
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in heterostructured cathodic electrocatalysts for non-aqueous Li–O2 batteries†
Qing
Xia
a,
Deyuan
Li
b,
Lanling
Zhao
 c,
Jun
Wang
*b,
Yuxin
Long
b,
Xue
Han
b,
Zhaorui
Zhou
b,
Yao
Liu
*b,
Yiming
Zhang
b,
Yebing
Li
b,
Abulgasim Ahmed Abbaker
Adam
b and
Shulei
Chou
*a
c,
Jun
Wang
*b,
Yuxin
Long
b,
Xue
Han
b,
Zhaorui
Zhou
b,
Yao
Liu
*b,
Yiming
Zhang
b,
Yebing
Li
b,
Abulgasim Ahmed Abbaker
Adam
b and
Shulei
Chou
*a
aInstitute for Carbon Neutralization, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325035, China. E-mail: chou@wzu.edu.cn
bKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (Ministry of Education), Shandong University, Jinan, 250061, China
cSchool of Physics, Shandong University, Jinan, 250100, China
First published on 22nd December 2021
Abstract
Developing efficient energy storage and conversion applications is vital to address fossil energy depletion and global warming. Li–O2 batteries are one of the most promising devices because of their ultra-high energy density. To overcome their practical difficulties including low specific capacities, high overpotentials, limited rate capability and poor cycle stability, an intensive search for highly efficient electrocatalysts has been performed. Recently, it has been reported that heterostructured catalysts exhibit significantly enhanced activities toward the oxygen reduction reaction and oxygen evolution reaction, and their excellent performance is not only related to the catalyst materials themselves but also the special hetero-interfaces. Herein, an overview focused on the electrocatalytic functions of heterostructured catalysts for non-aqueous Li–O2 batteries is presented by summarizing recent research progress. Reduction mechanisms of Li–O2 batteries are first introduced, followed by a detailed discussion on the typical performance enhancement mechanisms of the heterostructured catalysts with different phases and heterointerfaces, and the various heterostructured catalysts applied in Li–O2 batteries are also intensively discussed. Finally, the existing problems and development perspectives on the heterostructure applications are presented.
1. Introduction
Since the 21st century, the fossil energy crisis and environmental problems have continuously forced people to search for clean and effective energy storage and conversion technologies.1–4 Currently, the rapid developments of fuel cells,5–8 supercapacitors,9–11 and batteries12–15 have attracted more and more research attention. A comparison of different available and currently under-development energy storage devices is provided in Fig. 1 in terms of their theoretical energy densities. Metals (such as Al, Mg, Fe, and Zn) are appropriate for metal–air batteries in the aqueous environment.16 They are thermodynamically unstable in aqueous environments, but under specific conditions, their surfaces may be passivated by corresponding oxides or hydroxides, rendering them compatible with aqueous electrolytes to some extent. Zn–air batteries have been intensively assessed among these aqueous metal–air batteries. Zn–air batteries with a low cost of fabrication could deliver a theoretical energy density of 1086 W h kg−1 and theoretical working voltage of 1.65 V. However, the practically attainable energy density of Zn–air batteries is usually around 350 W h kg−1,17 making them more suitable for low-power applications, such as small hearing aids rather than future energy vehicles and large-scale grids. Fe-air batteries show a theoretical energy density of 1229 W h kg−1, comparable to that of Zn–air batteries, but their practical energy density is much lower (60–80 W h kg−1),18 and is only one fifth of Zn–air batteries. As for Al–air and Mg–air batteries, they exhibit a significant theoretical energy density of 8100 and 6815 W h kg−1, respectively. Nevertheless, due to the severe parasitic corrosion of their metallic anodes in contact with the aqueous electrolyte, only a very small fraction of the electrodes can be actually utilized.19,20 Among all metal–air batteries, nonaqueous Li–O2 batteries display the largest theoretical energy density of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 W h kg−1 (even close to that of gasoline). In addition, the anticipated realistic energy density of nonaqueous Li–O2 batteries matches the useable energy density of gasoline for automotive applications (about 1700 W h kg−1), and this means that the nonaqueous Li–O2 batteries hold great potential to be one of the most likely energy storage devices for future energy vehicles, which is considered as an important solution to deal with the problems of global warming and energy crisis.23–25 However, they still face many problems before practical use, such as high charging overpotentials, poor cycle stability, low coulombic efficiency, etc.26–28 Researchers found that the main reason for these problems is the sluggish dynamics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) during discharging and charging. In order to solve these obstacles, various ways have been used to accelerate the ORR/OER kinetics, including the addition of redox mediators, applying photo-assisted systems, and the development of various catalysts. The concept of using a photocatalyst-assisted charging strategy to alleviate the serious problem of high overpotentials in Li–O2 systems was introduced by Yu et al.,33 and it was found that the photogenerated holes in the photocathode might aid in the Li2O2 oxidation processes, thereby greatly reducing the overpotentials. Yu's group34 used cobalt-tetramino-benzoquinone (Co-TABQ) as a bifunctional catalyst for Li–O2 batteries under visible light conditions, exhibiting an overpotential gap of 0.2 V with a round-trip efficiency of 94.0%. Wang et al.35 established a novel photo-assisted Li–O2 system by applying a hierarchical TiO2/Fe2O3 heterojunction, which delivered an ultralow overpotential gap of 0.19 V. However, the key to develop these photo-assisted Li–O2 systems lies in the light itself, which would undoubtedly bring about difficulties to the practical application of Li–O2 batteries. So efficient cathode catalysts without photo-assistance are urgently needed. It is evident that noble metals and their oxides with high catalytic performance are undoubtedly the ideal choices.36–38 However, the scarcity in the Earth’s crust and high price of precious metals hinder their large-scale usage in Li–O2 batteries. Therefore, the rational design of cathode catalysts in Li–O2 batteries in recent years was mainly carried out through two pathways: (1) reducing the loading of precious metals via rationally constructing nanosized or single-atom catalysts.38–41 (2) Employing non-precious metal catalysts, including non-precious alloys, metal oxides,42,43 metal sulfides,44,45 metal nitrides,46,47 metal carbides,48,49 carbon materials, etc.50–52
400 W h kg−1 (even close to that of gasoline). In addition, the anticipated realistic energy density of nonaqueous Li–O2 batteries matches the useable energy density of gasoline for automotive applications (about 1700 W h kg−1), and this means that the nonaqueous Li–O2 batteries hold great potential to be one of the most likely energy storage devices for future energy vehicles, which is considered as an important solution to deal with the problems of global warming and energy crisis.23–25 However, they still face many problems before practical use, such as high charging overpotentials, poor cycle stability, low coulombic efficiency, etc.26–28 Researchers found that the main reason for these problems is the sluggish dynamics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) during discharging and charging. In order to solve these obstacles, various ways have been used to accelerate the ORR/OER kinetics, including the addition of redox mediators, applying photo-assisted systems, and the development of various catalysts. The concept of using a photocatalyst-assisted charging strategy to alleviate the serious problem of high overpotentials in Li–O2 systems was introduced by Yu et al.,33 and it was found that the photogenerated holes in the photocathode might aid in the Li2O2 oxidation processes, thereby greatly reducing the overpotentials. Yu's group34 used cobalt-tetramino-benzoquinone (Co-TABQ) as a bifunctional catalyst for Li–O2 batteries under visible light conditions, exhibiting an overpotential gap of 0.2 V with a round-trip efficiency of 94.0%. Wang et al.35 established a novel photo-assisted Li–O2 system by applying a hierarchical TiO2/Fe2O3 heterojunction, which delivered an ultralow overpotential gap of 0.19 V. However, the key to develop these photo-assisted Li–O2 systems lies in the light itself, which would undoubtedly bring about difficulties to the practical application of Li–O2 batteries. So efficient cathode catalysts without photo-assistance are urgently needed. It is evident that noble metals and their oxides with high catalytic performance are undoubtedly the ideal choices.36–38 However, the scarcity in the Earth’s crust and high price of precious metals hinder their large-scale usage in Li–O2 batteries. Therefore, the rational design of cathode catalysts in Li–O2 batteries in recent years was mainly carried out through two pathways: (1) reducing the loading of precious metals via rationally constructing nanosized or single-atom catalysts.38–41 (2) Employing non-precious metal catalysts, including non-precious alloys, metal oxides,42,43 metal sulfides,44,45 metal nitrides,46,47 metal carbides,48,49 carbon materials, etc.50–52
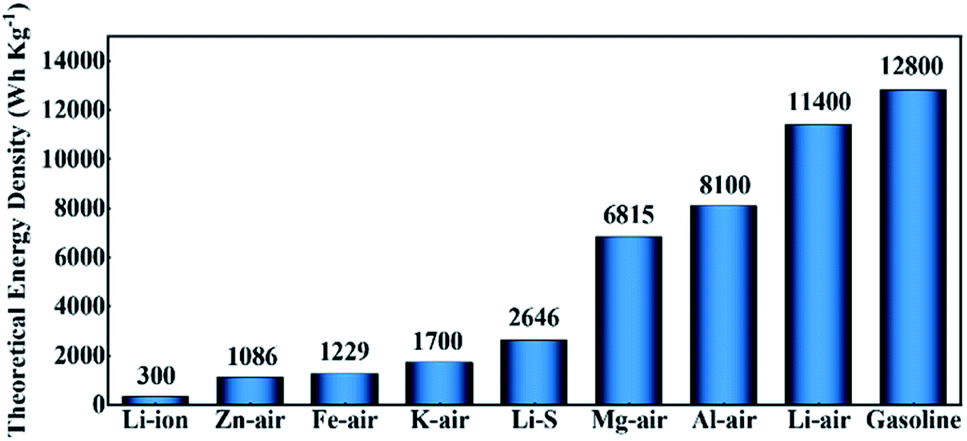 | ||
| Fig. 1 Gravimetric energy densities (W h kg−1) of typical rechargeable batteries and gasoline.16,21,22 | ||
However, most of these non-precious catalysts only show single catalytic activity for the OER or ORR, and the development of high-efficiency bifunctional catalysts is actually challenging and essential for the practical application of Li–O2 batteries.53–55 At present, various strategies including surface modification,56–58 strain engineering,59,60 phase modulation61–63 and heterostructure construction have been explored to realize the bifunctional ability for Li–O2 catalysis.64–66
Among them, heterostructures have been intensively investigated, and are usually defined as composite structures composed of interfaces formed by different solid materials in a broad sense, including conductors, insulators and semiconductors. The interface effect derived from the inherent electric fields among multiple components is beneficial for enhancing the electrocatalytic properties, which can remarkably promote surface reaction dynamics with fast mass and charge transfer.67–69
In this review, the construction and mechanisms of Li–O2 batteries were briefly introduced, and various functions of heterostructures in them were summarized and listed. Then, recent research reports on the application of heterostructures in Li–O2 batteries were provided. We focused on the summary of structure–function relationships between heterostructures and their catalytic properties in these reports, aiming to reveal the underlying mechanisms of their excellent ORR and OER performance. Finally, a personal view of the challenges and opportunities faced by various heterostructures in Li–O2 batteries was included.
2. Fundamentals of nonaqueous Li–O2 batteries
As shown in Fig. 2a, a typical non-aqueous Li–O2 cell consists of a Li metal anode, a porous air cathode and a non-aqueous electrolyte.70,71 The ideal operation of Li–O2 batteries is based on the electrochemical formation during discharging and decomposition during charging of lithium peroxide (Li2O2). The typical electrochemical reaction equation is as follows:| 2Li+ + 2e− + O2 ↔ Li2O2 (E0 = 2.96 V vs. Li/ Li+) | (1) |
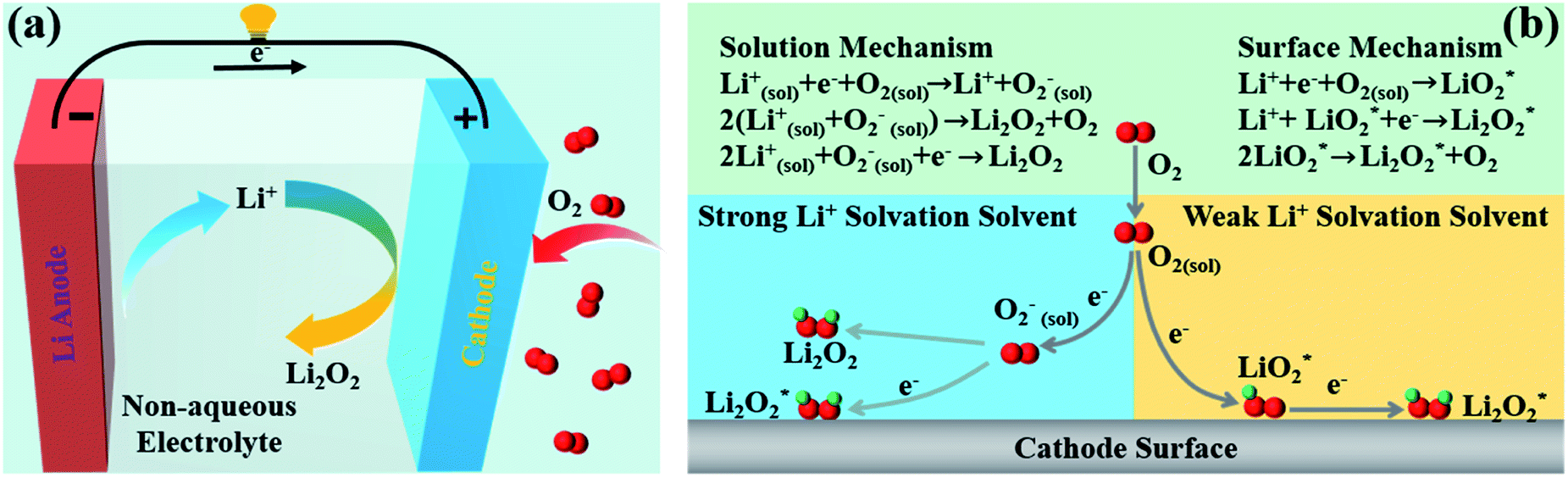 | ||
| Fig. 2 (a) Schematic of a nonaqueous Li–O2 cell. (b) Reduction mechanisms of Li–O2 batteries through different reaction routes.29–32 | ||
In 2014, Bruce's group elaborated on the formation mechanisms of discharge products in Li–O2 batteries,32 and they found that the formation pathways of Li2O2 are mainly controlled by the solubility of the LiO2 intermediate, which depends primarily on the solvation of the cations by the solvent molecules. In a strong Li+ solvating solvent, Li2O2 is usually formed through surface mechanisms, and in a weak Li+ solvating solvent, Li2O2 is normally generated through solution mechanisms (Fig. 2b).31,72,73
2.1. Surface mechanism and solution mechanism
In recent years, the Li2O2 mechanism has been intensively studied,31,32,74 and it is widely accepted that the ORR in Li+ containing non-aqueous solvents occurs starting from the one-electron reduction of O2 to generate superoxide (O2−), which combines with Li+ to form LiO2. In strong Li+ solvating solvents,75–77 such as dimethylsulfoxide and 1-methylimidazole, LiO2 is dissolved mainly in the electrolyte solution, and then it would get reduced or disproportionate to large-sized Li2O2 on the cathode surface.78–80 In weak Li+ solvating solvents including tetraethylene glycol dimethyl ether and acetonitrile, due to the low solubility of LiO2 in the electrolyte solvent, LiO2* is formed and deposits on the cathode surfaces, which is then reduced via an electrochemical process or disproportionation to produce a Li2O2 film.81–832.2. Current status for large-scale commercial applications
In fact, the practical application of Li–O2 batteries is not only limited to the problem of cathode catalysts. For example, the electrolytes in Li–O2 batteries are not only decomposed at high voltage during charging, but are also always easily reduced by superoxide. Another requirement characteristic of the electrolyte in Li–O2 cells is low volatility,73 which is very important in open systems through oxygen flows. Moreover, it is difficult to find corresponding substitutes for Li anodes, and as a result, safety issues such as explosions caused by Li dendritic short-circuits and blocking resulting from the production of thick insulating layers on the Li surface are also hard to avoid during cycling.84 Furthermore, Li–O2 cells currently studied in the laboratory are usually coin-cells,85–87 and few research studies were conducted on the implementation and evaluation of large-sized batteries for practical applications. In recent years, some groups have focused on the investigation of pouch Li–O2 cells for future commercial applications.88–90 Zhao's group90 has taken a crucial step to successfully construct a Li–O2 pouch cell, which shows a high energy density of 768.5 W h kg−1 (based on the weight of the entire battery). Actually, the key points before the large-scale commercialization of Li–O2 batteries are supposed to be the following aspects: (1) side reactions in the high-loading air electrodes, (2) pulverization of the Li-metal anodes and (3) large-scale inhomogeneity of the pouch cell. In general, although practical Li–O2 batteries still have many problems to be solved, it is expected that through unremitting efforts, high performance Li–O2 secondary batteries would eventually be fabricated in mass production. Once being successfully developed, Li–O2 batteries could broaden application prospects in many fields, such as digital electronics, medical and health, electric vehicles, aerospace, etc.3. Characteristics of heterostructures
3.1. Catalyst structure modulation
As shown in Fig. 3a, through heteroepitaxial growth, the growth orientation of active materials can be adjusted and the surfaces with high reactivity can be exposed, thereby affecting the diffusion of Li+ and the adsorption of O2 species to improve the ORR and OER kinetics.91,92 Moreover, the defect-free interfaces and structural connectivity between the epitaxial layer and the substrate are favorable for relaxing structural changes in the cathode caused by the generation and decomposition of Li2O2 during repeated cycling, preventing the active material from detaching.91,92 In fact, because perfect lattice matches are normally not realized, lattice distortions are found at the interfaces of the heterostructures, which significantly increase the number of active sites to enhance their electrocatalytic activities.93,94 Additionally, the disordered atomic arrangements can reduce the surface energies of the heterostructures and effectively change the adsorption capacity of the intermediate products, affecting the formation processes of Li2O2.95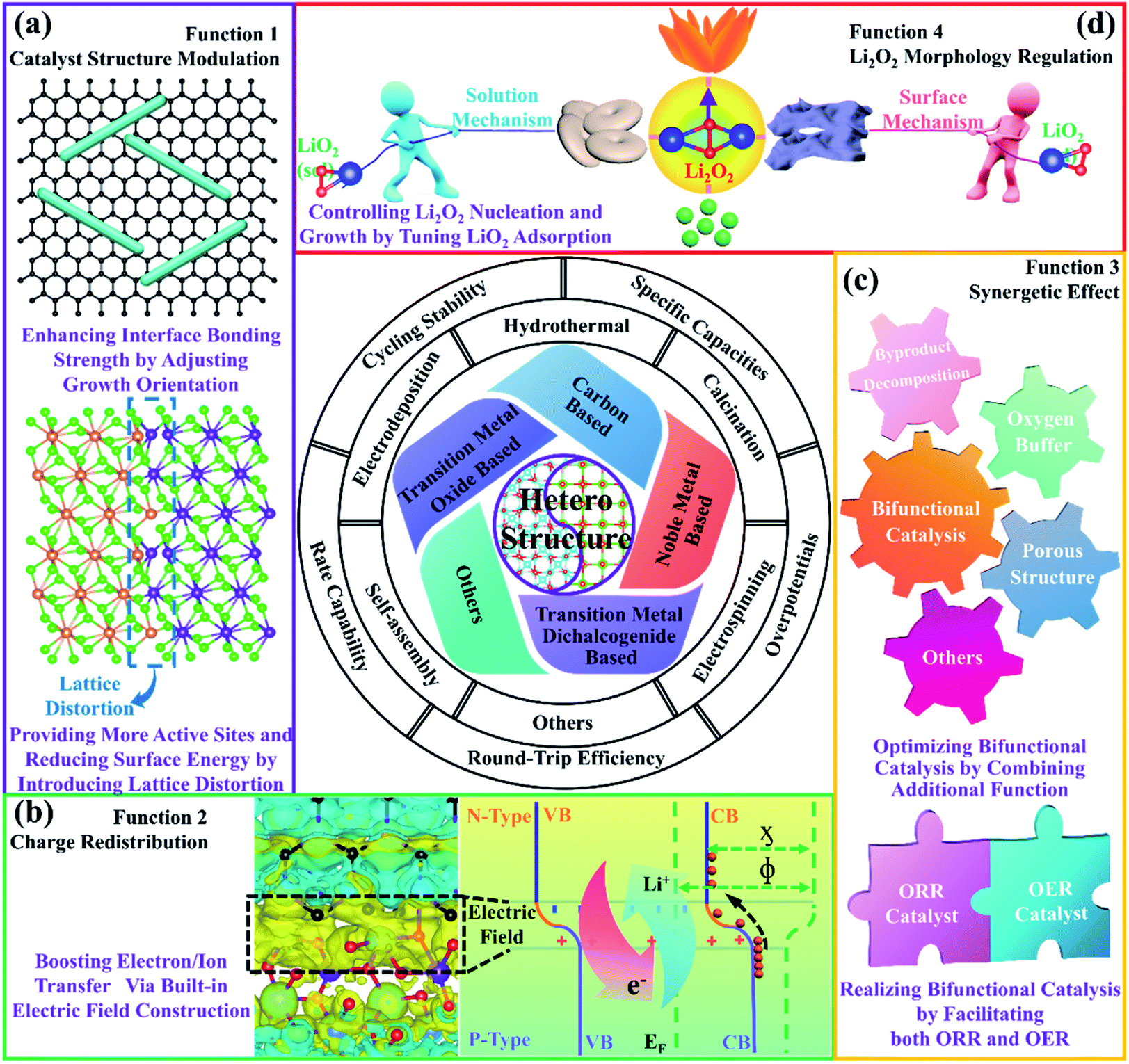 | ||
| Fig. 3 Summary of the typical heterostructures and their functions in Li–O2 batteries. | ||
3.2. Boosting electron/ion transport
The heterostructures we are talking about actually function at special interfaces formed by the contact of two different materials. We take the PN heterojunction as an example to illustrate the formation mechanism of the built-in electric field. As shown in Fig. 3b, because the difference in Fermi energy levels between the two materials will cause electron transfer across the hetero-interfaces, an equilibrium state of equal Fermi energy levels is thus established. In order to keep their original electron affinity (ϕ) and workfunction (χ) unchanged, a space charge region will be formed. Built-in field plants will appear on both sides of the hetero-interfaces.96,97 Due to the action of the built-in electric field, the transportation of Li+ and electrons will be accelerated, thereby promoting the ORR and OER kinetics.3.3. Synergetic effect
Different components of heterostructure composites play various roles in the electrocatalytic processes, notably exhibiting superior performance, compared with any one-component counterparts (Fig. 3c).98,99 For example, it is reported that graphene and CuGeO3 could respectively act as ORR and OER electrocatalytically active components, and their heterogeneous structures will exhibit bifunctional electrocatalytic properties with stronger ORR and OER activities.100 In addition, the combination of NiCo2O4 and NiO could deliver high electrocatalytic activity with effective decomposition of by-products during cycling, which is mainly due to the synergy effect from heterogeneous structures.101 Furthermore, it is evident that CeO2 as an O2 buffer combined with materials with high OER catalytic activity can exhibit larger specific capacity and lower overpotentials than those of single components.102–104 More importantly, porous nanostructured heterostructures also aid in accelerating the transportation of electrolyte and O2, as well as providing a large number of active sites to facilitate the electrocatalytic reactions.3.4. Facilitating O2 transfer and regulating the morphology of discharge products
In fact, the strong interactions of the heterostructures can effectively change the charge distribution of the active sites, thereby tuning the adsorption energy of the catalysts to O2 species (Fig. 3d).105,106 If the adsorption energy of O2 is perfected, O2 transport is largely promoted. Generally speaking, the change in the adsorption energy of O2 species will be linked to the morphologies of the discharge products, especially LiO2; as the adsorption energy of LiO2 increases, Li2O2 tends to form a film and small particles by the surface growth model.107 If it is reduced, Li2O2 usually becomes toroid via the solution growth model.108 For heterostructured composite materials, Li2O2 deposition mechanisms based on two components are usually combined, and special shapes are thus constructed via the competition of the two mechanisms, such as flower, pea, sheet, etc.99,107,1094. Developments and applications of heterostructures for nonaqueous Li–O2 batteries
4.1. Carbon supported heterostructures
Because of their low cost, environmental friendliness, high surface area, superior electrical conductivity, easy preparation, robust mechanical strength and good physicochemical stability,110–112 carbon materials have been widely studied and used in Li–O2 batteries. More importantly, they can also show excellent catalytic properties for boosting the ORR.113,114 Their OER catalytic activity, however, is evidently insufficient, largely limiting their practical Li–O2 battery performance.116,117 Therefore, it is necessary to employ bifunctional cathode catalysts with excellent performance towards both the ORR and OER by constructing heterogeneous structures containing carbon materials and active OER catalytic materials. Tan et al.115 prepared a dual-phase nitrogen-doped cobalt@graphene multi-capsule heterostructure via high-temperature synthesis and nitrogen and ammonia treatment, and Fig. 4a shows the electrocatalytic reaction processes of this heterostructure during cycling. N-doped graphene with a high surface area and excellent electrical conductivity functions as the porous and conductive support, on which the exposed Co and CoN nanoparticles and the N-doped defects together act as the active sites to enable strong binding interactions with Li2O2. The cathode catalyst thus delivered superior electrochemical catalytic activities, showing a low overpotential of 0.9 V at 0.1 mA cm−2, and there was almost no change in the charge and discharge overpotentials after 30 cycles with a fixed capacity of 1.0 mA h cm−2. Many studies focusing on carbon supported heterostructures also demonstrate a similar conclusion, such as Co4N nanorods anchored on CNF papers,89 Co9S8 nanorods on the porous carbon foil,118 (Mn1/3Co2/3)O on CNT microspheres,119etc.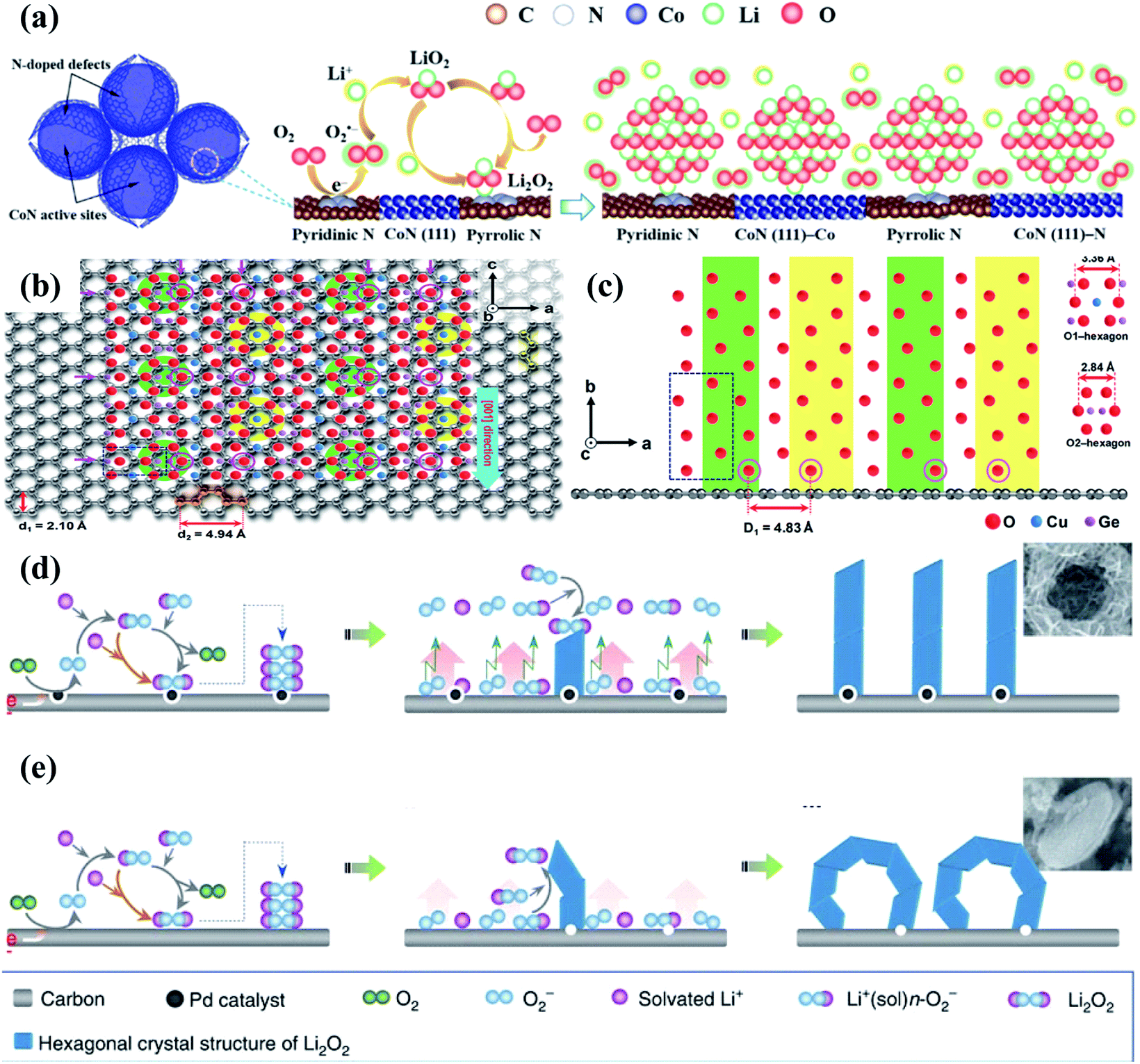 | ||
| Fig. 4 (a) Schematic of the biphasic N-doped cobalt@graphene multiple capsule heterostructure and the proposed reaction mechanism during the discharge process. Reproduced with permission: copyright 2017, American Chemical Society.115 (b) Top and (c) side views from the nanowire-axis direction. Reproduced with permission: copyright 2018, Wiley.100 Different formation mechanisms of Li2O2 aggregates on (d) P-HSC and (e) HSC cathodes. Reproduced with permission: copyright 2013, Springer Nature.52 | ||
Notably, carbon based materials are proven to tightly anchor the active catalyst on their surfaces, beneficial for improving the specific capacities and cycling stability of Li–O2 batteries.124,125 For example, Lee et al.100 successfully controlled the growth direction of 1D CuGeO3 nanowires with the specific crystal orientation on 2D conductive graphene by a facile hydrothermal route followed by thermal reduction. It is found that the lattice mismatch between CuGeO3 and graphene is only 2.2% (Fig. 4b and c, d2 = 4.94 Å, D1 = 4.83 Å), and the angles between them are 60 and 120°. This is because two hexagons (green and yellow circles), as shown in Fig. 4b, composed of oxygen atoms intersected in a zigzag pattern along the a-axis and c-axis directions of graphene, forming a zigzag line of oxygen atoms along the c-axis direction. Compared with individual graphene and CuGeO3 cathodes, the CuGeO3-graphene cathode shows better cycle stability of 100 cycles at 200 mA g−1 with a specific capacity limit of 1000 mA h g−1. The excellent catalytic performance of the CuGeO3–graphene heterostructured catalyst can be attributed to the bifunctional phase boundary synergy effect.
Besides, applying a heterogeneous structure in a cathode catalyst for the Li–O2 batteries can also effectively modulate the growth and morphology evolution of the discharge products, thereby largely reducing the OER overpotentials and enhancing the cycling performance. Xu et al.52 successfully synthesized a free-standing honeycomb-like palladium-modified hollow spherical carbon (P-HSC) deposited onto carbon paper through electrophoresis with template replication. The discharge products exhibit a unique architecture composed of Li2O2 nanosheets, and their relative formation mechanisms are depicted in Fig. 4d and e. Compared with the carbon surfaces, those of the Pd nanocrystals are less sticky to superoxide, which would weaken the binding interaction of superoxide and the carbon substrate. In response, superoxide molecules could easily diffuse away from the cathode. Benefiting from the mutual effect of spherical carbon and Pd nanoparticles, the nucleation and crystallization of Li2O2 preferentially occurs on prismatic crystal faces, resulting in nanosheet shaped Li2O2 growth. Kwak and co-workers127 dispersed molybdenum carbide particles on carbon nanotubes by ball milling and thermal treatment, and their catalytic properties were investigated in Li–O2 cells. It is worth noting that toroidal Li2O2 was formed on the carbon nanotube cathode in the discharge processes, while Li2O2 in the form of a well-dispersed layer covered the catalyst surfaces, which lead to a larger battery efficiency (88%) and better reversibility (over 100 cycles) with lower overpotentials.
In general, owing to multiple merits, it is a good choice to select carbon-based substrates for forming heterostructures. These highly conductive carbonaceous substrates with a large surface area can not only easily enable rapid electron transport and mass transfer, which aid in reducing the ORR and OER overpotentials, but also regulate the morphology of the discharge products, further improving Li–O2 battery performance.
4.2. Noble metal based heterostructures
Among various catalysts, noble metals have been intensively investigated and widely testified to feature superior catalytically active properties Li–O2 batteries. Due to their limited amount on earth and high cost, however, researchers have to look for alternatives or conduct structural modification of noble metals. Recent research studies show that constructing noble metal based heterostructures can not only greatly improve the catalytic performance of the cathode catalysts, but also make them more affordable for boosting practical applications of Li–O2 batteries. Liang et al.120 prepared a surface sulfur-vacancy modified Ru/ZnIn2S4 (RuZIS-Vs) Mott–Schottky heterojunction by hydrothermal and subsequent chemical reactions and used it as a cathode catalyst material for Li–O2 batteries, showing low ORR and OER overpotentials (0.39/0.38 V) and excellent cycling stability (321 cycles). They found that sulfur vacancies induced the interface defects of ZnIn2S4, which matched well with doped Ru, thereby establishing typical metal/semiconductor Mott–Schottky interfaces. By comparing the electronic structures of ZIS, ZIS-Vs and Ru-ZIS-Vs, it is clear from Fig. 5a and b that the Mott–Schottky interfaces could effectively reduce the bandgap of ZIS by varying its valence and conduction band values, which would greatly promote charge transfer on the heterostructure and advance the ORR and OER catalytic activity. Similarly, Agyeman et al.121 decorated Pd nanoparticles on NiCo2O4 nanosheets (Pd@NiCo2O4) via a simple hydrothermal and subsequent reduction processing, and their discharge products show a flower-like morphology. It is demonstrated that Pd@NiCo2O4 and NiCo2O4 exhibit different electronic structures caused by Pd doping and increased the oxygen vacancies in the heterostructure. As evidenced in Fig. 5c, it is mainly due to the fact that the electronegativity of O (3.44) is much higher than those of Pd (2.2), Co (1.88) and Ni (1.91), thereby attracting electrons from these metals into the O sites; notably, noble metal-based heterostructures could also contribute to accelerating the decomposition of Li2O2. For instance, Huang and co-workers128 prepared a core–shell Pd@Pd4S heterostructure decorated on a porous carbon matrix. It is evident that the charging voltage of the Pd@Pd4S–C cathode remains below 3.7 V with low overpotentials, which is much lower than those of other counterparts. In fact, during the charging process, the decomposition of Li2O2 was mainly divided into two parts, including decomposition of small Li2O2 particles to transform into Li2−xO2 through delithiation and subsequent decomposition of Li2−xO2 with the release of O2 at higher oxidation voltages. The stable charging voltage of about 3.7 V for the charge profile of the Pd@Pd4S–C cathode indicates that the Pd@Pd4S heterostructure can effectively promote Li2O2 delithiation and largely reduce OER overpotentials to achieve high battery efficiency. In fact, instead of directly accelerating the decomposition process of Li2O2, it is more common that noble metal-based heterostructures could induce the formation of a more easily decomposed Li2O2 structure during the ORR process. Cong's group122 applied a simple one-step electrospinning method to obtain a silver-modified perovskite La0.9FeO3-δ (Ag@LFO) as an effective cathode catalyst for Li–O2 batteries. As shown in Fig. 5d, the binding energies of O2 species on the surfaces of LFO (O2 is −2.84 eV and LiO2 is −6.01 eV) are much lower than those on Ag@LFO, indicating that they are more easily adsorbed on the Ag@LFO surfaces. It is also illustrated that the discharge product generation would prefer to follow the surface model on the Ag@LFO cathode, presenting the reason for the formation of thin-film Li2O2 on the surfaces of Ag@LFO and toroid Li2O2 on the surfaces of LFO. Actually, film-like Li2O2 is considered to be easier to decompose during charging, resulting in lower overpotentials and longer cycle life.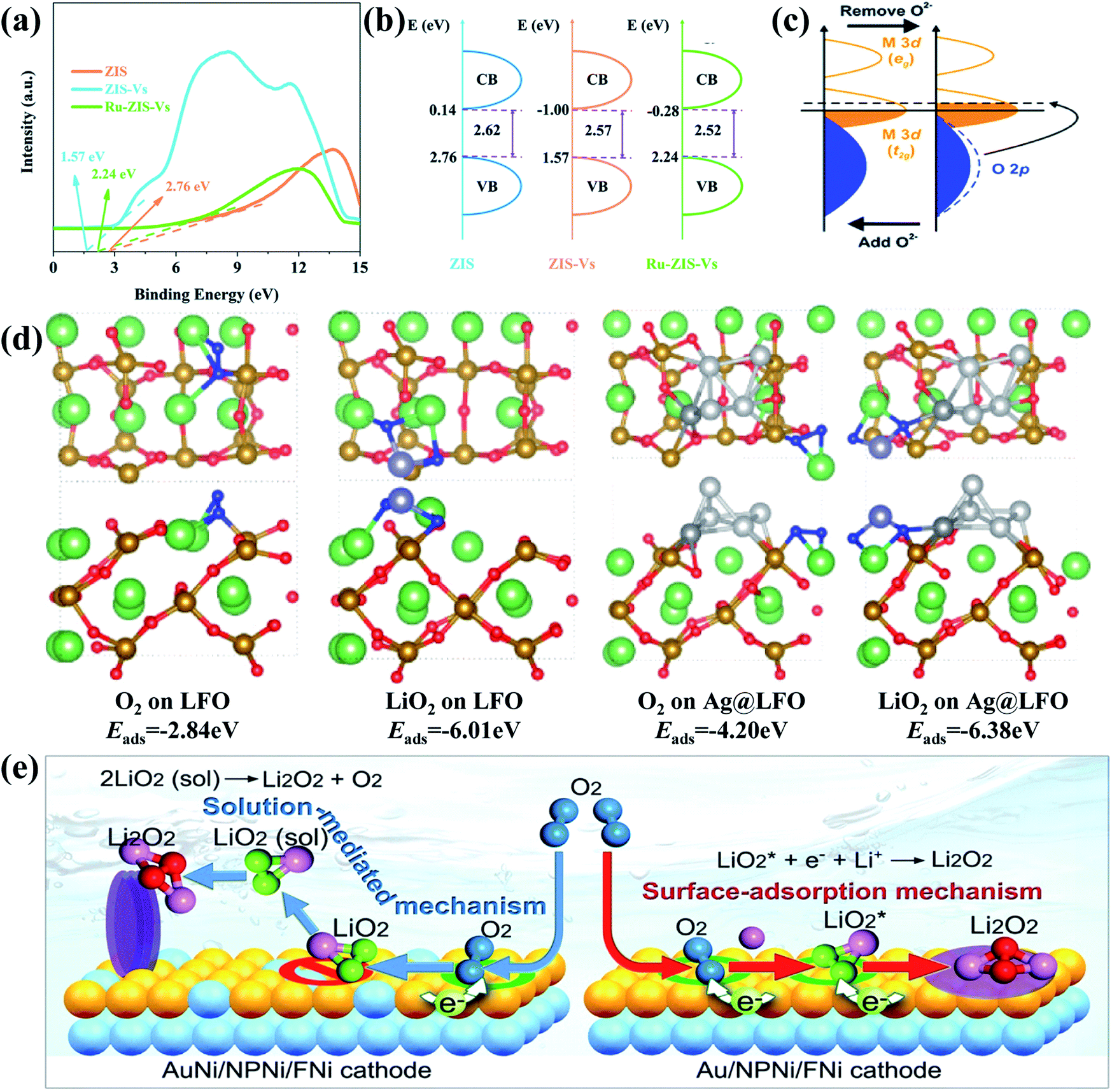 | ||
| Fig. 5 (a) XPS valence band spectra and (b) band diagram of ZIS, ZIS-Vs and RuZIS-Vs. Reproduced with permission: copyright 2020, Royal Society of Chemistry.120 (c) Molecular orbital structure change of Pd@NiCo2O4 as a result of Pd impregnation. Reproduced with permission: copyright 2017, Royal Society of Chemistry.121 (d) Optimized structures and the corresponding binding energies of O2 and LiO2 on LFO and Ag@LFO. Color codes: Li (violet), O (red), Fe (golden), Ag (light gray for Ag clusters and gray for replacing lanthanum in perovskite), lanthanum (green) and oxygen atom of O2 and LiO2 (blue). Reproduced with permission: copyright 2019, American Chemical Society.122 (e) The mechanisms of electrochemical growth of the film-like and nanosheet-like Li2O2. Reproduced with permission: copyright 2017, American Chemical Society.123 | ||
The activity of precious metal-based heterostructured catalytic materials is usually not as high as that of the precious metal itself. In recent years, to reduce costs, precious metals are often coupled with other materials in the form of nanosized structures, such as nanowires, nanoparticles, and nanoclusters. To fully exploit the catalytic properties of precious metal atoms, the application of precious metal single-atom catalysts has emerged as one of the most promising directions. Recently, Lu et al.129 successfully synthesized ultra-high-density single-atom catalysts containing metal contents up to 23 wt% for 15 metals (including Pd, Ru, Ir, and Pt) on chemically distinct carriers via a versatile approach combining impregnation and two-step annealing, and this approach could enable kilogram-scale material output, boosting the commercialization of precious metal-based single-atom catalysts.
4.3. Transition metal oxide based heterostructures
Owing to affordable cost, wide structural diversity, multiple valence states and relatively high electrocatalytic activity, transition metal oxides are considered as one of the alternative materials to noble metals as cathode catalysts for Li–O2 batteries. Their electrical conductivity, however, is normally too limited to enable fast reaction kinetics,130–132 and it is highly necessary to boost charge transfer and O2 adsorption on the surfaces for perfecting battery performance. Applying transition metal oxide based heterostructures as catalysts is proven to be one of the most effective strategies. Wu's group126 successfully prepared Mo2C/MoO2@ reduced graphene oxide heterostructures by in situ decoration of Mo2C between MoO2 and reduced graphene oxide through a hydrothermal-calcination method The atomic MoO2 (−111), Mo2C (101) and MoO2/Mo2C interface models are listed in Fig. 6a for revealing the O2 adsorption, and the DFT calculation data demonstrate that the Mo2C/MoO2 interfaces show greater O2 adsorption energy. This is due to the charge distribution imbalance around the interfaces caused by the accumulation of valence electrons, which could offer stable O2 chemical adsorption on the Mo atoms. Nucleation instead of crystal growth was thus preferred during cycling, and film-like amorphous discharge products tended to form on the composite catalyst, resulting in an ultra-low potential gap of 0.57 V, and a stable cycling performance with a charge potential of less than 4.0 V after 100 cycles at 200 mA g−1. Wang et al.107 reported, as shown in Fig. 6b, that the p–n heterojunction could be constructed from NiCo2S4 nanotubes and NiO nanoneedles with distinctly different bandgaps. This unique structure could introduce a built-in electric field at the hetero-interfaces, effectively facilitating interfacial charge transfer kinetics and fast ORR and OER kinetics. Boosted charge transfer could also be traced in PdO–Co3O4 heterostructures,134 which delivered a stable cycling performance of more than 90 cycles. Moreover, the synergetic effect was realized in urchin-like NiO–NiCo2O4 microspheres101 for perfecting their Li–O2 battery performance. As depicted in Fig. 6c, NiCo2O4 was believed to facilitate both the ORR and OER for Li–O2 batteries, and NiO was likely to boost the by-product decomposition. More importantly, fast transport of Li+ and electrons could be enabled to further promote the reaction kinetics, and superior specific capacities with low overpotentials and enhanced cycling stability were thus obtained. It is worth noting that the electrochemical performance is usually closely related to the formation and evolution of discharge products,135,136 which could be governed by the heterostructures. Zhang and co-workers99 deposited Co3O4 nanosheets and α-MnO2 nanorods on the carbon paper through a simple electro-deposition and calcination route. Compared with the granular morphology on the MnO2 cathode and nanosheet/film structure on the Co3O4 cathode, mooncake/large-sheet Li2O2 was homogeneously (Fig. 6d) grown on the composite cathode with an embedded architecture. This is mainly because α-MnO2 provided the growing sites, and Co3O4 nanosheets with evidently increased height further facilitated the formation of dissolved LiO2. Unique embedded growth of Li2O2 was thus realized, bringing about a large reversible specific capacity of 4850 mA h g−1 with almost 100% coulombic efficiency, and a low potential gap of 0.95 V at 103 mA g−1. 1-D RuO2/Mn2O3 hollow heterostructures were also fabricated through single-nozzle electrospinning followed by heating speed controlled annealing.140 Interestingly, their double-walled fiber structure could effectively trigger Li2O2 generation and decomposition inside and outside on the catalysts with close contact, displaying improved overpotential characteristics and stable cycle life of over 100 cycles in the Li–O2 cells.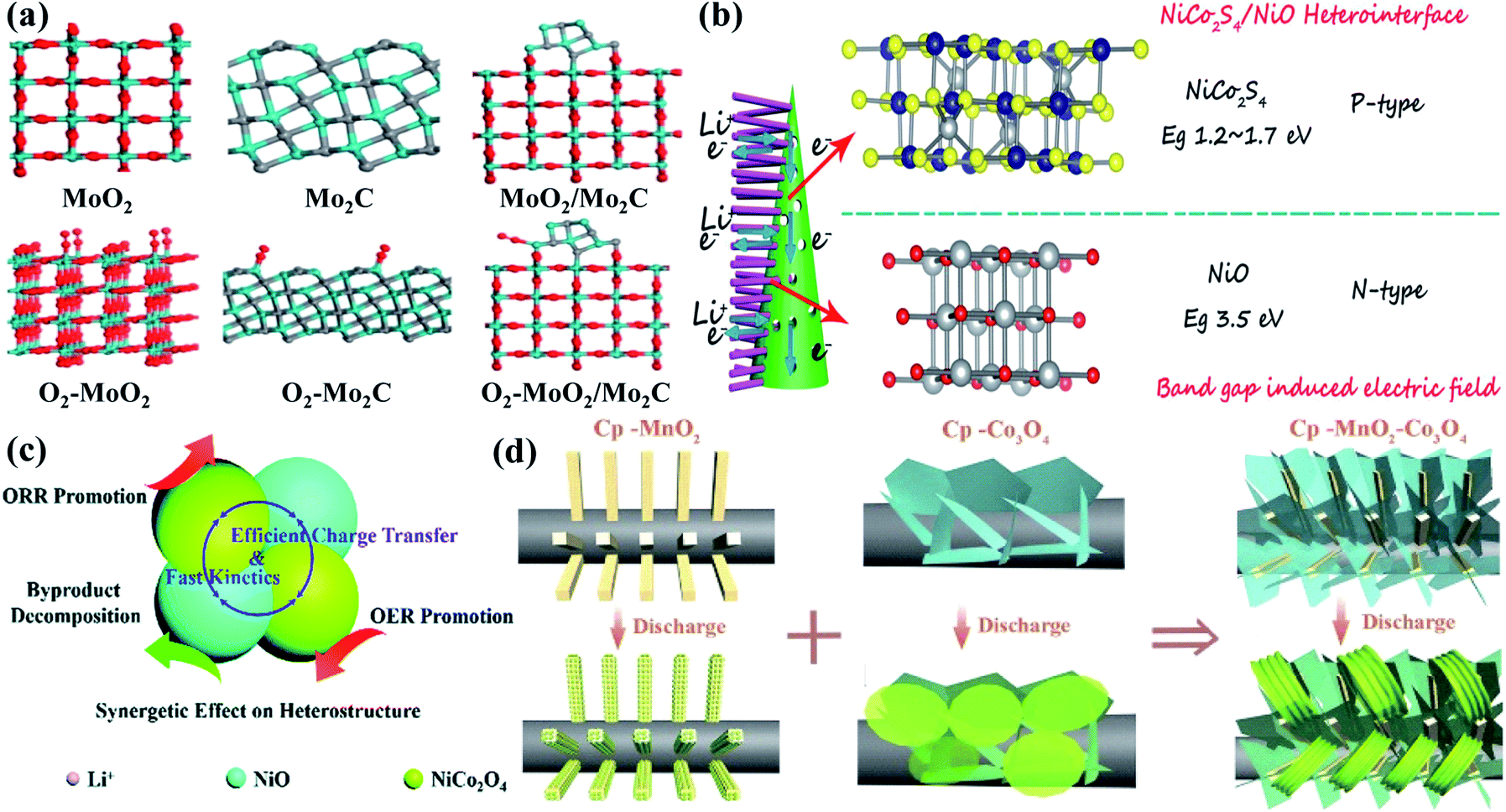 | ||
| Fig. 6 (a) Views of MoO2 (−111), Mo2C (101) and MoO2/Mo2C interfaces with corresponding O2 adsorption models (red, cyan and grey balls are O, Mo, and C atoms). Reproduced with permission: copyright 2020, Elsevier.126 (b) Summary of the enhanced high rate reaction mechanism with unique local electric fields from the NiCo2S4/NiO heterostructure. For the spheres in the models (gray, blue, yellow and red balls represent Ni, Co, S and O atoms). Reproduced with permission: copyright 2019, Wiley.107 (c) Specific roles of the NiO–NiCo2O4 heterostructure for Li–O2 catalysis during discharge and recharge processes. Reproduced with permission: copyright 2019, Royal Society of Chemistry.101 (d) Schematic illustration of different discharge product morphologies on carbon paper supported MnO2 and Co3O4 cathodes. Reproduced with permission: copyright 2017, Wiley.99 | ||
Although the TMO based heterostructures exhibit high redox activity in charge–discharge reactions, the overpotential depends strongly on the composite phase, and the heterostructures obtained from the combination of two transition metal oxides normally show high overpotentials, which are caused by the low electrical conductivity limiting the charge transport. The composite of transition metal oxides and high electrical conductivity materials can therefore effectively minimize the obstacles and maximize the activity of the catalytic sites.
4.4. Transition metal dichalcogenide based heterostructures
In recent years, transition metal dichalcogenides (TMDs), the chemical formula of which appears as MX2 (M and X represent the transition metal and chalcogen elements, respectively),141 have received broad research interest. Most of them with a unique X–M–X structure show typical 2D material properties and could expose more catalytically active sites.142,143 Besides, the weak van der Waals interaction and the missing of surface dangling bonds could effectively alleviate the strict thermal and lattice matching demand, contributing to easy heterostructure construction.144 Furthermore, TMDs could display different electrical characteristics, which are mainly affected by the polytype and the number of transition metal d electrons, and this could result in semiconducting or metallic behaviors,145 favorable for forming efficient heterostructures with various other catalysts for Li–O2 batteries. Hu et al.108 developed heterostructured NiS2/ZnIn2S4 nanosheets through a one-step hydrothermal method, whose density of electronic states close to the Fermi level at the heterogeneous interface increases significantly (Fig. 7a). On the one hand, the established built-in fields on the heterostructures are beneficial for fast interfacial charge transfer, providing numerous active interfaces for redox reaction kinetics. On the other hand, the heterostructures could induce strong Li+ and O2 adsorption and weak LiO2 adsorption (Fig. 7b), which boost LiO2 desorption on their surface and solvation in the electrolyte. This effectively promoted toroid-like Li2O2 deposition and decomposition in low Gutmann donor number (DN) solvents through the solution pathway, which guaranteed outstanding electrocatalytic activities. Similar findings also appeared in Ni3Se2/NiSe2 heterostructures,133 and it is also evident to realize advanced charge transfer by enabling distinct electron redistribution along the interfaces (Fig. 7c). Moreover, their 3D porous structure aided in barrier-free mass transport and urchin-like Li2O2 generation with intimate contact with the catalyst (Fig. 7d), leading to a large discharge specific capacity of 23092 mA h g−1 at 500 mA g−1 and reduced charging overpotential of 0.38 V, and a long cycling life of more than 500 cycles can also be achieved with a fixed capacity of 1000 mA h g−1 at 100 mA g−1. What is more, Liang's group95 conducted deliberate CoSe2@NiSe2 heterostructure engineering via the hydrothermal method combining heat treatment. As shown in Fig. 7e and f, the formation of a heterogeneous interface caused the distortion of local fine atom arrays. These distortions can not only serve as active sites, but also reduce the surface energies of the catalyst, thereby promoting the LiO2 and adsorption and reversible flower-shaped Li2O2 formation (Fig. 7g) with a low potential gap (1.2 V) during cycling.
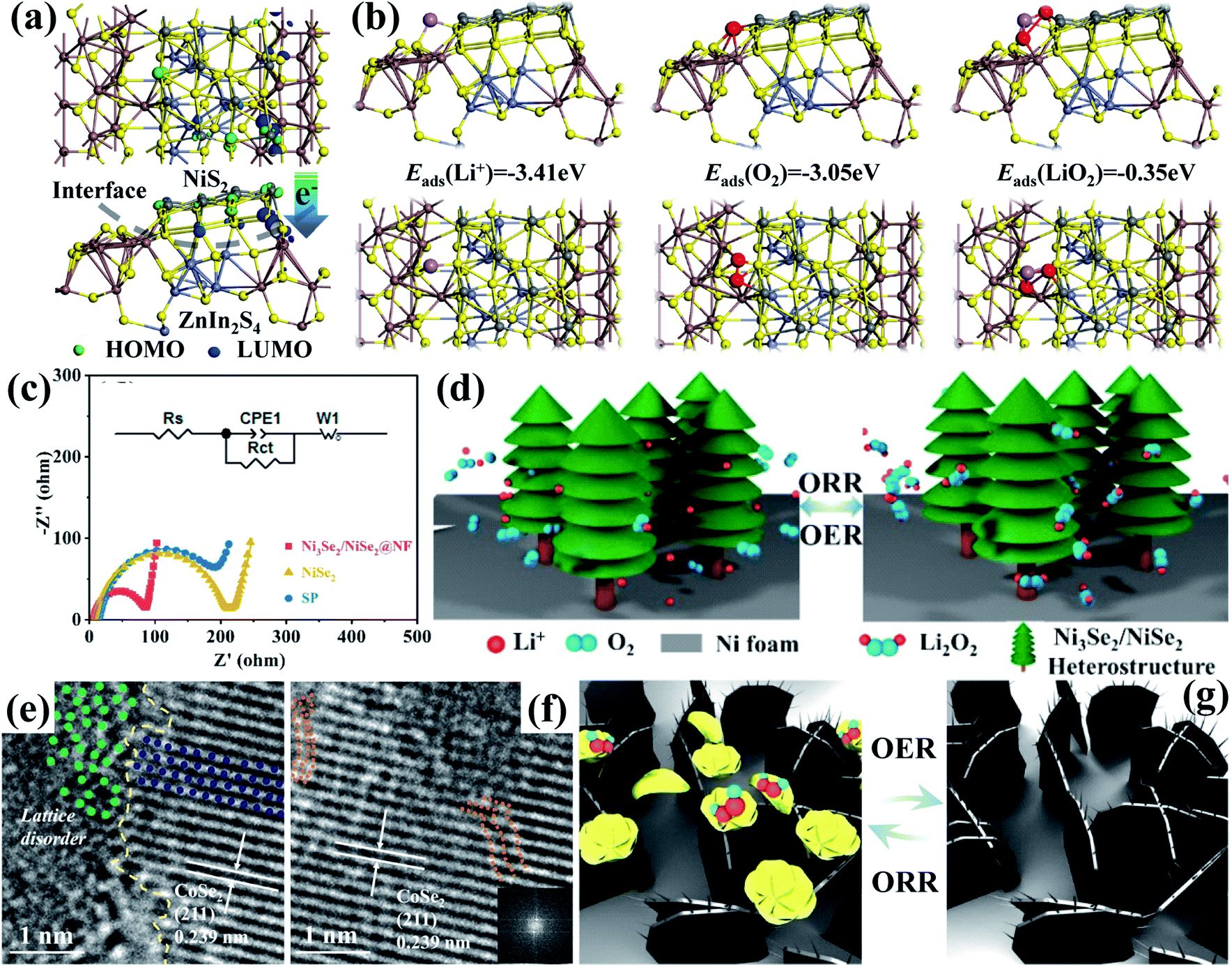 | ||
| Fig. 7 (a) Charge distribution at the interface of NiS2 and ZnIn2S4 with (b) adsorption energies (ΔEads) of Li+, O2 and LiO2 adsorbed on NiS2@ZnIn2S4 with optimized structures. Reproduced with permission: copyright 2020, American Chemical Society.108 (c) Nyquist plots of different cathodes with the inset showing the fitting equivalent circuits. (d) Schematic diagram of the reaction mechanisms of the Ni3Se2/NiSe2@NF cathode during discharge and charge processes. Reproduced with permission: copyright 2020, Elsevier.133 (e) HRTEM image of CoSe2@NiSe2 at the interface with the disordered edges. (f) HRTEM image of CoSe2@NiSe2 and inset shows the corresponding FFT pattern from the distortion region. (g) Schematic illustration of the oxygen electrode reactions on the CoSe2@ NiSe2 cathode during cycling. Reproduced with permission: copyright 2020, Elsevier.95 | ||
TMD based heterostructures show a high initial capacity and excellent cycling stability. To achieve high ion and electron transfer rates, the compositing of metallic phases for TMDs seems to be a favorable option. However, the problem of the conversion of pure metallic phases to semiconductor phases during charging and discharging has not yet been solved, and the combination of the two phases to obtain a heterogeneous structure has been conducted as a possible strategy in some studies,44,146 displaying the advantages of both high electrical conductivity and high stability.
4.5. Other heterostructures
In addition to the above heterostructures, there are also some other heterostructures applied in Li–O2 batteries. For example, Jin et al.137 synthesized Co and Fe layered double hydroxide (LDH)–RuO2 nanosheet heterostructures by the self-assembly technique (Fig. 8a). The strong interface electronic coupling between LDH and RuO2 nanosheets with oxygen vacancies greatly improved the diffusion of Li+ and reactants, which are responsible for the enhanced transfer kinetics towards the OER and ORR. Lee's group138 simulated the ORR pathway of the heterostructure of h-BN and Ni (111) as a cathode catalyst through DFT calculations; the structure of the heterostructure is shown in Fig. 8b, and they found that the heterostructure thermodynamically preferred to undergo the 2e− pathway (Fig. 8c), which is obvious different from most of the previous results with the 4e− pathway. The main reason is that the (Li2O2)2 adsorption is remarkably strong on the h-BN/Ni (111) surface, contributing to a maximum discharge potential of 1.93 V and a minimum charge potential of 3.83 V, comparable to those of noble metal based catalysts. In recent years, photocatalysis has also been developed rapidly and applied to the field of Li–O2 batteries. Veeramani and coworkers139 developed a photocathode nanocomposite of cadmium selenide/zinc sulfide quantum dots grown on carbon nanotubes (CdSe/ZnS QD@CNT), and it shows a low charge voltage within 4.0 V; this is because during charging, the photoexcited holes created by the photoelectrode absorbing photons from the light source on the heterostructure interfaces can assist to oxidize Li2O2 to oxygen gas and Li+ in the valence band (VB) position (Fig. 8d).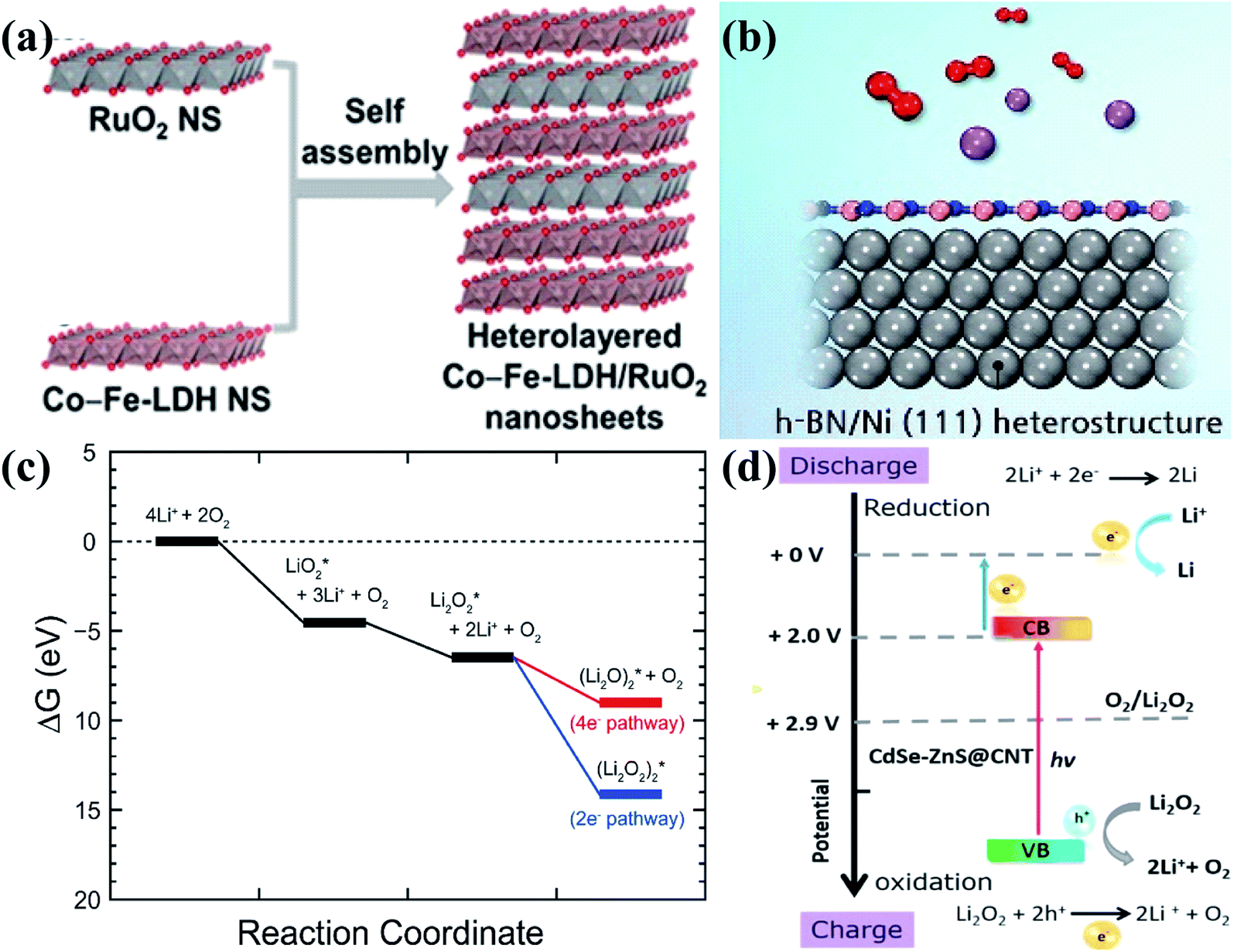 | ||
| Fig. 8 . (a) Schematic illustration of the synthesis of Co and Fe layered double hydroxide (LDH)–RuO2 nanosheets. Reproduced with permission: copyright 2019, Elsevier.137 (b) Schematic illustration of the h-BN/(111) heterostructure. (c) Free energy diagram of the ORR on the h-BN/Ni (111) surface. Reproduced with permission: copyright 2016, Elsevier.138 (d) The theoretical energy band diagram for the improved performance of rechargeable photoelectrochemical Li–O2 batteries. Reproduced with permission: copyright 2018, Elsevier.139 | ||
The rapid development of heterogeneous structures based on the stacking of two-dimensional materials (including graphene, hBN, MoS2, hydroxides, and MXenes) in recent years has also led to new directions in the field of energy storage and conversion,147–149 which offers the opportunity to build electrodes that will combine the advantages of each building block and eliminate the associated disadvantages. For example, hBN crystals have proved to be a similar substrate to graphene, allowing a tenfold increase in their electronic quality.
5. Summary and perspectives
In order to cope with the energy crisis, the development of Li–O2 batteries has attracted widespread attention from researchers, and it is essential to develop high-performance catalysts with high activity and long-term durability. In this review, we summarized the latest research progress of heterostructured catalysts used in Li–O2 batteries, and various strategies for battery performance improvement were proposed. Moreover, analysis of the mechanisms behind performance enhancement and the functions of hetero-interfaces for Li–O2 catalysis were emphasized, and the existing problems of the current heterostructure research studies and possible directions for future studies were finally presented, as depicted in Fig. 9.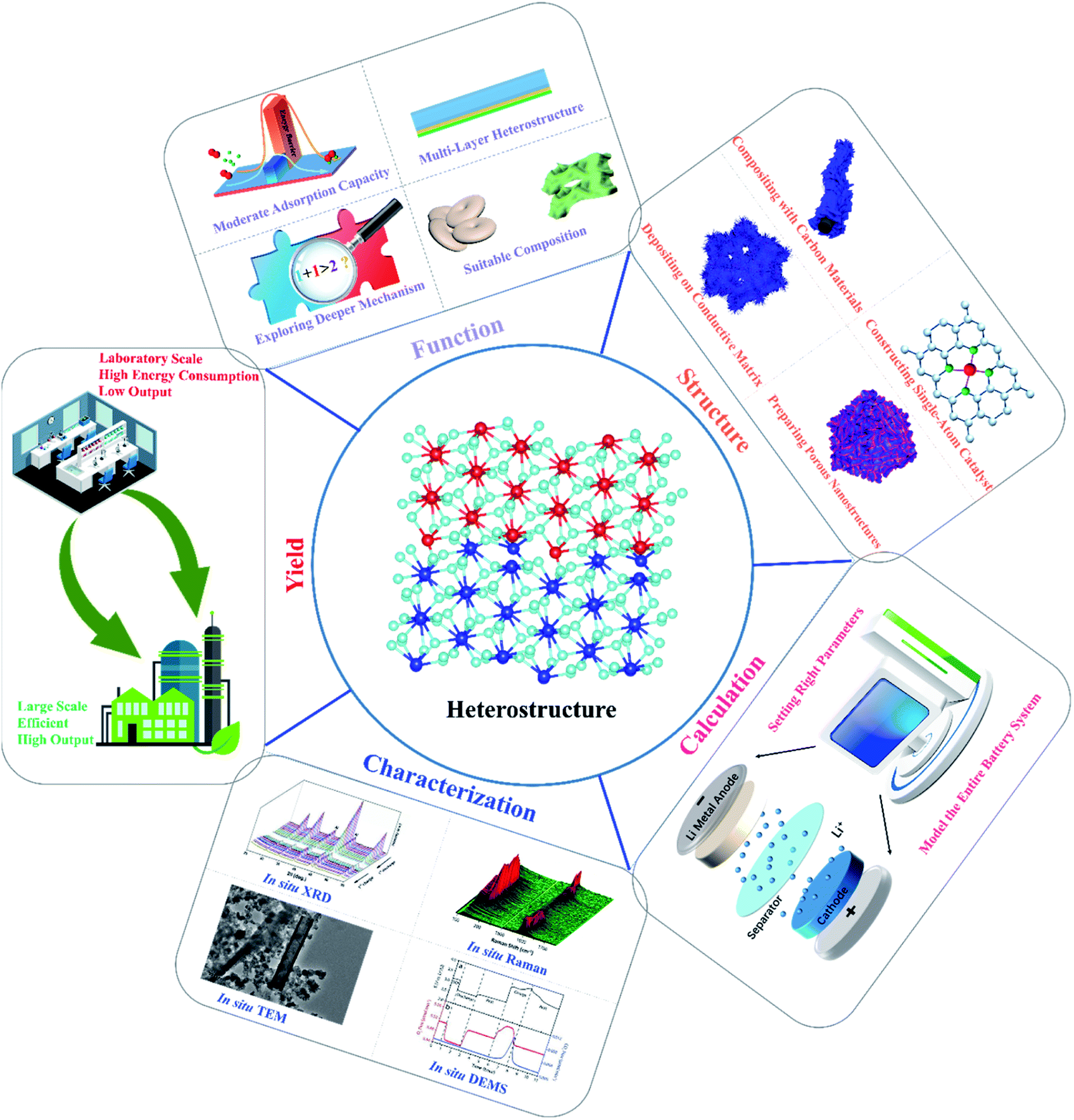 | ||
| Fig. 9 Directions and perspectives for future studies on heterostructured cathode catalysts for Li–O2 batteries. | ||
1. The critical roles of heterostructures for Li–O2 catalysis are mainly played in the following aspects: (1) the transportation of electrons and ions could be accelerated. Since the difference between the Fermi levels of the heterostructure components normally induces a built-in electric field, the migration of ions and electrons would be effectively promoted under the action of coulombic force to advance the reaction kinetics. To this end, it is believed that gradient hetero-interfaces with an ideal built-in interface potential should be induced for fast charge and mass transfer, such as multi-layer heterostructures, and even the construction of the built-in electric field inside the components through precise tuning of the composition of each component needs to be considered. It is worth noting that these strategies can also bring about new insights of designing high-current energy storage devices; (2) synergy effects are proposed to contribute to the excellent bifunctional catalytic activities of heterostructures, which refers to the outcomes of 1 + 1 > 2 under the actions of different components. In fact, the deeper intrinsic mechanisms for synergy effects have not been fully explored, and the as-presented synergy effects were comprehensive macroscopic properties largely affected by different components. Consequently, clearer evidence should be provided to illustrate the insightful effect of synergy in future research studies; (3) the adsorption behaviors of the catalysts are basically controlled by the electronic properties of the active sites, and regulating the adsorption energies of O2 species on heterostructures is thus essential for improving the Li–O2 battery performance. It is found that the electronic structures of the surrounding active sites on heterostructures could be effectively tuned, thereby changing the adsorption energies of O2 species and resulting in the alterations of ORR and OER pathways. Actually, too low or too high adsorption energies of O2 species tend to pose difficult adsorption or desorption during cycling, which demonstrates that the ideal heterostructured catalysts should show a moderate adsorption capacity to deliver the smallest reaction barrier in the adsorption and desorption processes. (4) Each component in the heterostructures affects the nucleation and growth of the discharge products via different formation mechanisms, and there is definitely a competition between them. There are currently more than 100 candidates for Li–O2 battery cathode catalysts (including graphene, molybdenum sulfide, Pt, etc.), and more than 10000 hetero structures composing them. How to select and use them to build heterostructures still remains challenging, and employing computational material pre-selection to build a genome for heterostructures for Li–O2 batteries is a good way to solve this problem. As a result of these research efforts, controlled access to small and readily decomposable Li2O2 or even highly conductive amorphous Li2O2 is possible, thus greatly extending the limits of current Li–O2 batteries.
2. Fig. 9 also shows the strategies for constructing high-performance carbon, noble-metal, and TMO and TMD-based heterostructures. Carbon-based heterostructures are favored by researchers as catalysts, mainly because of their low price and high electrical conductivity. However, in view of the fact that carbon materials themselves are vulnerable to superoxide attack and enable irreversible decomposition, the rational designs of carbon-based heterostructures are focused on composing heterostructures on the supported carbon substances, which can not only utilize the high electronic conductivity and large specific surface area of the carbon matrices, but also avoid the formation and accumulation of by-products to prevent the rapid death of Li–O2 batteries. In contrast, noble metal-based heterostructures usually deliver low overpotentials and stable cycle performance, and the purpose of preparing such heterostructures is to reduce the amount of noble metals in the catalysts with high catalytic performance close to those of noble metal catalysts. In this respect, guaranteeing uniform dispersion of noble metals throughout another component is of great significance, which is still a problem remained to be solved. Moreover, the rapid development of single-atom catalysts in recent years has paved the road for the embedding of noble metal atoms into the carbon matrix or other compounds. These heterostructures tend to feature superior electrochemical catalytic behaviors to those of traditional heterostructures containing nanoparticles with limited utilization of noble metals. Furthermore, directly depositing catalysts onto the conductive current collectors or gas diffusion layers is an effective way to enhance the electrical conductivities and restrain the side reactions of heterostructure cathodes, including carbon paper, nickel, titanium foams or meshes. TMD-based heterostructures are still less researched in Li–O2 batteries, but there has been a lot of progress in the field of HER catalysis and supercapacitors, which can bring some experience to the development of Li–O2 batteries. The main disadvantage of TMD materials is low conductivity as well as low specific surface area; the problem of low specific surface area can be solved by preparing porous nanostructures (nanoflower and hollow nanotube), and the low electrical conductivity can be enhanced by constructing heterostructures. The other component of the heterostructure should be carefully selected, such as high conductive composite (metal/carbon material/superconducting TMD), or phase modulation is recommended to obtain a mixed phase heterostructure (1T/2H) with improved electrical conductivity. In addition to the design of materials in terms of the function and category of the heterostructures, the dimensionality of the heterostructured material also has a significant impact on the electrochemical performance, and most heterostructured materials in Li–O2 batteries are currently of multiple dimensions; most of them are combinations of 3D + nD (n = 0, 1 and 3) materials. The large specific surface area of 1D or 2D materials increases the contact area with the electrolyte and improves the kinetics of ion transfer, in addition to the fact that they typically have a large number of exposed metal active sites and thus exhibit greater specific capacities. By combining the high electrical conductivity of single-walled carbon nanotubes, graphene, or MXene with TMOs or TMDs holding favorable redox activity, high catalytic performance can be achieved when individual shells or layers are assembled into stacked 1D or 2D heterogeneous structures.
3. At present, most research studies on the reaction mechanisms were carried out by means of theoretical calculations to obtain the reaction energy barrier, electronic state density distribution, charge distribution, with simulating the conversion kinetics of the corresponding device combined with the experimental results. It is essential to comprehend the mechanism of the catalytic process by theoretical calculations, while the parameter settings in the calculation processes determine the accuracy of the theoretical calculation results. The differences between the theoretical research models and the actual catalyst statements may lead to discrepancies, leading to inaccurate conclusions. Notably, the theoretical calculations are mainly based on the adsorption conditions and electrocatalytic reactions on the catalyst surfaces, but there are a lot of other factors that should also be involved, such as the dissolution of lithium in the anode, the heat transfer between the electrolyte and the electrodes, and even the changes in the electrolyte flow. Therefore, the entire Li–O2 battery system with relative influencing factors should be taken into consideration, and theoretical models and parameters need to be set as close as possible to actual conditions, reasonably offering the guidance for catalyst design and performance improvement.
4. In-depth and systematic characterization studies are highly demanded to intensively investigate Li–O2 battery reactions based on heterostructured catalysts, and typical advanced characterization techniques are listed in Fig. 9. In situ Raman could detect the changes in the types and contents of the deposited substances on the electrode surface during charge and discharge. In situ infrared is used to constantly detect the intermediate products of the catalytic cycle and the adsorption state of the real reaction. In situ XRD can qualitatively and quantitatively analyze the crystallization reaction product Li2O2 and other crystalline by-products. In situ TEM can also detect the change of the product structure to reveal the detailed nano-level mechanism at the atomic resolution and visualize the ORR and OER processes. In situ DEMS is used to analyze the gas consumption and emissions during battery cycling. In particular, these in situ technologies can help us determine the reaction pathway and the growth process of Li2O2 in the ORR and OER processes of Li–O2 batteries based on heterostructured catalysts, which can also be better compared with theoretical calculations.
5. Although heterostructures show great potential in Li–O2 batteries, some of the preparation procedures for the heterostructured catalysts consist of multiple steps, which largely affect the controllable modulation of the morphology of the materials. As shown in Table S1,† the synthesis routes reported in this review are based on the lab conditions rather than laboratory-scale production, and some of these methods require high energy consumption and cumbersome fabrication processes with low output (Fig. 9). Therefore, the development of green, mild and efficient preparation strategies is highly desired, and plays a vital role in realizing large-scale commercial applications of bi-functional electrocatalysts for Li–O2 batteries. In addition, the active centers of the homogeneous structural material with a single phase are assumed to be uniformly dispersed on the entire catalyst phases, while the heterostructure composed of multiple phases normally offers heterogeneous interfaces, making it more difficult to clarify the relative catalytic mechanism.
Author contributions
Jun Wang, Yao Liu, and Shulei Chou contributed to the conception of the study; Qing Xia contributed to the analysis and manuscript preparation; Deyuan Li, Lanling Zhao, Yuxin Long, Xue Han, Zhaorui Zhou, Yiming Zhang, Yebing Li and Abulgasim Ahmed Abbaker Adam performed the analysis with discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51971124), China Postdoctoral Science Foundation (2020M672054), Natural Science Foundation of Shandong Province (ZR2020QB122) and the Young Scholars Program of Shandong University (2019WLJH21).Notes and references
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- Y. J. Zhai, H. Tong, J. L. Deng, G. Y. Li, Y. Hou, R. H. Zhang, J. Wang, Y. Y. Lu, K. Liang, P. Chen, F. Dang and B. Kong, Energy Storage Materials, 2021, 43, 391–401 CrossRef.
- Y. Li, L. X. Wang, A. L. Song, M. R. Xia, Z. P. Li and G. J. Shao, Electrochim. Acta, 2018, 268, 268–275 CrossRef CAS.
- K. M. Naik, ACS Appl. Energy Mater., 2021, 4, 1014–1020 CrossRef CAS.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. S. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- Y. Wang, D. F. Ruiz Diaz, K. S. Chen, Z. Wang and X. C. Adroher, Mater. Today, 2020, 32, 178–203 CrossRef CAS.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. W. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- K. M. Naik, E. Higuchi and H. Inoue, J. Power Sources, 2020, 455, 227972 CrossRef CAS.
- S. L. Zhang and N. Pan, Adv. Energy Mater., 2015, 5, 1401401 CrossRef.
- F. Wang, X. Wu, X. Yuan, Z. Liu, Y. Zhang, L. Fu, Y. Zhu, Q. Zhou, Y. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- R. Wang, M. J. Yao and Z. Q. Niu, InfoMat, 2019, 2, 113–125 CrossRef.
- Z. Q. Yan, Z. H. Sun, K. C. Yue, A. Li, H. S. Liu, Z. H. Guo and L. Qian, Appl. Surf. Sci., 2021, 554, 149666 CrossRef CAS.
- D. Kim, K. Zhang, M. Cho and Y. M. Kang, Energy Environ. Sci., 2019, 12, 1326–1333 RSC.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- S. Kukunuri, K. Naik and S. Sampath, J. Mater. Chem. A, 2017, 5, 4660–4670 RSC.
- Y. G. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- T. P. Zhou, N. Zhang, C. Z. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC.
- S. R. Narayanan, G. K. S. Prakash, A. Manohar, B. Yang, S. Malkhandi and A. Kindler, Solid State Ionics, 2012, 216, 105–109 CrossRef CAS.
- D. R. Egan, C. P. de Leon, R. J. K. Wood, R. L. Jones, K. R. Stokes and F. C. Walsh, J. Power Sources, 2013, 236, 293–310 CrossRef CAS.
- T. R. Zhang, Z. L. Tao and J. Chen, Mater. Horiz., 2014, 1, 196–206 RSC.
- A. Zahoor, Z. K. Ghouri, S. Hashmi, F. Raza, S. Ishtiaque, S. Nadeem, I. Ullah and K. S. Nahm, ACS Sustainable Chem. Eng., 2019, 7, 14288–14320 CrossRef CAS.
- J. Lu, L. Li, J. B. Park, Y. K. Sun, F. Wu and K. Amine, Chem. Rev., 2014, 114, 5611–5640 CrossRef CAS PubMed.
- K. Song, D. A. Agyeman, M. Park, J. Yang and Y. M. Kang, Adv. Mater., 2017, 29, 1606572 CrossRef PubMed.
- B. Robert, A. Brian and L. F. Nazar, Adv. Energy Mater., 2012, 2, 801–815 CrossRef.
- Z. W. Chang, J. J. Xu, Q. C. Liu, L. Li and X. B. Zhang, Adv. Energy Mater., 2015, 5, 1500633 CrossRef.
- Y. J. Liu, P. He and H. S. Zhou, Adv. Energy Mater., 2018, 8, 1800089 CrossRef.
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS PubMed.
- A. Eftekhari and B. Ramanujam, J. Mater. Chem. A, 2017, 5, 7710–7731 RSC.
- F. J. Li and J. Chen, Adv. Energy Mater., 2017, 7, 1602934 CrossRef.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- Y. Wang, N. C. Lai, Y. R. Lu, Y. C. Zhou, C. L. Dong and Y. C. Lu, Joule, 2018, 2, 2364–2380 CrossRef CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J. M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- M. Yu, X. Ren, L. Ma and Y. Wu, Nat. Commun., 2014, 5, 5111 CrossRef CAS PubMed.
- M. Li, X. Wang, F. Li, L. Zheng, J. Xu and J. Yu, Adv. Mater., 2020, 32, 1907098 CrossRef CAS PubMed.
- Q. Lv, Z. Zhu, S. Zhao, L. Wang, Q. Zhao, F. Li, L. A. Archer and J. Chen, J. Am. Chem. Soc., 2021, 143, 1941–1947 CrossRef CAS PubMed.
- G. Wang, F. Tu, J. Xie, G. Du, S. Zhang, G. Cao and X. Zhao, Adv. Sci., 2016, 3, 1500339 CrossRef PubMed.
- C. T. Zhao, C. Yu, M. N. Banis, Q. Sun, M. D. Zhang, X. Li, Y. L. Liu, Y. Zhao, H. W. Huang, S. F. Li, X. T. Han, B. W. Xiao, Z. X. Song, R. Y. Li, J. S. Qiu and X. L. Sun, Nano Energy, 2017, 34, 399–407 CrossRef CAS.
- S. Song, W. Xu, J. Zheng, L. Luo, M. H. Engelhard, M. E. Bowden, B. Liu, C. M. Wang and J. G. Zhang, Nano Lett., 2017, 17, 1417–1424 CrossRef CAS PubMed.
- X. Hu, G. Luo, Q. Zhao, D. Wu, T. Yang, J. Wen, R. Wang, C. Xu and N. Hu, J. Am. Chem. Soc., 2020, 142, 16776–16786 CrossRef CAS PubMed.
- W. Zhao, J. Wang, R. Yin, B. Y. Li, X. S. Huang, L. L. Zhao and L. Qian, J. Colloid Interface Sci., 2020, 564, 28–36 CrossRef CAS PubMed.
- X. Y. Lu, L. Zhang, X. L. Sun, W. P. Si, C. L. Yan and O. G. Schmidt, J. Mater. Chem. A, 2016, 4, 4155–4160 RSC.
- X. M. Liu, L. L. Zhao, H. R. Xu, Q. S. Huang, Y. Q. Wang, C. X. Hou, Y. Y. Hou, J. Wang, F. Dang and J. T. Zhang, Adv. Energy Mater., 2020, 10, 2001415 CrossRef CAS.
- Y. Hou, J. Wang, C. X. Hou, Y. Q. Fan, Y. J. Zhai, H. Y. Li, F. Dang and S. L. Chou, J. Mater. Chem. A, 2019, 7, 6552–6561 RSC.
- Q. Xia, L. L. Zhao, D. Y. Li, J. Wang, L. L. Liu, C. X. Hou, X. M. Liu, H. R. Xu, F. Dang and J. T. Zhang, J. Mater. Chem. A, 2021, 9, 19922–19931 RSC.
- B. He, G. Y. Li, J. J. Li, J. Wang, H. Tong, Y. Q. Fan, W. L. Wang, S. H. Sun and F. Dang, Adv. Energy Mater., 2021, 11, 2003263 CrossRef CAS.
- S. Dong, X. Chen, K. J. Zhang, L. Gu, L. X. Zhang, X. H. Zhou, L. F. Li, Z. H. Liu, P. X. Han, H. X. Xu, J. H. Yao, C. J. Zhang, X. Y. Zhang, C. Q. Shang, G. L. Cui and L. Q. Chen, Chem. Commun., 2011, 47, 11291–11293 RSC.
- M. Idrees, S. Batool, J. Kong, Q. Zhuang, H. Liu, Q. Shao, N. Lu, Y. N. Feng, E. K. Wujcik, Q. Gao, T. Ding, R. B. Wei and Z. H. Guo, Electrochim. Acta, 2019, 296, 925–937 CrossRef CAS.
- J. T. Ren, L. Chen, C. C. Weng, G. G. Yuan and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2018, 10, 33276–33286 CrossRef CAS PubMed.
- C. X. Hou, J. Wang, W. Du, J. C. Wang, Y. Du, C. T. Liu, J. X. Zhang, H. Hou, F. Dang, L. L. Zhao and Z. H. Guo, J. Mater. Chem. A, 2019, 7, 13460–13472 RSC.
- H. D. Lim, K. Y. Park, H. Song, E. Y. Jang, H. Gwon, J. Kim, Y. H. Kim, M. D. Lima, R. Ovalle Robles, X. Lepro, R. H. Baughman and K. Kang, Adv. Mater., 2013, 25, 1348–1352 CrossRef CAS PubMed.
- H. Wang, X. Wang, M. Li, L. Zheng, D. Guan, X. Huang, J. Xu and J. Yu, Adv. Mater., 2020, 32, 2002559 CrossRef CAS PubMed.
- J. J. Xu, Z. L. Wang, D. Xu, L. L. Zhang and X. B. Zhang, Nat. Commun., 2013, 4, 2438 CrossRef PubMed.
- D. S. Geng, N. Ding, T. S. A. Hor, S. W. Chien, Z. L. Liu, D. Wuu, X. L. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef.
- P. Zhang, Y. Zhao and X. Zhang, Chem. Soc. Rev., 2018, 47, 2921–3004 RSC.
- K. X. Wang, Q. C. Zhu and J. S. Chen, Small, 2018, 14, 1800078 CrossRef PubMed.
- S. Hyun, B. Son, H. Kim, J. Sanetuntikul and S. Shanmugam, Appl. Catal., B, 2020, 263, 118283 CrossRef CAS.
- D. Wang, X. W. Mu, P. He and H. S. Zhou, Mater. Today, 2019, 26, 87–99 CrossRef CAS.
- X. Jin, M. Park, S. J. Shin, Y. Jo, M. G. Kim, H. Kim, Y. M. Kang and S. J. Hwang, Small, 2020, 16, 1903265 CrossRef CAS PubMed.
- R. Gao, D. Zhou, D. Ning, W. J. Zhang, L. Huang, F. Sun, G. Schuck, G. Schumacher, Z. B. Hu and X. F. Liu, Adv. Funct. Mater., 2020, 30, 2002223 CrossRef CAS.
- X. Y. Lu, Y. Yin, L. Zhang, S. Z. Huang, L. X. Xi, L. X. Liu, S. Oswald and O. G. Schmidt, Energy Storage Materials, 2019, 16, 155–162 CrossRef.
- M. H. Park, C. P. Liang, T. H. Lee, D. A. Agyeman, J. H. Yang, V. W. Lau, S. Choi, H. W. Jang, K. Cho and Y. M. Kang, Adv. Energy Mater., 2020, 10, 1903225 CrossRef CAS.
- C. Zhao, Y. Zhu, Q. Sun, C. Wang, J. Luo, X. Lin, X. Yang, Y. Zhao, R. Li, S. Zhao, H. Huang, L. Zhang, S. Lu, M. Gu and X. Sun, Angew. Chem., Int. Ed. Engl., 2021, 60, 5821–5826 CrossRef CAS PubMed.
- D. Cao, L. Zheng, Q. Li, J. Zhang, Y. Dong, J. Yue, X. Wang, Y. Bai, G. Tan and C. Wu, Nano Lett., 2021, 21, 5225–5232 CrossRef CAS PubMed.
- Y. Ma, J. T. Li, X. B. Liao, W. Luo, W. Z. Huang, J. S. Meng, Q. Chen, S. B. Xi, R. H. Yu, Y. Zhao, L. Zhou and L. Q. Mai, Adv. Funct. Mater., 2020, 30, 2005000 CrossRef CAS.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2020, 32, 1905679 CrossRef CAS PubMed.
- Y. Y. Guo, P. F. Yuan, J. A. Zhang, H. C. Xia, F. Y. Cheng, M. F. Zhou, J. Li, Y. Y. Qiao, S. C. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- J. Li, G. S. Li, J. H. Wang, C. L. Xue, X. S. Li, S. Wang, B. Q. Han, M. Yang and L. P. Li, Inorg. Chem. Front., 2020, 7, 191–197 RSC.
- H. P. Guo, W. B. Luo, J. Chen, S. L. Chou, H. K. Liu and J. Z. Wang, Adv. Sustainable Syst., 2018, 2, 1700183 CrossRef.
- P. Tan, M. L. Liu, Z. P. Shao and M. Ni, Adv. Energy Mater., 2017, 7, 1602674 CrossRef.
- B. Liu, Y. L. Sun, L. Liu, S. Xu and X. B. Yan, Adv. Funct. Mater., 2018, 28, 1704973 CrossRef.
- J. Lai, Y. Xing, N. Chen, L. Li, F. Wu and R. Chen, Angew. Chem., Int. Ed. Engl., 2020, 59, 2974–2997 CrossRef CAS PubMed.
- N. B. Aetukuri, B. D. McCloskey, J. M. Garcia, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- Q. Xiong, G. Huang and X. B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19311–19319 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- Z. Q. Peng, S. A. Freunberger and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- W. Yu, H. Wang, J. Hu, W. Yang, L. Qin, R. Liu, B. Li, D. Zhai and F. Kang, ACS Appl. Mater. Interfaces, 2018, 10, 7989–7995 CrossRef CAS PubMed.
- D. G. Kwabi, M. Tulodziecki, N. Pour, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2016, 7, 1204–1212 CrossRef CAS PubMed.
- M. Augustin, P. E. Vullum, F. Vullum-Bruer and A. M. Svensson, J. Power Sources, 2019, 414, 130–140 CrossRef CAS.
- Z. Y. Lyu, Y. Zhou, W. R. Dai, X. H. Cui, M. Lai, L. Wang, F. W. Huo, W. Huang, Z. Hu and W. Chen, Chem. Soc. Rev., 2017, 46, 6046–6072 RSC.
- Z. Y. Lyu, L. J. Yang, Y. P. Luan, X. R. Wang, L. J. Wang, Z. Hu, J. P. Lu, S. N. Xiao, F. Zhang, X. Z. Wang, F. W. Huo, W. Huang, Z. Hu and W. Chen, Nano Energy, 2017, 36, 68–75 CrossRef CAS.
- P. Wang, Y. Ren, R. Wang, P. Zhang, M. Ding, C. Li, D. Zhao, Z. Qian, Z. Zhang, L. Zhang and L. Yin, Nat. Commun., 2020, 11, 1576 CrossRef CAS PubMed.
- H. J. Shin, W. J. Kwak, D. Aurbach and Y. K. Sun, Adv. Funct. Mater., 2017, 27, 1605500 CrossRef.
- B. He, J. Wang, J. Q. Liu, Y. Li, Q. S. Huang, Y. Hou, G. Y. Li, J. J. Li, R. H. Zhang, J. J. Zhou, W. Tian, Y. Du, F. Dang, H. C. Wang and B. Kong, Adv. Energy Mater., 2020, 10, 1904262 CrossRef CAS.
- Y. Wang, J. Wang, Z. Mohamed, Q. S. Huang, T. T. Chen, Y. Y. Hou, F. Dang, W. B. Zhang and H. C. Wang, Appl. Mater. Today, 2020, 19, 100603 CrossRef.
- Y. B. Zhai, J. Wang, Q. Gao, Y. Fan, C. Hou, Y. Hou, H. Liu, Q. Shao, S. Wu, L. Zhao, T. Ding, F. Dang and Z. Guo, J. Catal., 2019, 377, 534–542 CrossRef CAS.
- G. Oh, S. Seo, W. Kim, Y. Cho, H. Kwon, S. Kim, S. Noh, E. Kwon, Y. Oh, J. Song, J. Lee and K. Ryu, ACS Appl. Mater. Interfaces, 2021, 13, 13200–13211 CrossRef CAS PubMed.
- K. R. Yoon, K. Shin, J. Park, S. H. Cho, C. Kim, J. W. Jung, J. Y. Cheong, H. R. Byon, H. M. Lee and I. D. Kim, ACS Nano, 2018, 12, 128–139 CrossRef CAS PubMed.
- S. Q. Zhao, L. Zhang, G. N. Zhang, H. B. Sun, J. Y. Yang and S. G. Lu, J. Energy Chem., 2020, 45, 74–82 CrossRef.
- X. K. Yu, X. Chen, D. B. Buchholz, Q. Q. Li, J. S. Wu, P. A. Fenter, M. J. Bedzyk, V. P. Dravid and S. A. Barnett, ACS Appl. Nano Mater., 2018, 1, 642–653 CrossRef CAS.
- S. Park, C. W. Lee, J. C. Kim, H. J. Song, H. W. Shim, S. Lee and D. W. Kim, ACS Energy Lett., 2016, 1, 216–224 CrossRef CAS.
- Y. H. Cao, X. R. Zheng, H. X. Zhang, J. F. Zhang, X. P. Han, C. Zhong, W. B. Hu and Y. D. Deng, J. Power Sources, 2019, 437, 226893 CrossRef CAS.
- L. An, Y. X. Li, M. C. Luo, J. Yin, Y. Q. Zhao, C. L. Xu, F. Y. Cheng, Y. Yang, P. X. Xi and S. J. Guo, Adv. Funct. Mater., 2017, 27, 1703779 CrossRef.
- R. X. Liang, C. Z. Shu, A. J. Hu, M. L. Li, Z. Q. Ran, R. X. Zheng and J. P. Long, Chem. Eng. J., 2020, 393, 124592 CrossRef CAS.
- T. A. Shifa, F. Wang, Y. Liu and J. He, Adv. Mater., 2019, 31, 1804828 CrossRef CAS PubMed.
- J. Su, G. D. Li, X. H. Li and J. S. Chen, Adv. Sci., 2019, 6, 1801702 CrossRef PubMed.
- Y. J. Ding, W. Y. Yang, S. Gao, W. Z. Sun, C. X. Sun and Q. Li, ACS Appl. Energy Mater., 2020, 3, 1328–1337 CrossRef CAS.
- P. Zhang, S. Zhang, M. He, J. Lang, A. Ren, S. Xu and X. Yan, Adv. Sci., 2017, 4, 1700172 CrossRef PubMed.
- G. H. Lee, M. C. Sung, J. C. Kim, H. J. Song and D. W. Kim, Adv. Energy Mater., 2018, 8, 1801930 CrossRef.
- W. Zhao, X. Li, R. Yin, L. Qian, X. Huang, H. Liu, J. Zhang, J. Wang, T. Ding and Z. Guo, Nanoscale, 2018, 11, 50–59 RSC.
- Y. Hou, J. Wang, J. Q. Liu, C. X. Hou, Z. H. Xiu, Y. Q. Fan, L. L. Zhao, Y. J. Zhai, H. Y. Li, J. Zeng, X. Gao, S. Zhou, D. W. Li, Y. Li, F. Dang, K. Liang, P. Chen, C. M. Li, D. Y. Zhao and B. Kong, Adv. Energy Mater., 2019, 9, 1901751 CrossRef CAS.
- C. H. Ahn, R. S. Kalubarme, Y. H. Kim, K. N. Jung, K. H. Shin and C. J. Park, Electrochim. Acta, 2014, 117, 18–25 CrossRef CAS.
- C. Cao, J. Xie, S. C. Zhang, B. Pan, G. S. Cao and X. B. Zhao, J. Mater. Chem. A, 2017, 5, 6747–6755 RSC.
- Q. C. Wang, Y. P. Lei, Y. C. Wang, Y. Liu, C. Y. Song, J. Zeng, Y. H. Song, X. D. Duan, D. S. Wang and Y. D. Li, Energy Environ. Sci., 2020, 13, 1593–1616 RSC.
- C. Zhao, Y. Li, W. Zhang, Y. Zheng, X. Lou, B. Yu, J. Chen, Y. Chen, M. Liu and J. Wang, Energy Environ. Sci., 2020, 13, 53–85 RSC.
- P. Wang, C. Li, S. Dong, X. Ge, P. Zhang, X. F. Miao, R. Wang, Z. Zhang and L. Yin, Adv. Energy Mater., 2019, 9, 1900788 CrossRef.
- A. Hu, W. Lv, T. Lei, W. Chen, Y. Hu, C. Shu, X. Wang, L. Xue, J. Huang, X. Du, H. Wang, K. Tang, C. Gong, J. Zhu, W. He, J. Long and J. Xiong, ACS Nano, 2020, 14, 3490–3499 CrossRef CAS PubMed.
- R. X. Liang, A. J. Hu, M. L. Li, Z. Q. Ran, C. Z. Shu and J. Long, Electrochim. Acta, 2019, 321, 134716 CrossRef CAS.
- W. Yang, X. Li, Y. Li, R. Zhu and H. Pang, Adv. Mater., 2019, 31, 1804740 Search PubMed.
- Z. H. Bi, Q. Q. Kong, Y. F. Cao, G. H. Sun, F. Y. Su, X. X. Wei, X. M. Li, A. L. Ahmad, L. J. Xie and C. M. Chen, J. Mater. Chem. A, 2019, 7, 16028–16045 RSC.
- G. Xue, Y. Xu, T. Ding, J. Li, J. Yin, W. Fei, Y. Cao, J. Yu, L. Yuan, L. Gong, J. Chen, S. Deng, J. Zhou and W. Guo, Nat. Nanotechnol., 2017, 12, 317–321 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. Zheng, H. Song, S. Chen, X. Yu, J. Zhu, J. Xu, K. A. I. Zhang, C. Zhang and T. Liu, Small, 2020, 16, 2004342 CrossRef CAS PubMed.
- G. Tan, L. Chong, R. Amine, J. Lu, C. Liu, Y. Yuan, J. Wen, K. He, X. Bi, Y. Guo, H. H. Wang, R. Shahbazian-Yassar, S. Al Hallaj, D. J. Miller, D. Liu and K. Amine, Nano Lett., 2017, 17, 2959–2966 CrossRef CAS PubMed.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 4394–4403 CrossRef CAS PubMed.
- X. D. Lin, R. M. Yuan, S. R. Cai, Y. H. Jiang, J. Lei, S. G. Liu, Q. H. Wu, H. G. Liao, M. S. Zheng and Q. F. Dong, Adv. Energy Mater., 2018, 8, 1800089 CrossRef.
- J. H. Kim, Y. J. Oh and Y. C. Kang, Carbon, 2018, 128, 125–133 CrossRef CAS.
- R. X. Liang, C. Z. Shu, A. J. Hu, C. X. Xu, R. X. Zheng, M. L. Li, Y. W. Guo, M. He, Y. Yan and J. P. Long, J. Mater. Chem. A, 2020, 8, 11337–11345 RSC.
- D. A. Agyeman, M. Park and Y. M. Kang, J. Mater. Chem. A, 2017, 5, 22234–22241 RSC.
- Y. Cong, Q. Tang, X. Wang, M. Liu, J. Liu, Z. Geng, R. Cao, X. Zhang, W. Zhang, K. Huang and S. Feng, ACS Catal., 2019, 9, 11743–11752 CrossRef CAS.
- J. J. Xu, Z. W. Chang, Y. B. Yin and X. B. Zhang, ACS Cent. Sci., 2017, 3, 598–604 CrossRef CAS PubMed.
- A. J. Hu, C. Z. Shu, X. M. Qiu, M. L. Li, R. X. Zheng and J. P. Long, ACS Sustainable Chem. Eng., 2019, 7, 6929–6938 CrossRef CAS.
- P. Wang, C. Li, S. Dong, X. Ge, P. Zhang, X. Miao, Z. Zhang, C. Wang and L. Yin, Small, 2019, 15, 1900001 CrossRef PubMed.
- C. Wu, Y. Y. Hou, J. C. Jiang, H. P. Guo, H. K. Liu, J. Chen and J. Z. Wang, J. Power Sources, 2020, 470, 228317 CrossRef CAS.
- W. J. Kwak, K. C. Lau, C. D. Shin, K. Amine, L. A. Curtiss and Y. K. Sun, ACS Nano, 2015, 9, 4129–4173 CrossRef CAS PubMed.
- Q. S. Huang, F. Dang, H. T. Zhu, L. L. Zhao, B. He, Y. Wang, J. Wang and X. M. Mai, J. Power Sources, 2020, 451, 227738 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Perez-Ramirez and J. Lu, Nat. Nanotechnol., 2021 DOI:10.1038/s41565-021-01022-y.
- T. Ling, P. F. Da, X. L. Zheng, B. H. Ge, Z. P. Hu, M. Y. Wu, X. W. Du, W. B. Hu, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2018, 4, eaau6261 CrossRef CAS PubMed.
- E. Lee, Y. S. Yoon and D. J. Kim, ACS Sens., 2018, 3, 2045–2060 CrossRef CAS PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed.
- Z. Q. Ran, C. Z. Shu, Z. Q. Hou, L. J. Cao, R. X. Liang, J. B. Li, P. Hei, T. S. Yang and J. P. Long, J. Power Sources, 2020, 468, 228308 CrossRef CAS.
- Y. Zhang, J. Ma, M. W. Yuan, Y. Li, R. A. Shen, W. C. Cheong, T. Han, G. B. Sun, C. Chen and C. Y. Nan, Chem. Commun., 2019, 55, 12683–12686 RSC.
- Y. Y. Dou, X. G. Wang, D. S. Wang, Q. M. Zhang, C. Y. Wang, G. Chen, Y. J. Wei and Z. Zhou, Chem. Eng. J., 2021, 409, 128145 CrossRef CAS.
- H. Cheng, J. Xie, G. S. Cao, Y. H. Lu, D. Zheng, Y. Jin, K. Y. Wang and X. B. Zhao, Energy Storage Materials, 2019, 23, 684–692 CrossRef.
- X. Y. Jin, D. A. Agyeman, S. Kim, Y. H. Kim, M. G. Kim, Y. M. Kang and S. J. Hwang, Nano Energy, 2020, 67, 104192 CrossRef CAS.
- M. Lee, Y. J. Hwang, K. H. Yun and Y. C. Chung, J. Power Sources, 2016, 307, 379–384 CrossRef CAS.
- V. Veeramani, Y. H. Chen, H. C. Wang, T. F. Hung, W. S. Chang, D. H. Wei, S. F. Hu and R. S. Liu, Chem. Eng. J., 2018, 349, 235–240 CrossRef CAS.
- K. R. Yoon, G. Y. Lee, J. W. Jung, N. H. Kim, S. O. Kim and I. D. Kim, Nano Lett., 2016, 16, 2076–2083 CrossRef CAS PubMed.
- P. Prabhu, V. Jose and J.-M. Lee, Matter, 2020, 2, 526–553 CrossRef.
- C. Xu, S. J. Peng, C. L. Tan, H. X. Ang, H. T. Tan, H. Zhang and Q. Y. Yan, J. Mater. Chem. A, 2014, 2, 5597–5601 RSC.
- H. Tang, K. P. Dou, C. C. Kaun, Q. Kuang and S. H. Yang, J. Mater. Chem. A, 2014, 2, 360–364 RSC.
- L. P. Pi, L. Li, K. L. Liu, Q. F. Zhang, H. Q. Li and T. Y. Zhai, Adv. Funct. Mater., 2019, 29, 1904932 CrossRef CAS.
- F. C. Liu, J. D. Zhou, C. Zhu and Z. Liu, Adv. Funct. Mater., 2017, 27, 1602404 CrossRef.
- X. Wang, Z. Wang, J. Zhang, X. Wang, Z. Zhang, J. Wang, Z. Zhu, Z. Li, Y. Liu, X. Hu, J. Qiu, G. Hu, B. Chen, N. Wang, Q. He, J. Chen, J. Yan, W. Zhang, T. Hasan, S. Li, H. Li, H. Zhang, Q. Wang, X. Huang and W. Huang, Nat. Commun., 2018, 9, 3611 CrossRef PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- B. Wang, T. T. Ruan, Y. Chen, F. Jin, L. Peng, Y. Zhou, D. L. Wang and S. X. Dou, Energy Storage Materials, 2020, 24, 22–51 CrossRef.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263–275 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05781b |
| This journal is © The Royal Society of Chemistry 2022 |