
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in noble metal complex based photodynamic therapy
Yanping
Wu
a,
Shumeng
Li
a,
Yuncong
Chen
 *ab,
Weijiang
He
*ab and
Zijian
Guo
*ab
*ab,
Weijiang
He
*ab and
Zijian
Guo
*ab
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Chemistry and Biomedicine Innovation Center (ChemBIC), Nanjing University, Nanjing 210023, China. E-mail: chenyc@nju.edu.cn; heweij69@nju.edu.cn; zguo@nju.edu.cn
bNanchuang (Jiangsu) Institute of Chemistry and Health, Nanjing 210000, China
First published on 1st April 2022
Abstract
Photodynamic therapy (PDT) utilizes light-activated photosensitizers (PSs) to generate toxic species for therapeutics. It has become an emerging solution for cancer treatment because of its specific spatiotemporal selectivity and minimal invasiveness. Noble metal (Ru, Ir and Pt) complexes are of increasing interest as photosensitizers for their excellent photophysical, photochemical, and photobiological properties. In this review, we highlight recent advancements in the development of noble metal complex photosensitizers for PDT during the last 5 years. We will summarize the design strategies of noble metal complexes for efficient and precise PDT, including increasing the light penetration depth, reducing the oxygen-dependent nature and improving target ability. Finally, we summarize recent efforts for the development of noble-based PSs and discuss the limitations of such PSs in clinical application and future perspectives in this field, such as the combination of PDT with other treatment modalities.
1. Introduction
Cancer is a leading cause of death in the world, and it is responsible for an estimated 19.3 million new cases and almost 10 million deaths in 2020.1 To date, the classical clinical treatments include surgery, chemotherapy, and radiotherapy. However, there are some drawbacks of these treatments such as invasiveness, a high recurrence rate, severe systemic toxicity and adverse effects.2–4 Photodynamic therapy (PDT) is a potential alternative cancer treatment, which usually requires three essential constituents: light, photosensitizers (PSs) and oxygen. Upon irradiation with the appropriate wavelength of light, the PSs will produce cytotoxicity by generation of reactive oxygen species (ROS) that oxidize biomolecules in cells, mainly nucleic acids, proteins, and lipids, leading to severe alteration in cell signaling cascades or gene expression regulation. This process will cause tumor cell damage including necrosis, apoptosis, autophagy or ferroptosis. Generally, most PSs have a degree of fluorescence which can also be incorporated for monitoring and treatment.5,6The precise photodynamic therapy mechanism is an ongoing hot topic of investigation. The possible mechanism may be divided into type I and type II by different photochemical reaction processes as illustrated in Fig. 1. After light absorption, the PS will be excited from the singlet ground state (S0) to singlet excited states (Sn, n ≥ 1). Then, the PS will relax from higher excited states to the lowest vibrational energy level (S1) by internal conversion (IC) or vibrational relaxation (VR). The PS in S1 may undergo intersystem crossing (ISC) to the triplet excited state (T1). In type I mechanism, PS in the singlet excited state or triplet excited state can generate radicals (OH•, O2−˙ and others) by proton or electron transfer with biological substrates, oxygen and water. For the type II PDT process, PS in the triplet excited state converts triplet oxygen into singlet oxygen (1O2) via energy transfer.7
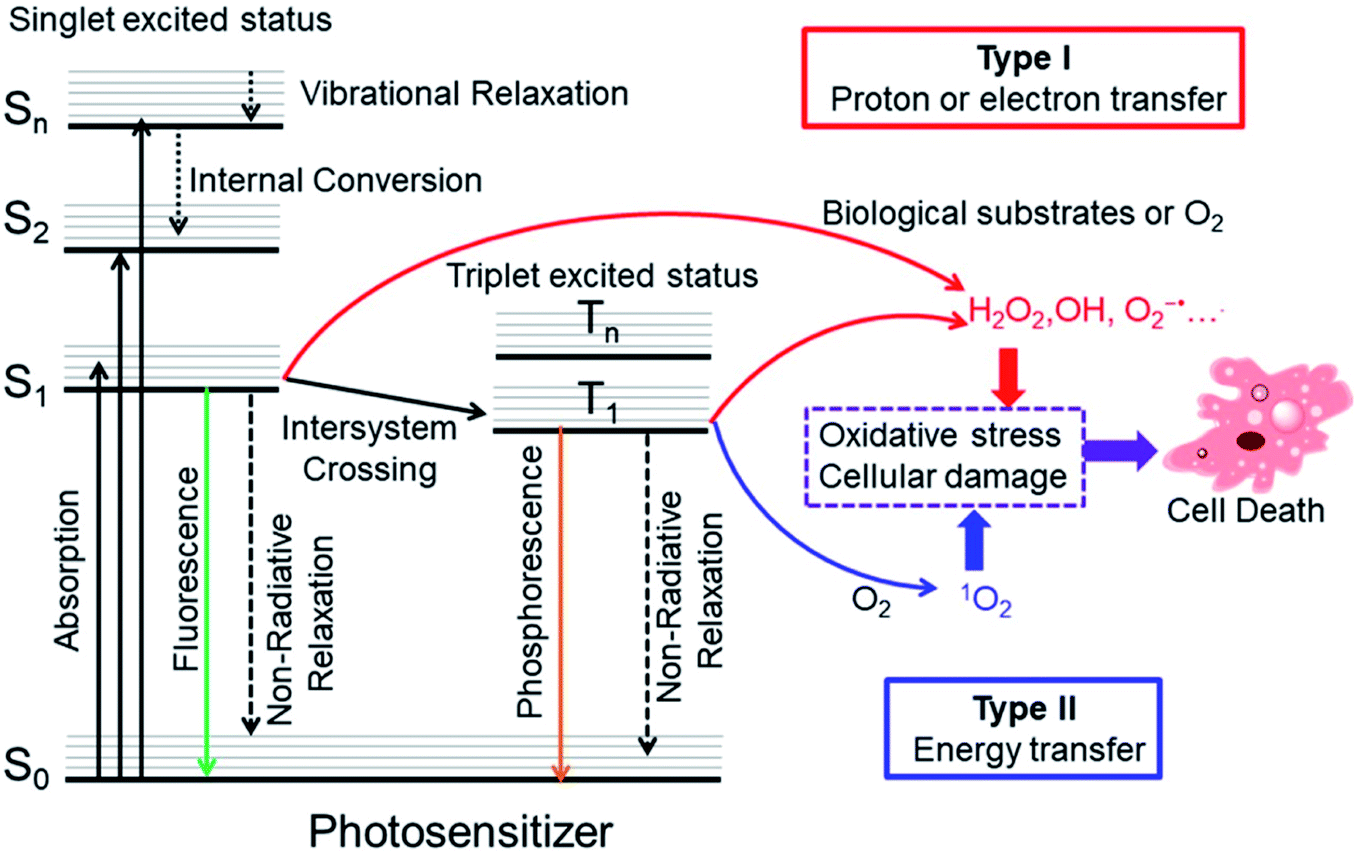 | ||
| Fig. 1 Modified Jablonski diagram of the photosensitization process and mechanism of action of PDT. Adapted with permission from ref. 7. Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Currently, three generations of PSs have been developed for anticancer PDT. The chemical structures of several PSs are presented in Fig. 2. The first generation of PSs is based on hematoporphyrin derivatives (HpD), a complex mixture of porphyrinic complexes. But its clinical applications have some limitations due to their low chemical purity or poor tissue penetration. The second generation of PSs includes 5-aminolevulinic acid, benzoporphyrin derivatives, and phthalocyanines. They have well-defined structures, high chemical purity, high yield of singlet oxygen formation and deep tissue penetration. However, their poor solubility in water and poor selectivity limited intravenous administration and application in therapeutics. The third-generation PSs introduced target groups of tumor receptors or surface markers to increase the selectivity for tumors and thus reduce the side effects on healthy tissues. With a higher affinity to the malignant tissue, third-generation photosensitizers can also be used as theranostic agents.8
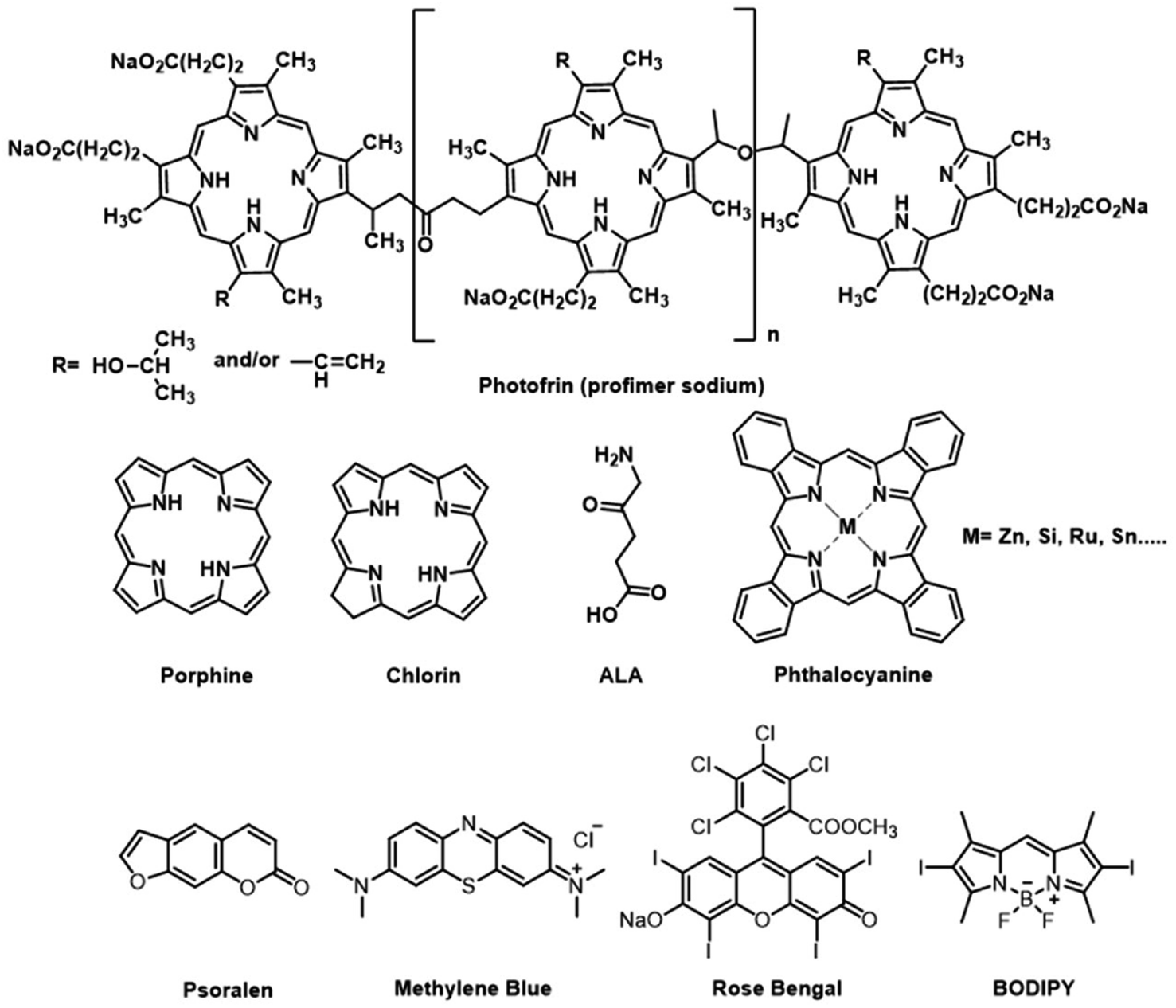 | ||
| Fig. 2 Chemical structures of some traditional PSs. | ||
To date, many different photosensitizers have been explored, for instance, nanoscale metal organic frameworks (NMOFs),9 engineering functional inorganic and metallic nanocomposites, and organic-molecule and transition metal complexes. Among them, noble metal center Ru(II), Ir(III), and Pt (II) complexes are of increasing interest as PSs due to their salient inherent feature of the heavy-atom effect that mediates strong spin-orbital coupling (SOC) promoting the rate of intersystem crossing, which provides more time for the triplet excited state (T1) to interact with dioxygen, biomolecular or other oxygen substrates.10 These metal complexes offer many more excited-state electronic configurations (Fig. 3) and can be exploited in both oxygen-dependent and -independent cytotoxic pathways.11 The characteristic reactivities of the different excited-state configurations provide the opportunity to design noble metal complexes with desirable photobiological mechanisms that are simply not easy for NMOFs and inorganic and metallic nanocomposite photosensitizers.12 In addition, most of the noble metal complexes have luminescence which is beneficial for the study of PS biodistribution, such as the subcellular location of the PSs can be visualized by confocal imaging.13,14 Their cellular uptake can also be quantified by inductively coupled plasma mass spectrometry (ICP-MS).15 Additionally, noble metal-based photosensitizers are of interest as their absorption and chemical binding properties can be modified via the alteration of ligands. For example, the ligands of a complex can be modified to rationally tune its solubility, lipophilicity, wavelength, targetability, and pharmacokinetics. Therefore, noble metal complexes could serve as efficient PS candidates for PDT. The Ru(II) complexes are 4d6 centers possessing a hexa-coordinated octahedral architecture with a different arrangement of ligands. They are widely used PSs for PDT due to their long-lived triplet excited-state properties and biocompatibility. The Ir(III) complexes are 5d6 center, and the electronic properties determine the octahedral coordination. As in other heavy transition metal complexes, the emissions may be produced from various excited states including MLCT, ILCT, LLCT, LMMCT, MMLCT, and MLLCT. Octahedral Ir(III) complexes exhibit great stability in biological media and are excellent phototherapeutic candidates.16 Pt(II) complexes are different from Ru(II) or Ir(III) complexes; they are 5d8 center with planar quadrilateral geometries. As photosensitizers, the Pt(II) center can increase the ROS generation due to the heavy atom effect. The photoinduced ROS can destroy the cell membranes and promote the cellular uptake of Pt(II) complexes. Some Pt(II) complexes also have DNA binding affinity to form cytolethal Pt-DNA cross-links or photoinduced DNA damage.17 As a chemotherapeutic agent, Ru(II) and Pt(II) complexes can interact non-covalently with nucleic acids, which can induce the unwanted toxic effect. These compounds have inevitable intrinsic toxicity. After the PDT treatment with visible light, the tested compounds showed higher photocytotoxicity, but no dark cytotoxicity was observed at the same concentration used for PDT cell treatment.
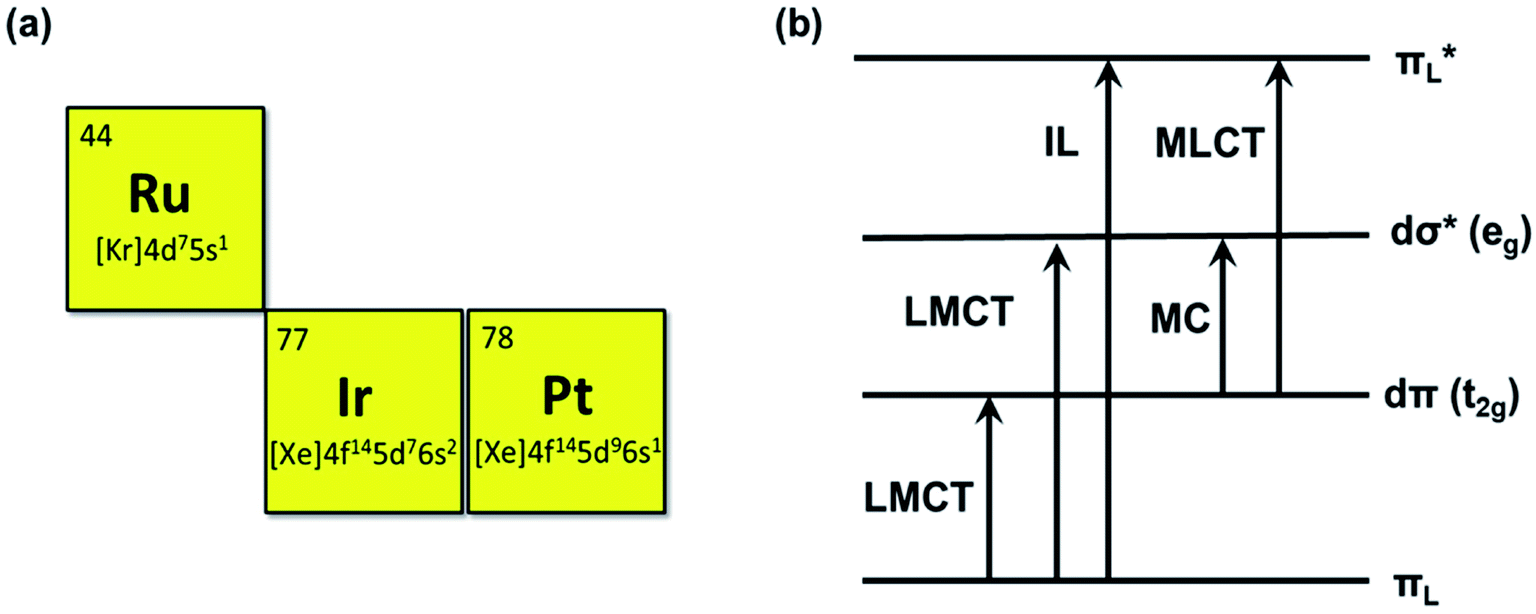 | ||
| Fig. 3 The atomic number and outer electron arrangement of ruthenium, iridium and platinum. Some of the electronic transitions, LMCT: ligand-to-metal charge transfer, IL: intraligand, MC: metal-centered, and MLCT: metal-to-ligand charge transfer. | ||
Compared with organic photosensitizers, the application of noble metal-based complexes for phototherapy is at an early stage, and it still has to face several challenges: (1) the tissue penetration limitation of excitation light hinders the widespread clinical use of PDT; (2) the noble metal-based photosensitizers are usually oxygen-dependent, which limits their efficiency against hypoxic tumors; (3) target ability of PSs for tumors or certain subcellular organelles is highly demanded and less reported. This minireview classifies the photosensitizers by different central metal atoms Ru, Ir, and Pt, and subclassifies according to the different strategies of noble metal photosensitizer design to address the above-mentioned issues: (1) develop long-wavelength near-infrared (NIR) or two-photon excitation (TPE) photosensitizers to increase the depth of tissue penetration; (2) explore more type I or O2-independent photosensitizers to overcome hypoxic tumors; (3) design tumor tissue or certain subcellular organelles targeting photosensitizers to avoid damage to surrounding healthy tissue. The aim of this review is not to provide a comprehensive introduction for the potential readers, and only a few representative studies are selected for each subcategory. At the end of this review, we have summarized ROS production, cellular targets, and dark- and photo-toxicity and the mode of cell death data for selected Ru, Ir and Pt photosensitizers in a table.
2. Ruthenium complexes in photodynamic therapy
Ru complexes have attracted great interest not only as chemotherapy agents but also as promising PDT PS candidates because of their favorable chemical and photophysical properties.9,10 In terms of the chemotherapy agent, Ru(II) complexes have been proven to damage DNA and inhibit tumor metastasis effectively.10 As photosensitizers, Ru(II) complexes exhibit long excited-state lifetimes, large quantum yields for the triplet-state formation and two-photon excitation. The electronic arrangement of Ruthenium is [Kr] 4d75s1(Fig. 3); therefore, most Ru(II) complexes usually have a hexa-coordinated octahedral architecture with a different arrangement of ligands, which can be modified for different functions including solubility, charge distribution, cellular uptake efficiency, targeting ability, electrochemical and photophysical properties, etc.18 Additionally, straightforward synthetic procedures for preparing coordination and Ru(II) complexes have been well-established; thus, Ru(II) complex PSs are easily available. In this section, some recent progress with new strategies based on Ru(II) complexes for effective PDT will be discussed in detail.2.1 The near-infrared (NIR) and two-photon excitation (TPE) Ru(II) based PSs
Many studies have been reported for PSs with deep tissue penetration. Chen et al. summarized deep PDT techniques, including direct (NIR OPE, NIR TPE and X-ray-excited PDT) and indirect excitation (self-illuminating PDT).19 PSs with long wavelengths can be excited by NIR light, which is one of the most effective ways to overcome the limitation of depth tissue penetration.Torres et al. developed a series of water-soluble Ru(II) phthalocyanines.20 These complexes have large and stable, conjugated π-systems that are suitable for efficient energy and electron-transfer processes. In particular, they display strong absorption in the visible region, with a maximum around 700 nm which enables greater tissue penetration. Therefore, one potent way to obtain long wavelengths for noble metal PSs is to extend the π-conjugation of the ligands.
To obtain long wavelengths, researchers attempted to increase the π-conjugation of the ligands. In 2017, the McFarland and Sun group reported five π-expansive heteroleptic tris-diimine Ru(II) complexes (1–5; Fig. 4) as PSs for PDT.21 The complex 5 showed the dppn ligand-based 3π–π* with a long triplet lifetime of 41.2 μs while complexes 1–4 displayed shorter triplet lifetimes (1–2 μs). The extended π-conjugation of the ancillary ligands affected the spin-forbidden transitions to the triplet excited states, and absorption spectral studies in the concentration range of 5 × 10−5 to 2 × 10−4 M showed a broad and weak band from 550–900 nm. The dark and light cytotoxicities of 1–5 were determined in human leukemia (HL60) and skin melanoma (SKMEL28) cell lines. In HL60 leukemia cells, the dark cytotoxicity decreased in the order of 4 > 5 ≈ 3 > 1 ≈ 2, and the order in the SKMEL28 melanoma cell line is 4 > 3 > 5 > 2 > 1. This new series of PSs presented minimal dark cytotoxicities. The author used broadband visible light and a red light-emitting diode at 625 nm light irradiation to study photocytotoxicity. Under visible light irradiation, EC50 values ranged from 8.2 to 48 μM in HL60 cells and 3.8 to 8.4 μM in SKMEL28 cells. Under red light irradiation, these values were 20–300 and 10–204 μM in HL60 and SKMEL28 cells, respectively. Thus, these PSs were highly photocytotoxic toward melanoma cell lines. Complex 5 exhibited the largest PIs for red light irradiation in SKMEL28 cells (PI = 12) and visible light irradiation in HL60 cells (PI = 14). Currently, red light is employed for clinical applications; complex 5 is as phototoxic with red light toward cells as Photofrin but with three-fold less dark toxicity and a larger therapeutic margin. In the studies of PDT, dppn was proved to be the critical ligand of complex 5 for exhibiting most potent photocytotoxicity and high photoselectivity for melanoma cells with red light irradiation. All of the PSs were taken up by cells and tracked by their intracellular luminescence before and after irradiation. These results indicated that this new series of PSs are useful as theranostic agents. Although these complexes showed a good PDT effect, the wavelengths of absorption and emission are not increased with the π-conjugation of the ancillary ligands extended.
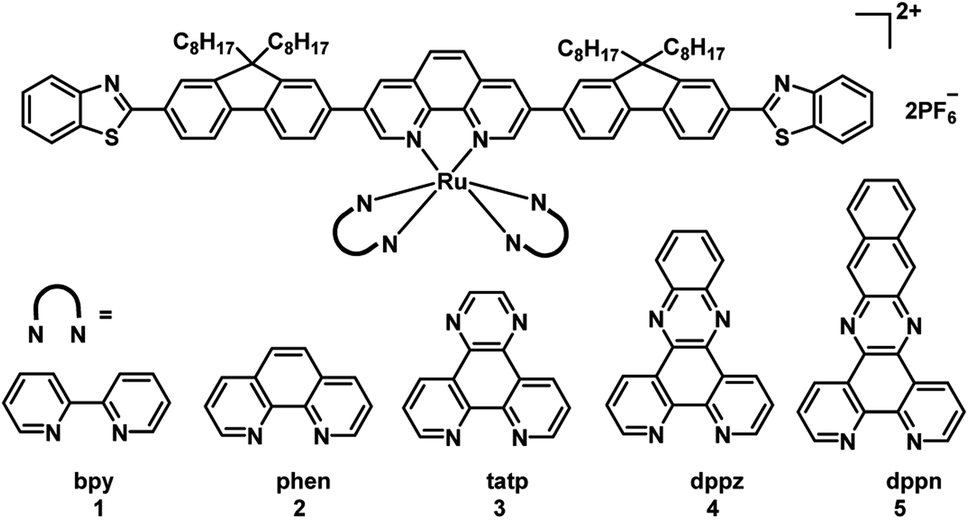 | ||
| Fig. 4 Chemical structures of complexes 1–5. | ||
In 2017, Wang et al. synthesized two new Ru(II) complexes 6–7 (Fig. 5), by incorporating a 5-chloro-8-oxyquinolate-based ligand into a merocyanine scaffold to extend the π conjugation system.22 Oxyquinolate ligands are characterized by a large π-overlap with the orbitals of the metal that diminished the HOMO–LUMO gap for Ru(II) complexes with oxyquinolate ligands. This method realized the bathochromic shift of the MLLCT absorption band (from 502 nm to 649 nm for complex 6 and 505 nm to 630 nm for 7) and endowed the complex with a high molar extinction coefficient. The complex 6 displayed PDT activity by efficient ˙OH-mediated DNA photocleavage that transformed pBR322 DNA from the supercoiled (SC) form into the nicked circular (NC) form. Compared with 6, complex 7 generated 1O2 more efficiently, but lower ˙OH, resulted in negligible DNA cleavage. Additionally, complex 6 exhibited more efficient PDT activity in E. coli bacterial cells over HeLa cells upon 635 nm (1.5 mW cm−2, 20 min) irradiation. However, these two complexes are not stable under irradiation, probably, owing to the merocyanine scaffold destroyed by the generated ˙OH.
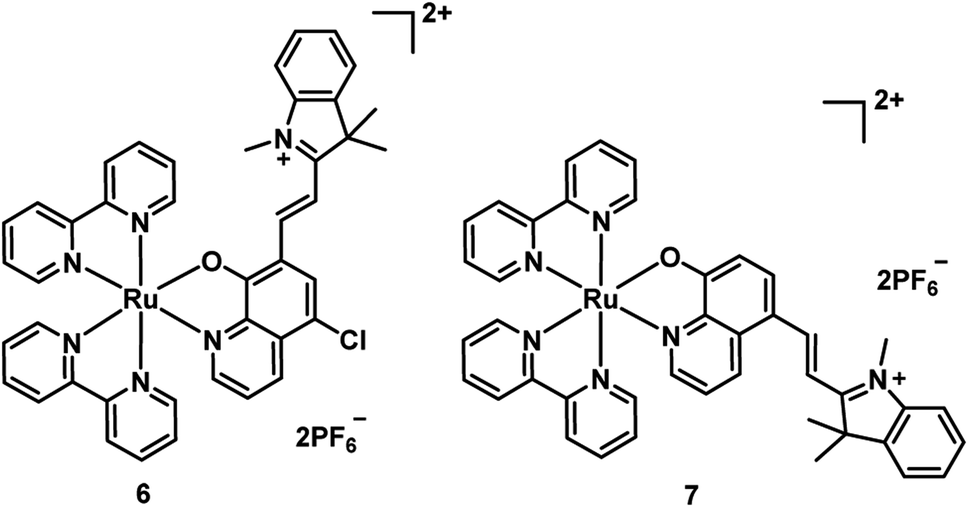 | ||
| Fig. 5 Chemical structures of complexes 6 and 7. | ||
Recently, with the advent of multiphoton lasers, the two-photon excitation (TPE) of PSs in the NIR window has been developed as an innovative approach compared with traditional one photo PDT. One of the advantages of TPE is significantly increased light penetration depth compared with UV or visible light. Another advantage of TPE is enhanced spatial resolution which depends on the square of luminescence intensity.23 Additionally, photodamage is reduced when using TPE, as the low energy NIR irradiation.
In 2020, the Chao group reported four Ru(II) polypyridyl complexes 8–11 (Fig. 6).24 All of the complexes exhibited significant two-photon (800 nm) absorption cross-sections up to ≈1600 GM and have a 1P absorption tail towards and 2P absorption within the biological spectral window (600–900 nm), potentially allowing the application for deep-seated or large tumors. These complexes have high 1O2 quantum yields in CH3CN between 43–92% and 2–14% in an aqueous solution. The dark cytotoxicity of phen-based complexes 8 and 9 for HeLa, CT26, and U373 cells is non-toxic in the dark (IC50 > 100 μM). And their phototoxic effect is very high in the low micromolar range (IC50 = 0.9–15.6 μM). However, bphen-coordinated complexes are cytotoxic in the low micromolar range (IC50 = 5.2–20.8 μM) in all tested cell lines. And their phototoxicity ranges from the high nanomolar to the low micromolar range (IC50 = 0.5–2.1 μM). Then they chose 800 μm HeLa 3D multicellular spheroids (MCSs) as a tumor model to test the PDT therapeutic outcome of four Ru(II) complexes. All complexes were able to fully penetrate the MCTSs and generate singlet oxygen in their hypoxic centers. The bphen-coordinated complexes (10 and 11) showed low dark cytotoxicity while the phen-coordinated complexes 8 and 9 showed no dark toxicity (IC50 > 300 μM) in 3D HeLa MCSs. All complexes exhibited phototoxic effects in the micromolar range (IC50,1P = 3.8–32.6 μM; IC50,2P = 0.8–27.8 μM). After light treatment, the MCTSs were eradicated, including in their large hypoxic centers. In addition, complex 9 was also able to eradicate an adenocarcinomic human alveolar basal epithelial tumor inside a mouse model upon clinically relevant 1P (500 nm) or 2P (800 nm) excitation. Four Ru(II) complexes exhibited much better PDT efficacy than the potential TP-PDT candidate H2TPP.
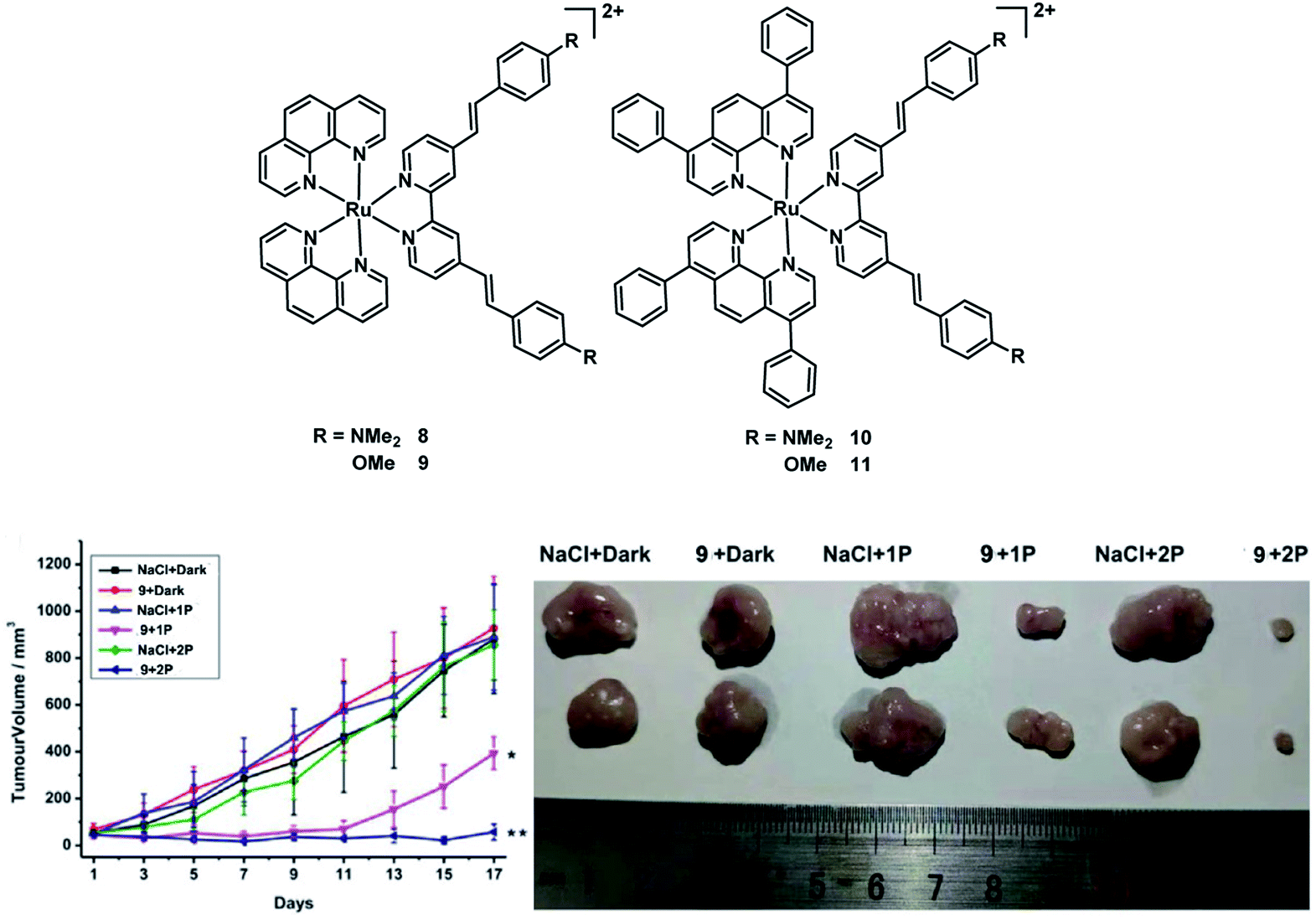 | ||
| Fig. 6 Chemical structures of complexes 8–11, and in vivo PDT study of 9 using 1P or 2P. (Left: tumor growth inhibition curves upon treatment; right: representative photographs of the tumor after different treatments on day 17. *p < 0.05, **p < 0.01) reproduced with permission from ref. 24. Copyright 2021 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The same group reported a glutathione-activatable dinuclear Ru(II)-azo complex 12 (Fig. 7) for TP-PDT.25 The ligand azobpy is strongly electron-withdrawing that quenched luminescence and singlet oxygen of the complex. After being reduced by GSH, the TPA value of complex 13 is 210 GM (810 nm), much larger than that of the clinical PS (tetraphenylporphyrin, 2.2 GM, 800 nm); luminescence increased 50-fold, and singlet oxygen quantum yield increased to 0.4. ICP-MS and colocalization indicated that mitochondria are the main targets of 12 and 13 in HeLa cells. From fluorescence microscopy, the uptake of complex 12 and 13 in HeLa cells is higher than in L02 cells(normal cells). The complex 12 displayed negative dark cytotoxicity (IC50 > 70 μM) for HeLa cell lines and (IC50 > 90 μM) for L02 cell lines. Under irradiation at 450 nm (20 mW cm2, 15 min), the cytotoxicity increased to about 5 μM for HeLa cells and 13 μM for L02 cells. The dark cytotoxicity of complex 12 was very low (IC50 > 100 μM) against HeLa MCTSs. Under two-photon irradiation (810 nm, 100 mW), the phototoxicity of complex 12 showed high toxicity to 3D MCTSs with an IC50 of about 5.71 μM (PI = 19). The calcein AM staining assay indicated that complex 12 effectively inhibited cancer growth and killed cancer cells in HeLa MCTSs.
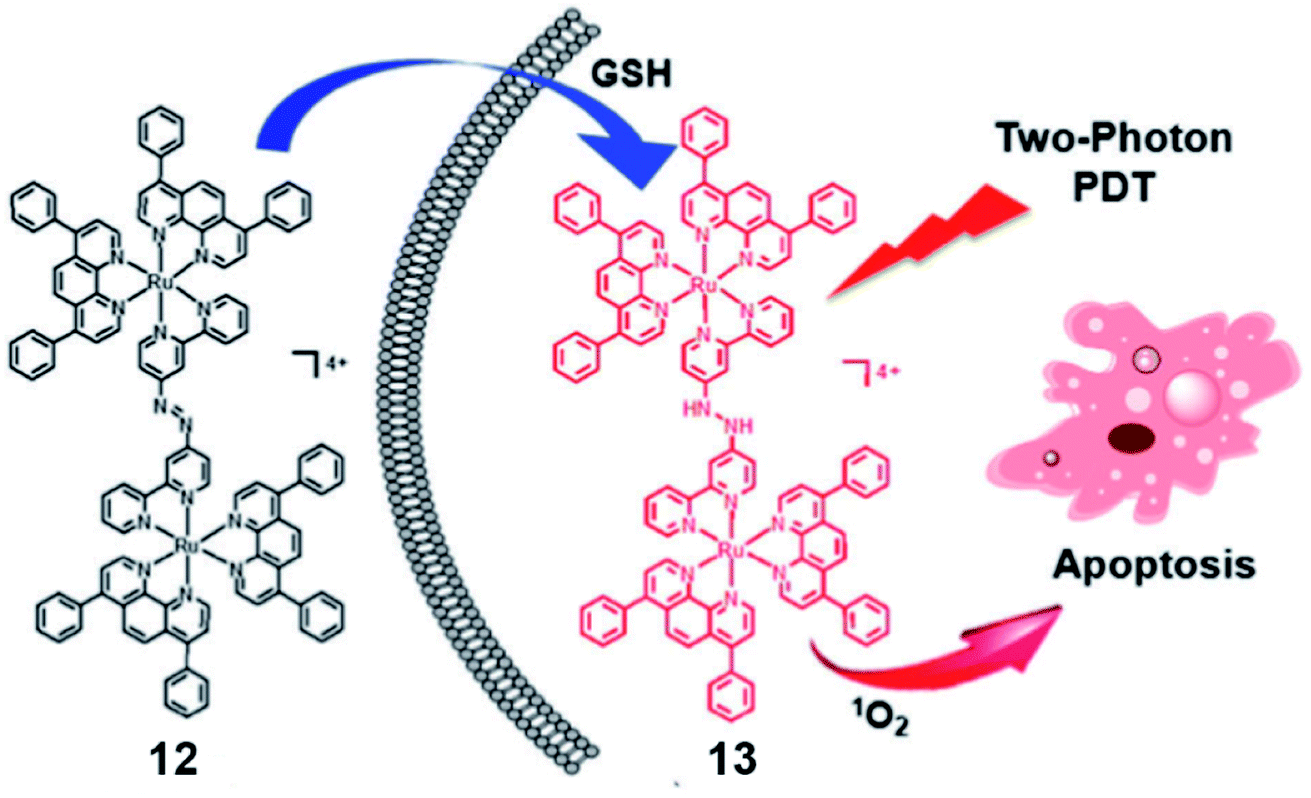 | ||
| Fig. 7 Schematic illustration of GSH activation and phototoxicity of complex 12. Reproduced with permission from ref. 25. Copyright 2017 The Royal Society of Chemistry. | ||
2.2 Ru(II) based PSs for PDT of hypoxic tumor cells
Due to the aggressive proliferation of cancer cells and insufficient oxygen supply, there is significant hypoxia in solid tumors.26 This is another important factor that reduced the efficiency of PDT, especially for the type II PDT pathway which is dependent on O2 significantly. Recently, many innovative strategies to overcome hypoxia have been reviewed by the Juyoung Yoon group.27 In these strategies, one of the simplest is diminished oxygen dependence using type I PSs which can break through the limitation of hypoxia in the type II PDT process. Thus, here we will introduce type I Ru(II) photosensitizers.TLD1433 (Fig. 8), the first Ru(II)-based photosensitizer in the treatment of non-muscle invasive bladder cancer to enter a human clinical trial in Canada, was developed by McFarland's laboratories.11,28 The researchers pointed out that the ligand in the structure of the complex might be responsible for both Type I electron exchange and Type II energy transfer mechanisms. Adamo et al. investigated in detail the theoretical exploration of type I/II dual photoreactivity of these Ru(II) dyads.12 Thiophene ligands were able to afford low-lying 3IL states which can contribute to the dual type I/II photoreactivity. The feasibility of the type I mechanism mediating photoinduced electron transfer reactions was established by computing the vertical electron affinity and ionization potentials for each molecule and molecular oxygen. The autoionization reactions generate a reduced form of the sensitizer through reduction of the T1 state of Ps by neighboring S0 or the T1 state of the PS itself. The reduced form of the sensitizer scavenged from oxygen leading to the superoxide anion.
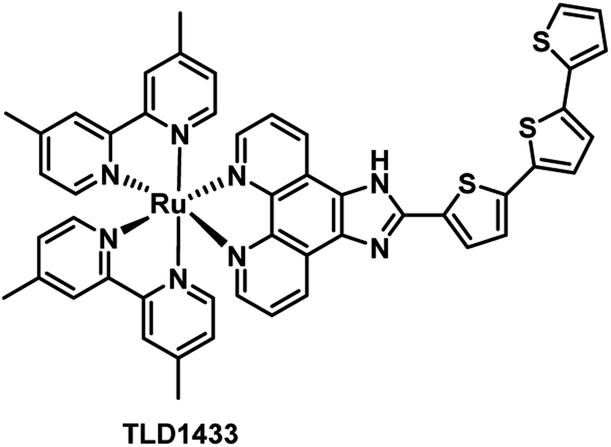 | ||
| Fig. 8 Chemical structure of TLD1433. | ||
It is considered that an ideal photosensitizer for type I should have low oxidation potential and good electron-donating ability.29 Recently, Huang et al. developed coumarin-modified cyclometalated Ru(II) complexes (14–15; Fig. 9), which displayed an excellent PDT effect on hypoxic solid tumors. The coumarin moiety enhanced the light-absorption ability and modulated the energy level of the Ru(II) complex due to its electron-donating and light-harvesting abilities. Compared with complex 14 (oxidation waves at 0.59 V), complex 15 has a lower oxidation potential (oxidation waves at 0.33 V). Thus, the ability of electron transfer to the substrates for 15 is stronger than that of 14, providing a possibility for the type I PDT pathway for 15. The quantum yields of singlet oxygen generation were calculated to be 0.14 for 14 and 0.16 for 15. After light irradiation, the EPR signal suggested that hydroxyl radicals were generated, and the intensity of 15 was much higher than that of 14. Therefore, complex 15 exhibits excellent PDT effects under both normoxia and hypoxia (white light: 400–800 nm, power: 35 mW cm−2, and irradiated time: 10 min 5% O2) in HeLa cells. In vivo experiment of complex 15 remarkably inhibited tumor growth after 14 days of treatment (injected intratumorally, dose: 5 mg kg−1, xenon lamp power: 250 mW cm−2, 15 min, and at 15 min post-injection). Complex 15 showed negligible dark toxicity towards the cancer cells and was non-toxic towards normal organs and has no cumulative effect on the body.
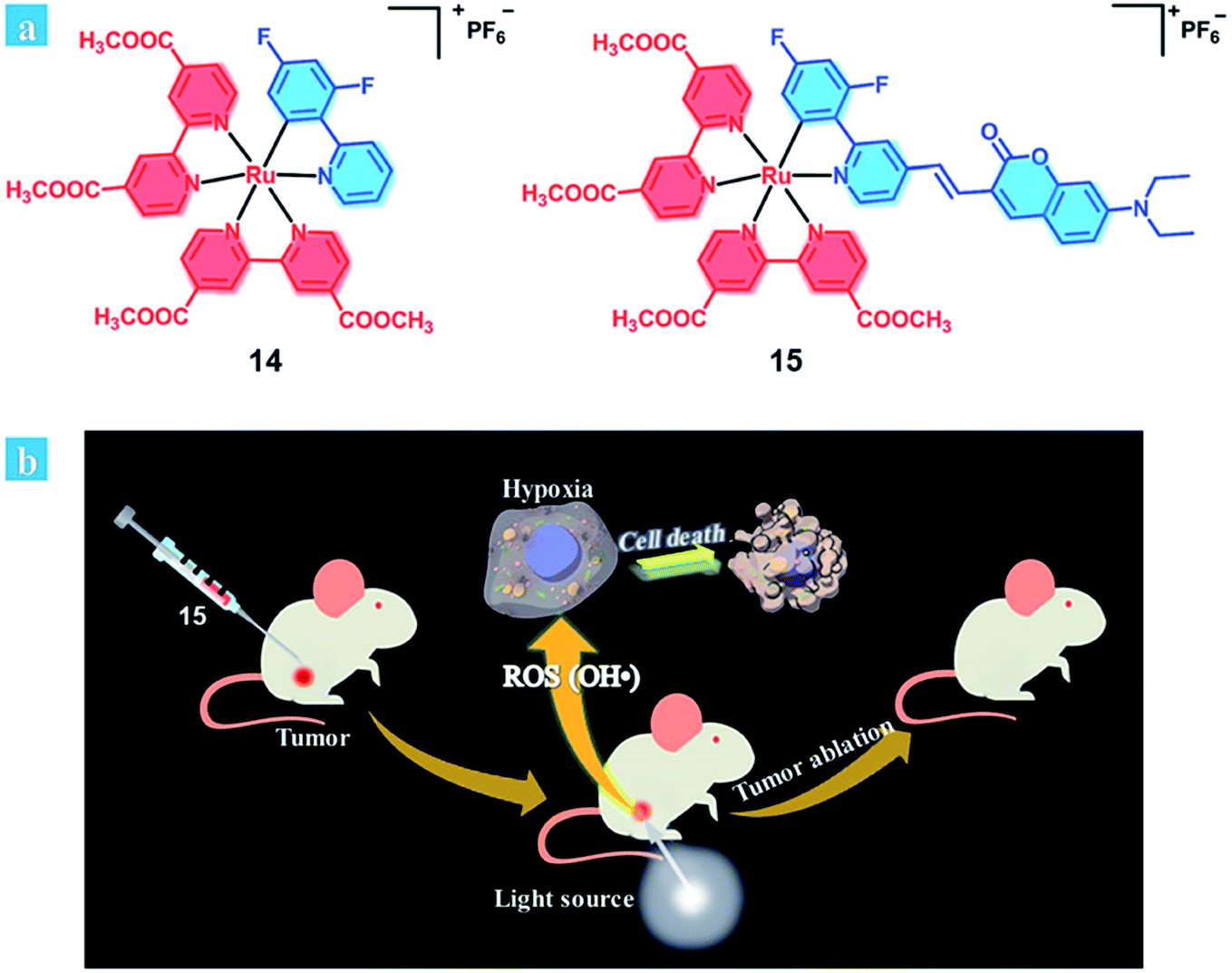 | ||
| Fig. 9 (a) Chemical structures of complexes 14 and 15. (b) Schematic illustration of 15 used as an efficient type I photosensitizer for photodynamic therapy. Reproduced with permission from ref. 29. Copyright 2018 The Royal Society of Chemistry. | ||
2.3 Targeted Ru(II) based PSs
The short half-life and low selectivity of ROS will limit the PDT effect. In this regard, the specific targeting ability towards tumor tissue or subcellular organelles of the tumor cells could be attractive for promoting the PDT effect and avoiding damage to healthy tissues.7,30 Biomolecules have been introduced as a targeting ligand to reduce the unwanted side effects, which allows for accumulation in specific tissues and cellular compartments. PSs with targeting groups have the potential to be used in clinical applications due to their better PDT performance, although their cost might be much higher.In 2019, the Chao group designed a biotin-modified Ru(II) complex 16 (Fig. 10) as a PS for PDT.34 Compared with biotin-free Ru-PhenNH2, complex 16 exhibited high accumulation and tumor selectivity in the biotin receptor overexpressing A549R cancer cells. Complex 16 has a large TPA value of 140.26 GM at 820 nm. After two-photon irradiation at 820 nm, complex 16 showed effective photocytotoxicity with an IC50 value of 3.3 μM and a PI value of 22.1 toward A549R cells. The excellent TPPDT was also demonstrated in the 3D MCS model based on A549R cells. Under light irradiation, complex 16 induced cell apoptosis via ROS generation and the activation of caspase-3/7 which accounts for its potency against cisplatin-resistant A549R cells.
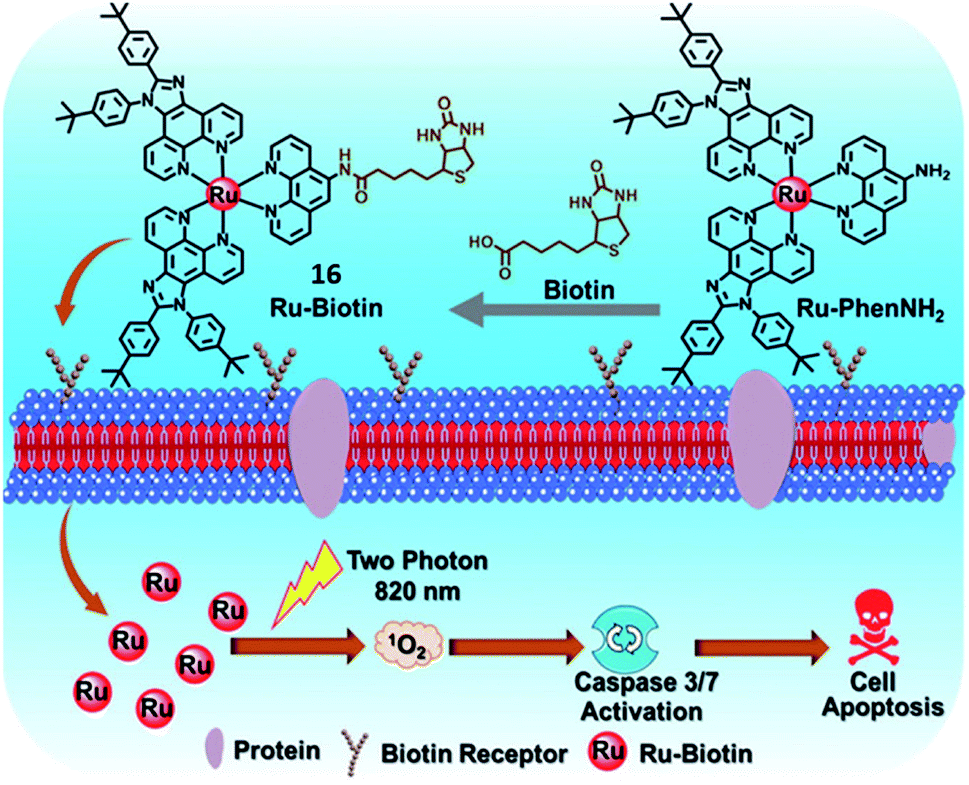 | ||
| Fig. 10 Schematic illustration of 16 for targeted-tumor TPPDT. Reproduced with permission from ref. 34. Copyright 2019 The Royal Society of Chemistry. | ||
In 2020, the Wang group developed an RGD-peptide conjugated Ru(II) complex 17 (Fig. 11) as a tumour-targeted PS for TPPDT.35 The ICP-MS results showed that 17 preferentially accumulated in the integrin positive U87MG cancer cells compared with the integrin negative MCF-7 cells. Complex 17 has a TPA value of 63.5 GM at 820 nm, which is much higher than that of H2TPP. The excellent TPPDT was confirmed in the 3D MCS model based on U87MG cells. Under irradiation at 820 nm, complex 17 showed effective photocytotoxicity with an IC50 value of 9 μM and a PI value of 24 toward 3D U87MG cells. The author examined in vivo the TPPDT anti-tumour on U87MG tumour-bearing Balb/c mice. Complex 17 was selectively accumulated in the tumours rather than in other organs. The TPPDT (50 mW, 120 s) was performed 2 h after the intravenous injection. The mice treated with 17 by TP irradiation showed a tumour inhibition rate (TIR) of 87%. These results showed that the rational design of complex 17 reduced side effects and offered sufficient PDT efficacy.
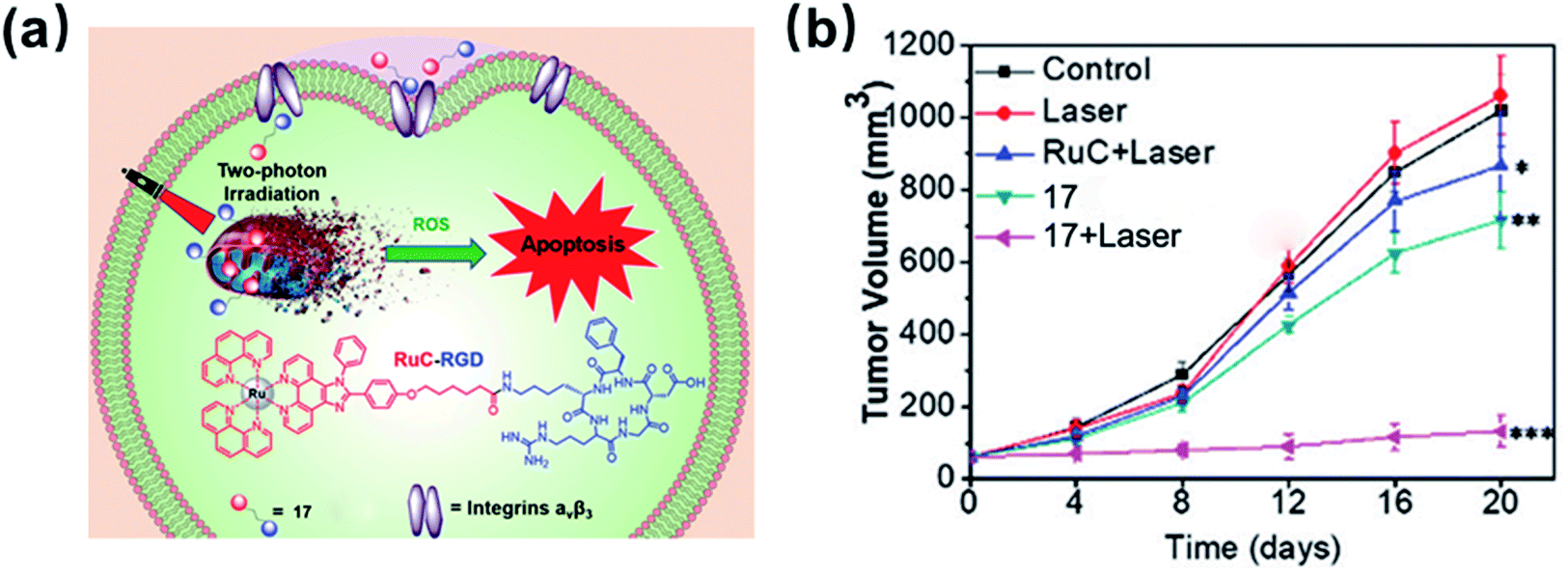 | ||
| Fig. 11 Schematic illustration of 17 for targeted-tumor TPPDT and in vivo inhibition of tumor growth. Reproduced with permission from ref. 35. Copyright 2020 The Royal Society of Chemistry. | ||
Mitochondria are the powerhouse of the cell that are involved in energy metabolism.38 Because of their significant role in mediating cell apoptosis, mitochondria became a main subcellular target for many PSs used in PDT.39 Due to the high mitochondrial membrane potential (−160 to −180 mV) of tumor cells, the cationic species are easy to realize mitochondrial selectivity.40,41 Most Ru(II) complexes are lipophilic cations that tend to accumulate in the mitochondria.10
Generally, the simplest approach is to incorporate mitochondria-targeting groups into PSs, such as lipophilic triphenylphosphonium (TPP). Ru(II) polypyridyl complexes 20–21 are effective mitochondria-targeted two-photon PDT anticancer agents. Colocalization and ICP-MS results indicated that complexes 18–21 partially localized in mitochondria of HeLa cells, with 21 showing a high correlation coefficient (R = 0.88) and a salient affinity of 85.3% for mitochondria(Fig. 12).
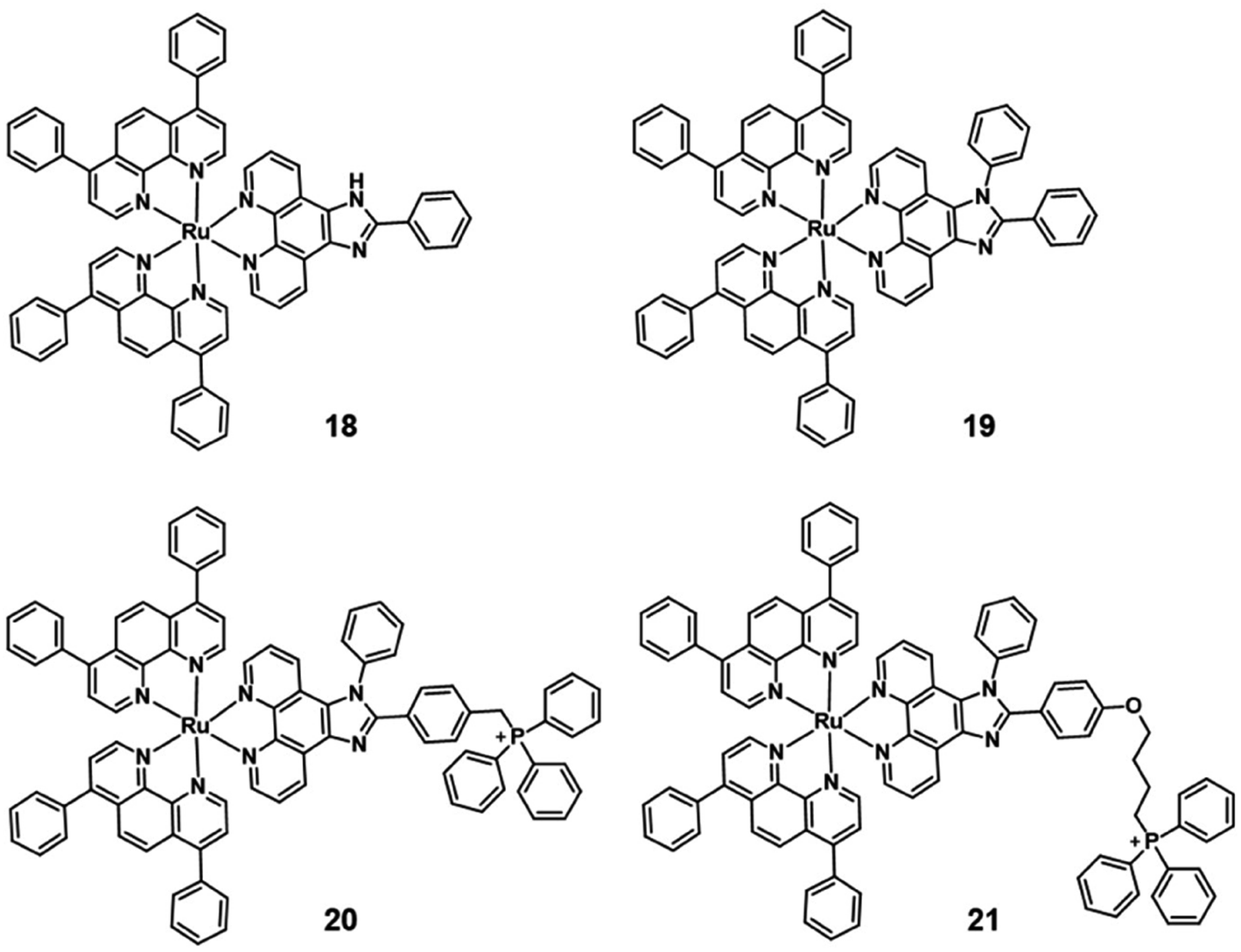 | ||
| Fig. 12 Structure of mitochondria targeting Ru(II) complexes 18–21. | ||
Additionally, many chloromethyl modified molecules displayed the mitochondria targeting ability.16 Three chloromethyl-modified Ru(II) complexes (Fig. 13) were designed as mitochondria targeting photosensitizers by Zhou et al. in 2019.42 The results of laser scanning confocal microscope colocalization experiments and ICP-MS confirmed their mitochondrial targeting abilities. Under visible light irradiation, these complexes can generate carbon radicals in the presence of nicotinamide adenine dinucleotide (NADH) which is abundant in mitochondria. The photoinduced carbon radicals will damage biomolecules such as DNA even under hypoxic conditions. This work provides guidelines for developing novel mitochondria targeting PSs against hypoxic tumor cells.
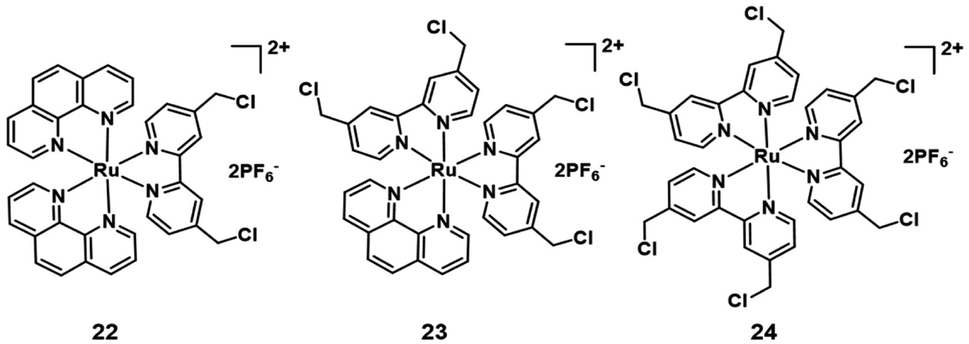 | ||
| Fig. 13 Structure of mitochondria targeting Ru(II) complexes. | ||
Lysosomes (pH 4.5–5.5) are known as the stomach of cells that contain a variety of hydrolytic enzymes, which can degrade almost all kinds of intracellular biomacromolecules.32 Disruption of the lysosomal integrity can induce cell death through lysosomal membrane permeabilization (LMP) that causes the leakage of hydrolases from the lysosomal lumen to the cytosol.43,44 Therefore, lysosomes are considered as attractive targets for effective PDT.
Chao et al. synthesized three highly positively charged Ru(II) complexes 25–27 (Fig. 14) that can specifically accumulate in lysosomes through endocytosis of HeLa cells.45 These complexes have a high TPA cross-section of around 800 nm with σ2 values between 185 and 250 GM, which are much higher than that of the clinical photosensitizer H2TPP (2.2 GM at 800 nm excitation). All complexes exhibit impressive 1O2 production quantum yields(92–99% in methanol and 49–67% in D2O). All complexes displayed low dark toxicity (IC50 > 500 μM), and 25 produced significant phototoxicity upon irradiation (800 nm, 10 J cm2) with an IC50 value of 1.9 μM and an PI value of 250 in 3D HeLa MCTSs. Thereby, complex 25 holds a large potential space in two-photon photodynamic therapy.
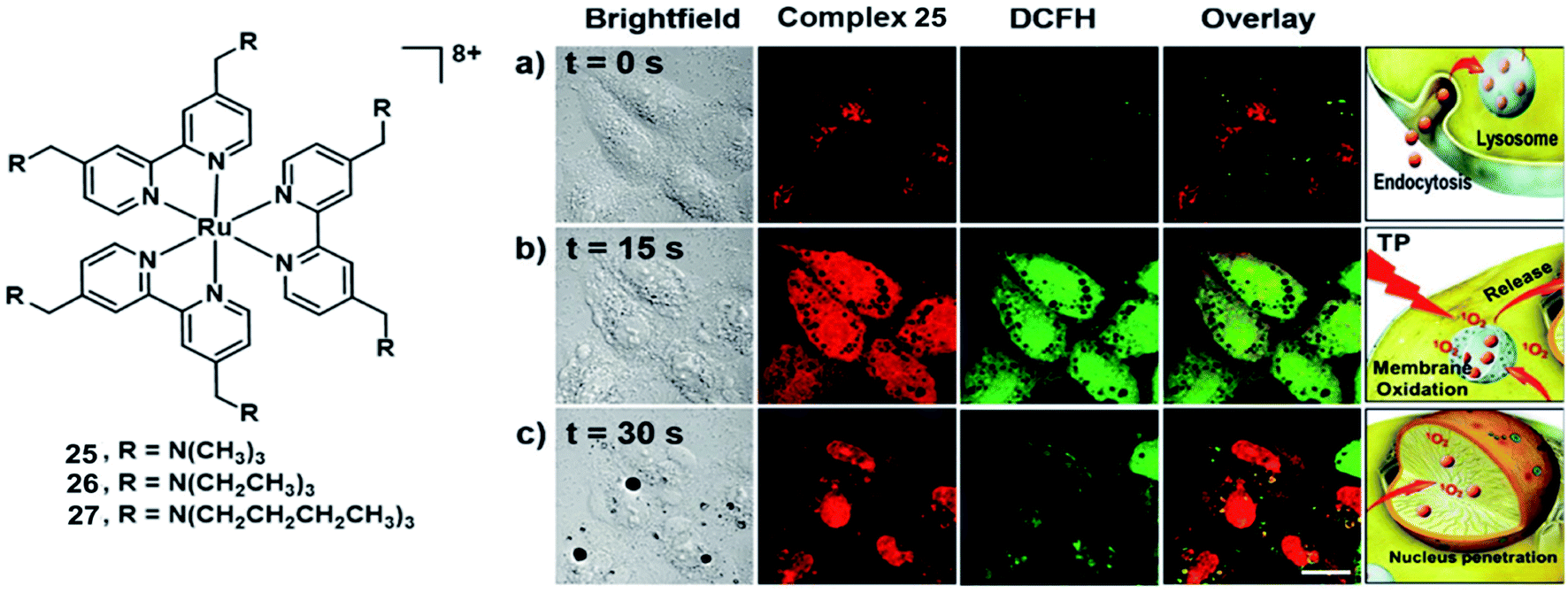 | ||
| Fig. 14 Structure of complex 25–27(left); micrographs of and ROS generation in HeLa cells incubated with 25 after irradiation with a two-photon confocal laser (800 nm) for different periods. (a) t = 0 s; (b) t = 15 s; (c) t = 30 s. Scale bar: 15 μm. Reproduced with permission from ref. 45. Copyright 2015 John Wiley & Sons. | ||
In 2021, Peng et al. reported a Ru(II) complex 28 (Fig. 15) for lysosome localization PDT.46 It internalized in the cells through an energy-dependent endocytosis mechanism. The distribution of 28 in MCF-7 almost overlaid with LysoTracker Red with a colocalization coefficient of 0.943. Under 660 nm irradiation, complex 28 demonstrated good phototoxicity with an IC50 value of 3.1 μM on MCF-7 cells. The author used acridine orange (AO) to monitor the integrity of cell lysosomes. After 660 nm irradiation, the complex generated 1O2 and destroyed the integrity of cell lysosomes. Hence, this Ru(II) complex 28 was able to efficiently induce lysosome damage and cause cancer cell apoptosis directly. Further, the author used the 4T1 tumor-bearing Balb/c mice for the in vivo PDT study. After intratumor injection with complex 28 (5 mg kg−1) for 2 hours, the tumour area was irradiated by 671 nm light (200 mW cm−2, 10 min) every two days. Compared with three control groups, the PDT treatment group almost completely inhibited the tumor growth.
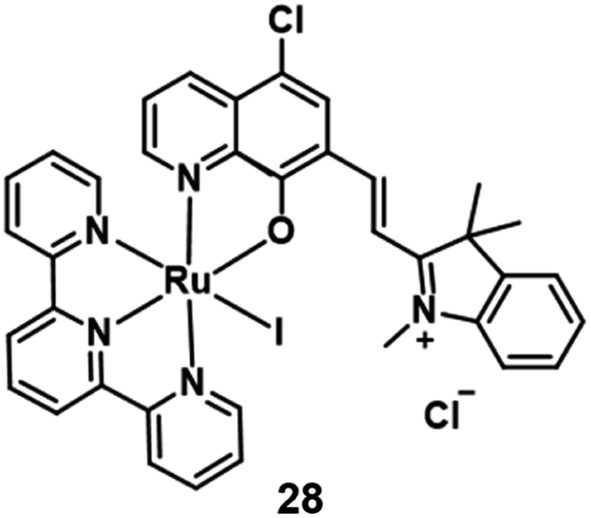 | ||
| Fig. 15 Chemical structure of complex 28. | ||
Another targeted organelle for PDT PSs is the nucleus. But targeting the nucleus is risky if the tumour targeting is not perfect as it may induce mutagenetic issues on healthy cells. The nucleus is more sensitive to ROS than other organelles. In the nucleus, ROS oxidative damage occurs on the DNA double-strand directly for cytotoxicity. Some studies manifested that dipyridophenazine ligands can intercalate within DNA.47,48 In 2019, Thomas et al. investigated two dinuclear Ru(II) complexes (29–30; Fig. 16).49 Two complexes predominantly localized in the nucleus in human melanoma (C8161) cells and showed high affinity towards duplex and quadruplex DNA. They displayed low cytotoxicity under dark conditions(>200 μM) against melanoma cells. However, the complexes are therapeutically activated by light to become highly phototoxic toward C8161. The excited state of complex 30 is quenched by quadruplex and duplex DNA, and this results in the generation of photooxidized guanine radical cation sites within the DNA. Therefore, complex 30 is capable of directly damaging biomolecules without the mediation of 1O2 or other ROS. The TP excition induces phototoxicity deep within hypoxic regions of C8161 spheroids. This indicates that 30 has the potential for application of in vivo models, because the spheroids represent a tumor microenvironment similar to that of solid tumors.
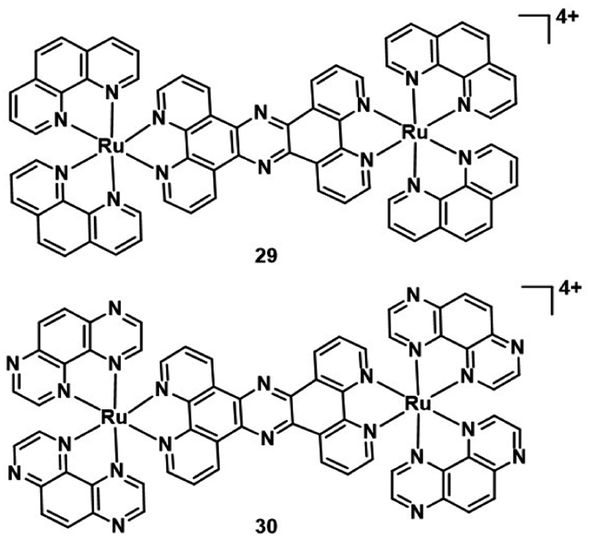 | ||
| Fig. 16 Structure of complex 29–30. | ||
The endoplasmic reticulum (ER) is a complex membranous network organelle that undertakes the synthesis, folding, transportation, and modification of secretory proteins and the maintenance of cellular Ca2+ homeostasis process.50 The disruption of ER functions leads to ER stress, such as the accumulation of unfolded or misfolded proteins. Severe ER stress can cause intrinsic apoptosis, resulting in cell death. The ER is considered to be an ideal target for effective PDT. There have been reported many Ru(II) complex anticancer agents with ER stress-mediated apoptosis pathways.51,52 In 2019, Chao et al. developed a Ru(II) complex containing a p-cresol group that induces apoptosis in human cervical carcinoma cells through ER stress and ROS production.53 As PDT agents, Ru(II) complexes that targeted the ER are relatively rare. A variety of iridium(III) complexes54,55 has been reported with ER-targeted abilities for PDT (see Section 3.3).
3. Iridium complexes in photodynamic therapy
Iridium is a third-row transition metal that belongs to precious metals. As anticancer metallodrugs, Ir(III) complexes have attracted much attention from researchers due to their excellent anticancer activity and potential to overcome cis-platinum resistance and side effects.56 But the use of Ir(III) complexes as PSs for PDT is still in its infancy. However, they have been promising PDT agents owing to their excellent photophysical and photochemical properties: large Stokes shifts reduced the interference between excitation and emission; high singlet oxygen quantum yield for triplet state formation; long luminescence lifetimes; and high chemical and photochemical stabilities.57 The properties of Ir(III) complexes can be tuned by modifying the coordination of different types of ligands.10 In this section, we summarize some recent advances in Ir(III) complexes as PSs for PDT.3.1 The NIR and TPE Ir(III) based PSs
Extension of the π-conjugation of ligands is also useful for obtaining long wavelength NIR Ir(III) complexes.58 In 2017, McFarland and Sun designed five NIR-emitting heteroleptic cationic iridium complexes 31–35 (Fig. 17) with a π-expansive cyclometalating 2,3-diphenylbenzo[g]quinoxaline (dpbq) ligand (C^N ligand) and diimine ligands with different degrees of π-conjugation as the co-ligands.59 All of the complexes displayed weak NIR phosphorescence, with the maximal emission spanning 700–1400 nm in deoxygenated CH2Cl2, and singlet oxygen quantum yields varied from 0.37–0.56 in MeCN. The cytotoxicities of the five Ir(III) complexes were measured in the melanoma cell line (SK-MEL-28) and normal skin fibroblasts (CCD-1064Sk). In the dark, the IC50 value toward melanoma cells varied from 230 nM to 18 μM, with the order: 33 > 32 > 34 > 35 > 31. Compared with normal skin fibroblasts, complexes 32 and 33 displayed 40-fold more cytotoxicity toward the melanoma cells, but complexes 30, 34, and 35 showed almost no selectivity for the cancer cells. Photocytotoxicity increased in the order 35 ≈ 32 ≈ 33 > 34 > 31 for visible PDT with IC50 values ranging from 12 to 252 nM, and 33 ≈ 32 ≈ 35 > 31 > 34 for red PDT with IC50 values spanning 150 nM to 2.1 μM in melanoma cells. Complex 35 exhibited the most effective PDT effects in this series, with a PI value of 273 for visible light irradiation and 16 for red light irradiation. And all of the complexes emitted bright NIR phosphorescence in melanoma cells. These excellent properties indicated their potential as theranostic PDT agents.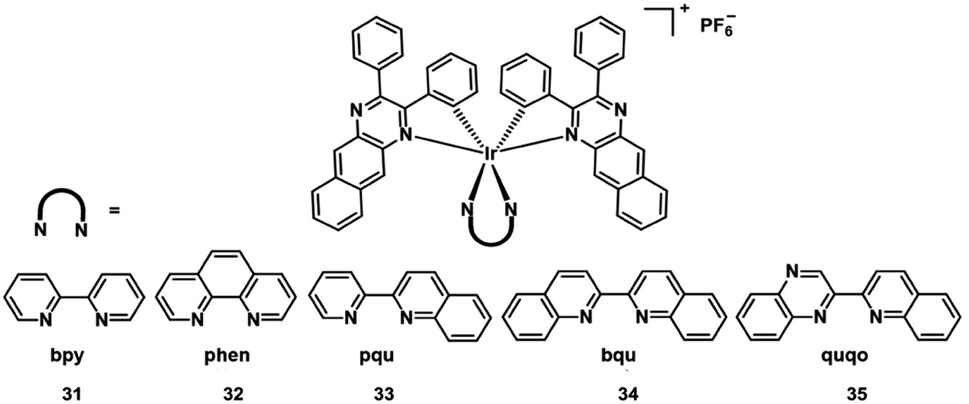 | ||
| Fig. 17 Chemical structures of the cationic iridium complexes 31–35. | ||
Recently, multi two-photon Ir(III) complexes as PSs for PDT have been reported.10 In 2017, Chao et al. developed a series of aggregation-induced emission(AIE) Ir(III) complexes 36–38 (Fig. 18) with selective lighting up of the photodynamic activity for TPA-PDT.60 The largest TPA cross-section (σ2) with an excitation wavelength at 730 nm is 214 GM for 36. And the σ2 of 37 (78.2 GM) and 38 (28.2 GM) is also much larger than that of clinical PDT agent H2TPP (2.8 GM). Complex 36 (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/w = 1.42) is more lipophilic than 37 and 38 (logPo/w = 1.06 and 0.77, respectively). The confocal laser scanning microscopy and ICP-MS results revealed that all Ir(III) complexes were taken up by endocytosis and selectively accumulated in mitochondria of HeLa and L02 (normal human hepatic) cells. Due to carcinoma cells exhibiting higher mitochondrial membrane potentials than normal cells, L02 showed lower uptake levels. In the dark, all of the complexes showed low cell toxicity towards both cell lines. Under irradiation at 405 nm using an LED area light (40 mW cm−2; light dose = 12 J cm−2), complex 36 displayed high light toxicity with an IC50 value of 0.40 μM (PI = 75) in HeLa cells and low light toxicity in L02 cells (IC50 = 2.4 μM; PI = 14). Strikingly, TPA-PDT (irradiated at 730 nm) of complex 36 exhibited impressive lethality (IC50 = 0.35 μM; PI = 110) in 400 μm 3D HeLa MCTSs. With large TPA cross-sections, effective ROS generation and high photocytotoxicity upon aggregation in mitochondria, complex 36 is a promising candidate for TPA-PDT.
Po/w = 1.42) is more lipophilic than 37 and 38 (logPo/w = 1.06 and 0.77, respectively). The confocal laser scanning microscopy and ICP-MS results revealed that all Ir(III) complexes were taken up by endocytosis and selectively accumulated in mitochondria of HeLa and L02 (normal human hepatic) cells. Due to carcinoma cells exhibiting higher mitochondrial membrane potentials than normal cells, L02 showed lower uptake levels. In the dark, all of the complexes showed low cell toxicity towards both cell lines. Under irradiation at 405 nm using an LED area light (40 mW cm−2; light dose = 12 J cm−2), complex 36 displayed high light toxicity with an IC50 value of 0.40 μM (PI = 75) in HeLa cells and low light toxicity in L02 cells (IC50 = 2.4 μM; PI = 14). Strikingly, TPA-PDT (irradiated at 730 nm) of complex 36 exhibited impressive lethality (IC50 = 0.35 μM; PI = 110) in 400 μm 3D HeLa MCTSs. With large TPA cross-sections, effective ROS generation and high photocytotoxicity upon aggregation in mitochondria, complex 36 is a promising candidate for TPA-PDT.
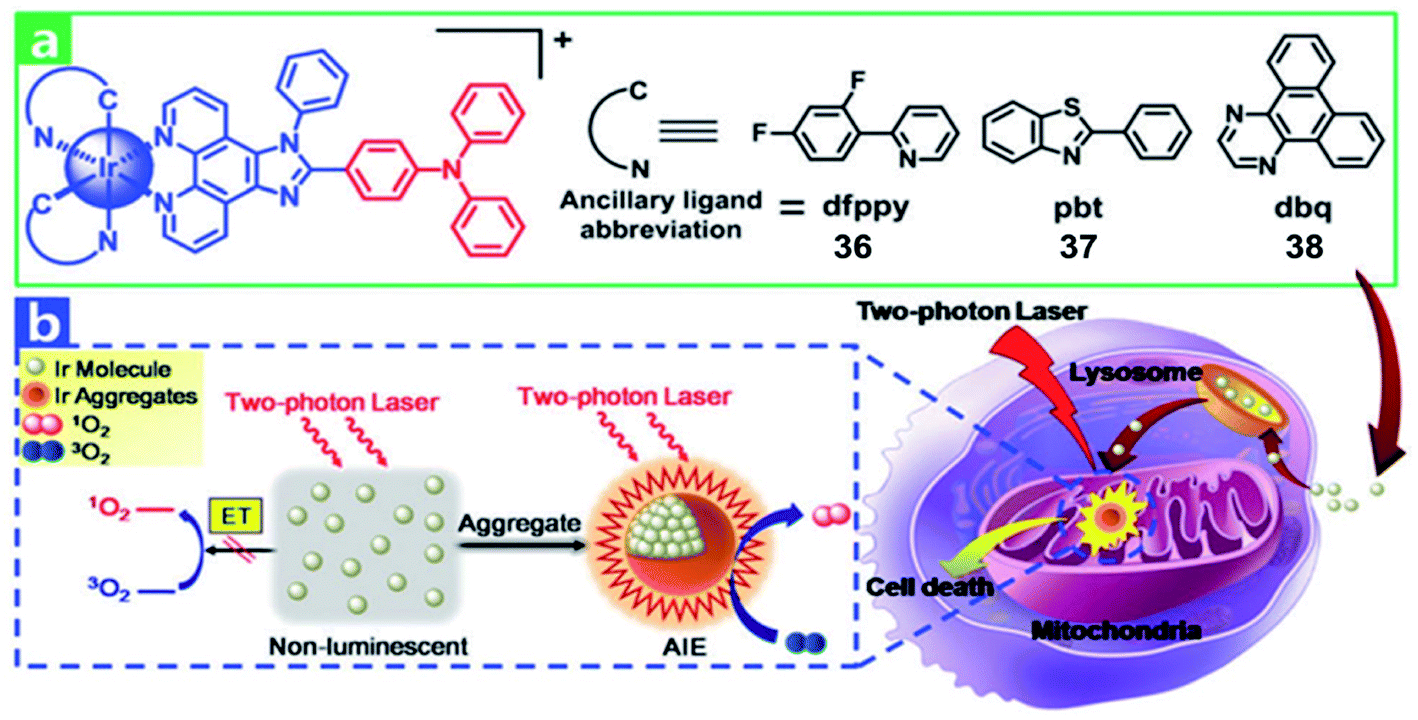 | ||
| Fig. 18 (a) Chemical structures of 36–38. (b) Schematic illustration of 36–38 for TPA-PDT. Reproduced with permission from ref. 60. Copyright 2017 The Royal Society of Chemistry. | ||
In 2021, Gou et al. constructed a donor–acceptor–donor structure-based Ir(III) complex 39 with evident NIR absorption in 600–1000 nm regions (Fig. 19).61 This complex shows highly efficient ROS and heat generation with a ROS quantum yield of 14.6% and a photothermal conversion efficiency of 27.5% under near-infrared (NIR) irradiation. This is the first report that an Ir(III) complex could be excited at 808 nm to generate ROS via the type I process. In addition, due to the long absorption wavelength, photothermal therapy was combined with PDT to inhibit tumor growth. To improve the biocompatibility, water solubility and tumor retention and accumulation of 39, 39 was conjugated with PEG and formulated into nanoparticles. The photocytotoxicities of 39 and 39-NPs against A549 cancer cells are with half maximal inhibitory concentrations (IC50) of 14.4 μM and 7.1 μM, respectively. The nanoparticles preferentially accumulated in the tumor area and exhibited a significant in vivo tumor regression of 96% in volume with a cure rate of 67% through synergistic PDT and PTT dual-modal therapy.
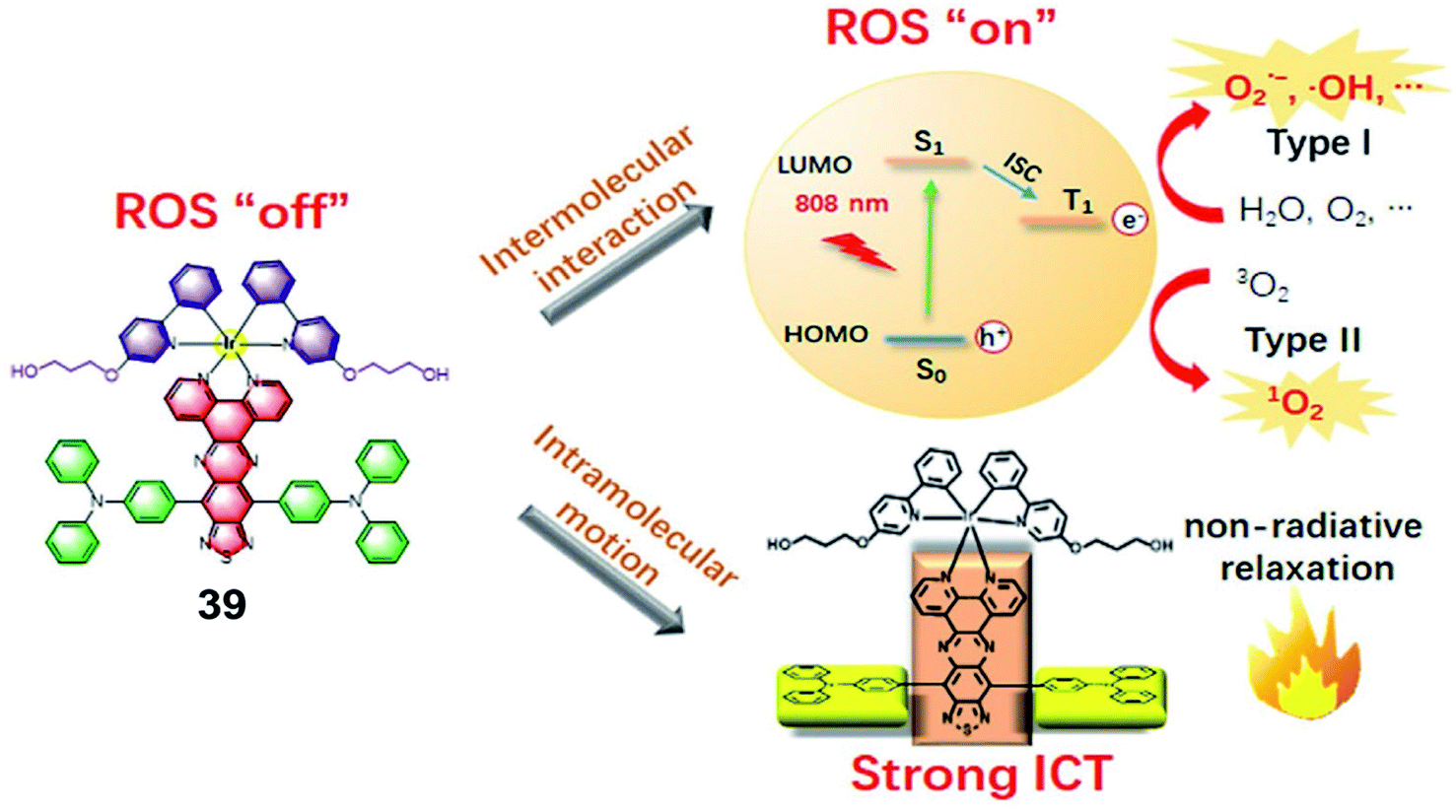 | ||
| Fig. 19 Illustration of the working mechanism of the Ir(III) complex bearing the donor–acceptor–donor type ligand for NIR-triggered phototherapy. Reproduced with permission from ref. 61. Copyright 2021 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.2 Ir(III) based PSs for PDT of hypoxic tumor cells
In 2019, Marchán et al. reported the first example of generation of superoxide anion radicals as a novel photodynamic anticancer agent that conjugated a cyclometalated Ir(III) complex (41; Fig. 20) to a coumarin-based fluorophore.62 The singlet oxygen quantum yields of complex 40 and 41 were below 0.01 in PBS. After visible-light irradiation, complex 41 produced more cytotoxic O2−˙ than complex 40 selectively. The cellular uptake experiments indicated that complex 40 and 41 were internalized by energy-dependent uptake pathways and accumulated in the cytoplasm. Under normoxic conditions (21% O2), the two complexes showed low dark cytotoxicity. Under visible-light irradiation (green and blue light), complex 41 displayed excellent photocytotoxicity with IC50 values of 2.51 μM (PI = 85) and 1.32 μM (PI = 161) in HeLa cells. Under hypoxic conditions (2% O2), the photocytotoxicity of complex 41 also displayed efficiency with IC50 values of 2.77 μM (PI = 79) and 1.43 μM (PI = 153) after irradiation with green and blue light, respectively. Hence, the conjugated coumarin improved the antitumor activity of the Ir(III) complex, leading to Ir(III)-fluorophore conjugates with promising applications in theranostic PDT against hypoxic tumors.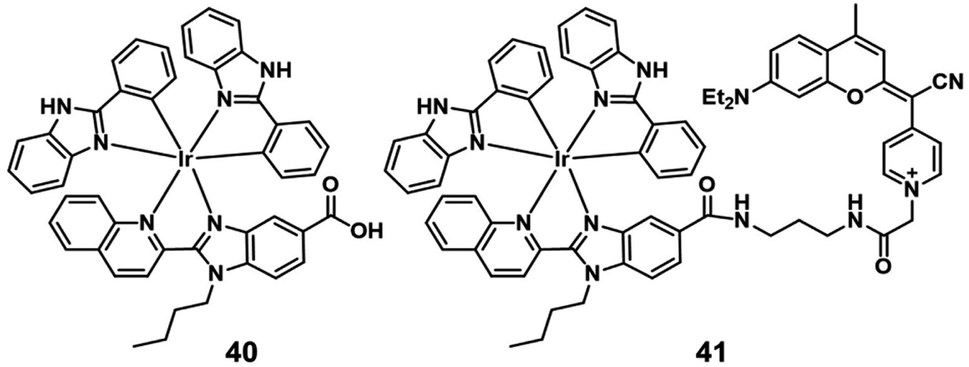 | ||
| Fig. 20 Chemical structures of 40 and 41. | ||
Sadler's group designed a novel, stable iridium photocatalyst complex 42 (Fig. 21) that showed almost equivalent photocytotoxicity under normoxia and hypoxia and low toxicity towards unirradiated normal cells.63 Compared with traditional photosensitizers, complex 42 had an unusually high excited-state reduction potential. It photocatalytically oxidized 1,4-dihydronicotinamide adenine dinucleotide (NADH) generating NAD˙ radicals with more than two orders of magnitude higher NADH oxidation turnover frequency (TOF) than organometallic half sandwich catalysts upon light irradiation in biological media. Complex 42 targeted the mitochondria and disrupts electron transport by reduction of Fe3+–cyt c, inducing redox imbalance in cancer cells. Moreover, the author proved π-stacking in adducts of complex 42 and NADH by density functional theory calculations, facilitating photoinduced single-electron transfer, and trapped and characterized NAD˙ radicals with detailed experimental and computational studies on photocatalytic NADH oxidation in aqueous media. This photocatalytic redox imbalance strategy offers a new approach for efficient cancer phototherapy.
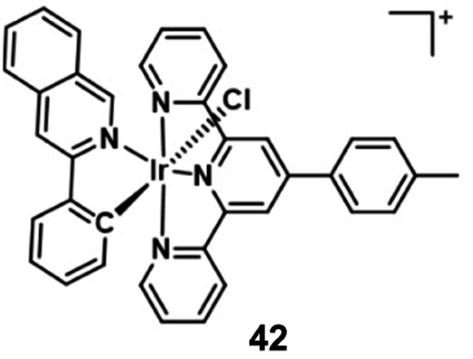 | ||
| Fig. 21 Chemical structures of 42. | ||
In 2019, Elias reported Ir(III)-based molecules which can oxidize biomolecules by type I processes under oxygen-free conditions. 43 and 44 are bis-cyclometalated Ir(III) complexes (Fig. 22), and they can form lipophilic cations characterized by rapid cellular uptake and tunable redox properties.64 The complexes were mainly observed in mitochondria via co-localization experiments with subcellular markers. While both complexes were inactive in the dark, cell viability decreased dramatically upon light excitation; interestingly, 44 showed a strong cleavage activity upon 30 min irradiation with 405 nm LEDs. The cytotoxic effect of the Ir(III) complexes had also been obtained from 3D cultures of FaDu cancer cells (diameter: 350–400 μm). Although both complexes possess the same absorption properties at the excitation wavelength (ε405 nm = ±800 M−1 cm−1 for both complexes), 44 showed a stronger photoactivity than 43 at the same concentration. Complex 44 (20 μM) can induce a complete destruction of the spheroidal structure, while 43 was actually limited to surface cell layers and does not vary a lot with the drug concentration and was a failure in inhibiting the spheroid growth. Actually, this phenomenon has been attributed to its rapid consumption of all the oxygen available in the spheroid, the complex 44 combined a low 1O2 quantum yield and the capacity to initiate the type I oxygen-independent processes. Nevertheless, the short activation wavelength (405 nm) of 44 might be an issue, which restricted its wider application when it comes to light penetration in living tissues.
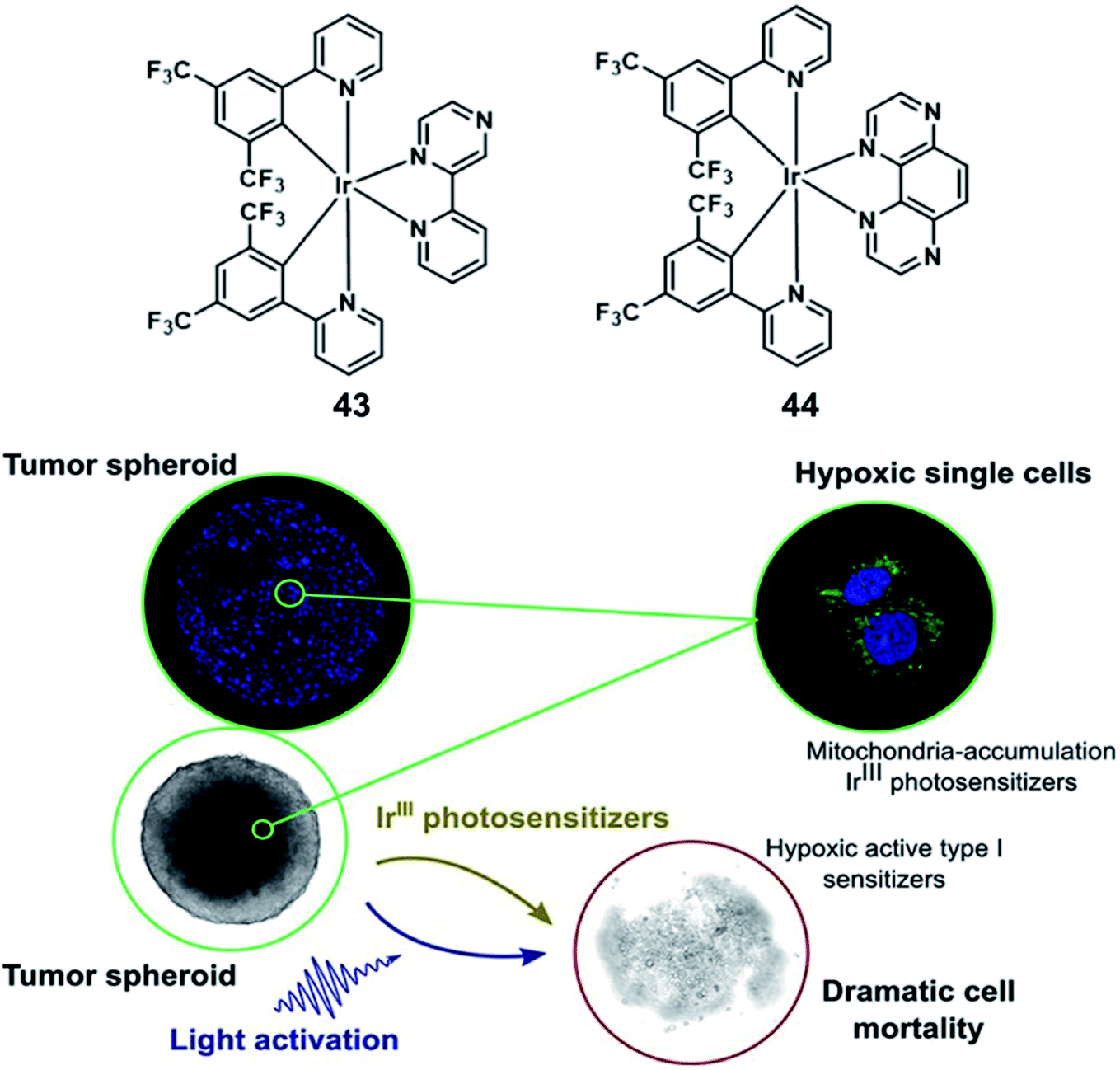 | ||
| Fig. 22 Chemical structures of 43 and 44 (top), and 3D FaDu tumor spheroids 24 h after the irradiation step (bottom). Reproduced with permission from ref. 64. Copyright 2019 American Chemical Society. | ||
Singlet oxygen released via endoperoxides is also a tool to overcome hypoxia. Chao's group reported a mitochondria-targeted Ir(III) endoperoxide complex 45 as a photoactivated prodrug (Fig. 23).65 Anthracene covalently connected the Ir(III) complex more effectively at the 2-position than the 9-position. Complex 45 could be generated by irradiating 2-IrAn at 405 nm (20 mW cm−2) under normoxic conditions, while 80% could release singlet oxygen and be photodecaged back in 2 minutes under hypoxic conditions. ESR spectroscopy verified the existence of singlet oxygen and alkoxide radicals. Based on this strategy, the metal complex had nanomolar phototoxicity towards hypoxic tumor cells and multicellular tumor spheres. Targeting mitochondria leads to the loss of mitochondrial membrane potential, which induces apoptosis. The author encapsulated complex 45 into a biotin functionalized polymer to improve cancer selectivity and pharmacological properties. When nanoparticles were made, mouse tumor models were also successfully treated by near-infrared two-photon irradiation. This Ir(III)-based endoperoxide prodrug for synergistic photodynamic therapy/photoactivated chemotherapy provides new ideas for design of PSs for the treatment of hypoxic tumors.
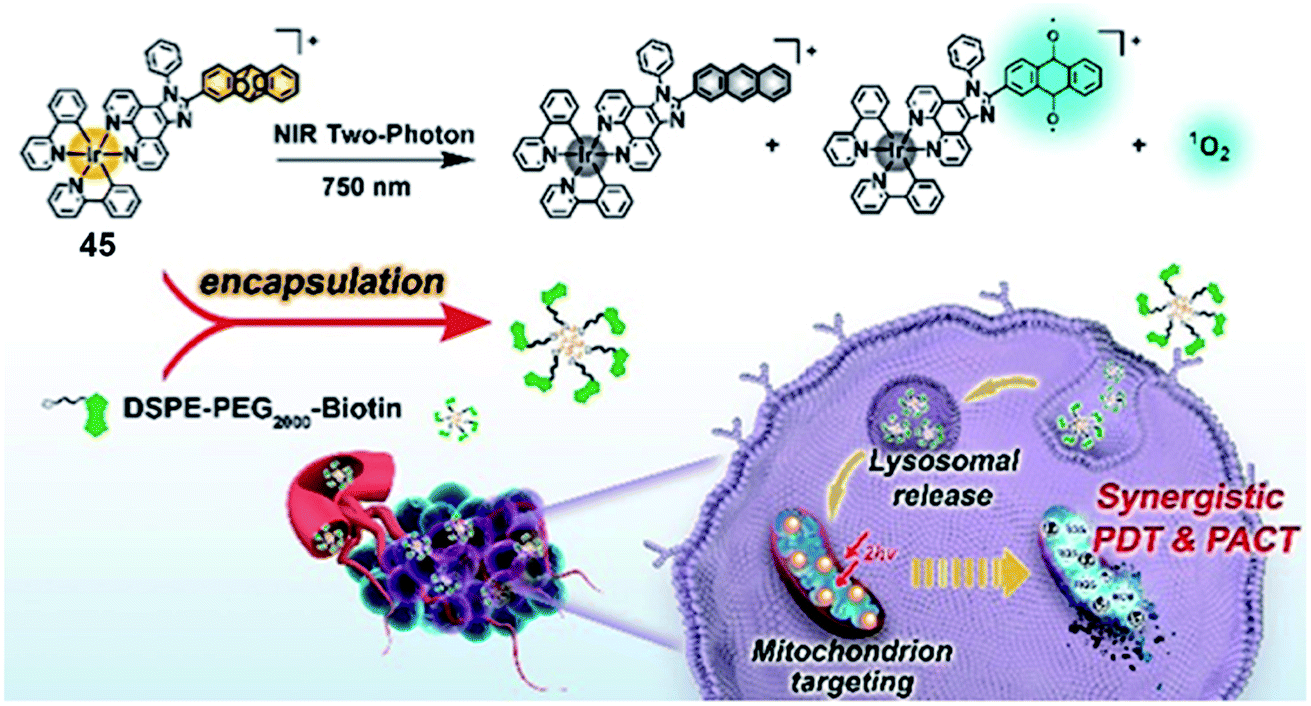 | ||
| Fig. 23 Chemical structure of 45, and schematic illustration of the PDT/PACT mechanism. Reproduced with permission from ref. 65. Copyright 2022 American Chemical Society. | ||
3.3 Targeted Ir(III) based PSs
Recently, our group reported two new benzothiophenylisoquinoline (btiq)-derived cyclometalated Ir(III) complexes 46 and 47 (Fig. 24) for effective PDT.65 Both complexes exhibited a type I photochemical process to form ˙O2− and ˙OH and caused photoinduced ferroptosis in tumor cells under hypoxia. This is the first example of Ir(III) based PSs that could induce ferroptosis, supported by the observations of lipid peroxide accumulation, glutathione peroxidase 4 (GPX4) down-regulation, mitochondrial shrinkage, and Fer-1-inhibited cell death. Complex 47 was modified with a mitochondria-targeting triphenylphosphonium (TPP) group. This modification introduced additional cell apoptosis, which displayed photoinduced mitochondrial membrane potential (MMP) collapse and ATP production suppression under hypoxia. The synergism of ferroptosis and apoptosis endows 47 with better PDT efficiency than 46. Under hypoxia, both complexes were capable of suppressing the growth of refractory cancer cells such as MCF-7 (IC50 = 0.41 μM, 0.51 μM for complex 47 and 46, respectively), PANC-1(IC50 = 4.77 μM, 7.91 μM for complexes 47 and 46, respectively), and MDA-MB-231 cells(IC50 = 1.45 μM, 4.23 μM for complexes 47 and 46, respectively). The synergism of ferroptosis and apoptosis provided a promising approach for combating hypoxic and apoptosis-resistant solid tumors through the type I PDT process and mitochondria targeting.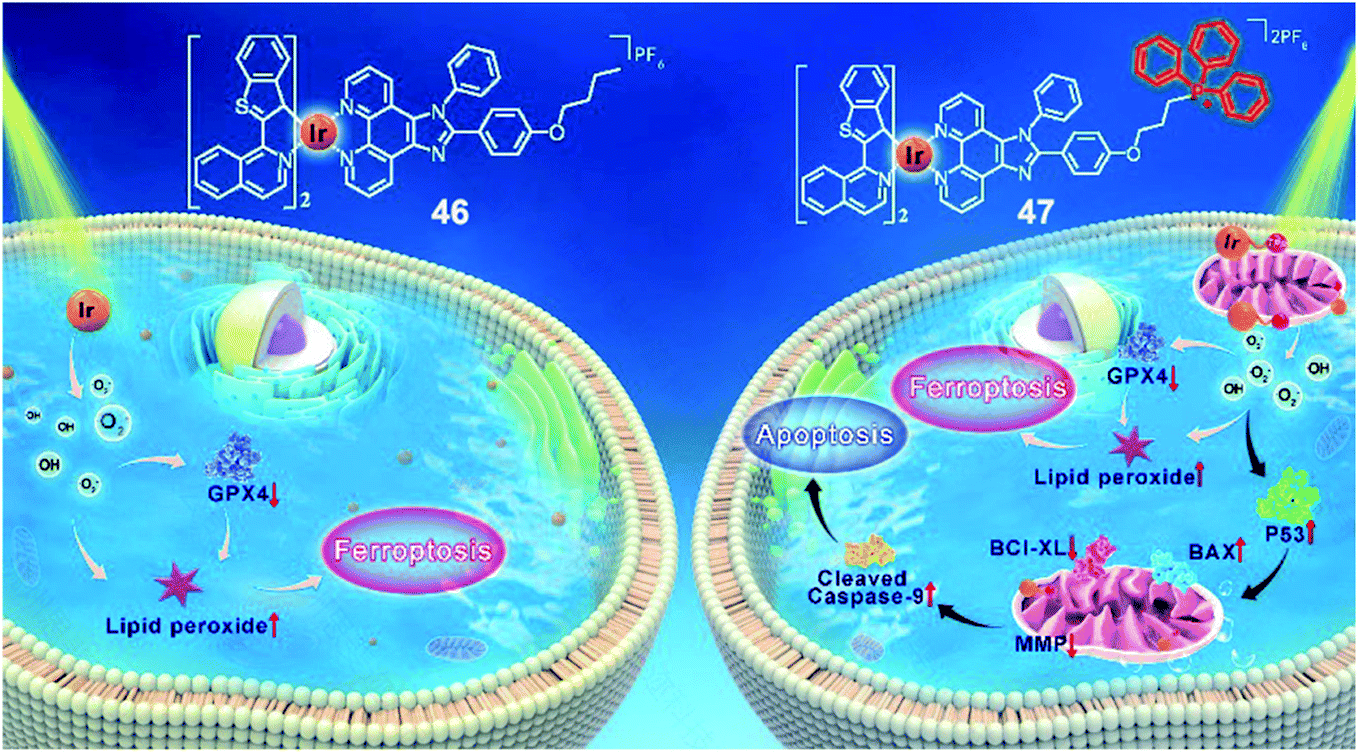 | ||
| Fig. 24 Chemical structures of 46 and 47, and the schematic illustration of cell death pathways induced by 46 and 47 upon photoirradiation under hypoxia. Reproduced with permission from ref. 66. Copyright 2021 John Wiley & Sons. | ||
NADH and NADPH are essential cellular reductants which play important roles in maintaining intracellular redox balance. In 2021, Huang and co-workers reported an Ir(III) photocatalyst 48 for the oxidation of NADPH and amino acids via a single electron transfer pathway (Fig. 25).67 Under photo-irradiation, complex 48 not only generates type II 1O2 but also generates type I O2−˙ and carbon-center radicals. Photo-irradiation enhanced intracellular uptake of complex 43, which then localized both in the lysosome and mitochondria. The photo-induced intracellular redox imbalance and change in mitochondrial membrane potential resulted in necrosis and apoptosis of cancer cells. This complex exhibited selective photo-cytotoxicity against sorafenib-resistant cell line, HepG2-SR, with an IC50 value of 0.37 μM and also photo-cytotoxicity towards cisplatin-resistant cell line A549R with an IC50 value of 1.6 μM, while it did not show any dark-toxicity up to 200 μM. The author confirmed that complex 48 has excellent biocompatibility and in vivo toxicity in zebrafish and the photo-catalytic anticancer therapeutic effect on the mouse CT26 colon carcinoma model. Photo-induced oxidation of these cellular reductants may lead to a significant change in the intracellular redox homeostasis and mitochondrial electron transport chain, leading to cell death. Additionally, the metal complex PSs with the synergistic effect of the multi cell death mechanism can improve anticancer efficiency.
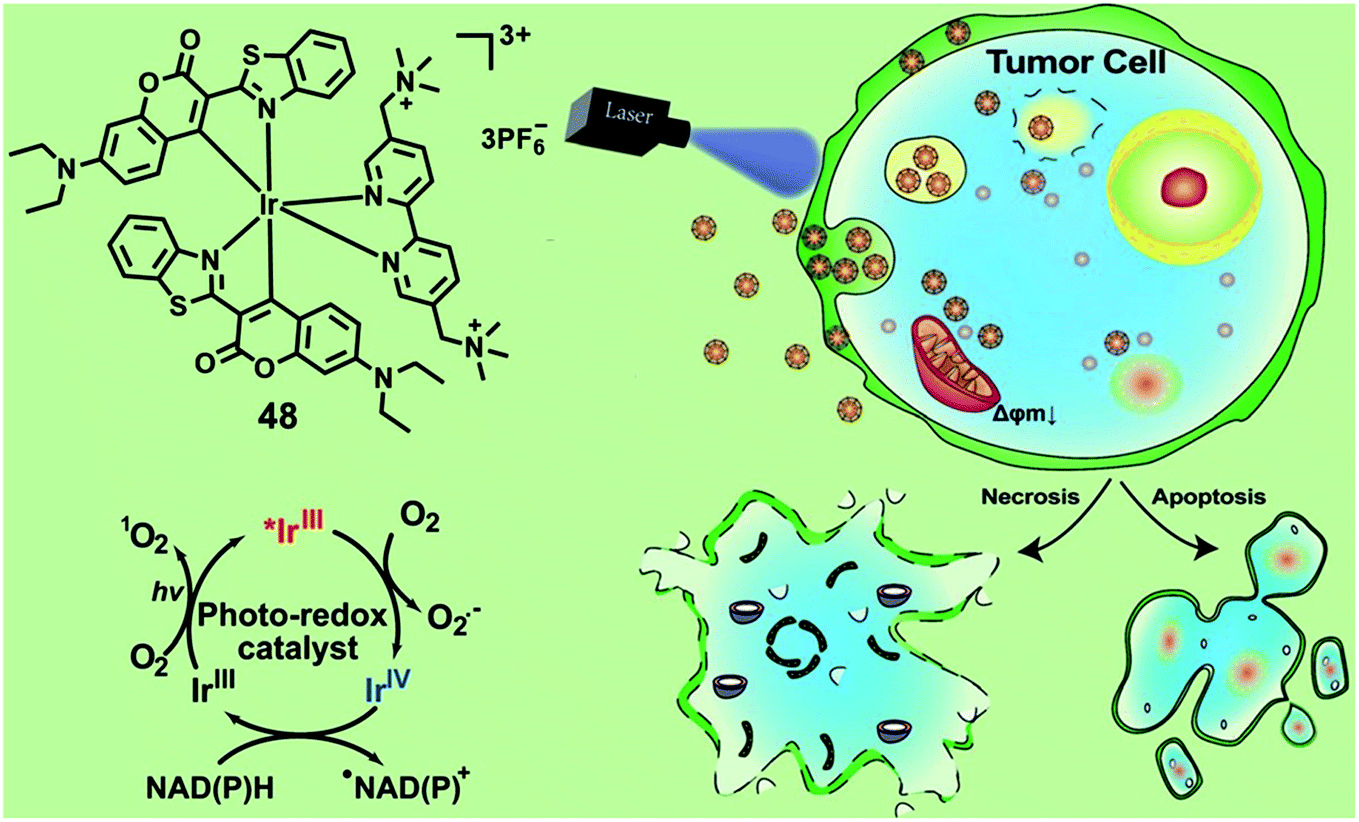 | ||
| Fig. 25 Chemical structures and schematic illustration of 48 induced photo-oxidation of NADPH and the cancer cell death mechanism. Reproduced with permission from ref. 67. Copyright 2021 John Wiley & Sons. | ||
In 2019, Chao reported three novel endoplasmic reticulum (ER)-targeted terpyridyl Ir(III) complexes (49–51; Fig. 26) as PDT photosensitizers.54 The distribution of 49–51 in A549 cells overlaid with ER tracker green with colocalization coefficients of 0.81,0.87, and 0.71, respectively. These complexes have a special conformation of cyclometalated Ir(III), coordinating to a series of bidentate ligands with a gradually expanded conjugating area. And these complexes can target the ER and effectively induce cell apoptosis after PDT therapeutics (405 nm, 6 J cm−2) by an ER stress mechanism, and both their singlet oxygen quantum yields and cytotoxicities increase as the conjugation area extends. Among them, complex 50 exhibited the highest PI value (94.3) against A549 cells with an IC50 down to 0.65 μM and was considered to be a promising candidate as an ER-targeted PDT photosensitizer.
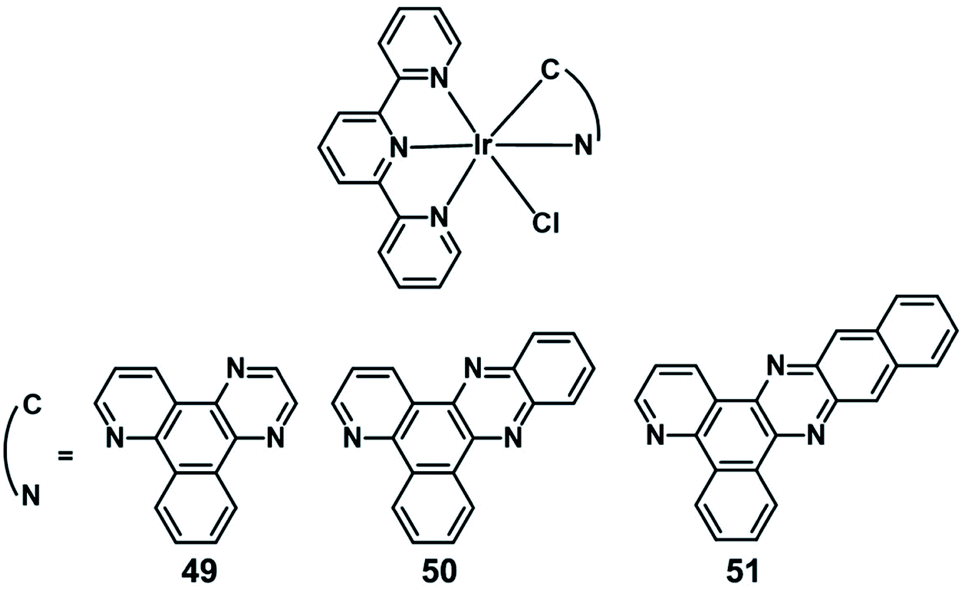 | ||
| Fig. 26 Chemical structures of 49–51. | ||
In 2019, Sadler reported the first example of an organo-iridium complex-HSA bioconjugate (Fig. 27) as a nucleus-targeted vehicle for anticancer photodynamic therapy.67 The octahedral organo-iridium(III) complex 52 contained two chelated phenylpyridine ligands and two monodentate pyridines functionalized with a maleimide substituent; an organoiridium–albumin bioconjugate 53 was synthesized by the reaction of a pendant maleimide ligand with human serum albumin. Compared to the complex 52, the phosphorescence of 53 was enhanced significantly that the quantum yield for 53 increased 36 fold (to 0.036) and its emission lifetime increased to 871.8 ns. The complex 53 mainly accumulated in the nucleus of living cancer cells and showed remarkable photocytotoxicity against a series of cancer cell lines and tumor spheroids (light IC50; 0.8–5 μM, photo-cytotoxicity index PI = 40–60), while remaining dormant in normal cells/spheroids, even after photo-irradiation. The discovery of complex 53 expands the range of Ir(III) complexes that are organelle-targeted photosensitizers and enables Ir(III) complexes to further have great potential for clinical PDT in the biomedical field.
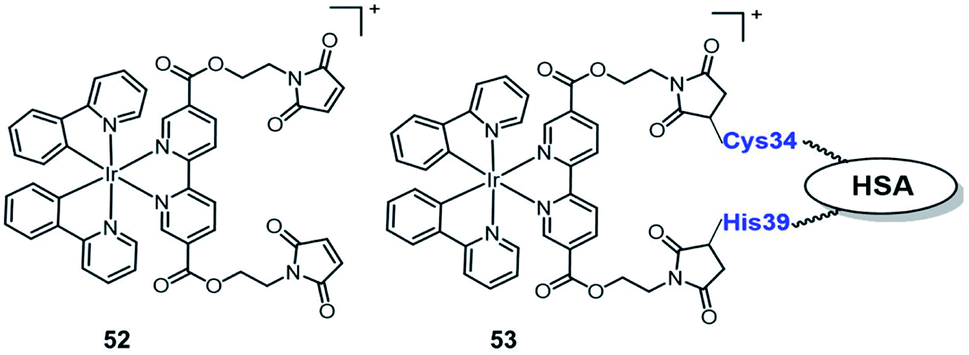 | ||
| Fig. 27 Chemical structures of 52–53. | ||
4. Platinum(II) complexes in photodynamic therapy
Platinum-based drugs have emerged as chemotherapeutic anticancer agents such as cisplatin carboplatin, and oxaliplatin.69 These classical Pt complexes are first-line agents used for the treatment of various types of cancer. Unfortunately, the severe side effects as well as intrinsic platinum-drug resistance are inevitable in clinical utility.70,71 However, PDT exhibits high selectivity by controllable light irradiation, which reduces the side effects on the adjacent healthy tissues. Targeting organelles are known to bypass the resistance mechanisms of conventional anticancer agents.38,72 Currently, one strategy was to develop Pt(II) complexes as promising photosensitizers for PDT with targeted organelles. Another was to integrate a photosensitizer with a Pt(II) moiety into a single platform that combines PDT treatment with classical Pt chemotherapy to achieve a synergistic effect.73 In this section, we summarize some recent advances in Pt(II) complexes as PSs and Pt(II) complex containing PSs for PDT.4.1 The NIR and TPE Pt(II) based PSs
In 2018, Hartman reported a Pt(II) complex of heptamethine cyanine as a NIR PDT agent.74 Complex 54 (Fig. 28) showed strong NIR absorption at 790 nm with a high molar extinction coefficient (4–5 × 104 L mol−1 cm−1). Confocal microscopy experiments showed that complex 54 localized in the mitochondria (Pearson correlation coefficient = 0.94) and lysosomes (P.C.C = 0.47) that are properties of the heptamethine cyanine backbone. And ICP-MS indicated light-promoted release and migration of active Pt(II) species to the nuclear DNA. Under NIR light irradiation, this complex displayed unprecedented Pt–O bond scission and significant singlet oxygen quantum yield (ΦΔ = 0.15). In the dark, complex 54 has lower cytotoxicity (IC50 = 8.4, 18.2 μM in C-33 A and MCF-7 cell lines, respectively) than cisplatin. When exposed to light (45 min, 720–740 nm, 3.5 ± 1.5 mW cm−2), the IC50 of complex 54 is 0.14 (PI = 60) and 0.65 μM (PI = 28) in C-33 A and MCF-7 cell lines, respectively. Therefore, complex 54 exhibited dual-modes of cytotoxicity through PDT and reactive Pt(II) species.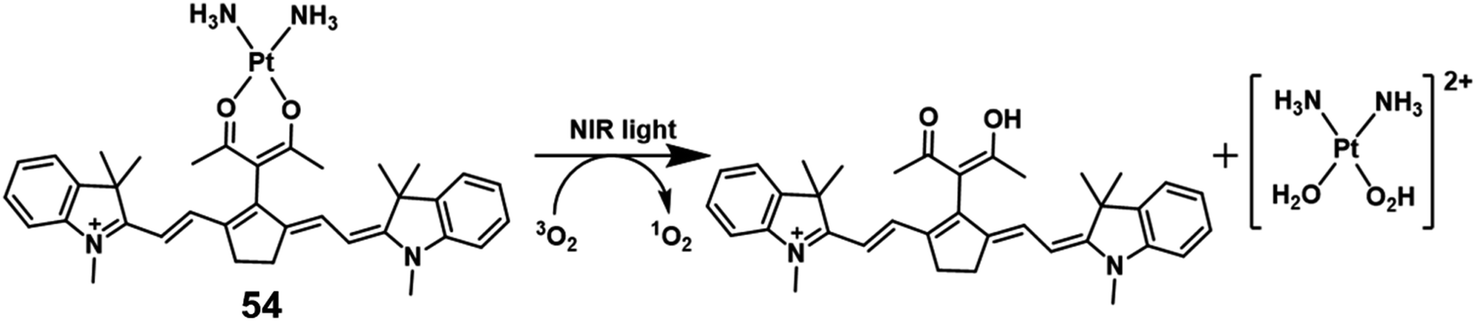 | ||
| Fig. 28 Chemical structures of the cationic Pt complexes. | ||
Triphenylamines (TPA) have been shown to be excellent materials for two-photon absorption.75 Mao et al. designed two photosensitive Pt(II)-based tripods as potent PDT agents which combined platinum(II) complexes (55–56; Fig. 29) with triphenylamine bridging ligands.76 The UV and fluorescence spectra showed an absorption maxima at 400 nm and an emission maxima at 550 nm in aqueous solutions for two complexes. The two-photon absorption maximum was at 810 nm, and the maximum two-photon action cross-section (Φδmax) was 45.7 GM and 30.6 GM for 55 and 56, respectively. Both complexes exhibited weak two-photon excited fluorescence. The PDT anticancer activities were not determined under two-photon excitation. However, complex 55 mainly accumulated in the cell nucleus and exhibited remarkable phototoxicity upon visible light irradiation (425 nm, 36 J cm−2), with IC50 values of 0.18 μM (PI = 538) in A549 cells and 0.33 μM (PI = 381) in A549cisR cells and low dark toxicity. In vivo PDT anticancer activity of complex 55 was investigated in HeLa xenograft-bearing nude mice. Through intratumoral injection of two repeated doses for 13 days, complex 56 (10 mg kg−1) can inhibit tumor growth with laser irradiation (430 nm, 360 mW cm−2, 15 min). One tumor in the light group was completely eradicated at the end point. Importantly, there is no mouse death or significant bodyweight after the treatment at this dose.
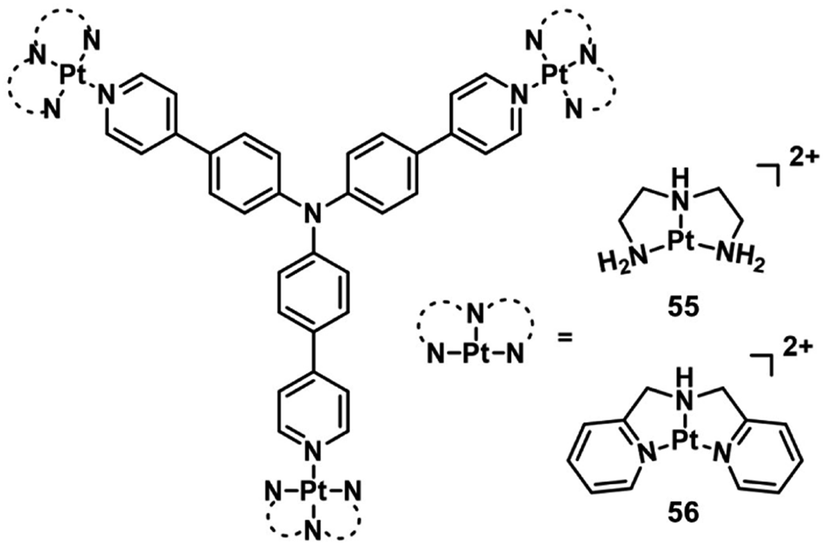 | ||
| Fig. 29 Chemical structures of 55 and 56. | ||
4.2 Pt(II) based PSs for PDT of hypoxic tumor cells
Zhao reported a series of hydrophilic phosphorescent starburst Pt(II) porphyrin complexes (57; Fig. 30) with four cationic fluorene oligomers as arms, which are bifunctional therapeutic agents for monitoring the hypoxia imaging and efficient PDT under hypoxia.77 Among them, complex 57 displayed good water solubility and a 3D architecture because of the increase of the length of cationic arms, which prevents the aggregation of starburst Pt(II) porphyrins. Furthermore, complex 57 showed outstanding oxygen-sensing performance with a phosphorescence intensity at 680 nm and singlet oxygen-generating ability (ΦΔ = 0.92). Therefore, the complex 57 with a deep red emission and long phosphorescence lifetime has been applied to tumor hypoxia imaging. The author investigated the therapeutic effect in vivo on the HeLa xenograft tumor-bearing mouse model. When the tumor volume reached a size of approximately 60 mm3, the HeLa tumor-bearing mice were randomly divided into six groups: the control group [phosphate buffered saline (PBS)], light group (PBS + light), dark group (57 only), HP-dark group, HP-light group, and PDT group (57 + light). The PDT group tumor-bearing mouse received intratumoral injection every 2 days (5 mg kg−1), and then the irradiated tumor area by using a 520 nm laser (160 mW cm−2, 10 min) at 30 min postadministration. In the PDT application, complex 57 can inhibit the tumor growth in the PDT treatment and exhibited nearly no dark toxicity towards tumor tissues.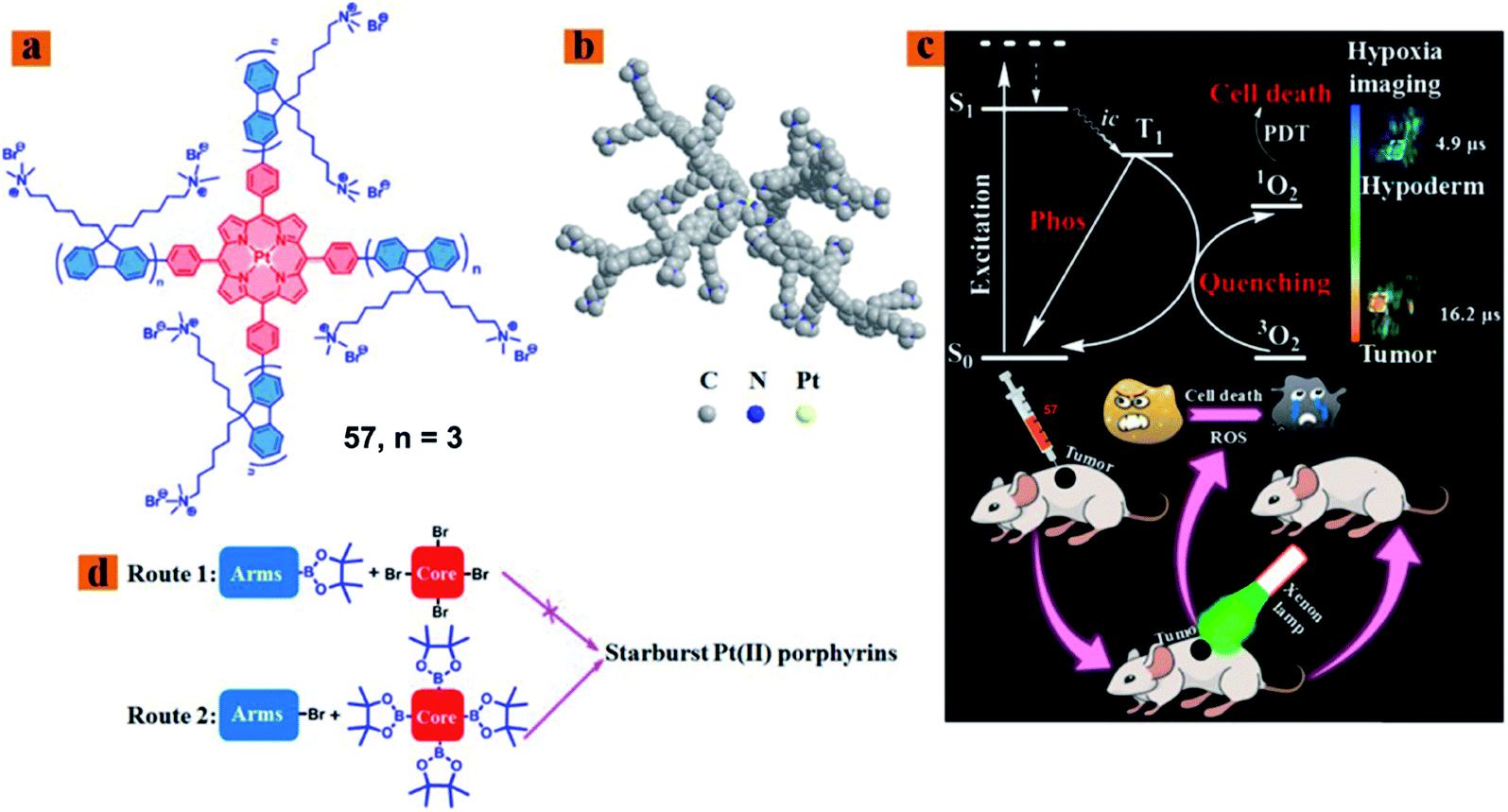 | ||
| Fig. 30 (a) Chemical structure of 57, (b) optimized 3D model of 57, (c) schematic illustration of 57 used as a bifunctional therapeutic agent for tumor hypoxia imaging and PDT, and (d) schematic illustration of synthetic routes 1 and 2. Reproduced with permission from ref. 77. Copyright 2018 American Chemical Society. | ||
4.3 Targeted Pt(II) based PSs
Due to the accelerated metabolism of tumor cells and high glucose requirements, glucose transporter GLUT-1 was overexpressed in a variety of tumors.78 Hence, appending a glucose moiety to the ligand will increase the targeting efficacy of PDT agents towards the tumor cell. In 2018, the Chakravarty group reported glucose-appended Pt(II)-BODIPY conjugates (58) for targeted PDT in red light (Fig. 31).79 Complex 58 showed an NIR absorption band at 715 nm (ε = 3.2 × 104 M−1 cm−1) and an emission at 825 nm in 10% DMSO–DMEM at pH = 7.2. The singlet oxygen generation is 0.58 for complex 58. By ICP-MS, complex 58 showed not only higher cellular uptake in cancer cells than a normal cell, but also high cellular uptake compared to the complex without the glucose moiety. The colocalization experiment showed that complex 58 localized significantly in the mitochondria with a Pearson's coefficient value of 0.72. The complex 58 exhibited significant photocytotoxicity with IC50 values of 2.6, 6.0, and 2.3 μM upon red light irradiation (λ = 600–720 nm; 30 J cm−2) in HeLa, MCF-7, and A549, respectively, while complex 58 has low toxicity in the dark (IC50 = 91.4, >100, >100 μM in HeLa, MCF-7, and A549, respectively). All the results indicated that complex 58 is a promising targeted PDT agent in NIR light.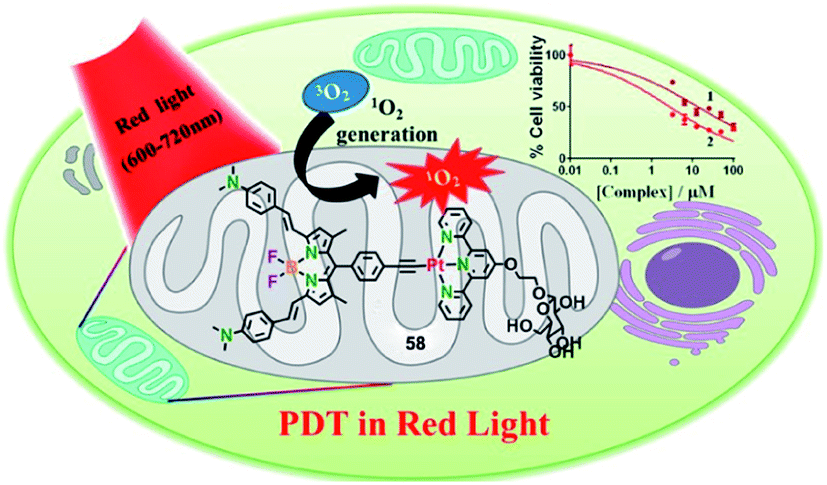 | ||
| Fig. 31 Chemical structure of 58, and a schematic illustration of 58 used as a targeted PDT agent in NIR light. Reproduced with permission from ref. 78. Copyright 2018 American Chemical Society. | ||
Lysosomes are the site of degradation for endocytosed poisonous species in the cell.80 It is reported that lysosomes can sequester specific Pt complexes, which is relevant to Pt drug resistance owing to nuclear DNA inaccessibility.71,81 In 2019, our group presented a promising strategy that utilizes lysosome Pt complex sequestration to silence Pt complexes as “prodrugs”, and specific activation released Pt complexes from lysosomes to the nucleus.82 Therefore, we designed a photosensitizing monofunctional Pt(II) complex 57 (Fig. 32) with a BODIPY chromophore bearing a Pt chelator. The heavy atom effect of the Pt(II) center can improve the photoinduced ROS production ability of BODIPY, which makes complex 57 an effective PS with a singlet oxygen quantum yield (ΦΔ) of 0.133. Confocal imaging indicated that complex 57 enters cells via energy-dependent endocytosis and accumulates in lysosomes for a long time in the dark. After photoirradiation, the sequestered complex generated ROS that can damage the integrity of the lysosome, and the complex 57 was released to the nucleus. The photoinduced ROS will consume intracellular GSH, promising the stability of complex 57 and its nuclear DNA accessibility. This complex showed poor dark cytotoxicity (IC50 > 50 μM) in both normal (HK-2, L02) and tumor cell lines. After irradiation, complex 57 exhibited distinct photocytotoxicity with IC50 values of 4.1, 5.8, and 3.8 μM in MCF-7, SGC-7901 and A549 cell lines, respectively. This work utilized the synergistic effect of PDT and Pt chemotherapy and provided a new method for precise therapy via selective photoactivated lysosomal escape to access the nucleus.
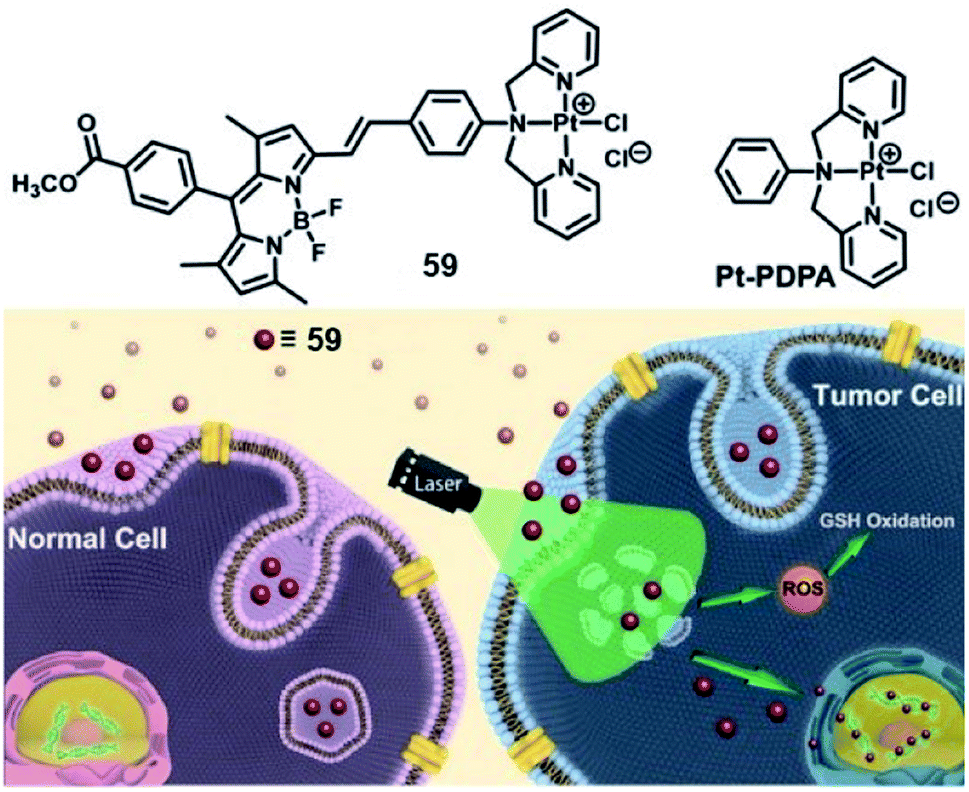 | ||
| Fig. 32 Chemical structure of 59, and schematic illustration of the photoactivated lysosomal escape of 59. Reproduced with permission from ref. 80. Copyright 2019 John Wiley & Sons. | ||
5. Conclusions and perspectives
Noble metal-based complexes as photosensitizers present excellent PDT activity, owing to their tunable photophysical and photochemical properties and high ROS generation. We highlighted a few of the most recent examples of Ru(II), Ir(III) and Pt(II) complexes in PDT. It is difficult to compare the data of PDT activities of different PSs reported by different laboratories, since different methods were adopted to detect PDT activities of PSs, such as the light dose (intensity and duration time), cell lines and so on. We summarized data of most noble metal-based PSs in Table 1, providing a reference for new PS design. Ru(II) complexes have been the most extensively investigated in recent years. In particular, McFarland's group developed TLD1433 which is the first Ru(II)-based PS to enter a human clinical trial for treating non-muscle invasive bladder cancer. Although Ir(III) complexes as PSs have been extensively studied and exhibited good performance on tumor inhibition, no Ir(III) based PSs have entered a clinical trial. Pt(II) complexes have been used less as PDT agents compared with Ru(II) complexes and Ir(III) complexes. In this minireview, we focused on novel strategies for design of noble metal-based PSs for effective PDT, which includes the development of long-wavelength NIR and TPE PSs to increase the depth of tissue penetration; type I PSs to overcome hypoxic tumors; tumor tissue or certain subcellular organelles targeted PSs with good selectivity.| Complex | Metal center | λ ir [nm] | Dose [J cm−2] | ROS/ΦΔ | IC50 [μM] | PI | Cell line | Cellular target | Cell death | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dark | Light | ||||||||||
| a Mito: mitochondria, Lyso: lysosome, Cyto: cytoplasm, Nuc: nucleus, ER: endoplasmic reticulum; Apo: apoptosis, Fer: ferroptosis, and Nec: necrosis. | |||||||||||
| Photofrin | — | 630 | 25 | — | — | ∼1.15 | — | ISOS-1 | — | Apo | 83 |
| — | 635 | 80 | — | >574 | 57.4 | >10 | HepG2 | Mito | Apo | 84 | |
| 5 | Ru | 625 | 100 | 1O2 | 123 | 9.96 | 12 | SKME28 | Nuc | — | 21 |
| 143 | 126 | 1.1 | HL60 | ||||||||
| 6 | Ru | 635 | 1.8 | 1O2, 0.033, ˙OH | — | — | — | HeLa | — | — | 22 |
| 7 | Ru | 635 | 1.8 | 1O2, 0.058, ˙OH | — | — | — | HeLa | — | — | 22 |
| 9 | Ru | 595 | 11.2 | 1O2, 0.92 | >100 | 2.4 | >41.7 | CT-26 | Cyto | Apo | 24 |
| 12 | Ru | 450 | 18 | 1O2, 0.92 | 75.46 | 5.21 | 14.48 | HeLa (MCTs) | Mito | Apo | 25 |
| 810 | 800 | 108.56 | 5.71 | 19 | |||||||
| 13 | Ru | 450 | 18 | 1O2, 0.4 | 76.71 | 5.01 | 15.31 | HeLa (MCTs) | Mito | Apo | 25 |
| 110.36 | 5.45 | 20 | |||||||||
| 14 | Ru | 400–800 | 18 | 1O2, 0.14 | — | — | — | HeLa | — | Apo | 29 |
| 15 | Ru | 400–800 | 18 | 1O2, 0.16 | — | — | — | HeLa | — | Apo | 29 |
| 16 | Ru | 820 | 0.032 | 1O2, 0.87 | 72.9 | 3.3 | 22.1 | A549R | — | Apo | 34 |
| 17 | Ru | 450 | 12 | 1O2, 0.52 | 230 | 28 | 8.2 | U87MG (MCTs) | Mito | Apo | 35 |
| 820 | 3 | 216 | 34 | 6.4 | |||||||
| 216 | 9 | 24 | |||||||||
| 25 | Ru | 800 | 10 | 1O2, 0.99 | 470 | 1.1 | 313 | HeLa (MCTs) | Lyso | Nec | 45 |
| >500 | 1.9 | 250 | |||||||||
| 28 | Ru | 660 | 60 | 1O2, ˙OH, O2−˙ | 43 | 3.1 | 13.9 | MCF7 | Lyso | Apo | 46 |
| 35 | Ir | 625 | 100 | 1O2, 0.56 | 3.27 | 0.2 | 16 | SKMEL28 | — | — | 59 |
| 36 | Ir | 405 | 12 | 1O2, 0.7 | 30.3 | 0.4 | 75 | HeLa (MCTs) | Mito | — | 60 |
| 730 | 38.6 | 0.35 | 110 | ||||||||
| 39 | Ir | 808 | 480 | 1O2, 0.15, ˙OH, O2−˙ | >100 | 14.4 | — | A549 | — | — | 61 |
| 41 | Ir | 420 | 28 | 1O2 < 0.01, O2−˙ | N:213 | N: 1.32 | 161 | HeLa | — | — | 62 |
| H: 1.43 | 153 | ||||||||||
| 550 | 21 | H: 219 | N:2.51 | 85 | |||||||
| H: 2.77 | 79 | ||||||||||
| 42 | Ir | N: 465 | N: 8.9 | 1O2, 0.71 | N: 16.6 | N: 1.0 | N: 16.6 | NCI–H460 | Mito | Apo | 63 |
| H: 450 | H: 10.0 | H: 31.6 | H: 8.7 | H: 3.6 | |||||||
| N: 35.4 | N: 1.9 | N: 18.6 | HeLa | ||||||||
| H: 43.6 | H: 5.2 | H: 8.3 | |||||||||
| N: 19.9 | N: 1.1 | N: 18.1 | SGC791 | ||||||||
| H: 23.6 | H: 5.8 | H: 4.0 | |||||||||
| N: > 50 | N:1.9 | N: >25 | Hep G2 | ||||||||
| H: > 50 | H: 3.5 | H: > 14 | |||||||||
| N: 43.6 | N: 1.6 | N: 27.2 | A549 | ||||||||
| H: 42.5 | H: 2.3 | H: 18.5 | |||||||||
| 43 | Ir | 405 | 2.83 | 1O2, 0.68 | N: 69.4 | N: 3.8 | N: 18.4 | FaDu | Mito | Apo | 64 |
| H: 79.6 | H: 18.1 | H: 4.4 | |||||||||
| N: >100 | N: 8.7 | N: >11.5 | HT-29 | ||||||||
| H: 96.2 | H: 28.6 | H: 3.4 | |||||||||
| 44 | Ir | 405 | 2.83 | 1O2, 0.08 | N: >100 | N: 5.4 | N: >18.5 | FaDu | Mito | Apo | 64 |
| H: >100 | H: 12.8 | H: >7.8 | |||||||||
| N: >100 | N: 12.0 | N: >8.3 | HT-29 | ||||||||
| H: >100 | H: 24.2 | H: >4.3 | |||||||||
| 45 | Ir | 405 | 12 | 1O2, 0.79 | N:49.42 | N < 0.052 | N: >959 | A549 | Mito | Apo | 65 |
| H: 41.42 | H: 0.06 | H: >690 | |||||||||
| 46 | Ir | 450 | 30 | 1O2, 0.81, ˙OH, O2−˙ | N: 20.48 | N: 0.51 | N: 40.39 | MCF-7 | — | Fer | 66 |
| H: 17.41 | H: 1.74 | H: 10.01 | |||||||||
| 47 | Ir | 450 | 30 | 1O2, 0.76, ˙OH, O2−˙ | N: 15.26 | N: 0.20 | N: 74.81 | MCF-7 | Mito | Apo | 66 |
| H: 14.76 | H: 0.41 | H: 36.27 | Fer | ||||||||
| 48 | Ir | 465 | 11.7 | 1O2, 0.76, O2−˙ | 63.8 | 0.37 | 172 | HepG2-SR | Mito–Lyso | Apo | 67 |
| Nec | |||||||||||
| 49 | Ir | 405 | 6 | 1O2, 0.39 | >100 | 24.3 | >4.1 | A594 | ER | — | 54 |
| 50 | Ir | 405 | 6 | 1O2, 0.45 | 56.6 | 0.6 | 94.3 | A594 | ER | Apo | 54 |
| 51 | Ir | 405 | 6 | 1O2, 0.49 | 22.3 | 0.5 | 44.6 | A594 | ER | — | 54 |
| 52 | Ir | 465 | 5.76 | 1O2, 0.06 | 89.6 | 53.3 | 1.7 | A594 | Nuc | — | 68 |
| 53 | Ir | 465 | 5.76 | 1O2, 0.83 | 62.3 | 1.1 | 56.6 | A594 | Nuc | — | 68 |
| 54 | Pt | 720–740 | 9.45 | 1O2, 0.15 | 8.4 | 0.14 | 60 | C-33 A | Mito–Lyso | Apo | 75 |
| 18.2 | 0.65 | 28 | MCF-7 | ||||||||
| 55 | Pt | 425 | 36 | 1O2, 0.31 | 96.8 | 0.18 | 538 | A594 | Nuc | Apo | 76 |
| 56 | Pt | 425 | 36 | 1O2, 0.28 | 49.2 | 4.6 | 11 | A594 | Lyso | — | 76 |
| 57 | Pt | 520 | 6 | 1O2, 0.92 | — | — | — | HeLa | — | Apo | 77 |
| 58 | Pt | 600–720 | 30 | 1O2, 0.58 | 91.4 | 2.6 | 35.2 | HeLa | Mito | — | 79 |
| >100 | 6.0 | >16.7 | A594 | ||||||||
| >100 | 2.3 | >43.5 | MCF-7 | ||||||||
| 59 | Pt | 532 | 1 | 1O2, 0.13 | >50 | 4.1 | >12 | MCF-7 | Lyso–Nuc | — | 82 |
| >50 | 5.8 | >8 | SGC791 | ||||||||
| >50 | 3.8 | >13 | A594 | ||||||||
Although noble metal-based complexes displayed significant PDT outcomes, there is still a long way to go for clinical applications. Limitations and drawbacks of these noble metal-based PSs for clinic application were less mentioned in most of these studies.85–88 For instance, safety and tolerability should be further evaluated in follow-up clinical trials, and most of the noble metal-based complexes showed the results only in 3D multicellular spheroids or short-range mouse testing. Most of the reported noble metal complexes (including TLD1433) showed relatively short excitation wavelengths, which limit their potential clinical applications. The ideal PSs should have long excitation wavelengths and strong absorption in the red or NIR regions (typically 600–1100 nm). Although a few reported noble metal-based PSs showed long wavelengths, the molar extinction coefficients need to be enhanced to improve light absorbing ability for a better PDT effect. Although two-photon excitation PSs could realize NIR excitation, their clinical application could be hindered due to the requirement of higher power lasers and proper devices. Utilizing chemiluminescence resonance energy transfer might be a promising strategy to solve the problem of limited light penetration depth, since external light irradiation is not needed for chemiluminescence systems. Future efforts should also be devoted to research studies such as comprehensive and indepth study on the structure–property relationship and detailed mechanistics of the type I PDT process, which will help the rational design of type I noble metal-based PSs. Meanwhile, developing noble metal-based PSs with optimized light toxicity with IC50 in the range of nM or pM and a PI value in the range of 103–105 is highly demanded. Metastasizing tumors also remains one of the largest challenges for PDT. Additionally, there are complicated processes for hit identification in different tiers, including synthesis, photophysics, in vitro 2D, in vitro 3D, in vivo zebrafish, in vivo murine, in vivo murine immunology, tier I/II pharmacodynamics screening, and drug discovery: pharmacokinetics screening and clinical evaluation of the lead PS candidate. Thus, noble metal-based photosensitizers still face many hurdles before entering clinical trials.
Further efforts should also be devoted to developing nobel metal complex based PSs capable of inducing multiple cell death pathways, which would be beneficial for improving PDT performance with a synergistic effect. The combination of PDT with other therapeutic modalities such as photothermal therapy (PTT), chemotherapy, and immune therapy offers exciting prospects that could utilize the synergy effects to improve the PDT efficiency and get better applicability in the clinic. PTT could elevate the temperature of tumor tissue to kill tumor cells and escalate blood supply to reduce tumor hypoxia. Nowadays, immunotherapy has gained intense attention in the field of cancer therapies.89 Many investigations have demonstrated that PDT can induce activation of the immune system and specific antitumor immunity by recruiting immune cells to release inflammatory mediators.90,91 On the one hand, more efforts should be devoted to designing noble metal-based PSs that can induce immunogenic cell death in cancer cells, which shows the potential to induce in vivo an “anticancer vaccine effect”, further enhancing the antitumor effect.92 On the other hand, we can combine PDT with immune checkpoint-blockade therapy to eliminate or inhibit immunosuppressive factors, such as metal-based complexes in cooperation with anti-PD-1/PD-L1 immune checkpoint inhibition.93
Author contributions
Yuncong Chen, Weijiang He and Zijian Guo conceived the idea of the review and supervised this project. Yanping Wu and Shumeng Li collected references and contributed to the graphic design; Yanping Wu prepared the initial draft; Yanping Wu, Shumeng Li and Yuncong Chen revised and edited the manuscript; all authors participated in the discussion of the draft and gave approval to the final version of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (22122701, 21731004, 21907050, 91953201, 21907044, and 92153303), the Natural Science Foundation of Jiangsu Province (BK20190282 and BK20202004), and the Excellent Research Program of Nanjing University (ZYJH004).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- H. Uramoto and F. Tanaka, Transl. Lung Cancer Res., 2014, 3, 242–249 Search PubMed.
- C. De Angelis, Curr. Oncol., 2008, 15, 198–199 CrossRef PubMed.
- W. P. Hogle, Semin. Oncol. Nurs., 2006, 22, 212–220 CrossRef PubMed.
- P. De Silva, M. A. Saad, H. C. Thomsen, S. Bano, S. Ashraf and T. Hasan, J. Porphyrins Phthalocyanines, 2020, 24, 1321–1360 CrossRef.
- C.-P. Tan, Y.-M. Zhong, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2021, 12, 2357–2367 RSC.
- Z. Meng, W. Hou, H. Zhou, L. Zhou, Ha. Chen and C. Wu, Macromol. Rapid Commun., 2018, 39, 1700614 CrossRef.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko and E. Kędzierska, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef CAS PubMed.
- Q. Zheng, X. Liu, Y. Zheng, K. W. K. Yeung, Z. Cui, Y. Liang, Z. Li, a S. Zhu, X. Wang and S. Wu, Chem. Soc. Rev., 2021, 50, 5086–5125 RSC.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS.
- S. Monro, K. L. Colon, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- M. E. Alberto, J. Pirillo, N. Russo and C. Adamo, Inorg. Chem., 2016, 55, 11185–11192 CrossRef CAS PubMed.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed.
- C. P. Tan, Y. M. Zhong, L. N. Ji and Z. W. Mao, Chem. Sci., 2021, 12, 2357–2367 RSC.
- M. R. Gill and K. A. Vallis, Chem. Soc. Rev., 2019, 48, 540–557 RSC.
- J. J. Cao, C. P. Tan, M. H. Chen, N. Wu, D. Y. Yao, Xi. G. Liu, L. Ni. Jia and Z. W. Mao, Chem. Sci., 2017, 8, 631–640 RSC.
- J. Mo, N. P. Mai Le and R. Priefer, Eur. J. Med. Chem., 2021, 225, 113770 CrossRef CAS PubMed.
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS.
- W. Fan, P. Huang and X. Chen, Chem. Soc. Rev., 2016, 45, 6488–6519 RSC.
- J. T. Ferreira, J. Pina, C. A. F. Ribeiro, R. Fernandes, J. P. C. Tome, M. S. Rodriguez-Morgade and T. Torres, Chem. – Eur. J., 2020, 26, 1789–1799 CrossRef CAS PubMed.
- L. Wang, H. Yin, M. A. Jabed, M. Hetu, C. Wang, S. Monro, X. Zhu, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2017, 56, 3245–3259 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhang, N. Tian, C. Li and X. Wang, Inorg. Chem., 2017, 56, 1865–1873 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- J. Karges, S. Kuang, Y. C. Ong, H. Chao and G. Gasser, Chem. – Eur. J., 2021, 27, 362–370 CrossRef CAS PubMed.
- L. Zeng, S. Kuang, G. Li, C. Jin, Li. Ji and H. Chao, Chem. Commun., 2017, 53, 1977–1980 RSC.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2008, 8, 56–61 CrossRef CAS PubMed.
- X. Li, N. Kwon, T. Guo, Z. Liu and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 11522–11531 CrossRef CAS PubMed.
- S. Lazic, P. Kaspler, G. Shi, S. Monro, T. Sainuddin, S. Forward, K. Kasimova, R. Hennigar, A. Mandel, S. A. McFarland and L. Lilge, J. Photochem. Photobiol., 2017, 93, 1248–1258 CrossRef CAS PubMed.
- Z. Lv, H. Wei, Q. Li, X. Su, S. Liu, K. Y. Zhang, W. Lv, Q. Zhao, X. Li and W. Huang, Chem. Sci., 2018, 9, 502–512 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- M. P. Gamcsik, M. S. Kasibhatla, S. D. Teeter and O. M. Colvin, Biomarkers, 2012, 17, 671–691 Search PubMed.
- J. Karges, M. Jakubaszek, C. Mari, K. Zarschler, B. Goud, H. Stephan and G. Gasser, ChemBioChem, 2020, 21, 531–542 CrossRef CAS PubMed.
- A. Stallivieri, F. Baros, G. Jetpisbayeva, B. Myrzakhmetov and C. Frochot, Curr. Med. Chem., 2015, 22, 1–23 CrossRef PubMed.
- J. Li, L. Zeng, K. Xiong, T. s. W. Rees, C. Jin, W. Wu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2019, 55, 10972–10975 RSC.
- Z. Zhao, K. Qiu, J. Liu, X. Hao and J. Wang, Chem. Commun., 2020, 56, 12542–12545 RSC.
- A. Juarranz, P. Jaén, F. Sanz-Rodríguez, J. Cuevas and S. González, Clin. Transl. Oncol., 2008, 10, 148–154 CrossRef CAS.
- A. Zamora, G. Vigueras, V. Rodríguez, M. D. Santana and J. Ruiz, Coord. Chem. Rev., 2018, 360, 34–76 CrossRef CAS.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- N. Denora a, R. M. Iacobazzi, G. Natile and N. Margiotta, Coord. Chem. Rev., 2017, 341, 1–18 CrossRef.
- X. Li, Y. Zhao, T. Zhang and D. Xing, Adv. Healthc. Mater., 2021, 10, 2001240 CrossRef CAS.
- J. S. Nam, M. Kang, J. Kang, S. Park, S. J. C. Lee, H. Kim, J. K. Seo, O. Kwon, M. H. Lim, H. Rhee and T. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef CAS PubMed.
- N. Tian, W. Sun, X. Guo, J. Lu, C. Li, Y. Hou, X. Wang and Q. Zhou, Chem. Commun., 2019, 55, 2676–2679 RSC.
- A. Serrano-Puebla and P. Boya, Biochem. Soc. Trans., 2018, 46, 207–215 CrossRef CAS PubMed.
- K. Qiu, Y. Wen, C. Ouyang, X. Liao, C. Liu, T. W. Rees, Q. Zhang, L. Ji and H. Chao, Chem. Commun., 2019, 55, 11235–11238 RSC.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 127, 14255–14258 CrossRef.
- G. He, N. Xu, H. Ge, Y. Lu, R. Wang, H. Wang, J. Du, J. Fan, W. Sun and X. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 19572–19580 CrossRef CAS PubMed.
- M. Jakubaszek, B. Goud, S. Ferrari and G. Gasser, Chem. Commun., 2018, 54, 13040–13059 RSC.
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Chem. Sci., 2016, 7, 6115–6124 RSC.
- A. Raza, S. A. Archer, S. D. Fairbanks, K. L. Smitten, S. W. Botchway, J. A. Thomas, S. MacNeil and J. W. Haycock, J. Am. Chem. Soc., 2020, 142, 4639–4647 CrossRef CAS.
- S. J. Healy, A. M. Gorman, P. Mousavi-Shafaei, S. Gupta and A. Samali, Eur. J. Pharmacol., 2009, 625, 234–246 CrossRef CAS PubMed.
- M. R. Gill, D. Cecchin, M. G. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512–4519 RSC.
- B. Purushothaman, J. Lee, S. Hong and J. M. Song, J. Nanobiotechnology, 2020, 18, 102 CrossRef CAS PubMed.
- L. Xu, P. Zhang, X. Fang, Y. Liu, J. Wang, H. Zhou, S. Chen and H. Chao, J. Inorg. Biochem., 2019, 191, 126–134 CrossRef CAS PubMed.
- B. Yuan, J. Liu, R. Guan, C. Jin, L. Ji and H. Chao, Dalton Trans., 2019, 48, 6408–6415 RSC.
- W. Lv, Z. Zhang, K. Y. Zhang, H. Yang, S. Liu, A. Xu, S. Guo, Q. Zhao and W. Huang, Angew. Chem., Int. Ed., 2016, 55, 9947–9951 CrossRef CAS PubMed.
- K. Qiu, H. Zhu, T. W. Rees, L. Ji, Q. Zhanga, H. Chao and H. Chao, Coord. Chem. Rev., 2019, 363, 113013 Search PubMed.
- H. Huang, S. Banerjee and P. J. Sadler, ChemBioChem, 2018, 19, 1574–1589 CrossRef CAS PubMed.
- P. Majumdar, X. Yuan, S. Li, B. L. Guennic, J. Ma, C. Zhang, D. Jacquemin and J. Zhao, J. Mater. Chem. B, 2014, 2, 2838–2854 RSC.
- L. Wang, H. Yin, P. Cui, M. Hetu, C. Wang, S. Monro, R. D. Schaller, C. G. Cameron, Bi. Liu, S. Kilina, S. A. McFarland and W. Sun, Dalton Trans., 2017, 46, 8091–8103 RSC.
- J. Liu, C. Jin, B. Yuan, X. Liu, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2017, 53, 2052–2055 RSC.
- J. Zhao, K. Yan, G. Xu, X. Liu, Q. Zhao, C. Xu and S. Gou, Adv. Funct. Mater., 2021, 31, 2008325 CrossRef CAS.
- V. Novohradsky, A. Rovira, C. Hally, A. Galindo, G. Vigueras, A. Gandioso, M. Svitelova, R. Bresolí-Obach, H. Kostrhunova, L. Markova, J. Kasparkova, S. Nonell, J. Ruiz, V. Brabec and V. Marchán, Angew. Chem., Int. Ed., 2019, 58, 6311–6315 CrossRef CAS PubMed.
- H. Huang, S. Banerjee, K. Qiu, P. Zhang, O. Blacque, T. Malcomson, M. J. Paterson, G. J. Clarkson, M. Staniforth, V. G. Stavros, G. Gasser, H. Chao and P. J. Sadler, Nat. Chem., 2019, 11, 1041–1048 CrossRef CAS PubMed.
- R. Bevernaegie, B. Doix, E. Bastien, A. Diman, A. Decottignies, O. Feron and B. Elias, J. Am. Chem. Soc., 2019, 141, 18486–18491 CrossRef CAS PubMed.
- S. Kuang, F. Wei, J. Karges, L. Ke, K. Xiong, X. Liao, G. Gasser, L. Ji and H. Chao, J. Am. Chem. Soc., 2022, 144, 4091–4101 CrossRef CAS PubMed.
- H. Yuan, Z. Han, Y. Chen, F. Qi, H. Fang, Z. Guo, S. Zhang and W. He, Angew. Chem., Int. Ed., 2021, 60, 8174–8181 CrossRef CAS PubMed.
- C. Huang, C. Liang, T. Sadhukhan, S. Banerjee, Z. Fan, T. Li, Z. Zhu, P. Zhang, K. Raghavachari and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 9474–9479 CrossRef CAS PubMed.
- P. Zhang, H. Huang, S. Banerjee, G. J. Clarkson, C. Ge, C. Imberti and P. J. Sadler, Angew. Chem., Int. Ed., 2019, 58, 2350–2354 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- X. Wang, X. Wang, S. Jin, N. Muhammad and Z. Guo, Chem. Rev., 2019, 119, 1138–1192 CrossRef CAS PubMed.
- A. V. Klein and T. W. Hambley, Chem. Rev., 2009, 109, 4911–4920 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- F. Ponte, M. E. Alberto, B. C. De Simone, N. Russo and E. Sicilia, Inorg. Chem., 2019, 58, 9882–9889 CrossRef CAS PubMed.
- K. Mitra, C. E. Lyons and M. C. T. Hartman, Angew. Chem., Int. Ed., 2018, 57, 10263–10267 CrossRef CAS PubMed.
- G. Bordeau, R. Lartia, G. Metge, C. Fiorini-Debuisschert, F. Charra and M. P. Teulade-Fichou, J. Am. Chem. Soc., 2008, 130, 16836–16837 CrossRef CAS PubMed.
- Y. F. Zhong, H. Zhang, W. T. Liu, X. H Zheng, Y. W. Zhou, Q. Cao, Y. Shen, Y. Zhao, P. Z. Qin, L. Ji and Z. W. Mao, Chem. – Eur. J. , 2017, 23, 16442–16446 CrossRef CAS PubMed.
- Z. Lv, L. Zou, H. Wei, S. Liu, W. Huang and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 19523–19533 CrossRef CAS PubMed.
- K. Tilekar, N. Upadhyay, C. V. Ianc, V. Pokrovsky, J. Choe and C. S. Ramaa, BBA, Rev. Cancer, 2020, 1874, 188457 CAS.
- V. Ramu, S. Gautam, A. Garai, P. Kondaiah and A. R. Chakravarty, Inorg. Chem., 2018, 57, 1717–1726 CrossRef CAS PubMed.
- F. M. Platt, Nat. Rev. Drug Discovery, 2018, 17, 133–150 CrossRef CAS PubMed.
- R. Safaei, B. J. Larson, T. C. Cheng, M. A. Gibson, S. Otani, W. Naerdemann and S. B. Howell, Mol. Cancer Ther., 2005, 4, 1595–1604 CrossRef CAS PubMed.
- X. Xue, C. Qian, H. Fang, H. K. Liu, H. Yuan, Z. Guo, Y. Bai and W. He, Angew. Chem., Int. Ed., 2019, 58, 12661–12666 CrossRef CAS PubMed.
- I. Jin, M. Yuji, N. Yoshinori, K. i. Makoto and M. Mikio, Arch. Dermatol. Res., 2008, 300, 161–166 CrossRef CAS PubMed.
- M. Atif, M. Fakhar-e-Alamb, S. S. Z. Zaidi and R. SulemancH, Laser Phys., 2011, 21, 1135–1144 CrossRef CAS.
- D. Straten, V. Mashayekhi, H. S. Bruijn, S. Oliveira and D. J. Robinson, Cancers, 2017, 9, 19 CrossRef PubMed.
- C. Fink, A. Enk and P. Gholam, J. Dtsch. Dermatol. Ges., 2015, 13, 15–22 Search PubMed.
- K. Wang, B. Yu and J. L. Pathak, Cancer, 2021, 12, 1154–1160 CrossRef CAS PubMed.
- J. Zhang, L. Ning, J. Huang, C. Zhang and K. Pu, Chem. Sci., 2020, 11, 618–630 RSC.
- Y. Zheng, Y. Han, Q. Sun and Z. Li, Exploration, 2022, 2, 20210166 CrossRef.
- A. Ribas and J. D. Wolchok, Science, 2018, 359, 1350–1355 CrossRef CAS PubMed.
- K. Pizova, K. Tomankova, A. Daskova, S. Binder, R. Bajgar and H. Kolarova, Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub., 2012, 156, 93–102 CrossRef CAS PubMed.
- E. Panzarini, V. Inguscio and L. Dini, Biomed Res. Int., 2013, 482160 Search PubMed.
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |