
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal encoding of next generation DNA-encoded chemical libraries†
Louise
Plais
 a,
Alice
Lessing
a,
Michelle
Keller
a,
Adriano
Martinelli
a,
Sebastian
Oehler
a,
Gabriele
Bassi
b,
Dario
Neri
*b and
Jörg
Scheuermann
*a
a,
Alice
Lessing
a,
Michelle
Keller
a,
Adriano
Martinelli
a,
Sebastian
Oehler
a,
Gabriele
Bassi
b,
Dario
Neri
*b and
Jörg
Scheuermann
*a
aDepartment of Chemistry and Applied Biosciences, Swiss Federal Institute of Technology (ETH Zürich), Vladimir-Prelog-Weg 4, CH-8093 Zürich, Switzerland. E-mail: joerg.scheuermann@pharma.ethz.ch
bPhilochem AG, Libernstrasse 3, CH-8112 Otelfingen, Switzerland. E-mail: dario.neri@philogen.com
First published on 11th January 2022
Abstract
DNA-encoded chemical libraries (DELs) are useful tools for the discovery of small molecule ligands to protein targets of pharmaceutical interest. Compared with single-pharmacophore DELs, dual-pharmacophore DELs simultaneously display two chemical moieties on both DNA strands, and allow for the construction of highly diverse and pure libraries, with a potential for targeting larger protein surfaces. Although methods for the encoding of simple, fragment-like dual-display libraries have been established, more complex libraries require a different encoding strategy. Here, we present a robust and convenient “large encoding design” (LED), which facilitates the PCR-amplification of multiple codes distributed among two partially complementary DNA strands. We experimentally implemented multiple coding regions and we compared the new DNA encoding scheme with previously reported dual-display DEL modalities in terms of amplifiability and performance in test selections against two target proteins. With the LED methodology in place, we foresee the construction and screening of DELs of unprecedented sizes and designs.
Introduction
DNA-encoded chemical libraries (DELs) are collections of small molecules, each connected to a unique encoding DNA sequence.1 As all compounds are individually encoded, this system allows for the efficient one-pot screening of an unprecedented number of compounds by affinity-based selections on a protein target of interest. The identity of the selected molecules can be retrieved by PCR amplification of the selected DNA tags and subsequent analysis by high-throughput DNA sequencing.2,3 DEL technology is increasingly recognized and implemented as an important tool for the de novo discovery of ligands.4–8 However, obtaining ligands for “difficult” targets – e.g., protein–protein interactions – may remain challenging when using Lipinski's rule of five compliant single-pharmacophore DELs, displaying typically compounds consisting of two or three sets of diversity elements.9–12 While some approaches are currently explored to create better performing single-pharmacophore DELs, for example by developing new DNA-compatible reactions,13–15 adopting diverse scaffolds and building blocks,16–18 or exploring different spatial building block arrangements,10,19,20 another promising option to tackle difficult targets may be exploring larger binding surfaces.21 This has been attempted, for instance, by generating macrocyclic DELs,16,22–25 but large libraries in general will require the introduction of several sets of building blocks, thereby lowering DEL library quality.The most widely used setup is the single-pharmacophore DEL, built on a “headpiece” linker on double-stranded-DNA, and it is well-suited for defined protein pockets. An attractive strategy to access larger protein surfaces may consist in the simultaneous display of two pharmacophores (“dual display”) as implemented in Encoded Self-Assembling Chemical (ESAC) libraries (Fig. 1A).26,27 Dual-display libraries feature the combinatorial assembly of independent sub-libraries, which allows to form large libraries containing multiple diversity elements.28 These large dual-display DELs can be obtained in high quality with rather little synthetic effort. Since building block coupling occurs separately on both strands of the DNA heteroduplex, the self-assembly of mutually complementary DELs may allow the construction of larger and purer combinatorial libraries, compared to conventional split-and-pool single-pharmacophore DELs.26,27,29–31 While dual-display technology requires the optimization of the linkage of the displayed pharmacophores,32 the technology may allow for the versatile combination of two single-stranded DELs. To this aim, recent work from our group has explored encoding modalities to assemble pre-existing single-stranded DELs.33
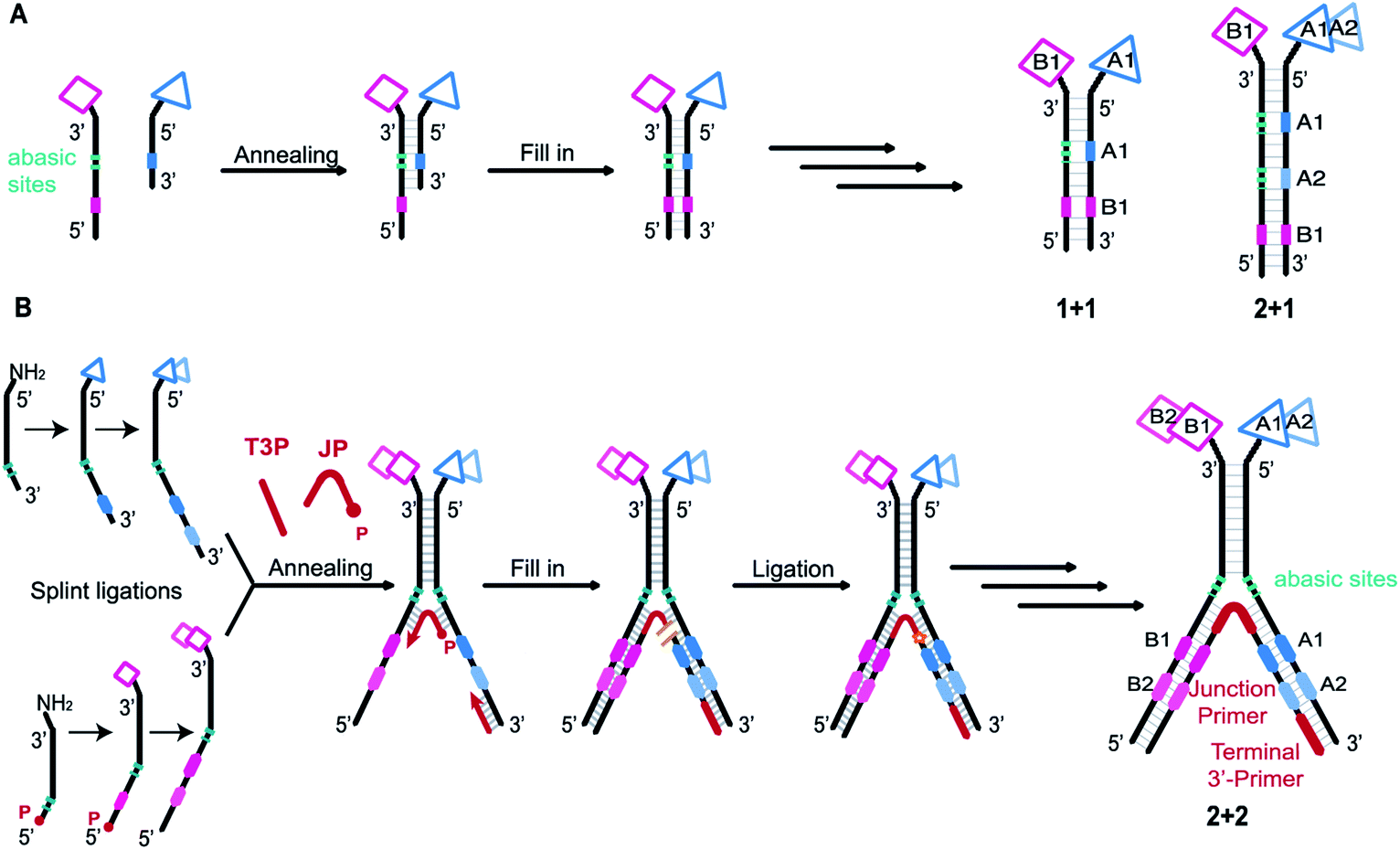 | ||
| Fig. 1 (A) Previous work. Schematic representation of a conventional ESAC library construction and examples of ESAC formats with “1 + 1” or “2 + 1” chemical building blocks. Two complementary sub-libraries each bearing one building block are annealed. The first coding region (in blue) is opposed to abasic sites while the second coding region (in pink) can be transcribed onto the blue strand by fill-in polymerisation, thus creating a unique DNA strand bearing all the coding information for subsequent PCR amplification and DNA sequencing. This scheme could be applied to “1 + 1” or “2 + 1” setups. (B) This work. Schematic representation of LED library construction and “2 + 2” LED setup used for this study. Two partially complementary sub-libraries are synthesized, hybridized and annealed with a junction primer (JP) and a terminal 3′-primer (T3P). Using T4 DNA polymerase the coding information is transferred onto two DNA strands which are subsequently ligated by T4 DNA ligase. P: phosphate; JP: junction primer; T3P: terminal 3′-primer. | ||
Initially, self-assembling dual-display DELs described the combination of two sets of building blocks in a “1 + 1” setting, one on each complementary DNA strand, as depicted in Fig. 1A.27,31 Then combinatorial display of small molecule fragments was investigated with DNA-templated synthesis34–37 but DNA-recorded synthesis eventually proved to be a more versatile strategy for the construction of large DELs. More recently, the ESAC construct was further developed with the combination of a conventional, single-stranded DEL of two diversity elements, paired with a sub-library of one building block on the complementary strand, in a “2 + 1” setting.38 For the creation of even larger dual-display DELs, this conventional encoding scheme cannot be easily adapted (e.g., for a “2 + 2” or any “m + n” setting) nor efficiently implemented.
We here propose a new encoding strategy named “Large Encoding Design” (LED), which offers a convenient solution for the construction of dual-display DELs of unprecedented size. We were inspired by the works of Xiaoyu Li's39–42 and Yixin Zhang's43,44 laboratories on Y-shaped dynamic self-assembling DELs featuring a three-way junction approach. In the dynamic setting, partially complementary sub-libraries bearing one set of building blocks each were allowed to pair in the presence of a target protein and the coding regions of both sub-libraries could be joined after selection into a unique, amplifiable DNA strand.
In contrast, LED features the stable display of two (or more) sets of diversity elements on complementary DNA strands, allowing to produce and store large combinatorial dual-display DELs which can then be submitted to affinity-based selections against the protein targets of interest.
We implemented LED with a unique set of four codes and subsequently tested the system in model selection experiments, while systematically studying selection conditions, in order to establish robust methods for DEL construction and screening. As high-quality DEL screenings depend on the reliable and sensitive amplification of the DNA after selections,45 we identified the minimum amount of DNA construct which could be confidently PCR amplified. We next investigated the minimum amount of DNA which is required for a proper selection experiment and we finally determined the highest possible dilution for a known binder to be retrieved from a dual-display library. To that aim we produced model libraries and compared the LED performance with conventional ESAC model libraries in selections against alpha-glycoprotein 1 (AGP) and carbonic anhydrase II (CAII) thereby demonstrating the potential of LED for future, ambitious dual-display DELs.
Results and discussion
An extended and versatile encoding system
As in the original ESAC approach, LED features the stable display of two combinatorially assembled pharmacophores on complementary DNA strands. Each strand contains a chemical moiety for building block attachment, a hybridization region, an abasic region and one or more coding regions (Fig. 1B). The codes can consecutively be added by splint ligation and encode for the building blocks displayed on their respective DNA strand only. As graphically depicted in Fig. 1B, the process for LED-based DEL library construction starts with the synthesis and encoding of each of the two sub-libraries on its respective DNA strand. Subsequently, the sub-libraries are annealed together with a terminal 3′-primer (T3P) and a junction primer (JP). DNA polymerase is used to transfer the DNA codes onto the growing primer strands and finally a DNA ligase links the two synthesized DNA strands to form a single amplifiable strand bearing all identification codes. After the affinity-based DEL selection, this strand can be PCR-amplified and sequenced for hit identification.Implementation
To demonstrate the feasibility of LED, we rehearsed the entire process for two identical sets of codes per DNA strand and we monitored the steps by LC-MS or analytical agarose gel electrophoresis. First, all the required DNA components were formed, starting with the strands II and IV (Fig. 2A). They were built from universal oligonucleotides which possess a hybridization region, four abasic d-spacer positions and an overhang of ten nucleotides. With the help of an adaptor, splint ligations were performed between this overhang and a first DNA coding sequence, to obtain strands I and III. Another splint ligation was performed to add the second code, in order to obtain II and IV, respectively. From this point, the sub-libraries were ready to be paired, so the full-length DNA strands II and IV were annealed together through their hybridization regions. The junction primer (JP) and the terminal 3′-primer (T3P) were allowed to anneal as well, to form X which was subsequently submitted to T4 DNA-polymerase mediated polymerization to yield product XI, and finally to T4 DNA ligase-mediated ligation, yielding XII (Fig. 2B). In XII, the strand depicted in orange bears all the necessary coding information and can be amplified to the PCR product XIII (Fig. 2C). The formation of DNA strands II and IV, as well as the amplification product XIII were easily monitored by analytical agarose gel, while X, XI and XII could be extensively characterized by LC-MS. It is noteworthy to mention that in an actual library setting, selection experiments are performed after the self-assembling step, i.e. after the stable formation of XII. With this first experiment we show that the LED encoding could be performed reliably and quantitatively.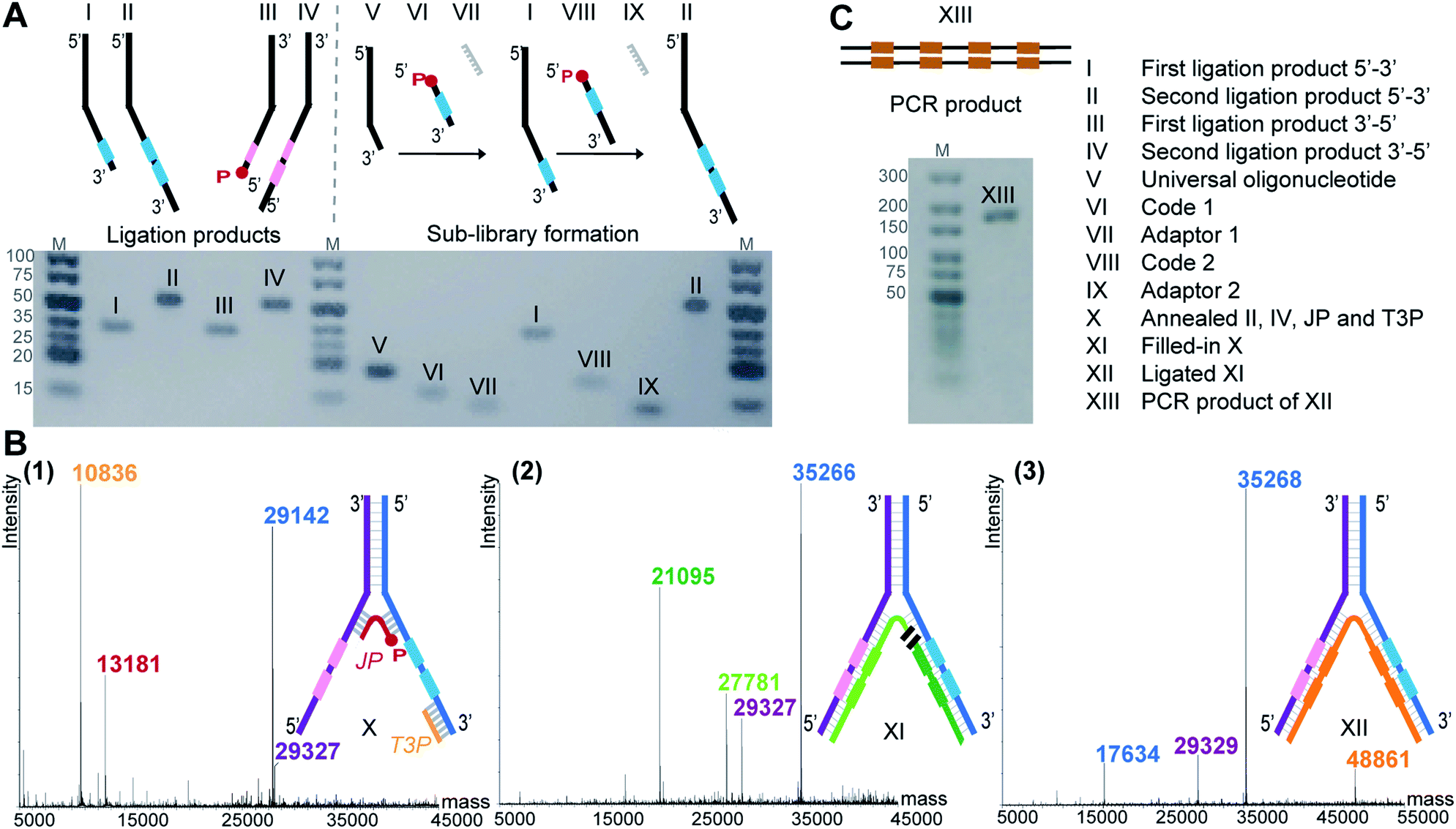 | ||
| Fig. 2 (A) Agarose gel image (3%) of individual ligation products (I–IV) and of the exemplary preparation of one sub-library (V–VI–VII–I–VIII–IX–II). (B) Formation of the amplifiable DNA strand containing all coding information. (1–3) Deconvoluted LC-MS spectra of (1) assembled strands II, IV, junction primer (JP) and terminal 3′-primer (T3P) forming X, (2) T4 DNA polymerase-mediated fill-in of X yields XI, (3) T4 DNA ligase-mediated ligation of the green XI fragments yields XII. (C) The orange fragment of XII is submitted to PCR yielding product XIII. Agarose gel image (2%). | ||
Optimization
In contrast to the dynamic setting proposed by Li's group39 we used a stable hybridization region, in line with the ESAC setup previously described by our group.27 Therefore, the LED components are annealed together and submitted to T4 DNA polymerase and T4 DNA ligase before the selection process. This allows for higher concentrations for the enzymatic conversions and for a proper assessment of the library homogeneity. In addition, four abasic nucleotides are added between the coding and the annealing regions in order to avoid the formation of side products when using the T4 DNA polymerase (Fig. 2B, (2)). Finally, a second code was added to each DNA strand, permitting the combinatorial assembly of “2 + 2” (and possibly even more) sets of diversity elements.The annealing length of the junction primer (JP) to strands II and IV proved to be critical, as the original ten nucleotides sequence led to the formation of side products (depicted in orange in Fig. 3A), since the T4 DNA polymerase was able to displace the JP. Such side product leads to a reduction of product XII, and hence to a critically reduced amount of DNA that can be sequenced after a DEL selection. In a first attempt to prevent the formation of this side product, two abasic nucleotides were added on strand II, just before the annealing region to the JP (Fig. 3B). Even though the formation of the impurity was suppressed, we found that the T4 DNA ligase was now unable to form XII, rendering the subsequent PCR amplification impossible. Finally, we decided to increase the length of the JP annealing sequence from ten to twenty bases. The concomitant increase in annealing strength was sufficient to prevent any JP displacement and to ultimately lead to the formation of desired XII (Fig. 3C).
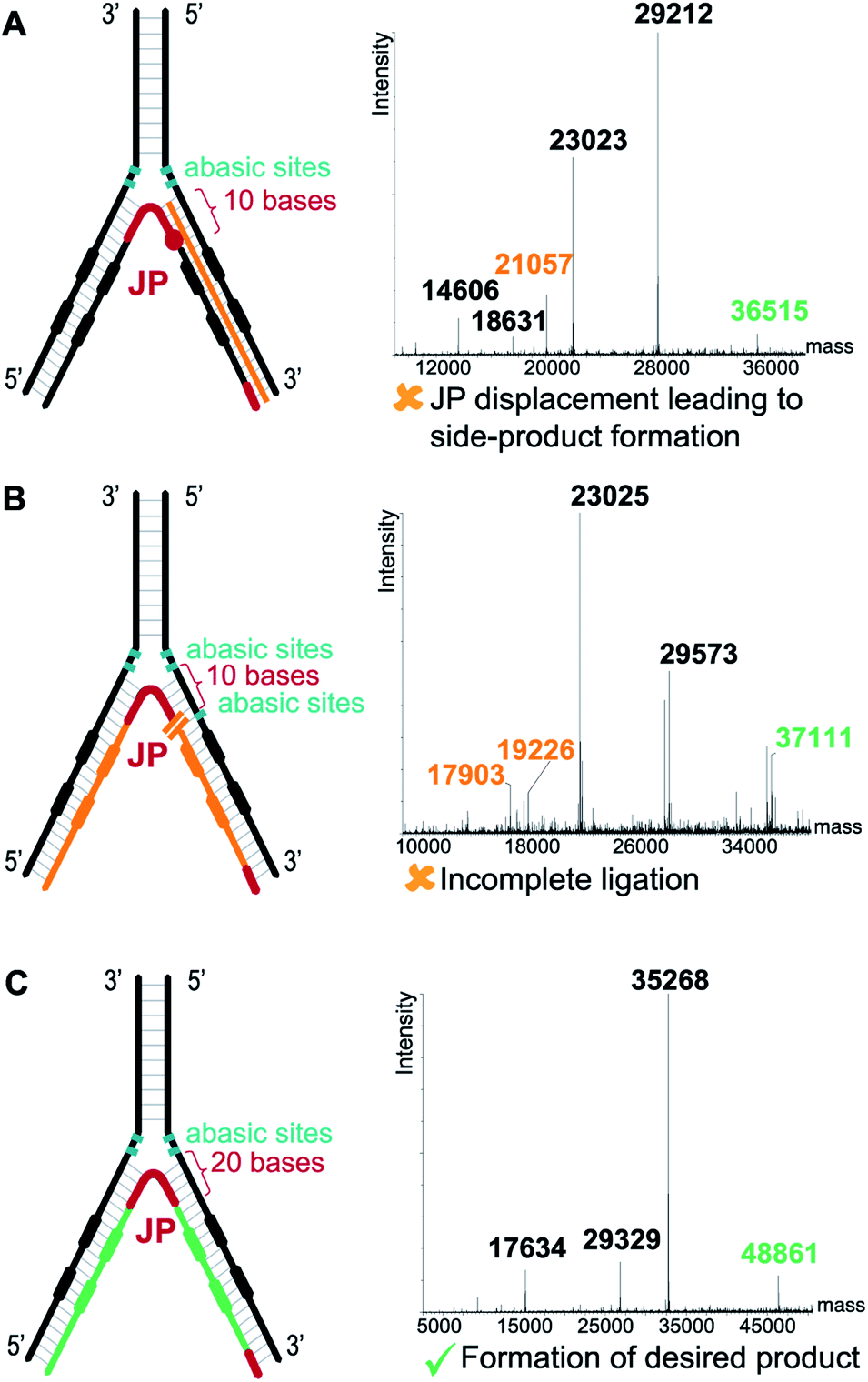 | ||
| Fig. 3 Critical optimization of the junction primer (JP) system. (A) With a 10-base annealing region the JP is displaced by the T4 DNA polymerase, leading to side product formation (orange strand). (B) Two abasic sites (in green) on the 5′ DNA strand prevent JP displacement by the T4 DNA polymerase but hampers the subsequent ligation step. (C) Increasing the JP annealing region to 20 bases prevents displacement. Right panels show the deconvoluted LC-MS spectra with the orange labels depicting the side products/non-ligated fragments, the green labels depicting the desired ligated products. | ||
Quality of the essential ligation step
If more than one set of codes is used, as in the case of an actual DEL, a quantitative ligation step is crucial to assure the correct decoding of simultaneously selected dual-pharmacophores. When, for instance, ligation is incomplete, non-ligated fragments can shuffle in the PCR steps following selections and lead to incorrect combinations of pharmacophores. We assessed this problem for two constructs with distinct sets of codes and hence different molecular masses. Each construct was assembled separately to produce the two distinct constructs represented in dark blue and light blue in Fig. 4. These constructs were then submitted to T4 polymerase and T4 ligase separately as well as a mixture (mimicking a LED library), yielding final constructs which were subsequently PCR-amplified. The PCR products were analyzed by LC-MS and could easily be distinguished for the separate constructs (Fig. 4A and B). The mixture of the constructs showed the expected masses of non-shuffled constructs, as expected from a quantitative ligation (Fig. 4C). However, in a control experiment without ligase, additional masses could be identified as resulting from the shuffling of the two constructs (Fig. 4D). These side-products could not be identified in the positive control (Fig. 4C), indicating that the ligation step proceeded quantitatively, thus reaffirming the integrity of the encoding process.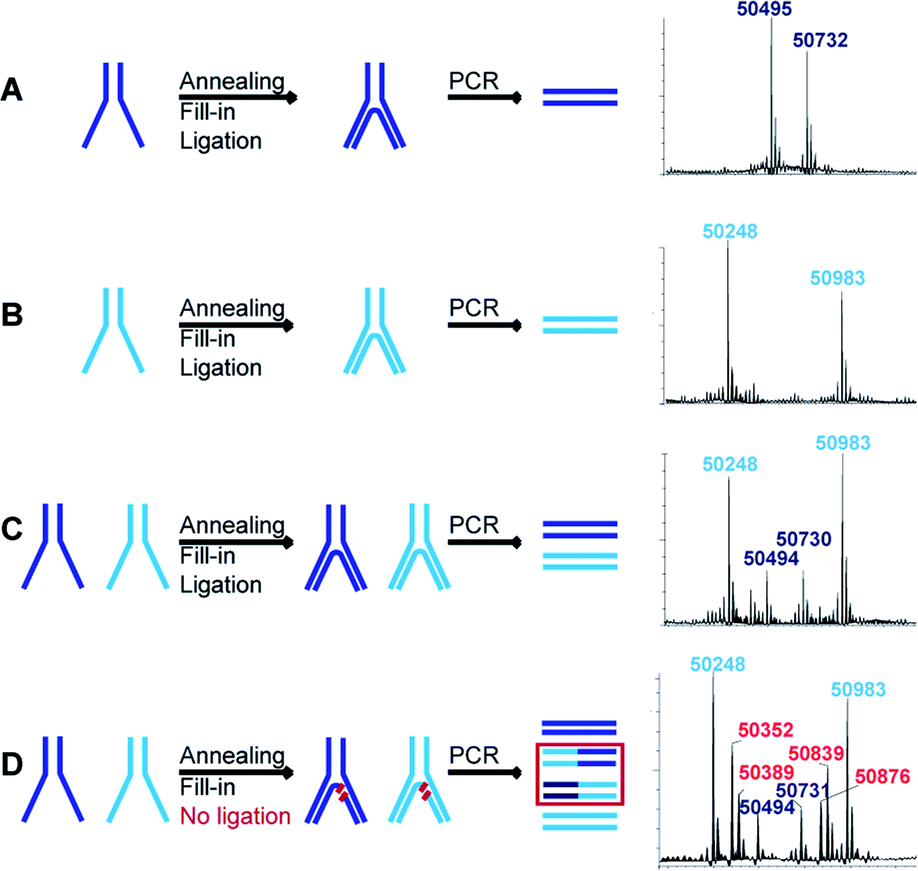 | ||
| Fig. 4 Assessment of ligation efficiency and concomitant code shuffling potential. Schematic representations of the assembly (annealing, fill-in and ligation) of LED constructs with two distinguishable sets of codes represented in light blue and dark blue. After assembly, the constructs are PCR-amplified and the respective deconvoluted LC-MS spectra of the PCR products are reported. (A) First set of codes assembly and PCR. (B) Second set of codes assembly and PCR. (C) The first and second sets of codes are assembled together and PCR-amplified. (D) Omitting the ligation step results in the production of shuffled side products during PCR amplification. | ||
This essential ligation step was additionally investigated before PCR amplification using fluorescence labeling (Fig. S1†). We repeated the LED construction with a fluoresceinated T3P or with a 5′-fluoresceinated IV strand. Polymerization and ligation were analyzed by denaturing SDS-PAGE. As expected, the fluorescent band completely shifted after polymerization and after ligation (Fig. S1,† transition to product 5 and 6), demonstrating quantitative polymerization and ligation.
Lowest level of detection
Since DNA input and recovery are essential parameters during a selection experiment,45–47 we wanted to evaluate the sensitivity of our method. To this extent, we performed a dilution series of a LED construct. Each dilution was individually PCR-amplified and analyzed on agarose gel (Fig. 5B), following our established protocol.2 The lowest level of detection was found for an input of DNA superior to approximately 3000 copies of LED construct. A negative control without DNA template was included to check for potential contamination. This experiment was additionally performed with single-stranded and double-stranded constructs previously used in our group for benchmarking with single-pharmacophore DEL technology and with a generic ESAC construct for a direct comparison with an already established dual-display system.27 | ||
| Fig. 5 (A) Representation of different DNA encoding systems, ss: single-stranded; ds: double-stranded. (B) Determination of lowest level of detection for PCR amplification of each DNA encoding system. The input (as number of DNA constructs) for PCR amplification is indicated. The amplification threshold is indicated with a blue dashed line. Negative controls (NC) were performed without DNA construct. Agarose gel image (2%). (C) Model selections results. Representation of positive control duplexes as a function of total number of duplex inputs in affinity-based selections against AGP (filled dots) and, as control, without selection (circles, theoretical value = 25%). Left: LED system, right: ESAC system. (D) Normalized enrichment of positive control duplexes as a function of the dilution of positive control duplexes present in the 1013 total DNA construct input, in AGP (left) or CAII (right) selections. Selections were done in triplicates; mean values are represented with corresponding standard error. | ||
For the other systems, at least 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 DNA molecules were necessary to obtain a satisfying PCR amplification. Since all DNA systems performed quite similarly in this lowest level of detection analysis, we concluded that LED should allow for robust PCR amplification after selection experiments despite the increase of complexity caused by the additional coding regions.
000 DNA molecules were necessary to obtain a satisfying PCR amplification. Since all DNA systems performed quite similarly in this lowest level of detection analysis, we concluded that LED should allow for robust PCR amplification after selection experiments despite the increase of complexity caused by the additional coding regions.
Minimum number of duplexes
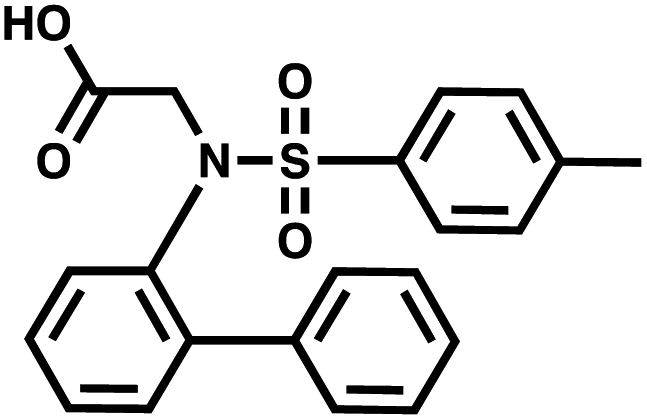
For small DEL quantities, determining the precise amount of DNA remaining after a selection experiment can be difficult. Instead, we determined the minimal quantity of library input that, post selection, would yield enough material for reliable PCR amplification. To this end, both for ESAC and LED systems, we prepared simple dual-pharmacophore mixtures containing a positive control against alpha-1-glycoprotein (AGP) and a negative control (Fig. S2 and S3†). AGP is one of the major acute phase proteins of human blood48 and was already tested in selection experiments with ESAC libraries by our group.27 A synergistic pair of fragments was identified and optimized, consisting of N-([1,1′-biphenyl]-2-yl)-N-tosylglycine, and of 3-(5-(2-(trifluoromethyl)phenyl)furan-2-yl)propanoic acid (Table 1).32 Affinity measurements of the optimized ligand displayed on locked nucleic acid yielded a dissociation constant of 108 nM, rendering this ligand a tool of choice to assess dual-display technologies.| DNA strand | Targeted protein | Building block | Chemical structure |
|---|---|---|---|
| 5′ → 3′ | AGP | N-([1,1′-Biphenyl]-2-yl)-N-tosylglycine |
|
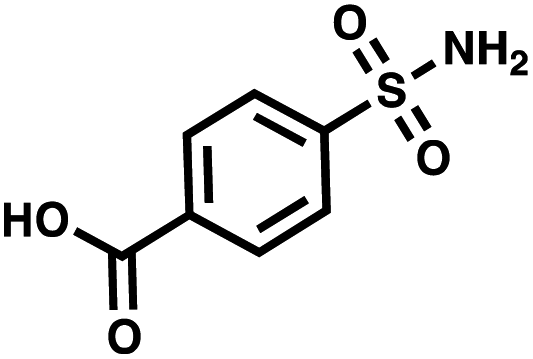
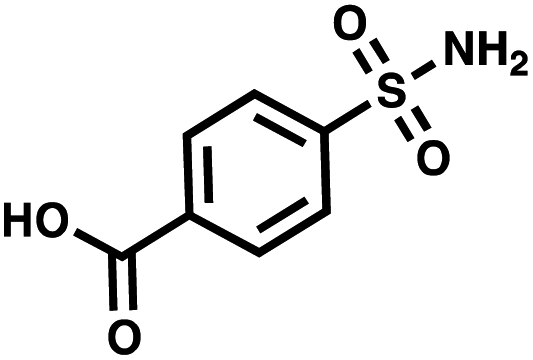
| CA II | 4-Sulfamoylbenzoic acid |
|
|
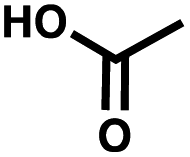
| Negative control | Acetic acid |
|
|
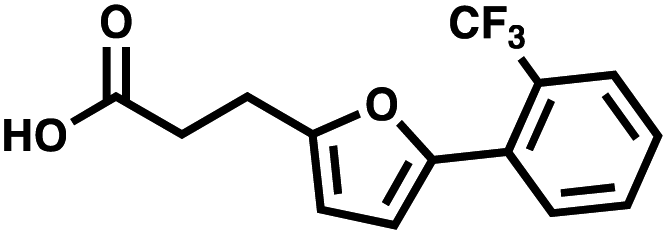
| 3′ → 5′ | AGP | 3-(5-(2-(Trifluoromethyl)phenyl)furan-2-yl)propanoic acid |
|
| CA II | 4-Sulfamoylbenzoic acid |
|
|
| Negative control | Acetic acid |
|
To synthesize the collections of duplexes for ESAC and LED, the corresponding 5′-strands were coupled to the glycine building block and the 3′-strands to the furanpropanoic acid building block. Acetic acid was used as a negative control for both strands (Fig. S2†). The building blocks were encoded on both coding positions of each sub-library, such that the individual readability of each of the four codes could be assessed. Positive and negative control strands were mixed in equimolar ratio to form duplexes. Amplifiable DNA was produced as previously described for LED and ESAC,27 and selections were performed with variable amounts of duplexes, ranging from 108 to 1012. Sequencing of the unselected mixtures showed equal repartition of all combinations of building blocks as expected (Fig. S5†). Selections were carried out in triplicate against immobilized AGP following an already described protocol,2 and the results are shown in Fig. 5C. The representation of the synergistic binding pair (i.e. the fraction of positive control counts against the total number of counts, expressed in percentage (formula (S1)†)), was calculated after selection, PCR amplification and DNA sequencing and is displayed for LED in red and for ESAC in blue. The representation of the three other duplexes was very low, indicating successful selection experiments, and is presented in Fig. S7.† After selection, the representation of the positive control duplex rises with increasing amounts of DNA in the selection experiment. From the plotted results, we concluded that 109 is the minimum number of duplexes needed for reliable selections.
Maximum dilution of positive controls
The last parameter we wanted to investigate in order to establish good selection conditions was the maximum dilution of a positive control in the total of all library members. We performed another set of model selections in which we varied the amount of positive controls while keeping constant the total number of duplexes at 1013, thus introducing different dilutions of the positive controls from 1:104 to 1:109. Once again, we used AGP and its pair of synergistic fragments as a first positive control. We selected carbonic anhydrase II (CAII) as a second test system, a protein of pharmaceutical relevance,49 and as positive control 4-sulfamoylbenzoic acid (SABA), a known CAII ligand. SABA was displayed on both 5′ and 3′ DNA strands of the duplexes, to assess potential avidity effects (Table 1 and Fig. S4†). Again, acetic acid modification was used as a negative control and each building block was encoded with the two sub-library coding positions. For LED and ESAC, encoded sub-libraries of positive and negative controls were mixed together to produce five model libraries with pre-defined amounts of positive control duplexes, ranging from 104 to 108 (Table S4†). Sequencing of the unselected model libraries confirmed that the desired dilutions of positive controls were obtained (Fig. S6†). Selections were carried out in triplicate against AGP and CAII following the same protocol2 and PCR amplification was performed. The results of these selections are given in Fig. 5D, where normalized enrichments (formula (S3)†) of the known pairs of binders are plotted against the number of positive duplexes present in each of the model libraries. Results for the other duplexes are reported in Fig. S8 and S9.†
In these selections, we could successfully retrieve the positive controls and, importantly, their enrichments displayed comparable trends for LED (in red) and ESAC (in blue). Significant enrichment was detected when at least 107 copies of the positive control duplexes were present, corresponding to a dilution of 1:1000000 with the negative controls. Interestingly, good enrichment values were observed only for the positive control duplexes consisting of either the full AGP binder duplex or the double-positive SABA duplex. For AGP selections, this means that only the combination of the previously identified simultaneously binding fragments was able to efficiently bind to the protein, as was already reported for the conventional ESAC setup.32 For CAII selections, since monovalent SABA has an 0.6 μM affinity for CAII, this means that avidity was greatly helping to retrieve the SABA moiety.50
Conclusion
In the present work, we have established a new large encoding design, LED, to widen the potential of DEL technology for more complex library construction and to facilitate approaching more difficult protein targets, such as protein–protein interactions. For this purpose, we designed and implemented a LED construct and we optimized our system until a reliable transfer of four codes partitioned between two sub-libraries onto a unique, amplifiable DNA strand was achieved. PCR sensitivity was investigated and found to be comparable or even superior to already established dual-pharmacophore systems. The system was validated through model selections and it allowed retrieving known pairs of binders against AGP and CAII, by synergistic and avidity effects, respectively. Thorough comparison with conventional ESAC demonstrated the potential of LED in selections and subsequent PCR amplification. Furthermore, optimal affinity-based selection conditions were established in terms of DNA input and dilution of the positive controls within a pool of DNA-encoded compounds. The minimum total DNA amount needed in selections was determined to be 109 duplexes both for LED and ESAC. This minimal input permits the retrieval of enough DNA construct after selection to perform reliable PCR amplification and observe enrichment of binders. As we are aware that concentration of potential ligands in the DEL pool is a key factor for determining an appropriate final library size, we determined the maximum dilution of a given small molecule in a model library. A maximal ratio of 1:1000000 between positive and negative controls allowed us to recover known binders, both in case of LED and of the conventional ESAC system. Knowledge of both minimum input and limiting dilution will help to design and interrogate novel types of very large dual-display libraries and enable DEL technology to become a more reliable source of specific, small- and medium-sized ligands to difficult protein targets. We described the utility of LED for the construction of “2 + 2” DELs, however, as it is very robust and versatile, the new LED encoding scheme may also well serve the design of a whole range of structurally diverse libraries of several diversity elements, and spanning from linear to branched, or macrocyclic structures. Using LED we foresee the versatile construction of single-pharmacophore DELs and of dual-pharmacophore DELs of unprecedented sizes.
Data availability
The data supporting the findings of this study are given within this article and its ESI.† Further raw DNA sequencing data can be obtained from the corresponding authors upon reasonable request.Author contributions
LP, DN and JS developed the encoding strategy, designed the validation experiments and wrote the manuscript. LP performed the experiments for implementing the encoding strategy. AM optimized the software for deconvoluting data from NGS sequencing. LP, AM and JS analyzed the data. LP, AL, MK, SO and GB performed the selection experiments and PCR amplifications.Conflicts of interest
Dario Neri is a co-founder and shareholder of Philochem AG (Otelfingen, Switzerland).Acknowledgements
Financial support from ETH Zürich, Innosuisse – Swiss Innovation Agency, (48350.1 IP-LS), Swiss National Science Foundation (CRSII5_198673), Krebsliga Schweiz (KSF-5012-02-2020) is gratefully acknowledged. MK is funded by a Horizon 2020 Marie Skłodowska-Curie Innovative Training Network (no. 861316 Magicbullet Reloaded). We would like to thank the Genetic Diversity Centre (GDC) of ETH Zürich and the Functional Genomics Center Zürich (FGCZ) for access to their services. We further thank Martina Bigatti and Dr Florent Samain for fruitful discussions and Timon Gradinger for help with PCR procedures and selections.References
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- W. Decurtins, M. Wichert, R. M. Franzini, F. Buller, M. A. Stravs, Y. Zhang, D. Neri and J. Scheuermann, Nat. Protoc., 2016, 11, 764–780 CrossRef CAS PubMed.
- Y. Song and X. Li, Acc. Chem. Res., 2021, 54, 3491–3503 CrossRef CAS PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci., 2006, 89, 5381–5383 CrossRef PubMed.
- R. A. Goodnow and C. P. Davie, DNA-Encoded Library Technology: A Brief Guide to Its Evolution and Impact on Drug Discovery, Elsevier Inc., 1st edn, 2017, vol. 50 Search PubMed.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- N. Favalli, G. Bassi, J. Scheuermann and D. Neri, FEBS Lett., 2018, 592, 2168–2180 CrossRef CAS PubMed.
- V. Kunig, M. Potowski, A. Gohla and A. Brunschweiger, Biol. Chem., 2018, 399, 691–710 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- R. M. Franzini and C. Randolph, J. Med. Chem., 2016, 59, 6629–6644 CrossRef CAS PubMed.
- T. Kodadek, N. G. Paciaroni, M. Balzarini and P. Dickson, Chem. Commun., 2019, 55, 13330–13341 RSC.
- N. Favalli, G. Bassi, C. Pellegrino, J. Millul, R. De Luca, S. Cazzamalli, S. Yang, A. Trenner, N. L. Mozaffari, R. Myburgh, M. Moroglu, S. J. Conway, A. A. Sartori, M. G. Manz, R. A. Lerner, P. K. Vogt, J. Scheuermann and D. Neri, Nat. Chem., 2021, 41557 Search PubMed.
- J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed.
- J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404–E6410 CrossRef CAS PubMed.
- L. Su, J. Feng, T. Peng, J. Wan, J. Fan, J. Li, J. O'Connell, D. R. Lancia, G. J. Franklin and G. Liu, Org. Lett., 2020, 22, 1290–1294 CrossRef CAS PubMed.
- Y. Li, R. de Luca, S. Cazzamalli, F. Pretto, D. Bajic, J. Scheuermann and D. Neri, Nat. Chem., 2018, 10, 441–448 CrossRef CAS PubMed.
- P. Dickson and T. Kodadek, Org. Biomol. Chem., 2019, 17, 4676–4688 RSC.
- C. Zambaldo, S. N. Geigle and A. L. Satz, Org. Lett., 2019, 21, 9353–9357 CrossRef CAS PubMed.
- N. Favalli, S. Biendl, M. Hartmann, J. Piazzi, F. Sladojevich, S. Gräslund, P. J. Brown, K. Näreoja, H. Schüler, J. Scheuermann, R. Franzini and D. Neri, ChemMedChem, 2018, 13, 1303–1307 CrossRef CAS PubMed.
- C. J. Gerry and S. L. Schreiber, Curr. Opin. Chem. Biol., 2020, 56, 1–9 CrossRef CAS PubMed.
- G. Zhang, J. Zhang, Y. Gao, Y. Li and Y. Li, Expert Opin. Drug Discovery, 2021, 1–15 Search PubMed.
- D. L. Usanov, A. I. Chan, J. P. Maianti and D. R. Liu, Nat. Chem., 2018, 10, 704–714 CrossRef CAS PubMed.
- Z. Zhu, A. Shaginian, L. C. Grady, T. O'Keeffe, X. E. Shi, C. P. Davie, G. L. Simpson, J. A. Messer, G. Evindar, R. N. Bream, P. P. Thansandote, N. R. Prentice, A. M. Mason and S. Pal, ACS Chem. Biol., 2018, 13, 53–59 CrossRef CAS PubMed.
- L. Plais and J. Scheuermann, RSC Chem. Biol., 2022, 3, 7–17 RSC.
- Q. Nie, S. Zhong, Y. Li, G. Zhang and Y. Li, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.09.041.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- Á. Furka, MedComm, 2021, 2, 481–489 CrossRef PubMed.
- K. I. Sprinz, D. M. Tagore and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2005, 15, 3908–3911 CrossRef CAS PubMed.
- R. M. Franzini, D. Neri and J. Scheuermann, Acc. Chem. Res., 2014, 47, 1247–1255 CrossRef CAS PubMed.
- J. Scheuermann and D. Neri, Dual-Pharmacophore DNA-Encoded Chemical Libraries, 2014 Search PubMed.
- M. Bigatti, A. Dal Corso, S. Vanetti, S. Cazzamalli, U. Rieder, J. Scheuermann, D. Neri and F. Sladojevich, ChemMedChem, 2017, 12, 1748–1752 CrossRef CAS PubMed.
- S. Oehler, L. Plais, G. Bassi, D. Nario and J. Scheuermann, Chem. Commun., 2021, 12289–12292 RSC.
- K. Gorska, K.-T. Huang, O. Chaloin and N. Winssinger, Angew. Chem., 2009, 121, 7831–7836 CrossRef.
- J. P. Daguer, M. Ciobanu, S. Alvarez, S. Barluenga and N. Winssinger, Chem. Sci., 2011, 2, 625–632 RSC.
- H. Eberhard, F. Diezmann and O. Seitz, Angew. Chem., 2011, 123, 4232–4236 CrossRef.
- J. P. Daguer, C. Zambaldo, M. Ciobanu, P. Morieux, S. Barluenga and N. Winssinger, Chem. Sci., 2015, 6, 739–744 RSC.
- G. Bassi, N. Favalli, M. Vuk, M. Catalano, A. Martinelli, A. Trenner, A. Porro, S. Yang, C. L. Tham, M. Moroglu, W. W. Yue, S. J. Conway, P. K. Vogt, A. A. Sartori, J. Scheuermann and D. Neri, Adv. Sci., 2020, 7(22), 2001970 CrossRef CAS PubMed.
- Y. Zhou, C. Li, J. Peng, L. Xie, L. Meng, Q. Li, J. Zhang, X. D. Li, X. Li, X. Huang and X. Li, J. Am. Chem. Soc., 2018, 140, 15859–15867 CrossRef CAS PubMed.
- Y. Deng, J. Peng, F. Xiong, Y. Song, Y. Zhou, J. Zhang, F. S. Lam, C. Xie, W. Shen, Y. Huang, L. Meng and X. Li, Angew. Chem., Int. Ed., 2020, 59, 14965–14972 CrossRef CAS PubMed.
- Y. Zhou, J. Peng, W. Shen and X. Li, Biochem. Biophys. Res. Commun., 2020, 533, 215–222 CrossRef CAS PubMed.
- Y. Zhou, W. Shen, J. Peng, Y. Deng and X. Li, Bioorg. Med. Chem., 2021, 45, 116328 CrossRef CAS PubMed.
- F. V. Reddavide, W. Lin, S. Lehnert and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7924–7928 CrossRef CAS PubMed.
- F. V. Reddavide, M. Cui, W. Lin, N. Fu, S. Heiden, H. Andrade, M. Thompson and Y. Zhang, Chem. Commun., 2019, 55, 3753–3756 RSC.
- A. Sannino, E. Gabriele, M. Bigatti, S. Mulatto, J. Piazzi, J. Scheuermann, D. Neri, E. J. Donckele and F. Samain, ChemBioChem, 2019, 20, 955–962 CrossRef CAS PubMed.
- Q. Chen, X. Cheng, L. Zhang, X. Li, P. Chen, J. Liu, L. Zhang, H. Wei, Z. Li and D. Dou, SLAS Discovery, 2020, 25, 523–529 Search PubMed.
- A. L. Satz, R. Hochstrasser and A. C. Petersen, ACS Comb. Sci., 2017, 19, 234–238 CrossRef CAS PubMed.
- T. Fournier, N. Medjoubi-N and D. Porquet, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1482, 157–171 CrossRef CAS.
- C. T. Supuran, Nat. Rev. Drug Discovery, 2008, 7, 168–181 CrossRef CAS PubMed.
- L. Prati, M. Bigatti, E. J. Donckele, D. Neri and F. Samain, Biochem. Biophys. Res. Commun., 2020, 533, 235–240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05721a |
| This journal is © The Royal Society of Chemistry 2022 |