alkyl tetrahydroisoquinolines with [Ru(bpy)3]2+ photocatalysis11b in batch (entry 7) gave no reaction, and when the more potent photooxidizing SET catalyst [Ru(bpz)3]2+ was used only traces of 3a were observed. As such and due to cost of the catalysts, we did not examine these any further in flow.
|
|
||||
|---|---|---|---|---|
| Entry | PC | Deviation from condition |
2a:3aa |
% Yield of 2aa |
| a Selectivity and yields determined by 1H NMR of the crude reaction mixture using 1,3,5-trimethoxybenzene (TMB) as internal standard. b Single pass. c T = 40 °C. d For exact details of conditions including terminal oxidants attempted, see ESI. e 1 mol% of Ru(bpy)3Cl2 or Ru(bpz)3(PF6)2. PC = photocatalyst, n.r. = no reaction, n.d. = not detected, c.r.m. = complex reaction mixture. | ||||
| 1 | DCAS | Dry air, O2 or N2/O2 (1:1) |
4:1 |
25 |
| 2 | — | Dry air | n.r. | n.d. |
| 3 | DCAS | N2 | n.r. | n.d. |
| 4b | DCAS | O2, RT = 20 min, no LiClO4 | >30:1 |
25 |
| 5b,c | DCAS | O2, RT = 20 min, no LiClO4 | >30:1 |
40 |
| 6b,c | DCA | O2, RT = 20 min, no LiClO4 | >30:1 |
15 |
| 7d | DCA | Batch, hν > 420 nm | — | c.r.m. |
| 8d | [Ru] | Batch, 459 nm | — | Traces 3a |
When under N2 protection (entry 3), a purple coloration in the post-reactor flowing reaction mixture was observed (see ESI†) which hinted at formation of DCAS˙−. We note that the related parent structure DCA˙− is well-known to be purple in color.44 When the purple post-reactor reaction mixture was collected and exposed to air, immediate discoloration back to yellow was observed. From these observations, we had initially assumed a reductive SET quenching of 1DCAS* by the amine, as originally proposed by Santamaria and co-workers (Fig. 2B).36,37 However, this was later refuted (see the Mechanistic studies for details).
Based on this mechanistic assumption, we reasoned that formation of 2a reached its upper limit due to limiting oxygen solubility at ambient conditions in the tubular reactor, preventing catalyst turnover. The solubility of O2 in an O2-saturated solution of MeCN is 8.1 mM,45 and considering the theoretical requirement of 2 equiv. O2 to remove 2 electrons from the trialkylamine, mass transfer limits full conversion of a reaction mixture containing 12.0 mM trialkylamine (later in the revised mechanism, we find that [O2] is still a limiting factor for the reaction yield).
Studies using a gas–liquid flow photoreactor
Considering the abovementioned observations, we opted for a photoreactor designed for biphasic gas–liquid reactions. A commercial microfluidic continuous flow photoreactor (Corning Lab Photoreactor©) designed for excellent mixing via turbulent slug flow allowed us to safely operate up to 60 °C and 8 bar backpressures. The hazard of the flammable reaction mixture was safely contained by the thermal isolation of the flow path and the small volume of reaction mixture (2.7 mL) at any given time. A summary of reaction condition optimization is shown in Table 2 (see ESI† for full optimization). Transferring conditions from the previous tubular reactor (Table 1, entry 5), 2a was afforded in 22% yield (Table 2, entry 1), as expected since the decreased yield exactly consists with (is proportional to) the decreased residence time (RT). However, the yield almost doubled when 395 nm LEDs were used (entry 2), which accorded with a higher extinction coefficient of DCAS's UV-vis band at ca. 395 nm compared to its 420 nm band (vide infra). At 24 mM 1a and double the residence time, the yield increased to 44% (entry 7). At 48 mM of 1a the yield decreased to 24% (entry 8), presumably again due to the limiting [O2]. At T = 60 °C and 24 mM 1a, the yield of 2a marginally improved to 46% (entry 9). The inherent back pressure on the flow by the microfluidic module was sufficient to ensure precise, reproducible, low flow rates (down to 0.1 mL min−1) up to 60 °C. To our delight, tropine 1b afforded 2b in 60% under reaction conditions at T = 40 °C and RT = 27 min (entry 10) despite its free 2° alcohol typically prone to oxidation under similar oxidative conditions.23–28,46 Decreasing catalyst loading decreased the yield (entries 11 and 12). Like the case of substrate 1a, a marginal increase of yield to 61% occurred at 60 °C (entry 13). At this stage, we explored the effect of a back pressure (8 bar) to evaluate higher O2 solubility (entries 14–16). At lower backpressures, the flow was heterogeneous slug flow but at 7–8 bar, homogenous flow was observed indicating full solubilization of O2 and higher dissolved [O2]. At 7–8 bar, doubling the concentration to 48 mM or using a residence time as short as RT = 6.8 min negatively impacted the yield of 2b (entries 14 and 15), but we found that yield (61%) was preserved at RT = 13.5 min (entry 16 vs. 13). This doubled productivity of 2b to 0.65 g per day which was the upper limit of the gas–liquid organophotocatalytic flow reaction in this system.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Amine | Conc. (mM) | R T (min) | hν (nm) | T (°C) | % Yieldd |
| a Unless otherwise stated, reaction conditions: DCAS (5 mol%), O2 (ambient pressure), at 40 °C. b T = 25 °C. c R T = residence time = (2.7 mL)/(flow rate). d Yield determined by 1H NMR using 1,3,5-TMB as internal standard. e T = 60 °C. f DCAS (3 mol%). g DCAS (1 mol%). h At 7–8 bar back pressure. MFC = mass flow controller (O2), BPR = back pressure regulator, n.r. = no reaction. | ||||||
| 1 | 1a | 12 | 13.5 | 420 | 40 | 22 (2a) |
| 2 | 1a | 12 | 13.5 | 395 | 40 | 40 (2a) |
| 3 | 1a | 12 | 5.4 | 395 | 40 | 12 (2a) |
| 4 | 1a | 12 | ∼1.0 | 395 | 40 | Trace (2a) |
| 5 | 1a | 12 | ∼1.0 | None | 40 | n.r. |
| 6 | 1a | 24 | 13.5 | 395 | 40 | 40 (2a) |
| 7 | 1a | 24 | 27.0 | 395 | 40 | 44 (2a) |
| 8 | 1a | 48 | 27.0 | 395 | 40 | 24 (2a) |
| 9e | 1a | 24 | 27.0 | 395 | 60 | 46 (2a) |
| 10 | 1b | 24 | 27.0 | 395 | 40 | 60 (2b) |
| 11f | 1b | 24 | 27.0 | 395 | 40 | 53 (2b) |
| 12g | 1b | 24 | 27.0 | 395 | 40 | 42 (2b) |
| 13e | 1b | 24 | 27.0 | 395 | 60 | 61 (2b) |
| 14e,h | 1b | 48 | 13.5 | 395 | 60 | 48 (2b) |
| 15e,h | 1b | 24 | 6.8 | 395 | 60 | 31 (2b) |
| 16e,h | 1b | 24 | 13.5 | 395 | 60 | 61 (2b) |
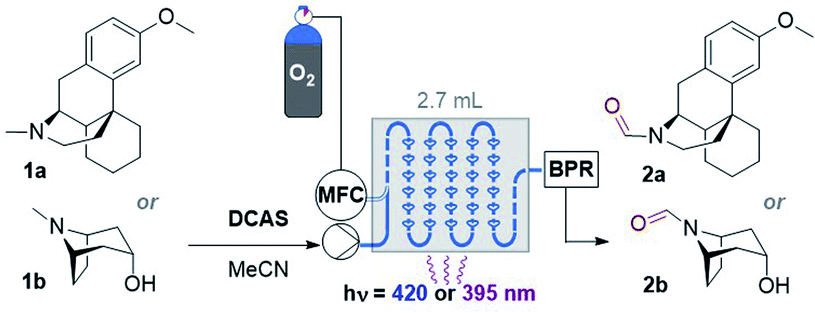
Next, we tested the scope of the reaction (Table 3). Since isolations of polar formamides were oftentimes challenging due to the N-formyl group being a weak chromophore, the following discussion deems 1H NMR yields more representative of reaction efficiency. Compounds 2c (59%) and 2d (67%) were obtained from natural products tropane and (free alcohol-bearing) atropine. Even scopolamine, which has a free alcohol, an ester, and an epoxide, afforded 2e in 62% yield with no nor-scopolamine detected, albeit requiring 2 passes through the reactor (total RT = 27 min). This contrasts with Santamaria and co-workers’ conditions using DCA and without LiClO4, which afforded a 1:1 mixture of 2e:nor-scopolamine.36,37 Compared to 2b, the yield of 2f was lower (34%) presumably due to the presence of the Si protecting group known to stabilize radicals and quench excited photosensitizers via different pathways.47 Benzoyl-containing compound 2g was afforded in good (73%) yield. Electron-poor (–CF3) and electron-rich (–OMe) substituents on the benzoyl group were tolerated equally, affording 2h (56%) and 2i (58%) respectively. We note both 2g and 2i are natural products; novel tropanoid compound 2g was recently isolated from Pellacalyx saccardianus and our method corroborated its proposed structure.48 Compound 2g (confoline) was isolated from Convolvulus subhirsutus and our method accessed it from convolvamine in a single step (in the literature, semi-synthesis of 2i was achieved by formylation of nor-convolvamine, hence a demethylation step from convolvamine culminates in a two-step process).17 Compounds 2j to 2o were obtained from piperazines as common API fragments (such as those present in sildenafil and danofloxaxin).49 Despite having 3 possible sites for functionalization (one exocyclic N–CH3 and two endocyclic N–CH2–R sites), selective oxidation at the N–CH3 (exo-:endo- = 5.7:1 for 2j, 3.4:1 for 2k, 3.7:1 for 2l, and 6.5:1 for 2p, see ESI†) was apparent, affording N-formyl compounds in respectable yields. Despite the modest yields of products 2l (55%), 2m (21%), and 2n (30%) (as well as 2h), we were surprised by the tolerance of halogen-bearing substrates under the reaction conditions. Especially, given the aforementioned putative presence of DCAS˙−via reductive quenching of 1DCAS* by trialkylamines (well known for DCA's case)50,51 and given that photoexcited radical anions are known to reductively cleave aryl halides and other strong bonds.50–53 C–F bonds and N–Ts groups are also prone to reductive cleavage under reductive photocatalysis54 or by photoexcited super electron donors.55
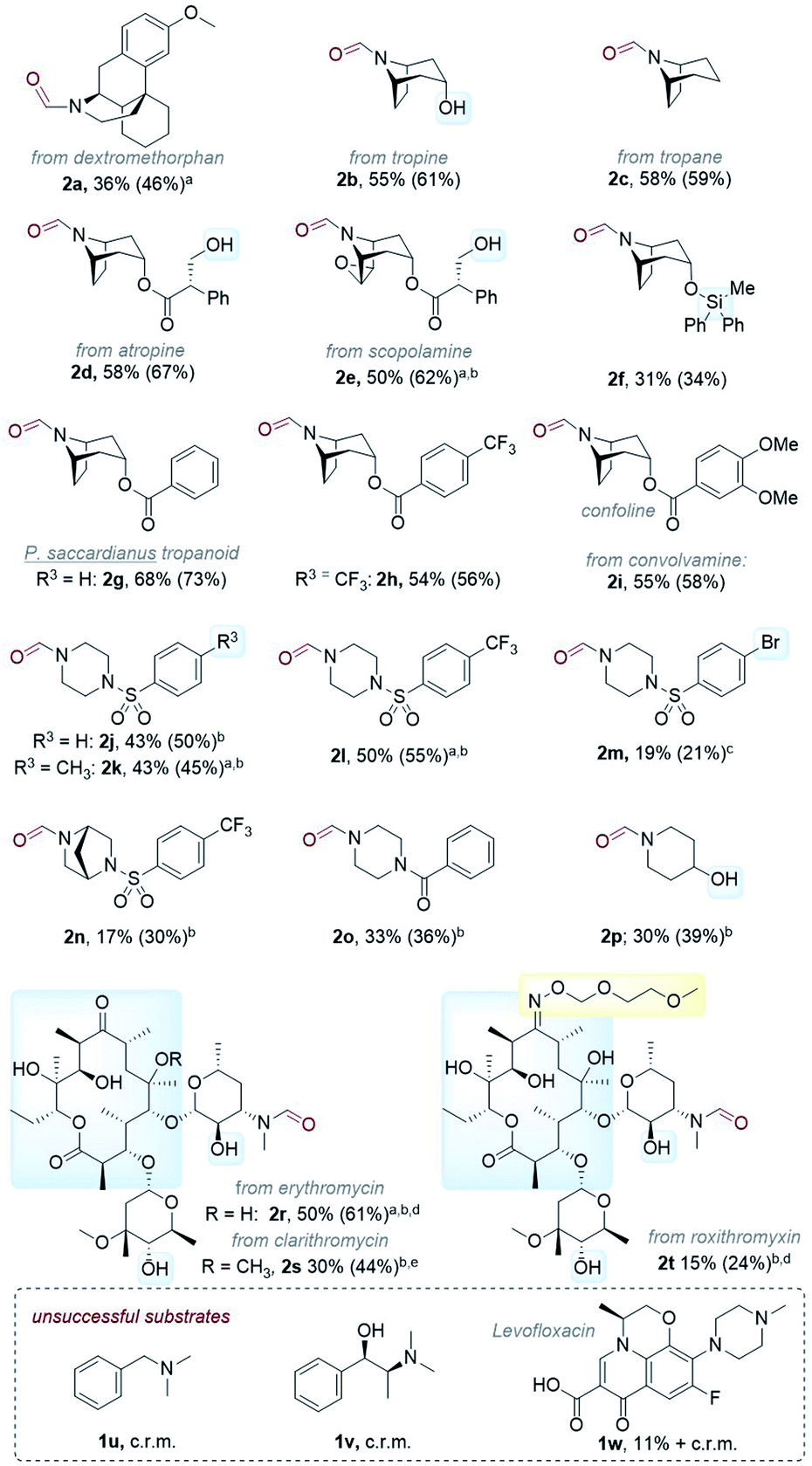
A simple piperidine 2p (39%) was also tolerated. Our success with 2b, 2d, 2e, and 2p whose precursors bore free alcohol groups encouraged us to explore more complex molecules. Gratifyingly, conditions were successfully applied to macrolide antibiotics with dense functionalities (free alcohols, an oxime ether, and a ketone). Erythromycin, clarithromycin and roxithromycin afforded 2r, 2s, and 2t in 61%, 44%, and 24% yields, respectively. However, benzylic amines and trialkylamines containing benzylic alcohols or free carboxylic acids such as 1u, 1v and 1w, were unsuccessful. Benzaldehyde formation (C–N cleavage, possibly via endocyclic iminium ion formation and then hydrolysis) and intractable complex reaction mixtures were observed for these substrates.
Mechanistic studies
Cyclic voltammetry (CV) revealed DCAS (E1/2[DCAS/DCAS˙−] = −0.59 V vs. SCE) is substantially easier to reduce than DCA (E1/2[DCA/DCA˙−] = −0.98 V vs. SCE), due to the electron-withdrawing sulfonamide groups at the 2,6-positions (Fig. 4, left). UV-vis absorption and emission spectra were measured for DCA and DCAS (Fig. 4, right) and their comparison revealed that the 2,6-sulfonamides hardly affect the absorptive or emissive profiles of the dicyanoanthracene core. In both cases, overlap of the longest wavelength absorption band (λmax = 422 nm) and shortest wavelength emission band (λmax = 435 nm) allows to approximate E0–0 for the singlet excited state (≈2.90 eV). Taking this value together with measured redox potentials, the photocatalyst excited state oxidation potentials were approximated by a derivative of the Rehm–Weller equation.561DCAS* (E1/2[1DCAS*/DCAS˙−] = +2.31 V vs. SCE) is a notably more potent photooxidant than 1DCA* (E1/2[1DCA*/DCA˙−] = +1.93 V vs. SCE). Our initial hypothesis thus continued to align with the SET mechanism proposed by Santamaria (Fig. 5).36,37 In this premise, 1DCAS* was assumed to behave like 1DCA* which underwent reductive quenching by trialkylamine 1, and oxidation of DCAS˙− by O2 regenerated DCAS. Deprotonation of radical cation 1′ and radical combination of the α-amino radical and superoxide would ultimately afford N-formyl product 2. We note SET reactions were also proposed as the main pathways for trialkylamine activations by thiazine and fluorescein organophotocatalysts, either via oxidation to N-oxides or via N-demethylations.57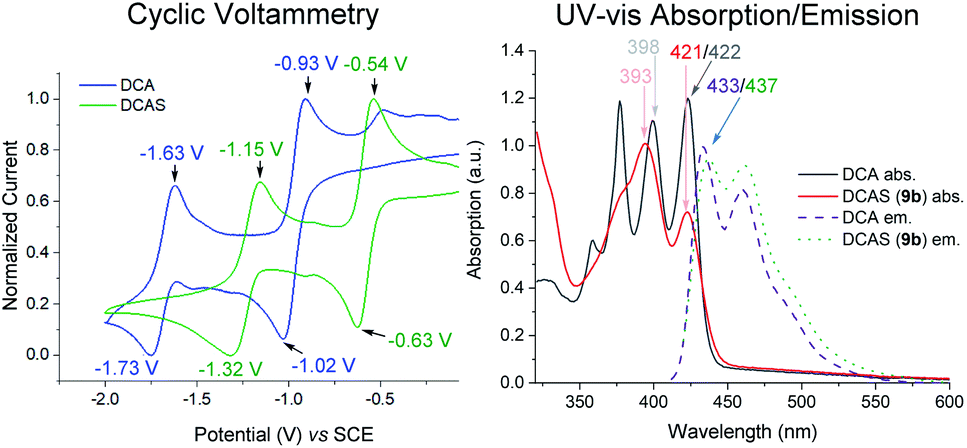 | ||
| Fig. 4 Left: cyclic voltammetry of catalysts. Conditions: 0.01 M DCA/DCAS in 0.1 M nBu4NPF6/MeCN, scan rate 50 mV s−1. Right: UV-vis and emission spectra. | ||
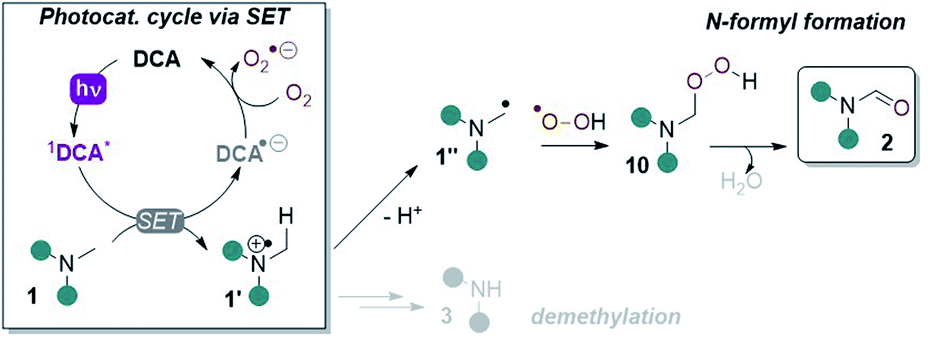 | ||
| Fig. 5 SET reductive quenching mechanism of DCA proposed by Santamaria and co-workers leading to demethylation and N-formyl products. | ||
To test this initial hypothesis, two control batch experiments with stoichiometric (2.0 equiv.) DCA and DCAS were conducted under strict N2 protection in PhCN solvent to promote solubility. Both afforded clean conversion of 1a to a 1:1 mixture of 1a:3a (Fig. 6A), although DCA's reaction required >3.5× reaction time due to poorer solubility. Upon irradiation, the reaction mixtures changed from a pale yellow color to dark purple (Fig. 6B). Removal the light and exposing to air, the colors of reaction solutions quickly reverted to yellow (consistent with aforementioned observations of the flow reaction under N2). The UV-vis spectra of DCA˙− is well studied in the literature,51,53 and it is known to be purple in color.44 We confirmed the presence of DCAS˙− spectroscopically by matching the spectra of a sample of DCAS treated by cathodic electrolysis to that treated photochemically in the presence of a trialkylamine reductive quencher (see ESI† for details). Both gave a new, broad absorption spectrum at the visible-green region (λmax ≈ 544 nm, Fig. 6C), thus an apparent purple color.
 | ||
| Fig. 6 Experiments suggesting the generation of cyanoanthracene radical anions. (A) aDue to stoichiometric PC loadings, PhCN was chosen as solvent for improved solubility. bIn the absence of additional base, 1a deprotonates 1a˙+ to afford 1a′′, meaning the reaction fundamentally could never exceed 50% conversion. (B) Observed color changes. (C) cIn MeCN, λmax at the visible region when dgenerated by photochemical reductive quenching of 1DCAS* with a trialkylamine; egenerated electrochemically; or fcalculated using TD-DFT at CAM-B3LYP/6-31++g(2d,p), CPCM(MeCN) level of theory (for details, see ESI†). | ||
A Time-Dependent Density Functional Theory (TD-DFT) calculation of the UV-vis transitions of DCAS˙−consisted with this green absorption peak (λmax = 547 nm). The detection of these radical anions together with N-demethylation reaction confirmed that the SET oxidation of 1 to 1′ by the organophotocatalysts was possible, at least under anaerobic conditions. Considering the preceding discussion supportive of an SET mechanism in Fig. 5 and catalysts' redox potentials, one would expect 1DCAS* to undergo more rapid fluorescence quenching than 1DCA* by trialkylamines. Very surprisingly, the opposite was clearly true (Fig. 7). The Stern–Volmer rate constant for quenching of 1DCAS* by 1a (kq = 1.44 × 109 M−1 s−1) was two orders of magnitude smaller than that of 1DCA* (kq = 1.69 × 1011 M−1 s−1).58,59 Presumably, either (i) the 2-methoxyethyl groups of DCAS affects the kinetics of bimolecular quenching by sterically obstructing the approach of trialkylamine, or (ii) aggregation of DCA accelerates reductive quenching by trialkylamines60 where DCAS exhibits a different kind of aggregation in solution.61
 | ||
| Fig. 7 Stern–Volmer quenching experiments of DCA (right) and DCAS (left) with various amine quenchers (under N2) or O2. | ||
To probe the mechanistic role of the structural changes on the catalyst, and to rationalize the unexpected trend between the order of redox potentials of 1DCA* and 1DCAS* vs. their fluorescence quenching rates, DFT and TD-DFT calculations were performed. The activation energies (ΔG‡SET) for the photoinduced single electron transfer (SET) from 1b to photoexcited dicyanoanthracenes were determined using Marcus theory (Table 4). Aside from free energy (ΔGSET), another key parameter of Marcus theory is the reorganization energy (λ) which accounts for the properties of the solvent, the size of, and the distance between reacting species. The calculated vertical excitation energies (ΔGEx ≈ 3.3 eV) were close to E0–0 value obtained from optical spectroscopy (vide supra). As expected from the experimentally-determined photocatalyst redox potentials, SET of 1b with 1DCAS* is 1.3× more exergonic (ΔGSET = −49.9 kcal mol−1) than with 1DCA* (ΔGSET = −37.3 kcal mol−1). However, the kinetic barrier is notably (3×) higher for 1DCAS* (ΔG‡SET = 13.7 kcal mol−1) than 1DCA* (ΔG‡SET = 4.4 kcal mol−1). This agreed with the relatively slower fluorescence quenching of 1DCAS* by trialkylamines. However, this is juxtaposed with the greater synthetic efficiency of the reaction catalysed by DCAS compared to DCA. Taken together, these results show that although SET between excited cyanoanthracenes and trialkylamines can occur under anaerobic conditions, an alternative mechanism must operate for DCAS under aerobic conditions in order for it to deliver higher synthetic efficiencies.
|
|
||||
|---|---|---|---|---|
| 1 PC* | k q | ΔGExc | ΔGSETd | ΔG‡SETe |
| a Geometry optimization, molecular radius (r) and free energies calculated using DFT (ground state) or TD-DFT (excited state) at CAM-B3LYP (or ωB97X-D in parentheses)/6-31++g(2d,p), CPCM(acetonitrile) level of theory (see ESI). b From Stern–Volmer analyses (Fig. 7), in M−1 s−1. c Vertical excitation energy. d Photoinduced-SET free energy. e Photoinduced-SET activation energy. All free energy units in kcal mol−1. PC = photocatalyst. For further details, see ESI. | ||||
| DCA | 1.69 × 1011 | 75.4 (75.9) | −37.3 (−37.7) | 4.4 (5.2) |
| DCAS | 1.44 × 109 | 77.2 (76.8) | −49.9 (−47.7) | 13.7 (13.4) |
| DCAS/DCA | ∼0.01 | ∼1 (∼1) | 1.3 (1.3) | 3.1 (2.6) |

Elsewhere, 1DCA* is also known as an efficient singlet oxygen sensitizer (kq = 4.3 × 109 M−1 s−1) via a photosensitized energy transfer (EnT) mechanism.62 The high reported quantum yield (reaching 2.0) supports the generation of 2× 1O2 molecules per 1× 1DCA*.62 This quenching rate constant of 1DCA* by EnT is more than double that of the reductive SET quenching of 1DCAS* by trialkylamines. Thus, as [O2] increases and approaches that of the trialkylamine ([O2] ≈ [trialkylamine]), singlet oxygen generation dominates in the case of 1DCAS*. This consists with the increase in yields observed at higher back pressures, temperatures and thus higher dissolved [O2]. The kq for quenching of 1DCAS* by O2 was slightly higher (4.89 × 109 M−1 s−1Fig. 7, left) than that reported for 1DCA*.62a Taken together with the fluorescence quenching rates (kq) with trialkylamines, this points to a photochemical mechanistic switchover: 1DCA* is quenched faster (39×) by 1a than O2, while 1DCAS* is quenched faster (3×) by O2 than 1a. Thus, under aerobic reaction conditions, 1DCA* favors an SET mechanism while 1DCAS* favors an EnT mechanism.
We then studied the behaviour of the excited cyanoanthracenes under the aerobic reaction conditions (i.e., catalyst, trialkylamine and O2 (from air) are all present). This was done by comparing the relative intensity change of light (420 nm) transmitted through the coil of the tubular flow reactor. The principle is as follows: the faster the excited state catalyst can relax to the ground state, the greater the steady-state population of ground state photocatalyst is, leading to more absorption of light (therefore less transmission). The aerated, flowing reaction mixture of DCA (5 mol%) + 1a (12 mM), under the conditions of Table 1, entry 6, gave minimal light absorption (Fig. 8A). As discussed earlier, the reductive quenching of 1DCA* by 1a is even faster than quenching by O2 and does not directly afford DCA but affords DCA˙− whose absorption51,53 is shifted far into the visible green region and thus is not detected by the probe. Regeneration of the ground-state catalyst relies on the oxidation of DCA˙− by O2, which is comparatively slow. In the absence of 1a, the aerated solution of DCA (Fig. 8B) gave strong light absorption (decrease of transmitted light intensity to roughly half). O2 no longer competes with 1a and is now the exclusive quencher, regenerating and sustaining a large steady-state concentration of absorbing DCAvia rapid EnT quenching of 1DCA*. In contrast, the reaction mixture (Table 1, entry 4) of DCAS (5 mol%) in MeCN gave notable light absorption even when 1a (12 mM) was present (Fig. 8C), since quenching of 1DCAS* by O2 now outcompetes reductive quenching by 1a, ensuring a larger steady-state concentration of absorbing DCAS. This agrees with the aforementioned differences in quenching rate constants. Finally, the light absorption of an aerated solution of DCAS in the absence of 1a (Fig. 8D) was greater in the absence of competing 1a, and was more pronounced than in the case of DCA (Fig. 8B). This reflects the enhanced fluorescence quenching of the former with O2 (for light transmission measurements under N2 or with 380 nm, see ESI†).
 | ||
| Fig. 8 Transmission intensity of light through the tubular reactor. (A) DCA (5 mol%) + 1a (12 mM) in aerated MeCN; (B) DCA only in aerated MeCN; (C) DCAS (5 mol%) + 1a (12 mM) in aerated MeCN; (D) DCAS only in MeCN. Before/After = light transmission before or after the reaction mixture passed through the tubular coil reactor, i.e. flowing MeCN only. During = light transmission while the reaction mixture slug is flowing through the coil and upon reaching a steady value. | ||
The lifetimes of 1DCA* and 1DCAS* as measured by Time-correlated Single Photon Counting (TSCPC) in MeCN under Ar were similar, at 14.5 and 13.8 ns, respectively (Table 5).
The lifetime of 1DCA* was 1.8 ns lower in presence of air, while the lifetime of 1DCAS* was 4.7 ns lower, confirming the slight enhancement of quenching by O2 (and consistent with the Stern–Volmer kqs of 1DCAS* and 1DCA*, vide supra). Further experiments supported the photosensitized EnT quenching of 1DCAS* as the dominant mechanism, rather than photoinduced SET to afford O2˙− (Fig. 9A and B). Firstly, when α-terpinene was employed as the substrate, ascaridole was formed in 65% yield as quantified by 1H NMR. Endoperoxide formation is a hallmark reporter for 1O2 through its Diels–Alder [4 + 2]-cycloaddition with dienes (Fig. 9A, left),63 thus confirming 1DCAS* is capable of 1O2 generation. Secondly, the presence of DABCO as an additive inhibited conversion in 1b's reaction (Fig. 9B, right). Despite DABCO's low oxidation potential (Epox = +0.66 V),13 this inhibition was not due to its competitive SET reductive quenching of 1DCAS*, since the quenching rate constant (kq = 7.28 × 108 M−1 s−1) confirmed it was even less efficient as a quencher of 1DCAS* than O2 or 1a/1b (Fig. 7). Rather, DABCO is a well-known physical quencher of 1O2.64,65 This was confirmed by a linear correlation (R2 = 0.997) between the reciprocal relative rate and [DABCO], an experiment designed by Lapi and co-workers. Finally, as proof of the direct fixation of oxygen atoms from O2 gas into trialkylamines, 18O-2b was detected by HRMS when isotopically-enriched oxygen (18O2) gas was employed in the batch reaction of 1b (Fig. 9B).
 | ||
| Fig. 9 Experiments evidencing (i) an 1O2 mechanism, (ii) fixation of oxygen atoms from O2 and (iii) breakup of π-stacking for DCAS. (A) Left: 1O2 trapping via [4 + 2] cycloaddition. Reaction conditions: α-terpinene (12 mM) DCAS (5 mol%), O2 (8 bar), RT = 13.5 min, hν = 395 nm; right: effect of increasing [DABCO] on the (relative rate)−1 of N–CH31b oxidation. Reaction conditions: 1b (12 mM), DABCO (0 to 9 mM), DCAS (5 mol%), O2 (8 bar), RT = 13.5 min, hν = 395 nm. Relative rate = (yield of 2b)/(yield of 2b with DABCO). (B) Isotope labeling batch reaction with 18O2 gas. (C) XRD crystal structure of 2 molecules of DCAS with the distance between the anthracene cores. Thermal ellipsoids are set at the 50% probability level. H atoms are omitted for clarity, C atoms (grey), N atoms (blue), O atoms (red) S atoms (yellow). | ||
In summary, increased efficiency of DCAS over DCA in the reaction is not only attributed to the former's enhanced solubility. The sulfonamide substituents at the 2,6-positions of the dicyanoanthracene markedly decrease the reductive quenching of 1DCAS* by trialkylamines, compared to that of 1DCA*. This observation may be explained by a change in the aggregation state of the organophotocatalyst,61 where the ordered π-stacking of DCA aggregates creates a large effective volume for collisions with amines, while DCAS behaves differently. The distance of π-sandwich planes for DCA = 3.37 Å and the usual range for 2 interacting planes is 3.3 to 3.8 Å.66 In the X-ray diffraction (XRD) structure of DCAS (Fig. 9C), the distance between π-planes of anthracene = 13.60 Å and considering that 2r = distance between molecules, this value agrees with 2× the calculated spherical radii of DCAS in MeCN (Table 4). From this, we tentatively propose that the bulky, freely-rotating sulfonamide substituents sterically inhibit bimolecular (or unimolecular)59 quenching events with trialkylamines. The smaller O2 molecules outcompete larger trialkylamines to reach the cyanoanthracene core, diverting the mechanism to 1O2 sensitization. This consists with the need for constrained trialkylamine substrates with protruding N–CH3 groups herein, and may rationalize DABCO's inefficiency as a reductive quencher on steric grounds.13 A similar “steric-bulk” strategy was recently employed using tert-butyl substituents to prevent an unproductive EDA complexation in a catalytic reaction.67
In light of all the above, we propose the following mechanism (Fig. 10). Photoexcitation of DCAS affords 1DCAS* which undergoes EnT with 3O2. The generated 1O2 interacts with the trialkylamine via a well-studied exciplex,62,64,68,69 which can undergo one of two pathways: SET or HAT. Redox potentials dictate SET between trialkylamines (Epox > +0.9 V vs. SCE)13 and 1O2 (Epred > +0.1 V vs. SCE)68 is endergonic, consistent with our DFT calculations of an endergonic free energy (ΔG = 6.0 kcal mol−1). Thus, we deemed SET within the exciplex as the minor pathway. Conversely, HAT within the exciplex was slightly exergonic (ΔG = −0.1 kcal mol−1) suggesting this is the major pathway. Combination of 1′′ with proximally-generated peroxyl radical affords 10 (which could also be accessed by SET oxidation of α-amino radical 1′′ by 1O2 followed by combination of 11 with O2˙− and subsequent protonation is also possible). Finally, liberation of H2O from 10 affords 2 and DCAS is regenerated by the reported triplet–triplet annihilation of 3DCAS* with a second molecule of 3O2.62
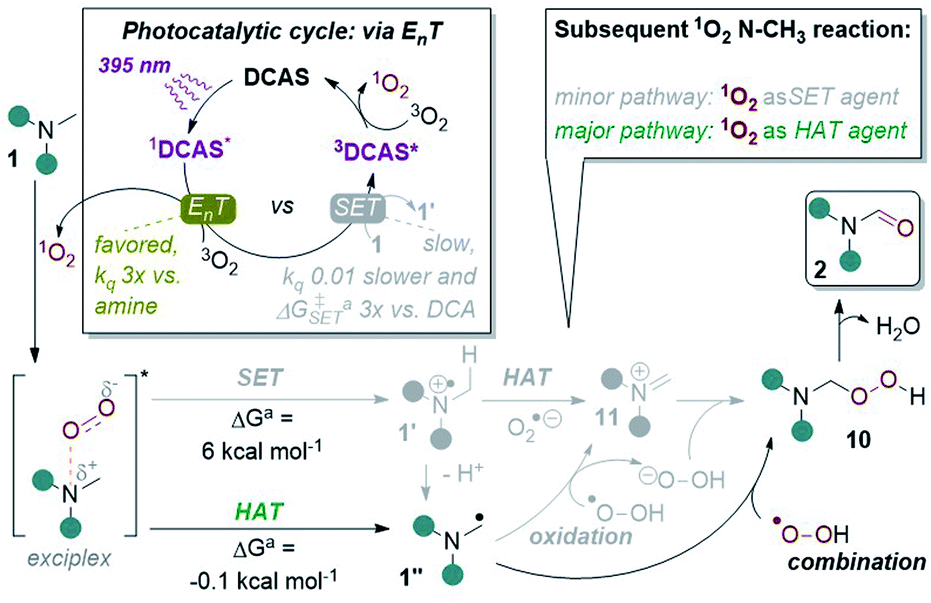 | ||
| Fig. 10 Proposed reaction mechanism with key experimental and computational evidence. aDFT calculations at CAM-B3LYP/6-31++g(2d,p), CPCM(acetonitrile) level of theory. | ||
In a recent study by Rovis, Schoenebeck and co-workers on the photocatalytic functionalizations of cyclic trialkylamines,14e they proposed that a reversible and fast HAT is responsible for their endocyclic selectivity. Our computational studies point to a rapid, irreversible HAT in the 1O2-trialkylamine exciplex, thus steric factors must govern the selectivity (i.e. at the less sterically demanding N–CH3 position). In the case of less-constrained trialkylamines (1u, 1v), the 1O2-bound exciplex can react promiscuously in HAT with endocyclic/non-N–CH3 positions (e.g. benzylic groups, free alcohols) leading overall to degradation.
Conclusions
Herein, we report DCAS as a new organophotocatalyst for late-stage N–CH3 to N-formyl oxidations of complex trialkylamine-containing natural products and pharmaceuticals, using molecular oxygen and continuous flow. Redox sensitive functionalities were tolerated, allowing the LSF post-modification of alkaloids and macrolide antibiotics to their N-formyl derivatives in good yields with excellent chemo- and regioselectivities, all in a continuous manner. The safe handling of O2 under increased back pressures and temperatures via gas–liquid continuous flow in turn promoted mass transfer of O2 to the reaction, increasing yields, shortening reaction (residence) times to several minutes and unleashing synthetically useful productivities (0.65 g per day). Mechanistic insights demonstrate how seemingly minor structural variations in an organophotocatalyst can not only increase solubility, but profoundly divert the excited state mechanism from photoinduced SET to EnT, followed by a downstream HAT mechanism. Precious metal photocatalysts of Ru- and Ir-based polypyridyl complexes are well known to participate in both EnT and SET, where structural tuning of ligands can affect switching between the divergent pathways. To our knowledge, such a concept has rarely been exploited in organophotocatalysis on the same core, privileged organophotocatalyst structures are typically developed either for SET or EnT pathways. Switching the mechanism offers opportunities to control selectivity, as indicated by the tolerance of reductively-labile groups herein. With the generation of 1O2 revealed, our study showcases one of few successful applications of 1O2 as a reagent in complex natural product synthesis.31c,70 Further investigations on the selectivity of 1O2's reactions with trialkylamines and the nature of interactions between DCAS, O2 and trialkylamine quenchers are ongoing.71Data availability
Respectfully, all experimental and computational data is adequately available and retreivable from the ESI file.†Author contributions
M. J. P. M. contributed the major effort on the optimization of the organophotocatalytic reaction in flow (microfluidic reactor), synthesized substrates, synthesized and purified all products and performed experimental mechanistic studies and computation; J. Ž developed an efficient synthetic route to new catalyst DCAS and synthesized gram quantities for the study; C. I. M. contributed preliminary studies on the optimization of the organophotocatalytic reaction in flow (tubular reactor); L. J. E. supervised and guided J. P. B. and C. I. M. in preliminary studies, measured light source emissions and transmission spectroscopy; A. S. undertook Stern–Volmer quenching studies and measured UV-vis spectroscopy of radical anion catalyst forms; L. d'H. contributed to the synthesis of complex trialkylamine substrates. P. Y. measured catalyst lifetimes and steady state emission spectroscopy. D. J. S. B. supervised and guided P. Y.; T. G. contributed to the separation of highly polar products by preparative HPLC; J. H. supervised and guided T. G.; M. P. J. supervised and guided J. P. B. in preliminary studies; J. P. B. conceptualized the project, conducted first investigations of photocatalytic trialkylamine activations, first synthesized new catalyst DCAS, measured cyclic voltammetry and UV-vis spectroscopy, guided and supervised C. I. M., M. J. P. M., J. Ž, A. S. and L. d'H. in their contributions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. J. P. M., J. Ž., A. S., L. d'H. and J. P. B. thank the Alexander von Humboldt Foundation, within the framework of a Sofja Kovalevskaja Award endowed by the German Federal Ministry of Education and Research, for funding the main investigation. C. I. M., L. J. E., M. P. J. and J. P. B. thank GlaxoSmithKline for funding preliminary investigations. P. Y. and D. J. S. B. thank the Scottish Funding Council for a SUPA INSPIRE research studentship. The authors thank Prof. Dr Robert Wolf's group for access to a Glovebox; Katrin Kuck and Mirjam Abu Salah for technical assistance on preparative HPLC; Prof. Dr Oliver Reiser's group and Dr Christian Fischer for providing the resources to generate 18O2 gas; and Prof. Dr Julia Rehbein for advice on DFT computations. We thank Regina Hoheisel for support in online irradiation/UV-vis measurements. The authors thank Philippe M. C. Roth and Marc Winter (Corning) for technical support. This paper is dedicated to the memory of Matthew P. John.Notes and references
- M. Moir, J. J. Danon, T. A. Reekie and M. Kassiou, Expert Opin. Drug Discovery, 2019, 14, 1137–1149 CrossRef CAS PubMed.
- G. Wu, T. Zhao, D. Kang, J. Zhang, Y. Song, V. Namasivayam, J. Kongsted, C. Pannecouque, E. D. Clercq, V. Poongavanam, X. Liu and P. Zhan, J. Med. Chem., 2019, 62, 9375–9414 CrossRef CAS PubMed.
- (a) L. Guillemard, N. Kaplaneris, L. Ackermann and M. J. Johansson, Nat. Rev. Chem., 2021, 5, 522–545 CrossRef CAS; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC; (c) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed.
- (a) J. P. Barham, S. Tamaoki, H. Egami, N. Ohneda, T. Okamoto, H. Odajima and Y. Hamashima, Org. Biomol. Chem., 2018, 16, 7568–7573 RSC; (b) J. P. Barham, T. N. J. Fouquet and Y. Norikane, Org. Biomol. Chem., 2020, 18, 2063–2075 RSC; (c) M. J. P. Mandigma, M. Domański and J. P. Barham, Org. Biomol. Chem., 2020, 18, 7697–7723 RSC.
- (a) J. Kim, J. A. Ashenhurst and M. Movassaghi, Science, 2009, 324, 238–241 CrossRef CAS PubMed; (b) J. Kaur, A. Shahin and J. P. Barham, Org. Lett., 2021, 23, 2002–2006 CrossRef CAS PubMed; (c) J. Kim and M. Movassaghi, Acc. Chem. Res., 2015, 48, 1159–1171 CrossRef CAS PubMed; (d) S. Das, K. Murugesan, G. J. V. Rodríguez, J. Kaur, J. P. Barham, A. Savateev, M. Antonietti and B. König, ACS Catal., 2021, 11, 1593–1603 CrossRef CAS; (e) B. S. Bhakuni, A. Yadav, S. Kumar, S. Patel, S. Sharma and S. Kumar, J. Org. Chem., 2014, 79, 2944–2954 CrossRef CAS PubMed.
- K. L. Kohnen-Johannsen and O. Kayser, Molecules, 2019, 24, 796 CrossRef PubMed.
- (a) P. Le Couteur and J. Burreson, Napoleon's buttons. 17 molecules that changed history, Penguin, New York, 2004 Search PubMed; (b) Macrolide antibiotics: chemistry, biology, and practice, ed. S. Omura, Academic Press, Amsterdam, Boston, 2nd edn, 2002 Search PubMed; (c) Dextromethorphan: pharmacology, clinical uses and health effects, ed.X. Wang, Nova Biomedical, New York, 2016 Search PubMed; (d) WHO, Annex 1 19th WHO Model List of Essential Medicines, WHO, Geneva, Switzerland, 2015 Search PubMed.
- (a) A. P. Feinberg, I. Creese and S. H. Snyder, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 4215–4219 CrossRef CAS PubMed; (b) M. Spetea and H. Schmidhammer, Curr. Med. Chem., 2012, 19, 2442–2457 CrossRef CAS PubMed; (c) T. Kaserer, A. Lantero, H. Schmidhammer, M. Spetea and D. Schuster, Sci. Rep., 2016, 6, 1–15 CrossRef PubMed; (d) E. Prommer, Support. Care Cancer, 2006, 14, 109–115 CrossRef PubMed; (e) T. R. Deer, M. S. Leong, A. Buvanendran, V. Gordin, P. S. Kim, S. J. Panchal and A. L. Ray, Comprehensive Treatment of Chronic Pain by Medical, Interventional, and Integrative Approaches, Springer, New York, 1st edn, 2013 CrossRef.
- (a) Z. Dong and P. J. Scammells, J. Org. Chem., 2007, 72, 9881–9885 CrossRef CAS PubMed; (b) G. B. Kok, C. C. Pye, R. D. Singer and P. J. Scammells, J. Org. Chem., 2010, 75, 4806–4811 CrossRef CAS PubMed; (c) A. Mary, D. Z. Renko, C. Guillou and C. Thal, Tetrahedron Lett., 1997, 38, 5151–5152 CrossRef CAS; (d) K. McCamley, J. A. Ripper, R. D. Singer and P. J. Scammells, J. Org. Chem., 2003, 68, 9847–9850 CrossRef CAS PubMed; (e) R. A. Olofson, R. C. Schnur, L. Bunes and J. P. Pepe, Tetrahedron Lett., 1977, 18, 1567–1570 CrossRef; (f) L. Werner, A. Machara, D. R. Adams, D. P. Cox and T. Hudlicky, J. Org. Chem., 2011, 76, 4628–4634 CrossRef CAS PubMed; (g) W. Shu, A. Genoux, Z. Li and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 10521–10524 CrossRef CAS PubMed.
- C. R. J. Stephenson and T. P. Yoon, Visible light photocatalysis in organic chemistry, Wiley-VCH, Weinheim, 1st edn, 2018 Search PubMed.
- Selected examples: (a) G. Bergonzini, C. S. Schindler, C. J. Wallentin, E. N. Jacobsen and C. R. J. Stephenson, Chem. Sci., 2013, 5, 112–116 RSC; (b) J. P. Barham, M. P. John and J. A. Murphy, Beilstein J. Org. Chem., 2014, 10, 2981–2988 CrossRef PubMed ; for a representative review, see:; (c) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- (a) A. S. H. Ryder, W. B. Cunningham, G. Ballantyne, T. Mules, A. G. Kinsella, J. Turner-Dore, C. M. Alder, L. J. Edwards, B. S. J. McKay, M. N. Grayson and A. J. Cresswell, Angew. Chem., Int. Ed., 2020, 59, 14986–14991 CrossRef CAS PubMed; (b) H. E. Askey, J. D. Grayson, J. D. Tibbetts, J. C. Turner-Dore, J. M. Holmes, G. Kociok-Kohn, G. L. Wrigley and A. J. Cresswell, J. Am. Chem. Soc., 2021, 143, 15936–15945 CrossRef CAS PubMed; (c) J. H. Blackwell, G. R. Harris, M. A. Smith and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 15946–15959 CrossRef PubMed.
- J. P. Barham, M. P. John and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 15482–15487 CrossRef CAS PubMed.
- (a) J. He, L. G. Hamann, H. M. L. Davies and R. E. J. Beckwith, Nat. Commun., 2015, 6, 1–9 Search PubMed; (b) J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046–6050 CrossRef CAS PubMed; (c) T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC; (d) Y. Shen and T. Rovis, J. Am. Chem. Soc., 2021, 143, 16364–16369 CrossRef CAS PubMed; (e) Y. Shen, I. Funez-Ardoiz, F. Schoenebeck and T. Rovis, J. Am. Chem. Soc., 2021, 143, 18952–18959 CrossRef CAS PubMed .; for a review, see:; (f) J. Kaur and J. P. Barham, Synthesis, 2021 DOI:10.1055/a-1677-6619.
- (a) W. Ahmad, M. Z. Rehan, A. Gupta and J. Iqbal, J. Saudi Chem. Soc., 2016, 20, 543–546 CrossRef CAS; (b) C. Köppel and J. Tenczer, Biomed. Mass Spectrom., 1985, 12, 499–501 CrossRef PubMed; (c) R. E. McMahon, H. W. Culp and J. C. Occolowitz, J. Am. Chem. Soc., 1969, 91, 3389–3390 CrossRef CAS.
- (a) N. A. Razzakov and S. F. Aripova, Chem. Nat. Compd., 2004, 40, 54–55 CrossRef CAS; (b) E. G. Sharova, S. F. Aripova and S. Y. Yunusov, Chem. Nat. Compd., 1980, 16, 487–490 CrossRef; (c) Natural compounds: plant sources, structure and properties, ed. S. S. Azimova, Springer, New York, confoline, 2013 Search PubMed; (d) H. Z. Zhang, Y. F. Wang, M. L. Zhang, M. Dong, C. H. Huo, F. Sauriol, Q. W. Shi, Y. C. Gu and H. L. Wang, Chem. Nat. Compd., 2013, 48, 1035–1038 CrossRef CAS.
- In contrast to secondary amines which provide nucleophilic activity, N-formamides react as electrophiles.
- M. A. Ganiek, M. R. Becker, G. Berionni, H. Zipse and P. Knochel, Chem.–Eur. J., 2017, 23, 10280–10284 CrossRef CAS PubMed.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 124, 9360–9371 CrossRef.
- Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 5070–5071 CrossRef CAS PubMed.
- N. V. Reddy, G. S. Kumar, P. S. Kumar, M. L. Kantam and K. R. Reddy, Synlett, 2014, 25, 2133–2138 CrossRef CAS.
- G. S. Kumar, R. A. Kumar, P. S. Kumar, N. V. Reddy, K. V. Kumar, M. L. Kantam, S. Prabhakar and K. R. Reddy, Chem. Commun., 2013, 49, 6686–6688 RSC.
- L. M. Berkowitz and P. N. Rylander, J. Am. Chem. Soc., 1958, 80, 6682–6684 CrossRef CAS.
- T. Endo and J. Zemlicka, J. Org. Chem., 1979, 44, 3652–3656 CrossRef CAS.
- S. W. Pelletier, H. K. Desai, J. Finer-Moore and N. V. Mody, Tetrahedron Lett., 1982, 23, 4229–4232 CrossRef CAS.
- R. Perrone, G. Carbonara and V. Tortorella, Arch. Pharm., 1984, 317, 635–639 CrossRef CAS.
- R. Perrone, G. Carbonara and V. Tortorella, Arch. Pharm., 1984, 317, 21–27 CrossRef CAS.
- (a) S. Nakai, T. Yatabe, K. Suzuki, Y. Sasano, Y. Iwabuchi, J. Y. Hasegawa, N. Mizuno and K. Yamaguchi, Angew. Chem., Int. Ed., 2019, 58, 16651–16659 CrossRef CAS PubMed; (b) For comments on the cost comparisons of nitroxyl radical catalysts and DCAS, see ESI file.
- J. Su, X. Ma, Z. Ou and Q. Song, ACS Cent. Sci., 2020, 6, 1819–1826 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed; (b) N. Emmanuel, P. Bianchi, J. Legros and J. C. M. Monbaliu, Green Chem., 2020, 22, 4105–4115 RSC; (c) Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489–495 CrossRef CAS PubMed; (d) S. Mazzanti, G. Manfredi, A. J. Barker, M. Antonietti, A. Savateev and P. Giusto, ACS Catal., 2021, 11, 11109–11116 CrossRef CAS; (e) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed; (f) A. Pulcinella, D. Mazzarella and T. Noël, Chem. Commun., 2021, 57, 9956–9967 RSC; (g) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. Koolman, M. Fagnoni, S. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS PubMed.
- (a) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed; (b) C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS; (c) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS; (d) V. Klöpfer, R. Eckl, J. Floß, P. M. C. Roth, O. Reiser and J. P. Barham, Green Chem., 2021, 23, 6366–6372 RSC.
- (a) J. A. Ripper, E. R. T. Tiekink and P. J. Scammells, Bioorg. Med. Chem. Lett., 2001, 11, 443–445 CrossRef CAS PubMed; (b) Y. Chen, G. Glotz, D. Cantillo and C. O. Kappe, Chem.–Eur. J., 2020, 26, 2973–2979 CrossRef CAS PubMed.
- Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C. H. Tan, Green Chem., 2011, 13, 3341–3344 RSC.
- (a) Y. Zhang, D. Riemer, W. Schilling, J. Kollmann and S. Das, ACS Catal., 2018, 8, 6659–6664 CrossRef CAS ; for a related transformation of ureas, see:; (b) H. Wang, Y. Man, K. Wang, X. Wan, L. Tong, N. Li and B. Tang, Chem. Commun., 2018, 54, 10989–10992 RSC.
- J. Santamaria, Pure Appl. Chem., 1995, 67, 141–147 CAS.
- J. Santamaria, R. Ouchabane and J. Rigaudy, Tetrahedron Lett., 1989, 30, 3977–3980 CrossRef CAS.
- F. Glöcklhofer, A. J. Morawietz, B. Stöger, M. M. Unterlass and J. Fröhlich, ACS Omega, 2017, 2, 1594–1600 CrossRef PubMed.
- M. F. Acquavella, M. E. Evans, S. W. Farraher, C. J. Nevoret and C. J. Abelt, J. Org. Chem., 1994, 59, 2894–2897 CrossRef CAS.
- (a) M. L. Crossley, J. Am. Chem. Soc., 1915, 37, 2178–2181 CrossRef CAS; (b) E. Schwenk, Angew. Chem., 1931, 44, 912–913 CrossRef CAS.
- T. Hinoue, Y. Shigenoi, M. Sugino, Y. Mizobe, I. Hisaki, M. Miyata and N. Tohnai, Chem.–Eur. J., 2012, 18, 4634–4643 CrossRef CAS PubMed.
- Y. Mizobe, T. Hinoue, A. Yamamoto, I. Hisaki, M. Miyata, Y. Hasegawa and N. Tohnai, Chem.–Eur. J., 2009, 15, 8175–8184 CrossRef CAS PubMed.
- Theoretical solubilities were gauged using calculated logP and total polar surface area. Calculations were made using: Molinspiration Cheminformatics, Slovensky Grob, Slovakia, https://www.molinspiration.com/. for further details, see ESI.
- (a) T. Shida, Electronic Absorption Spectra of Radical Ions. Physical Sciences Data, Elsevier, 1988, vol. 34 Search PubMed; (b) D. T. Breslin and M. A. Fox, J. Phys. Chem., 1994, 98, 408–411 CrossRef CAS.
- (a) J. M. Achord and C. L. Hussey, Anal. Chem., 1980, 52, 601–602 CrossRef CAS; (b) D. Dvoranová, Z. Barbieriková and V. Brezová, Molecules, 2014, 19, 17279–17304 CrossRef PubMed.
- R. Takeda, S. Y. Ryu, J. H. Park and K. Nakanishi, Tetrahedron, 1990, 46, 5533–5542 CrossRef CAS.
- T. Karatsu, K. Kanayama, M. Takahashi, N. Ishigohoka, K. Fukui and A. Kitamura, Heteroat. Chem., 2001, 12, 269–275 CrossRef CAS.
- Z. Y. Chan, P. Krishnan, S. M. Modaresi, L. W. Hii, C. W. Mai, W. M. Lim, C. O. Leong, Y. Y. Low, S. K. Wong, K. T. Yong, A. Z. X. Leong, M. K. Lee, K. N. Ting and K. H. Lim, J. Nat. Prod., 2021, 84, 2272–2282 CrossRef CAS PubMed.
- (a) P. J. Dunn, Org. Process Res. Dev., 2005, 9, 88–97 CrossRef CAS; (b) P. Lees and F. Shojaee Aliabadi, Int. J. Antimicrob. Agents, 2002, 19, 269–284 CrossRef CAS PubMed.
- J. Castellanos-Soriano, J. C. Herrera-Luna, D. Díaz Díaz, M. C. Jiménez and R. Pérez-Ruiz, Org. Chem. Front., 2020, 7, 1709–1716 RSC.
- M. Neumeier, D. Sampedro, M. Májek, V. A. D. O‘Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem.–Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- (a) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (b) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed; (c) X. Tian, T. A. Karl, S. Reiter, S. Yakubov, R. de Vivie-Riedle, B. König and J. P. Barham, Angew. Chem., Int. Ed., 2021, 60, 20817–20825 CrossRef CAS PubMed; (d) A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- For representative reviews on radical ion photocatalyst reactions and physical properties, see: S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202107811 and ref. 44b..
- (a) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed; (b) I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed; (c) J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed; (d) C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed.
- E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934–10937 CrossRef CAS PubMed.
- (a) S. P. Pitre, C. D. McTiernan and J. C. Scaiano, Acc. Chem. Res., 2016, 49, 1320–1330 CrossRef CAS PubMed; (b) D. Rehm and A. Weller, Ber. Bunsen. Phys. Chem., 1969, 73, 834–839 CAS; (c) D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS; (d) For a general review about the importance of mechanistic studies in photocatalysis, see: L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- R. F. Bartholomew and R. S. Davidson, J. Chem. Soc. C, 1971, 2347–2351 RSC.
- In MeCN, the diffusion-controlled rate constant is expected to be ca. 2 × 1010 M−1 s−1 at rt. See: S. Blanc, T. Pigot, C. Cugnet, R. Brown and S. Lacombe, Phys. Chem. Chem. Phys., 2010, 12, 11280–11290 RSC.
- Bimolecular quenching of DCA by photoinduced electron transfer is known to proceed beyond the diffusion limit in MeCN. Rates as high as 1012 M−1 s−1 can be still rationalized within Marcus theory. See: A. Rosspeintner, G. Angulo and E. Vauthey, J. Am. Chem. Soc., 2014, 136, 2026–2032 CrossRef CAS PubMed.
- For recent reviews on the effect of aggregation on photoredox properties see: (a) A. Bhattacharyya, S. de Sarkar and A. Das, ACS Catal., 2021, 11, 710–733 CrossRef CAS; (b) D. Bialas, E. Kirchner, M. Röhr and F. Würthner, J. Am. Chem. Soc., 2021, 143, 4500–4518 CrossRef CAS PubMed.
- (a) DCA molecules are known to π-stack, a phenomenon that is receiving attention in photoredox catalysis giving rise to self-assembled organic dyes with enhanced redox properties (see ref. 60). The XRD of DCAS clearly shows a disruption of π-stacking due to a large π-plane distance of 13.60 Å. While we recognize aggregation can differ in solution and solid-state, our DFT calculated radius of DCAS using an implicit solvation model gave ca. 6.7 Å (thus 13.4 Å between two ‘spherical’ molecules), see ESI.† Therefore, bulky substituents inhibit π-stacking aggregation of solvated DCAS. Moreover, fluorescence lifetime decay profiles of the two catalysts indicated a different aggregation for DCAS. Consistent with literature: ref. 58; (b) A. F. Olea, D. R. Worrall, F. Wilkinson, S. L. Williams and A. A. Abdel-Shafi, Phys. Chem. Chem. Phys., 2002, 4, 161–167 RSC , we observed monoexponential decay profiles for DCA (see ESI†). For DCAS (alone, under Ar), a multiexponential decay was observed indicating a static self-quenching that points to a different kind of aggregation in solution vs.DCA. We recognize the possibility the electron transfer event between 1DCAS* and amines occurs at the Marcus inverted region, accounting for the unintuitive relationship between the SET driving force (ΔGSET) and kinetics (kq). Overall, we postulate the Marcus inverted region and different aggregation pattern may work together to suppress reductive quenching in the case of 1DCAS*..
- Singlet oxygen generation via EnT from dicyanoanthracenes is known, see: (a) C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1758 CrossRef CAS PubMed . It was reported that singlet oxygen can be produced from both singlet and triplet excited states of dicyanoanthracenes, see:; (b) R. C. Kanner and C. S. Foote, J. Am. Chem. Soc., 1992, 114, 678–681 CrossRef CAS.
- (a) X. Han, R. A. Bourne, M. Poliakoff and M. W. George, Chem. Sci., 2011, 2, 1059–1067 RSC; (b) T. B. Truong and J. Santamaria, J. Chem. Soc., Perkin Trans. 2, 1987, 1–5 RSC; (c) K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- (a) E. Baciocchi, T. Del Giacco and A. Lapi, Org. Lett., 2004, 6, 4791–4794 CrossRef CAS PubMed; (b) S. K. Silverman and C. S. Foote, J. Am. Chem. Soc., 1991, 113, 7672–7675 CrossRef CAS.
- The fact that trialkylamines themselves can act as physical quenchers of 1O2 supports the need for sufficient [O2] to counteract this toward productive reactions, thus further justifying the use of continuous flow.
- (a) J. Xiao, Z. Yin, B. Yang, Y. Liu, L. Ji, J. Guo, L. Huang, X. Liu, Q. Yan, H. Zhang and Q. Zhang, Nanoscale, 2011, 3, 4720–4723 RSC; (b) T. Imagawa, M. Nakamoto, R. Shang, Y. Adachi, J. Ohshita, N. Tsunoji and Y. Yamamoto, Chem. Lett., 2020, 49, 1022–1025 CrossRef CAS; (c) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. Alfonzo and A. B. Beeler, Chem. Sci., 2019, 10, 7746–7754 RSC.
- A. P. Darmanyan, W. S. Jenks and P. Jardon, J. Phys. Chem. A, 1998, 102, 7420–7426 CrossRef CAS.
- I. Saito, T. Matsuura and K. Inoue, J. Am. Chem. Soc., 1981, 103, 188–190 CrossRef CAS.
- Catalytically-generated 1O2 finds utility in LSF modification of many natural products containing alkene and diene moieties. Far fewer examples are known for amines due to their physical quenching of 1O2, see: A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed . We envisage the method herein may inspire further development of catalytically generated 1O2 as an LSF strategy for amines..
- (a) M. Rueping, C. Vila, A. Szadkowska, R. M. Koenigs and J. Fronert, ACS Catal., 2012, 2, 2810–2815 CrossRef CAS; (b) D. B. Ushakov, M. B. Plutschack, K. Gilmore and P. H. Seeberger, Chem.–Eur. J., 2015, 21, 6528–6534 CrossRef CAS PubMed; (c) X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem.–Eur. J., 2012, 18, 2624–2631 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2109522. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05840a |
| This journal is © The Royal Society of Chemistry 2022 |