
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heavy atom oriented orbital angular momentum manipulation in metal-free organic phosphors†
Wenhao
Shao
 ,
Hanjie
Jiang
,
Ramin
Ansari
,
Paul M.
Zimmerman
* and
Jinsang
Kim
*
,
Hanjie
Jiang
,
Ramin
Ansari
,
Paul M.
Zimmerman
* and
Jinsang
Kim
*
Department of Chemistry, Department of Chemical Engineering, Department of Materials Science and Engineering, Macromolecular Science and Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA. E-mail: paulzim@umich.edu; jinsang@umich.edu
First published on 24th December 2021
Abstract
Metal-free purely organic phosphors (POPs) are emerging materials for display technologies, solid-state lighting, and chemical sensors. However, due to limitations in contemporary design strategies, the intrinsic spin–orbit coupling (SOC) efficiency of POPs remains low and their emission lifetime is pinned in the millisecond regime. Here, we present a design concept for POPs where the two main factors that control SOC—the heavy atom effect and orbital angular momentum—are tightly coupled to maximize SOC. This strategy is bolstered by novel natural-transition-orbital-based computational methods to visualize and quantify angular momentum descriptors for molecular design. To demonstrate the effectiveness of this strategy, prototype POPs were created having efficient room-temperature phosphorescence with lifetimes pushed below the millisecond regime, which were enabled by boosted SOC efficiencies beyond 102 cm−1 and achieved record-high efficiencies in POPs. Electronic structure analysis shows how discrete tuning of heavy atom effects and orbital angular momentum is possible within the proposed design strategy, leading to a strong degree of control over the resulting POP properties.
1. Introduction
Organic phosphors are the functional components in modern technologies such as displays, solid-state lighting, and chemical sensors. While conventional organometallic phosphors suffer from metal ion dislocation,1,2 device longevity problems, and toxicity issues, metal-free purely organic phosphors (POPs) have many advantageous properties such as large design windows, easy processability, economic material cost, and lower toxicity.3–5 However, POPs typically exhibit long emission lifetimes due to the involvement of spin-forbidden transitions, which should be facilitated by spin–orbit coupling (SOC).6 In the design of contemporary POPs, SOC is promoted mostly by heavy atom effects and the El-Sayed rule. Fig. 1 listed a few representative POPs with halogen or chalcogen heavy atoms: Br,7–19 I,20–22 or Se.23–28 The El-Sayed rule explains the necessity of orbital angular momentum change in promoting SOC,29 and the utilization of carbonyl,7,8,10–12,14,30,31 heterocyclic rings32–34 (e.g. triazine in DPhCzT32), and other moieties having rich non-bonding electrons (e.g. sulfoxide in Cs–Br35) to stimulate low energy (n,π*) states and thus create (π,π*)–(n,π*) transition channels. Due to the forbidden nature of singlet–triplet transitions and consequently slow triplet emission, matrix engineering to effectively suppress collisional quenching (the major non-radiative decay route) is essential to achieve POPs with decent quantum efficiency.36 For that purpose, crystal design and strong intermolecular bonding between POPs and rigid matrix have been implemented.10,30,35,37–44 The slow decay nature of POPs, on the other hand, has been explored to create persistent emitters with long lifetimes in the 10−1 to 100 second regime.3 | ||
| Fig. 1 Contemporary POP design vs. POPs designed with the heavy atom oriented angular momentum manipulation (HAAM) concept implemented. | ||
However, current design strategies have reached their limit. The intrinsic SOC efficiencies of POPs remain low (e.g. T1–S0 SOC is usually 100 to 101 cm−1) with their emission lifetime stuck in the millisecond to second regime. The lack of synergistic interactions between the SOC-promoting components is the major deficiency of contemporary design strategies. On one hand, the El-Sayed rule hasn't been fully exploited to capture the detailed picture of molecular features leading to angular momentum changes between spin states. On the other hand, the interplay between the heavy atom effect and orbital angular momentum is missing, i.e., heavy atoms are not rationally positioned in such a way that their orbitals strongly interact with those undergoing angular momentum change. Such delicate manipulations are not required in organometallic complexes, where the angular momentum changes are centered on the metal atom. In POPs, however, this connection is not trivial to achieve but could have a great impact on increasing SOCs. An additional factor that has hindered creation of better design strategies is that contemporary computational tools—while able to quantify SOCs—have provided little insight into the electronic origins of the SOC within chromophores.
Here, we report a novel molecular design concept to manipulate SOC in POPs that synergistically uses the heavy atom effect in close connection with orbital angular momentum to overcome SOC limits of organic phosphors. To emphasize the core of our design concept that uses heavy atoms to directly stimulate ΔL, we named it “heavy atom oriented orbital angular momentum manipulation” (HAAM) (see Fig. 1). This rational design strategy enhances SOCs to over 102 cm−1 and pushes the lifetime limit of organic phosphors to below ms regime, as demonstrated in a series of prototype POPs using chalcogen heavy atoms. HAAM is confirmed to be operational through a novel natural transition orbital (NTO)-based computational method to visualize the molecular orbital origins of SOC. In total, this work introduces the HAAM design concept and tests its relevance using theory, computation, and experiment.
2. Results
2.1. Theory behind SOC
Before moving to the molecular design, it's worth revisiting the theory behind SOC that motivates the HAAM concept. Under Fermi's golden rule, the transition rate (state 1 to state 2) for intersystem crossing is given as k12 ∝ |ψ2|HSO|ψ1|2ρ(E12).45 Here, ρ(E12) denotes the joint density of states of the initial and final wavefunctions, ψ1 and ψ2; HSO is the transition Hamiltonian, and the term ψ2|HSO|ψ1 is the SOC transition matrix element (SOCME). For one-electron systems under relativistic conditions, the main-part of the spin–orbit Hamiltonian is ,46 where r is the orbital radius, L and S are the orbital and spin angular momentum, respectively. For many-electron systems, the Hamiltonian is expressed in terms of Breit-Pauli (BP) Hamiltonian47 as
,46 where r is the orbital radius, L and S are the orbital and spin angular momentum, respectively. For many-electron systems, the Hamiltonian is expressed in terms of Breit-Pauli (BP) Hamiltonian47 as | (1) |
Intuitively, the coupling of electrons' spin and angular momentums in eqn (1) follows the conservation of total angular momentum rule—that since S changes with a spin change, the orbital angular momentum, L, has to change as well. The El-Sayed rule29 qualitatively elucidated the angular momentum conservation law, for example in a typical case the (π,π*)–(n,π*) transition carries a change in orbital angular momentum (ΔL). On the other hand, large ΔL has been observed in other types of transitions,24,48 even for (π,π*)–(π,π*) transitions.
More importantly, it is clear from eqn (1) that orbital and spin angular momenta are coupled through the interaction with atomic number. Importantly, due to the form of eqn (1), the heavy atom must be in close proximity with the spin orbital transition, otherwise the effective Z from that atom is suppressed by the r−3 dependence of HSO. While our discussion here makes these principles transparent, in practice the combination of heavy atom effects with orbital angular momentum change has rarely been addressed in the design of POPs. For instance, Sarkar and Hendrickson et al.49 examined benzaldehydes with Br substitution at various positions and revealed extraordinarily boosted SOC when Br is at the para position relative to aldehyde. However, the orbital angular momentum influence was not discussed although it was clear from their results that Br's contribution to ΔL was drastically enhanced at the para position. In our recent reports,24,48 we elucidated the origin of efficient SOC from the perspective of ΔL and expanded the ΔL-promoting mechanism beyond traditional (π,π*)–(n,π*) transitions, but the interaction of heavy atom with ΔL was not reviewed. In short, HAAM strategy hasn't been well established in the systematic molecular design of POPs, which gives an open design space to explore the HAAM method for creating novel, tunable organic triplet emitters.
To help instantiate the HAAM strategy, ab initio simulations can provide insight into proposed chromophore designs by revealing their specific electronic structures and SOC elements. In particular, the restricted active space-spin flip (RAS-SF) method50–54 is a wave function theory that is well-suited for treating electronically excited states55,56 of photoactive molecular systems. RAS-SF has been shown to accurately treat a variety of challenging electronic structure problems, including polyradicals, singlet fission mechanisms, and charge transfer processes.57–61 Recent work has enabled RAS-SF to predict accurate SOC elements, making it particularly useful to complement the HAAM design concept. Furthermore and vital to instantiating the HAAM strategy, RAS-SF can produce natural transition orbitals (NTO) that couple pairs of spin states, revealing the specific changes in electronic structure that give rise to spin–orbit interactions. In this work, RAS-SF will show how the HAAM concept applies in practice to novel organic phosphors. This study therefore provides quantitative predictions of SOC and direct visualization of the interactions leading to SOC.
2.2. Implementation of HAAM strategy for molecular design
Increasing the involvement of heavy atoms in ΔL is critical to the HAAM concept, and the best amplification should be achieved where heavy atom orbitals are involved in the electronic transition of interest. Thus, the utilization of non-bonding electrons from heavy atoms is critically important. Chalcogen was selected as the heavy atom since, unlike halogens, it could be incorporated in the core of POPs to activate ΔL channels on its own, as shown in our previous work.24 To access the non-bonding p electrons from Se, we designed N-methylated phenylselenazine (Se–N, 1) carrying a bent geometry enabled by sp3 hybridization at the nitrogen (Fig. 2a). Accordingly, p electrons from Se are expected to be partially decoupled from the surrounding π-conjugated system and the non-bonding electrons could participate in ΔL.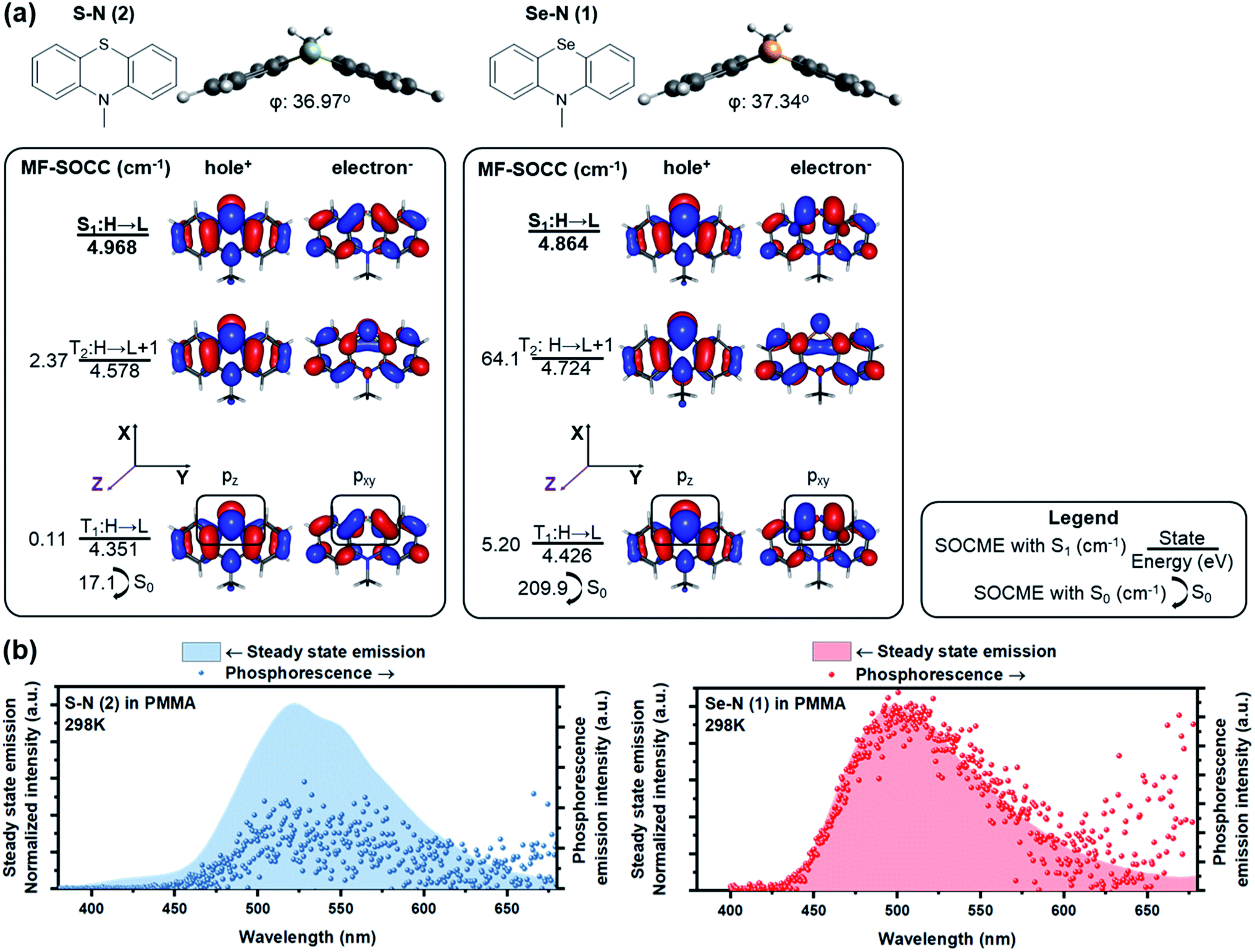 | ||
| Fig. 2 Computational and experimental results of Se–N (1) and Se–N (2). (a) Chemical structures of S–N and Se–N. Electronic structure results from RAS-SF showing the ground state optimized structure with bent geometry and dihedral angle and the NTOs of S1, T1, T2 states. Energy of each state and the selected mean field SOCMEs are shown as well. (b) Steady state emission spectra (filled graph, left axis) and gated phosphorescence emission spectra (dot, right axis, 0.5–5 ms for Se–N and 0.5–20 ms for S–N) of the two emitters doped in atactic PMMA (1 wt%, spin-cast) measured at room temperature in vacuum. | ||
RAS-SF calculations of Se–N showed a large SOCME of 209.9 cm−1 between T1 and the ground state S0, which is the critical transition affecting phosphorescence rate. A 64.1 cm−1 SOCME for the S1–T2 transition was also calculated, for the critical ISC process to populate triplet excitons. A detailed analysis on the NTOs of Se–N was performed to elucidate the high SOC efficiency of Se–N and its connections with the HAAM concept. As shown in Fig. 2a, pz electrons of Se populated the hole orbital of T1 while pxy (linear combination of px and py, denoting orbitals in the xy plane in general) populated the electron orbital. Since orbital angular momentum rotates 90° between pz and pxy orbitals, electron migration between the hole and electron orbitals of T1 would carry a large “heavy atom oriented” ΔL, enabled by the heavy Se atom. In other words, the T1–S0 transition of Se–N follows the HAAM concept, leading to its large SOCME. Similarly, HAAM is also manifested in the S1–T2 SOC: the transition occurs through the excited electron orbitals of the S1 and T2 states, which are populated by pxy and pz electrons of Se, respectively.
To further test the novel design principle, Se–N was synthesized and embedded in polymeric matrixes (see methods for details†). In anoxic environment, fast room-temperature phosphorescence decay was observed (Fig. 2b), with measured τph of 385 μs and Φph of 19.2%. These results are consistent with the computed SOCME of 102 cm−1, which is a record-high in POPs,3 and is comparable to that of some organometallic phosphors.62–64 This large SOCME enabled by the HAAM concept shows that POPs can have similar emissive properties compared to their organometallic counterparts, where SOCMEs are typically in the range of 102–103 cm−1.62–64
While the NTO analyses above provide qualitative visualization for how non-bonding p-electrons of Se directs ΔL, this phenomenon could also be quantified. Intuitively, if ΔL is dominated by a pz–pxy transition, by using the right-hand rule, ΔL should be parallel to the xy plane. In other words, if we reduce the angular momentum change operator ![[L with combining circumflex]](https://www.rsc.org/images/entities/i_char_004c_0302.gif) using cartesian coordinates into x/y (i.e. x or y) and z, SOC efficiency will be more pronounced with the x/y operator, since the z operator only performs an orbital rotation around the z axis.
using cartesian coordinates into x/y (i.e. x or y) and z, SOC efficiency will be more pronounced with the x/y operator, since the z operator only performs an orbital rotation around the z axis.
RAS-SF based NTO analyses not only produce accurate representation of SOC mechanism, but also provide insights on the reduced SOCME in selected orientations, which reveal the contributions of each angular momentum operator.62 In practice, according to the matrix representation of the angular momentum in the basis of p-orbitals, Se pz–pxy transition in T1–S0 of Se–N should produce a considerable pz|x/y|pxy matrix element compared to that from the pz|z|pxy operator. This is confirmed by RAS-SF results (Table 1) showing major contributions to SOC from the reduced components in L−(Lx) and L+(Ly) orientations. Most importantly, the HAAM concept is directly supported since the majority of SOC is facilitated by ΔL on Se heavy atom. The benefit from RAS-SF methods is substantial in our discussion.
| Orientation | S–N | Se–N |
|---|---|---|
| L x or L− | −10.50 − 6.02i | −137.56 + 0.01i |
| L z or L0 | 0.00 | −78.77i |
| L y or L+ | −10.50 + 6.02i | −137.56 − 0.01i |
The HAAM strategy was further examined by discretely tuning the heavy atom effect and ΔL. First, heavy atom effect was measured by replacing Se in Se–N with S while keeping ΔL relatively consistent. Frontier excited states of the designed molecule, S–N, have similar NTOs to those of Se–N, suggesting similar ΔL is present in the relevant electronic transitions. As expected, the SOCMEs of S–N were much smaller than those of the Se counterpart (e.g., 17.1 vs. 209.9 cm−1 T1–S0 SOCME). Table 1 also indicates largely reduced SOCME of S–N for the reduced components in all three orientations.
Secondly, Se was reintroduced and the contribution of heavy atom orbitals in ΔL was examined over a range of angular momentum changes. This was achieved via simulation by measuring the SOC for T1–S0 along various bending angles (Fig. 3a). The contribution of Se pxy orbitals in the T1 state gradually increased as the bending angle was enlarged from 10 to 60° since pxy electrons are gradually decoupled from the π-conjugated system, leading to the increased ΔL and T1–S0 SOCME (Fig. 3b). Similar trends also exist in the reduced SOCMEs in L−, L0, and L+ orientations (Fig. S1 and Table S2†).
 | ||
| Fig. 3 Computational results of Se–N (1) scanned through the dihedral angle. (a) T1–S0 NTOs of Se–N optimized with dihedral angle (φ) fixed; λ represents the contribution of the NTO pair in SOC (max. 1). (b) RAS-SF one-electron and mean-field SOCMEs of T1–S0 transition vs. dihedral angle, φ(°). | ||
To probe the dihedral degree of freedom via experiment, the functional group opposite to Se provides a possible handle. Thus, a series of molecules was designed where the nitrogen in Se–N is replaced by oxygen, boron, or carbonyl (Fig. 4a). With these substitutions, the dihedral angle was reduced from ∼37° to 0° (Fig. 4d) due to a change in orbital hybridization from sp3 (N) to sp2 (CO). Accordingly, ΔL is expected to decrease since non-bonding electrons of Se experience a higher degree of conjugation with the nearby ring system. Since Se is a much heavier atom than oxygen, boron, or carbonyl, the heavy atom effect should remain approximately constant. In line with expectations, the calculated T1–S0 SOCMEs show a decreasing trend upon reducing the bending angle from Se–N through Se–B to Se–CO (Fig. 4b and d). A similar trend is observed in the primary SOCME component in the L± direction (Fig. 4c and Table S1†). Besides the bending angle, which affects the rotation of L, the induction effect of the substituents controls the electron density on Se and consequently the absolute value of L. This could explain the larger T1–S0 SOCME value of Se–O than that of Se–N, despite its smaller dihedral angle.
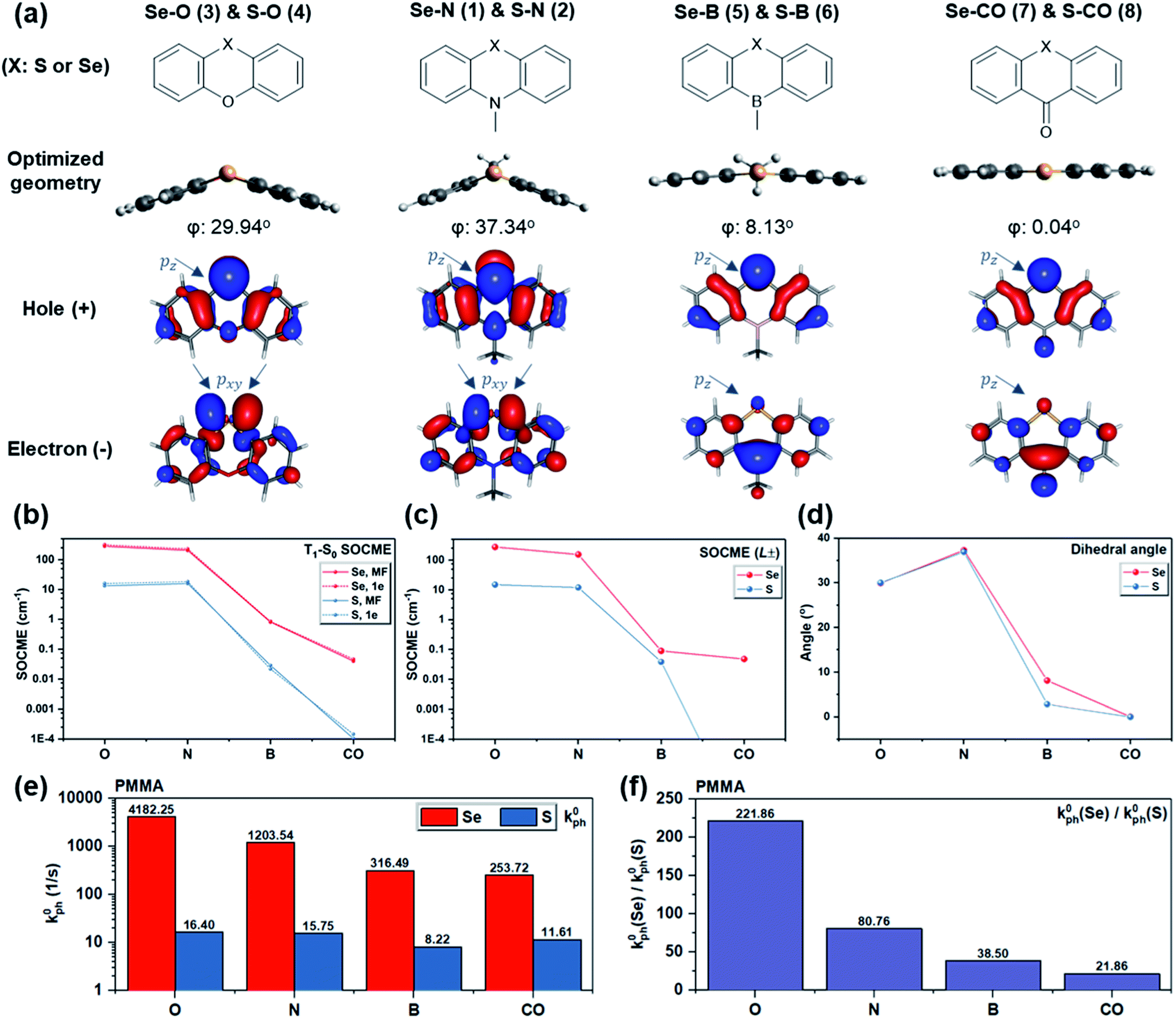 | ||
| Fig. 4 Computational and experimental results for S/Se–O, S/Se–N, S/Se–B, and S/Se–CO. (a) Molecular structures of the oxygen, nitrogen, boron, and carbonyl derivatives (X = S or Se), their optimized ground state geometry with dihedral angle marked, and RAS-SF NTOs of T1 states with Se pz and pxy orbitals marked; (b) RAS SF one-electron (1e) and mean-field (MF) SOCMEs of T1–S0 transition, (c) the reduced 1e SOCME in L− or L+ orientations (modulus) vs. functional groups; (d) the dihedral angles of molecules studied vs. their functional groups; (e) the experimental intrinsic phosphorescence rate k0ph measured in doped PMMA matrix at 78 K in vacuum, and (f) the k0ph(Se derivative)/k0ph(S derivative) value of each functional group. | ||
The computational results along with the molecular design rationale were experimentally checked by analyzing the intrinsic phosphorescence rate (k0ph) measured at 78 K. As expected, k0ph decreased dramatically as the dihedral flattens in the series from Se–O to Se–CO (Fig. 4e). However, this observation alone only indirectly supports that the dihedral angle, or ΔL variation, leads to k0ph variation. To gain further insight, k0ph of each Se compound is compared with its S counterpart (e.g. Se–N vs. S–N). The experimental k0ph of the S compounds doesn't experience a sharp decreasing trend from S–O to S–CO, but generally follows the prediction except for S–CO. We hypothesize that inevitable non-radiative decay for such long-lived triplet excitons in the sulfur compounds, as well as small mismatch between predicted and experimental dihedral angle could lead to unexpectedly larger k0ph in the S compounds (see section V in ESI for details†).
Generally, while all four derivatives showed remarkable enhancement in k0ph by replacing S with Se in the same molecular frame (Fig. 4e), the degree of k0ph “boost”, characterized by k0ph(Se-derivative)/k0ph(S-derivative), varies with each molecular frame. It is worth addressing that the degree of k0ph boost follows the dihedral angle, since a larger ΔL would enhance the contribution of heavy atom orbitals, and thus lead to more prominent heavy atom effects. Experimental results suggested the same trend (Fig. 4f) in the degree of k0ph boost, which increased from Se–CO (ca. 22-fold), through Se–B, Se–N, to Se–O (a remarkable ca. 222-fold), strongly implying the beneficial effect of the increased dihedral angle and T1–S0 ΔL. These experimental results therefore demonstrate that the “heavy atom oriented” ΔL can be effective in practice, as motivated and expected by the HAAM concept.
3. Discussion
The analysis above implements the “heavy atom oriented orbital angular momentum manipulation” (HAAM) as a novel design feature for POPs and tested the concept using simulations and experiments. This HAAM strategy enables a powerful use of SOC theory in molecular design, and shows the potential to give control over SOC in triplet emitters. Highly efficient POPs with fast emission were realized with this strategy, having k0ph over 103 s−1 and promising room-temperature phosphorescence (RTP) properties.This advance was supported by a novel computational method, RAS-SF, which can accurately quantify SOC in triplet emitters. RAS-SF also provides detailed NTO analysis to provide direct computational supports for the HAAM concept by revealing the electronic changes from triplet to singlet state that come with SOC. Importantly, the connection between experimental findings and systematic RAS-SF-assisted molecular design gave strong evidence that the tuning of heavy atom effects and orbital angular momentum—in synergy—is vital to increasing SOC in POPs. In other words, the combined quantitative and qualitative aspects of RAS-SF allowed the HAAM concept to be successfully translated from theory to practice for the design of POPs.
This combination of HAAM strategy with the RAS-SF SOC method can be extended to other organic triplet-based emitter scaffolds. Whereas the HAAM concept is enacted in practice to the Se series in this work, the key to efficient HAAM-based POPs is activating the contribution of heavy atom orbitals in ΔL, and the key to activate efficient ΔL channels is utilizing non-bonding electrons of heavy atoms. The “bent geometry” investigated here is expected to be just one efficient molecular scaffold to fulfill this task. A related, but less powerful method is to place heavy atoms adjacent to conventional El-Sayed rule satisfying moieties, so that they could participate in the (π,π*)−(n,π*) type ΔL. This design rationale employs halogen-containing POPs,48,49 since halogen only has one available bonding site and could not create efficient ΔL channels on their own.
In addition, while the fused-ring motif of the presented article appears to efficiently utilize the HAAM concept by connecting the heavy atom within the ΔL-producing channel, heavy chalcogen atoms could also be placed in attachable pendent groups to create ΔL channels, such as coupling them with ester or anhydride groups, or directly replace the oxygen in carbonyl with heavier chalcogens. However, these moieties haven't been systematically explored yet.
Further exploration of the HAAM strategy will focus on expanding the library of HAAM-based POPs in order to further break the SOC limits of POPs. Another promising direction is to create new OLEDs that outperform their metal-organic counterparts.
Author contributions
W. S. synthesized the materials and conducted the photophysical analyses, and wrote the manuscript; H. J. performed all computation analyses and contributed to the computation sections of the article; R. A. provided the selenium precursor for the compound Se–N; J. K. and P. M. Z. supervised the research and revised this article.Conflicts of interest
The authors declare no competing financial interests. Further information and requests for the data that support the findings of this study should be directed to and will be fulfilled by the lead contact, Jinsang Kim (E-mail: jinsang@umich.edu).Acknowledgements
We acknowledged the financial support from National Science Foundation (DMREF DMR 1435965) and Samsung Global Research Outreach.References
- S. Schmidbauer, A. Hohenleutner and B. König, Chemical Degradation in Organic Light-Emitting Devices: Mechanisms and Implications for the Design of New Materials, Adv. Mater., 2013, 25(15), 2114–2129 CrossRef CAS PubMed.
- C. Jeong, C. Coburn, M. Idris, Y. Li, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Understanding Molecular Fragmentation in Blue Phosphorescent Organic Light-Emitting Devices, Org. Electron., 2019, 64, 15–21 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Recent Advances in Purely Organic Phosphorescent Materials, Chem. Commun., 2015, 51(55), 10988–11003 RSC.
- A. D. Nidhankar, A. Goudappagouda, V. C. Wakchaure Ab and S. Santhosh Babu, Efficient Metal-Free Organic Room Temperature Phosphors, Chem. Sci., 2021, 12, 4216 RSC.
- Z. Wu, J. Nitsch and T. B. Marder, Persistent Room-Temperature Phosphorescence from Purely Organic Molecules and Multi-Component Systems, Adv. Opt. Mater., 2021, 9(20), 2100411 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. ScaianoModern Molecular Photochemistry of Organic Molecules, First Indi.; University Science Books, Sausalito, CA, 2017 Search PubMed.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Achieving Persistent Room Temperature Phosphorescence and Remarkable Mechanochromism from Pure Organic Luminogens, Adv. Mater., 2015, 27(40), 6195–6201 CrossRef CAS PubMed.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Versatile Room-Temperature-Phosphorescent Materials Prepared from N-Substituted Naphthalimides: Emission Enhancement and Chemical Conjugation, Angew. Chem. Int. Ed., 2016, 55(34), 9872–9876 CrossRef CAS PubMed.
- J. L. Ma, H. Liu, S. Y. Li, Z. Y. Li, H. Y. Zhang, Y. Wang and C. H. Zhao, Metal-Free Room-Temperature Phosphorescence from Amorphous Triarylborane-Based Biphenyl, Organometallics, 2020, 39(23), 4153–4158 CrossRef CAS.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Activating Efficient Phosphorescence from Purely Organic Materials by Crystal Design, Nat. Chem., 2011, 3(3), 205–210 CrossRef CAS PubMed.
- B. Song, W. Shao, J. Jung, S. J. Yoon and J. Kim, Organic Light-Emitting Diode Employing Metal-Free Organic Phosphor, ACS Appl. Mater. Interfaces, 2020, 12(5), 6137–6143 CrossRef CAS PubMed.
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Phosphorescence from a Pure Organic Fluorene Derivative in Solution at Room Temperature, Chem. Commun., 2013, 49(76), 8447–8449 RSC.
- P. She, Y. Yu, Y. Qin, Y. Zhang, F. Li, Y. Ma, S. Liu, W. Huang and Q. Zhao, Controlling Organic Room Temperature Phosphorescence through External Heavy-Atom Effect for White Light Emission and Luminescence Printing, Adv. Opt. Mater., 2020, 8(4), 1–7 Search PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, White Light Emission from a Single Organic Molecule with Dual Phosphorescence at Room Temperature, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Pavanello, A. Previtali, S. Righetto and E. Cariati, Cyclic Triimidazole Derivatives: Intriguing Examples of Multiple Emissions and Ultralong Phosphorescence at Room Temperature, Angew. Chem. Int. Ed., 2017, 56(51), 16302–16307 CrossRef CAS PubMed.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Versatile Room-Temperature-Phosphorescent Materials Prepared from N-Substituted Naphthalimides: Emission Enhancement and Chemical Conjugation, Angew. Chem. Int. Ed., 2016, 55(34), 9872–9876 CrossRef CAS PubMed.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Achieving Persistent Room Temperature Phosphorescence and Remarkable Mechanochromism from Pure Organic Luminogens, Adv. Mater., 2015, 27(40), 6195–6201 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Intermolecular Electronic Coupling of Organic Units for Efficient Persistent Room-Temperature Phosphorescence, Angew. Chem. Int. Ed., 2016, 55(6), 2181–2185 CrossRef CAS PubMed.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Organic Nanocrystals with Bright Red Persistent Room-Temperature Phosphorescence for Biological Applications, Angew. Chem. Int. Ed., 2017, 56(40), 12160–12164 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, A. Lien and J. Xu, Linearly Tunable Emission Colors Obtained from a Fluorescent-Phosphorescent Dual-Emission Compound by Mechanical Stimuli, Angew. Chem. Int. Ed., 2015, 54(21), 6270–6273 CrossRef CAS PubMed.
- G. D. Gutierrez, G. T. Sazama, T. Wu, M. A. Baldo and T. M. Swager, Red Phosphorescence from Benzo[2,1,3]Thiadiazoles at Room Temperature, J. Org. Chem., 2016, 81(11), 4789–4796 CrossRef CAS PubMed.
- L. Xiao, Y. Wu, J. Chen, Z. Yu, Y. Liu, J. Yao and H. Fu, Highly Efficient Room-Temperature Phosphorescence from Halogen-Bonding-Assisted Doped Organic Crystals, J. Phys. Chem. A, 2017, 121(45), 8652–8658 CrossRef CAS PubMed.
- A. Kremer, C. Aurisicchio, F. Deleo, B. Ventura, J. Wouters, N. Armaroli, A. Barbieri and D. Bonifazi, Walking Down the Chalcogenic Group of the Periodic Table: From Singlet to Triplet Organic Emitters, Chem.–Eur. J., 2015, 21(43), 15377–15387 CrossRef CAS PubMed.
- D. R. Lee, K. H. Lee, W. Shao, C. L. Kim, J. Kim and J. Y. Lee, Heavy Atom Effect of Selenium for Metal-Free Phosphorescent Light-Emitting Diodes, Chem. Mater., 2020, 32(6), 2583–2592 CrossRef CAS.
- S. Wang, H. Shu, X. Han, X. Wu, H. Tong and L. Wang, A Highly Efficient Purely Organic Roomtemperature Phosphorescence Film Based on a Selenium-Containing Emitter for Sensitive Oxygen Detection, J. Mater. Chem. C, 2021, 9(31), 9907–9913 RSC.
- T. Weng, G. Baryshnikov, C. Deng, X. Li, B. Wu, H. Wu, H. Ågren, Q. Zou, T. Zeng and L. Zhu, A Fluorescence–Phosphorescence–Phosphorescence Triple-Channel Emission Strategy for Full-Color Luminescence, Small, 2020, 16, 1906475 CrossRef CAS PubMed.
- M. Jiang, J. Guo, B. Liu, Q. Tan and B. Xu, Synthesis of Tellurium-Containing Ï€-Extended Aromatics with Room-Temperature Phosphorescence, Org. Lett., 2019, 8328–8333 CrossRef CAS PubMed.
- D. de Sa Pereira, D. R. Lee, N. A. Kukhta, K. H. Lee, C. L. Kim, A. S. Batsanov, J. Y. Lee and A. P. Monkman, The Effect of a Heavy Atom on the Radiative Pathways of an Emitter with Dual Conformation, Thermally-Activated Delayed Fluorescence and Room Temperature Phosphorescence, J. Mater. Chem. C, 2019, 7(34), 10481–10490 RSC.
- M. A. El-Sayed, Spin-Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics, J. Chem. Phys., 1963, 38(12), 2834–2838 CrossRef CAS.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, Room Temperature Phosphorescence of Metal-Free Organic Materials in Amorphous Polymer Matrices, J. Am. Chem. Soc., 2013, 135(16), 6325–6329 CrossRef CAS PubMed.
- L. Zang, W. Shao, M. S. Kwon, Z. Zhang and J. Kim, Photoresponsive Luminescence Switching of Metal-Free Organic Phosphors Doped Polymer Matrices, Adv. Opt. Mater., 2020, 8, 23 Search PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Stabilizing Triplet Excited States for Ultralong Organic Phosphorescence, Nat. Mater., 2015, 14(7), 685–690 CrossRef CAS PubMed.
- A. Lv, W. Ye, X. Jiang, N. Gan, H. Shi, W. Yao, H. Ma, Z. An and W. Huang, Room-Temperature Phosphorescence from Metal-Free Organic Materials in Solution: Origin and Molecular Design, J. Phys. Chem. Lett., 2019, 10(5), 1037–1042 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Carbazole Isomers Induce Ultralong Organic Phosphorescence, Nat. Mater., 2021, 20(2), 175–180 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, The Influence of the Molecular Packing on the Room Temperature Phosphorescence of Purely Organic Luminogens, Nat. Commun., 2018, 9(1), 1–10 CrossRef PubMed.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Suppressing Molecular Motions for Enhanced Roomerature Phosphorescence of Metal-Free Organic Materials, Nat. Commun., 2015, 6, 8947 CrossRef PubMed.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Tailoring Intermolecular Interactions for Efficient Room-Temperature Phosphorescence from Purely Organic Materials in Amorphous Polymer Matrices, Angew. Chem., 2014, 126(42), 11359–11363 CrossRef.
- X. Yang and D. Yan, Long-Afterglow Metal-Organic Frameworks: Reversible Guest-Induced Phosphorescence Tunability, Chem. Sci., 2016, 7(7), 4519–4526 RSC.
- B. Zhou and D. Yan, Hydrogen-Bonded Two-Component Ionic Crystals Showing Enhanced Long-Lived Room-Temperature Phosphorescence via TADF-Assisted Förster Resonance Energy Transfer, Adv. Funct. Mater., 2019, 29, 4 Search PubMed.
- Z. Cheng, H. Shi, H. Ma, L. Bian, Q. Wu, L. Gu, S. Cai, X. Wang, W. W. Xiong, Z. An and W. Huang, Ultralong Phosphorescence from Organic Ionic Crystals under Ambient Conditions, Angew. Chem. Int. Ed., 2018, 57(3), 678–682 CrossRef CAS PubMed.
- E. Hamzehpoor and D. F. Perepichka, Crystal Engineering of Room Temperature Phosphorescence in Organic Solids, Angew. Chem. Int. Ed., 2020, 59(25), 9977–9981 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. Chen and Y. Liu, Efficient Room-Temperature Phosphorescence of a Solid-State Supramolecule Enhanced by Cucurbit[6]Uril, Angew. Chem. Int. Ed., 2019, 58(18), 6028–6032 CrossRef CAS PubMed.
- Z. Y. Zhang and Y. Liu, Ultralong Room-Temperature Phosphorescence of a Solid-State Supramolecule Between Phenylmethylpyridinium and Cucurbit[6]Uril, Chem. Sci., 2019, 10(33), 7773–7778 RSC.
- O. Bolton, D. Lee, J. Jung and J. Kim, Tuning the Photophysical Properties of Metal-Free Room Temperature Organic Phosphors via Compositional Variations in Bromobenzaldehyde/Dibromobenzene Mixed Crystals, Chem. Mater., 2014, 26(22), 6644–6649 CrossRef CAS.
- S. R. Forrest, Organic Electronics: Foundations to Applications, Oxford Scholarship Online, 2020 Search PubMed.
- S. P. McGlynn, T. Azumi and M. Kinoshita, Molecular Spectroscopy of the Triplet State, Prentice-Hall, Englewood Cliffs, N. J., 1969 Search PubMed.
- D. R. Yarkony, Spin-Forbidden Chemistry within the Breit-Pauli Approximation, Int. Rev. Phys. Chem., 1992, 11(2), 195–242 Search PubMed.
- B. Song, W. Shao, J. Jung, S. J. Yoon and J. Kim, Organic Light-Emitting Diode Employing Metal-Free Organic Phosphor, ACS Appl. Mater. Interfaces, 2020, 12(5), 6137–6143 CrossRef CAS PubMed.
- S. Sarkar, H. P. Hendrickson, D. Lee, F. Devine, J. Jung, E. Geva, J. Kim and B. D. Dunietz, Phosphorescence in Bromobenzaldehyde Can Be Enhanced through Intramolecular Heavy Atom Effect, J. Phys. Chem. C, 2017, 121(7), 3771–3777 CrossRef CAS.
- A. D. Chien and P. M. Zimmerman, Recovering Dynamic Correlation in Spin Flip Configuration Interaction Through a Difference Dedicated Approach, J. Chem. Phys., 2017, 146, 014103 CrossRef PubMed.
- F. Bell, P. M. Zimmerman, D. Casanova, M. Goldey and M. Head-Gordon, Restricted Active Space Spin-Flip (RAS-SF) with Arbitrary Number of Spin-Flips, Phys. Chem. Chem. Phys., 2013, 15(1), 358–366 RSC.
- P. M. Zimmerman, F. Bell, M. Goldey, A. T. Bell and M. Head-Gordon, Restricted Active Space Spin-Flip Configuration Interaction: Theory and Examples for Multiple Spin Flips With Odd Numbers of Electrons, J. Chem. Phys., 2012, 137(16), 164110 CrossRef PubMed.
- H. Jiang and P. M. Zimmerman, Charge Transfer via Spin Flip Configuration Interaction: Benchmarks and Application to Singlet Fission, J. Chem. Phys., 2020, 153, 064109 CrossRef CAS.
- D. Casanova and M. Head-Gordon, Restricted Active Space Spin-Flip Configuration Interaction Approach: Theory, Implementation and Examples, Phys. Chem. Chem. Phys., 2009, 11(42), 9779–9790 RSC.
- A. I. Krylov, Spin-Flip Configuration Interaction: An Electronic Structure Model That Is Both Variational and Size-Consistent, Chem. Phys. Lett., 2001, 350(5–6), 522–530 CrossRef CAS.
- A. I. Krylov, Size-Consistent Wave Functions for Bond-Breaking: The Equation-of-Motion Spin-Flip Model, Chem. Phys. Lett., 2001, 338(4–6), 375–384 CrossRef CAS.
- H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vázquez, T. Goodson and P. M. Zimmerman, Enacting Two-Electron Transfer from a Double-Triplet State of Intramolecular Singlet Fission, J. Am. Chem. Soc., 2018, 140(25), 7760–7763 CrossRef CAS PubMed.
- H. Kim and P. M. Zimmerman, Coupled Double Triplet State in Singlet Fission. Physical Chemistry Chemical Physics, Royal Society of Chemistry, 2018, pp. 30083–30094 Search PubMed.
- P. M. Zimmerman, F. Bell, D. Casanova and M. Head-Gordon, Mechanism for Singlet Fission in Pentacene and Tetracene: From Single Exciton to Two Triplets, J. Am. Chem. Soc., 2011, 133(49), 19944–19952 CrossRef CAS PubMed.
- A. D. Chien, A. R. Molina, N. Abeyasinghe, O. P. Varnavski, T. Goodson and P. M. Zimmerman, Structure and Dynamics of the 1(TT) State in a Quinoidal Bithiophene: Characterizing a Promising Intramolecular Singlet Fission Candidate, J. Phys. Chem. C, 2015, 119(51), 28258–28268 CrossRef CAS.
- A. E. Rudenko, N. E. Clayman, K. L. Walker, J. K. Maclaren, P. M. Zimmerman and R. M. Waymouth, Ligand-Induced Reductive Elimination of Ethane from Azopyridine Palladium Dimethyl Complexes, J. Am. Chem. Soc., 2018, 140(36), 11408–11415 CrossRef CAS PubMed.
- P. Pokhilko and A. I. Krylov, Quantitative El-Sayed Rules for Many-Body Wave Functions from Spinless Transition Density Matrices, J. Phys. Chem. Lett., 2019, 10(17), 4857–4862 CrossRef CAS PubMed.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Recent Progresses on Materials for Electrophosphorescent Organic Light-Emitting Devices, Adv. Mater., 2011, 926–952 CrossRef CAS PubMed.
- K. Mori, T. P. M. Goumans, E. van Lenthe and F. Wang, Predicting Phosphorescent Lifetimes and Zero-Field Splitting of Organometallic Complexes with Time-Dependent Density Functional Theory Including Spin-Orbit Coupling, Phys. Chem. Chem. Phys., 2014, 16(28), 14523–14530 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods of prototype molecules, computational details, reduced SOCME in the selected orientations, excitation and emission profile at room temperature and 78 K. See DOI: 10.1039/d1sc05689a |
| This journal is © The Royal Society of Chemistry 2022 |