
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise fabrication of porous polymer frameworks using rigid polyisocyanides as building blocks: from structural regulation to efficient iodine capture†
Xun-Hui
Xu
,
Yan-Xiang
Li
,
Li
Zhou
 ,
Na
Liu
and
Zong-Quan
Wu
*
,
Na
Liu
and
Zong-Quan
Wu
*
Department of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, Anhui Key Laboratory of Advanced Catalytic Materials and Reaction Engineering, Hefei University of Technology, Hefei 230009, Anhui Province, China. E-mail: zqwu@hfut.edu.cn
First published on 12th January 2022
Abstract
Porous materials have recently attracted much attention owing to their fascinating structures and broad applications. Moreover, exploring novel porous polymers affording the efficient capture of iodine is of significant interest. In contrast to the reported porous polymers fabricated with small molecular blocks, we herein report the preparation of porous polymer frameworks using rigid polyisocyanides as building blocks. First, tetrahedral four-arm star polyisocyanides with predictable molecular weight and low dispersity were synthesized; the chain-ends of the rigid polyisocyanide blocks were then crosslinked, yielding well-defined porous organic frameworks with a designed pore size and narrow distribution. Polymers of appropriate pore size were observed to efficiently capture radioactive iodine in both aqueous and vapor phases. More than 98% of iodine could be captured within 1 minute from a saturated aqueous solution (capacity of up to 3.2 g g−1), and an adsorption capacity of up to 574 wt% of iodine in vapor was measured within 4 hours. Moreover, the polymers could be recovered and recycled for iodine capture for at least six times, while maintaining high performance.
Introduction
Porous polymers, which combine the advantages of polymers and porous materials, have attracted ever increasing research attention on account of their structural diversity and broad applications, for instance, in the fields of gas separation and storage, chemical catalysis, pollutant removal, energy storage, and irradiative iodine adsorption.1–9 Porous polymers were commonly fabricated by using rigid small molecules as building blocks.10–12 Thanks to the structural diversity of organic frameworks and connecting methods, a variety of porous polymers have been readily developed.13–16 However, due to the limited dimensions of the available building blocks, the pores present in the reported porous polymers are intrinsically quite small, usually less than 10 nm in size.17–24 Indeed, constructing porous polymers with pores of large and tunable size is quite difficult,25 given that the preparation of large and rigid macromolecules as frame blocks is very hard and usually requires tedious and complicated synthetic processes.26 Therefore, developing macromolecules or polymers as rigid building blocks for fabricating porous polymers with large and pre-determined pore sizes is an interesting research goal. In fact, these kinds of porous polymers should have distinct properties and functions that are inaccessible by the traditional porous polymers fabricated from small building blocks.The recently developed living polymerization techniques yielded polymers with well-defined structures, desired molar mass (Mn), and low dispersity (Mw/Mn). However, most of the reported polymers have a flexible backbone.27,28 These polymers are unsuitable to be used as frame blocks in the construction of porous polymers because such building blocks do not have sufficient rigidity to support the desired frameworks.29 An exception to the described situation is polyisocyanides, which possess a rigid rod-like backbone because they are composed of carbon–carbon single bonds and bear substituents on every backbone atom.30–32 The steric effect of the pendants results in polyisocyanides adopting a rod-like structure, which twists into a helical shape. Recently, we reported a new kind of alkyne–Pd(II) complex that can efficiently catalyze the living polymerization of a diverse range of isocyanide monomers and afforded the synthesis of stereoregular polyisocyanides with predicted Mn and very low Mw/Mn.33 Taking advantage of these catalysts, a variety of topological polyisocyanides were facilely prepared.34–39 We, therefore, envisioned that rigid rod-like helical polyisocyanides with a defined chain length and low dispersity may be ideal building blocks for constructing porous polymer frameworks. The well-defined polyisocyanide blocks can produce semi-crystalline frameworks, which are distinct from amorphous porous organic polymers.13–17 Owing to the helical structure of polyisocyanide backbones and porosity of the frameworks, these materials may have great application potentials in many areas, for instance, the efficient capture of iodine. Radioactive iodine compounds are usually discharged as a result of the nuclear fission process, and are deemed severe radioactive pollutants.40–42 Among them, 129I is characterized by a half-life of ∼107 years, and it has to be captured and stored safely. On the other hand, 131I has a very short half-life (∼8.02 days), and it should thus be rapidly trapped because it may cause damage to the human metabolism.43–46 Given the high volatility of iodine and the fact that it is characterized by a degree of water solubility, a large amount of radioactive iodine could be present in nuclear waste water and in air, which would result in serious damage to the environment.47–49 Therefore, the search for robust adsorbents that quickly capture iodine in aqueous and vapor phases and exhibit large adsorption capacity is a very worthwhile research effort.50–52 Recently, Yang et al. reported conjugated macrocycle polymers based on macrocyclic arenes, which have high iodine affinity with an uptake capacity of 2.1 g g−1.53 Wang and coworkers reported porous polymers based on calix[4]pyrroles. The iodine uptake value was up to 3.4 g g−1 in iodine vapor.54 Koner et al. synthesized a sp2/sp3-N-rich polymeric network with an iodine adsorption capacity of 2.9 g g−1.55 However, to the best of our knowledge, polymer-based porous materials for iodine adsorption have rarely been reported. We postulated that using rigid polymers as building blocks can help in obtaining high-efficiency and large-capacity iodine adsorption porous materials.
In this article, we report the construction of polymer frameworks using rod-like polyisocyanides as building blocks and also explored the application of the frameworks to iodine capture. First, a tetrahedral four-arm Pd(II)-catalyst was prepared, which successfully initiated the living polymerization of an isocyanide monomer, tert-butyl (R)-2-(((4-isocyanobenzoyl)oxy)methyl)pyrrolidine-1-carboxylate (1), to afford four-arm helical polyisocyanides 4-poly-1ms (the footnote indicates the ratio of [1]0/[Pd]0) with the desired Mn value and low Mw/Mn ratio. Importantly, by crosslinking the chain-ends of the polyisocyanide segments, the desired porous frameworks were generated. Interestingly, the polymers with a smaller pore size exhibited excellent performance in iodine capture in water as well as in the vapor phase. Indeed, these polymers were observed to be able to take up >98% of the iodine present in a saturated aqueous iodine solution within 1 minute with only 2.5 mg mL−1 polymer loading and to adsorb 5.74 g g−1 iodine present in vapor within 4 hours; in fact, these polymers acted much faster and adsorbed much larger quantities of iodine than most materials reported to date.40–52
Results and discussion
As can be evinced from Scheme 1, tetrakis-(4-ethynylphenyl)methane was first prepared and made to react with trans-dichlorobis(triethylphosphine)palladium via catalysis with cuprous chloride.33 The thus generated four-functional Pd(II)-based catalyst (4-Pd(II)) was characterized by 1H NMR, 13C NMR, Fourier-transform infrared (FT-IR) spectroscopy, and mass spectrometry analyses, as well as by microanalysis (Fig. S1–S3 in the ESI†). The isocyanide monomer 1 was tentatively polymerized by 4-Pd(II) at 55 °C in CHCl3 at a [1]0/[Pd(II)]0 ratio of 200. The polymer thus generated was precipitated using n-hexane and isolated via filtration in 83% yield. Size exclusion chromatography (SEC) analysis of 4-poly-1200 (see Scheme 1) resulted in a unimodal and narrow elution peak. The Mn of 4-poly-1200 was determined to be 73.5 kDa by SEC, with Mw/Mn = 1.20 (Fig. 1a). As indicated by low Mw/Mn, the reaction probably followed a living polymerization mechanism. Thus, a series of polymerization reactions were conducted at different concentration ratios of 1 to 4-Pd(II). As expected, the generated 4-poly-1m polymers all exhibited single modal SEC curves, and the Mw/Mn values were smaller than 1.22 (Fig. 1a). Furthermore, Mn showed a linear relationship with a feed ratio of 1 to 4-Pd(II) (Fig. 1b), underlining the living nature of the polymerization. Therefore, a family of 4-poly-1m polymers with different Mn values and narrow Mw/Mns were prepared just by tuning the ratio of 1 to the 4-Pd(II) catalyst (Table S1, ESI†).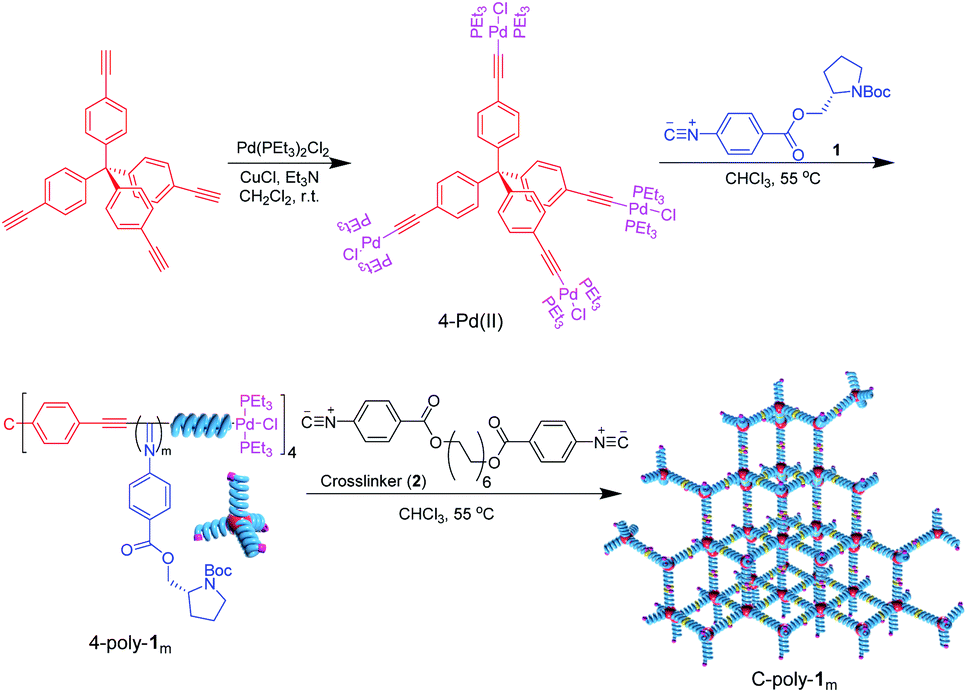 | ||
| Scheme 1 Fabrication of porous organic frameworks using polyisocyanides as frame blocks. | ||
 | ||
| Fig. 1 (a) SEC curves of 4-poly-1m polymers with different Mns. (b) Plot of Mn and Mw/Mn values of 4-poly-1m polymers as a function of the 1-to-Pd(II) ratio. (c) 31P NMR spectra of 4-Pd(II), 4-poly-120, and C-poly-120. (d) CD and UV-visible spectra of 4-poly-1m polymers measured in THF at 20 °C. | ||
The 1H NMR spectrum of 4-poly-120 is similar to that of its linear analog.30 Resonance signals attributed to the phenyl ring and CO2CH2 groups of the pendants were clearly discerned at 7.86–6.91 ppm (ArH), 6.20–5.34 ppm (ArH), and 4.80–3.58 ppm (CH and CH2) (Fig. S4, ESI†). The 31P NMR spectrum of 4-Pd(II) was characterized by a singlet resonance at 17.9 ppm, which disappeared completely after the polymerization of 1 (Fig. 1c). Concomitantly, a new resonance at 13.6 ppm was observed in the 31P NMR spectrum of 4-poly-120, which corresponds to the 31P resonance of the terminal –Pd(PEt3)2Cl complex (see Scheme 1). These data suggest that the four Pd(II) units on the 4-Pd(II) catalyst have the same activity and all participated in the living polymerization of 1; evidence also suggests that the Pd(II) units resided on the terminals of the yielded four-arm polyisocyanides. The 13C NMR showed characteristic resonances of the polyisocyanide backbones (Fig. S5, ESI†). The FT-IR spectrum of 4-poly-120 comprised a sharp absorption peak at 1600 cm−1, which is attributable to the vibration mode of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the polyisocyanide backbone (Fig. S6, ESI†). The optical activity of 4-poly-1ms was verified by circular dichroism (CD) spectroscopy. The 4-poly-1m polymers characterized by different chain lengths (or different degrees of polymerization (DP)) exhibited intense CD bands in the polyisocyanide absorption area, with the maximum intensity recorded for the polymer with a DP value larger than 100 (Fig. 1d). The negative CD signals around 364 nm that reflect the helix-sense of the polyisocyanide backbone suggest polyisocyanides were twisted into a left-handed preferred helix due to the asymmetric induction of the chiral monomer.39 Temperature-dependent CD spectra were recorded from −10 to 60 °C in THF. However, no obvious change could be observed, indicating that the helicity was quite stable (Fig. S7, ESI†). This also confirms that the CD signal was from the helical backbone and not due to the self-assembly or the aggregate.34
N bond in the polyisocyanide backbone (Fig. S6, ESI†). The optical activity of 4-poly-1ms was verified by circular dichroism (CD) spectroscopy. The 4-poly-1m polymers characterized by different chain lengths (or different degrees of polymerization (DP)) exhibited intense CD bands in the polyisocyanide absorption area, with the maximum intensity recorded for the polymer with a DP value larger than 100 (Fig. 1d). The negative CD signals around 364 nm that reflect the helix-sense of the polyisocyanide backbone suggest polyisocyanides were twisted into a left-handed preferred helix due to the asymmetric induction of the chiral monomer.39 Temperature-dependent CD spectra were recorded from −10 to 60 °C in THF. However, no obvious change could be observed, indicating that the helicity was quite stable (Fig. S7, ESI†). This also confirms that the CD signal was from the helical backbone and not due to the self-assembly or the aggregate.34
Given the rigidity of polyisocyanide segments, 4-poly-1m polymers can be regarded as tetrahedra composed of 4-arm polymer ‘match sticks’. The active Pd(II) complexes at the living chain-ends were crosslinked through the insertion of bifunctional isocyanide (2) at 55 °C in CHCl3 ([2]0/[Pd]0 = 1/2). Although linker 2 is flexible, the length is very short as compared to the four-arm building blocks. Thus, intramolecular crosslinking is hard to take place due to the limited length, which is also supported by the molecular modeling (Fig. S8, ESI†). The porous polymers thus generated were precipitated into methanol and collected in high yield via centrifugation (Table 1). Note that 4-poly-1ms have good solubility in methanol, while the resulting crosslinked C-poly-1ms are insoluble in this solvent and have higher viscosity. The 1H and 13C NMR and FT-IR spectra of the crosslinked polymer C-poly-120 were similar to those of the 4-poly-120 precursor (Fig. S6 and S9–S14, ESI†), which revealed that the chemical structures of the polyisocyanide segments were maintained. After crosslinking, the 31P resonance of 4-poly-120 at 13.6 ppm completely disappeared and a new resonance at 14.1 ppm was discerned for the resulting C-poly-120. Based on the 31P NMR analyses, the intermolecular crosslinking density is higher than 95%. Thermal gravimetric analysis (TGA) data indicated that the thermal decomposition of the crosslinked polymers occurred at around 230 °C, in a temperature range similar to that of the 4-poly-1m precursors (Fig. S15a, ESI†). However, the differential scanning calorimetry curves of the porous polymers and of the four-arm 4-poly-1m precursors were remarkably different from each other. The glass transition temperature (Tg) of 4-poly-1200 was ∼75 °C; by contrast, no such transition was identified for crosslinked C-poly-1200. The possible reason for this observation is that the crosslinked structure restricts the slipping of molecular segments, causing the Tg of the polymer networks to not be detected (Fig. S15b, ESI†). Powder X-ray diffraction (PXRD) profiles of 4-poly-1ms precursors showed only a broad diffraction peak with the maximum 2θ around 19° (Fig. S16a, ESI†), while PXRD of the resultant C-poly-1ms showed a sharp diffraction peak with a 2θ around 3–6°, in addition to the broad diffraction peak (Fig. S16b, ESI†). Interestingly, a clear trend was observed from the PXRD profiles. The 2θ values of the sharp diffraction peaks of C-poly-1ms decreased with the increasing length of the polyisocyanide backbone. These results support the formation of ordered porous polymers owing to the intermolecular linkage of the rigid polyisocyanide termini and also suggest that the pore size was determined by the polyisocyanide length.
| Run | Polymer | Yieldb (%) | S BET (m2 g−1) | V pore (cm3 g−1) | Pore-sizec (nm) | Adsorption capacityd (g g−1) |
|---|---|---|---|---|---|---|
| a The porous polymers were fabricated according to Scheme 1. b Isolated yields. c These data were estimated from N2 adsorption isotherms. d Iodine gravimetric capacity = m(I2)/m(polymer). | ||||||
| 1 | C-poly-15 | 87 | 96.16 | 1.3 × 10−1 | 2.21 | 5.74 |
| 2 | C-poly-120 | 93 | 29.72 | 8.2 × 10−2 | 4.03 | 4.31 |
| 3 | C-poly-150 | 95 | 14.19 | 7.9 × 10−2 | 8.25 | 3.97 |
| 4 | C-poly-180 | 92 | 9.46 | 7.4 × 10−2 | 10.21 | 3.75 |
| 5 | C-poly-1100 | 90 | 8.89 | 7.1 × 10−2 | 14.08 | 3.59 |
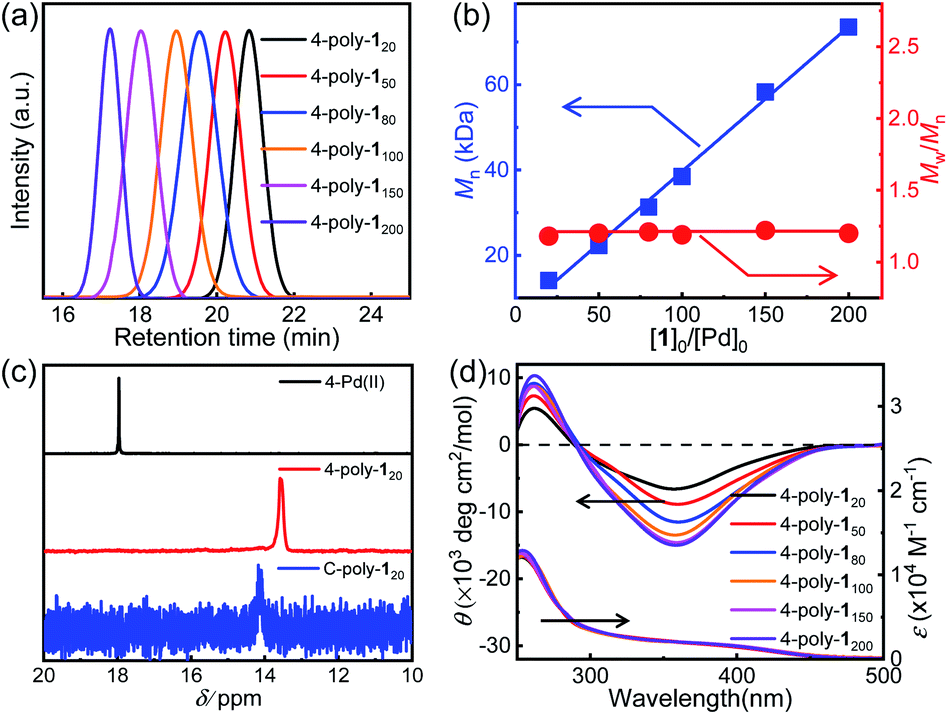
In order to investigate the porosity of the synthesized polymers, nitrogen adsorption isotherms were recorded (Fig. 2a). The isotherm of C-poly-15 exhibited N2 adsorption at low pressure (P/P0 < 0.2), which is indicative of the presence of micro- or mesopores. C-poly-120 adsorbed N2 in the medium pressure region, indicating that C-poly-120 has larger pores than C-poly-15. A relatively sharp rise in N2 adsorption in the high pressure region (P/P0 = 0.8–1.0) was observed for C-poly-150, C-poly-180, and C-poly-1100, suggesting that these polymers mainly comprise meso- or macropores. The Brunauer–Emmett–Teller (BET) specific surface area (SBET) of C-poly-15 was 96.16 m2 g−1 and its pore volume (Vpore) was 0.13 mL g−1, as calculated using a nonlocal density functional theory approach (Table 1). The value for the SBET was reduced to 29.72 m2 g−1 for C-poly-120, 14.19 m2 g−1 for C-poly-150, 9.46 m2 g−1 for C-poly-180, and 8.89 m2 g−1 for C-poly-1100, as the length of polyisocyanide blocks progressively increased (Table 1). The pore size was also observed to depend on the length of the polyisocyanide arms. For instance, the pore size was 2.21 nm for C-poly-15, and it increased to 4.03, 8.25, 10.21, and 14.08 nm for C-poly-120, C-poly-150, C-poly-180, and C-poly-1100, respectively; in all cases, the pore sizes had narrow distributions (Table 1 and Fig. 2b). Interestingly, detailed analyses revealed that the pore size of the mentioned species was linearly related to the length of the polyisocyanide blocks (Fig. S17, ESI†). This can be ascribed to that the pores were formed through the intermolecular crosslinking of the polyisocyanide termini, and thus the pore size directly relies on the length of the rigid polyisocyanide blocks. Furthermore, the pore volume was also dependent on the length of the polyisocyanide arm. It increased with the decrease of the polyisocyanide length (Table 1). Because of the large pore size, surface areas of the polymers with long polyisocyanide backbones such as C-poly-150, C-poly-180, and C-poly-1100 are relatively low.56 It is worthy of note that BET analysis of the uncrosslinked 4-poly-15 suggests it has no porous structure (Fig. S18, ESI†). These investigations demonstrated that the crosslinked polymeric frameworks comprise permanent pores and that the porosity can be controlled by varying the length of the polyisocyanide blocks. Consequently, a series of polymer frameworks with different pore sizes and narrow pore size distributions were facilely obtained.
 | ||
| Fig. 2 Nitrogen sorption isotherms (a) and pore size distributions (b) of the herein synthesized porous organic frameworks recorded at 77 K. Notably, in (a) the filled symbols denote gas adsorption while the empty symbols denote gas desorption. | ||
The microstructures of the porous polymers were investigated by scanning electron microscopy (SEM). As can be evidenced from the SEM images in Fig. 3, C-poly-1ms with different polyisocyanide lengths have different morphologies. C-poly-15 and C-poly-120, which were synthesized with relatively short polyisocyanide blocks, exhibited rough surfaces decorated with pores (Fig. 3a and b). As the length of the polyisocyanide increased, the surface of C-poly-150 tends to be flat (Fig. 3c). Additionally, C-poly-1100, which is characterized by even longer polyisocyanide segments, acquired a sheet-like morphology (Fig. 3d). The SEM image of uncrosslinked 4-poly-15 is smoother than that of crosslinked C-poly-15 (Fig. S19, ESI†). These observations suggested that the morphology of these porous polymers could be controlled by regulating the length of the polyisocyanide segments. Because of the limited resolution, it is hard to directly determine the intrinsic pore structure of the polymer frameworks by SEM.
 | ||
| Fig. 3 Scanning electron microscopy images of the porous organic networks of C-poly-15 (a), C-poly-120 (b), C-poly-150 (c), and C-poly-1100 (d). | ||
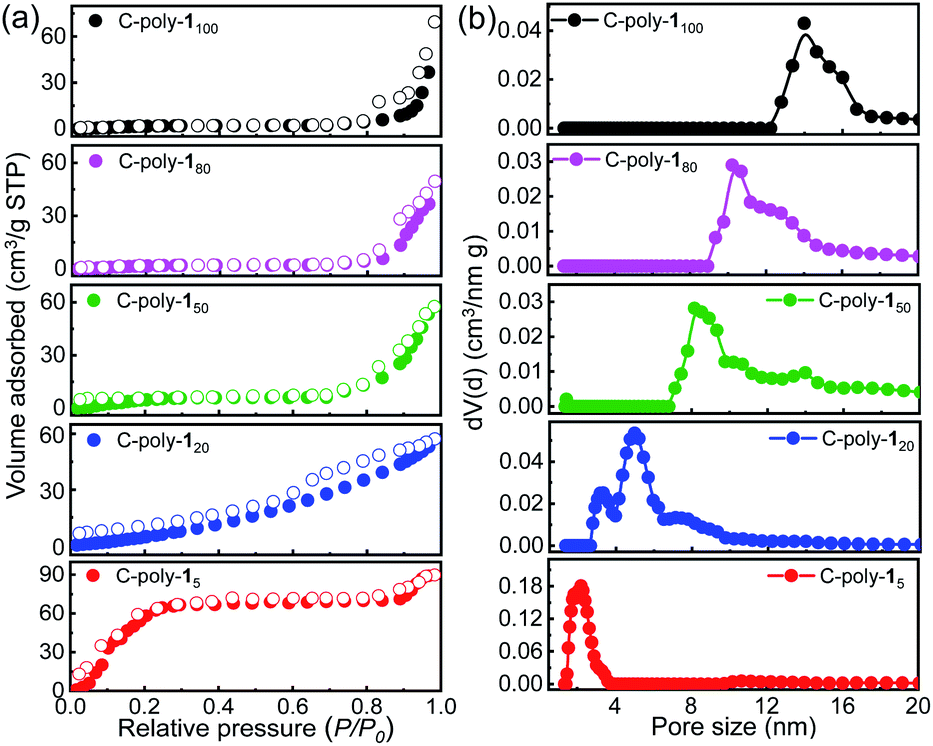
Since these polymers exhibited poor solubility in water and in common organic solvents, their ability to adsorb iodine was tested in water and n-hexane. Although some metal–organic frameworks (MOFs) have been employed in experiments whereby iodine was adsorbed from the vapor phase, given the instability of MOFs in water, experiments in which iodine was captured from aqueous solution have been rarely reported.57 A preliminary experiment revealed that C-poly-1m polymers could quickly capture iodine from a saturated aqueous solution (1.18 mM). Upon the addition of C-poly-15 (polymer loading: 2.5 mg mL−1, Video 1 in the ESI†), the yellow iodine solution immediately became colorless. The iodine concentration was reduced to 2.99 ppm in 1 minute, based on UV-visible (UV-vis) absorption spectroscopy analysis, suggesting that over 98% of iodine had been removed from water (Fig. S20a, ESI†). In order to obtain quantitative details, the polymer loading was reduced to 0.25 mg mL−1, and the process of iodine capture was followed by measuring the absorption spectra of the residual iodine in water. As can be evidenced from the data in Fig. 4a, the UVvis absorption of iodine decreased dramatically after the addition of C-poly-15, and the iodine concentration was reduced to 42.8 ppm after 1 minute and to less than 6.5 ppm after 20 minutes, suggesting a fast iodine capture. The iodine adsorption could be accelerated by increasing the loading of C-poly-15 (Fig. S20b–f,† ESI). Other C-poly-1m polymers also exhibited a good iodine capture performance; in fact, the color of the iodine solution faded rapidly after addition of the polymers (insets in Fig. 4b–e). After 20 minutes from the polymer addition, the residual iodine concentration was 14.1 ppm in the case of C-poly-120, 34.8 ppm in the case of C-poly-150, 67.3 ppm in the case of C-poly-180, and 86.5 ppm in the case of C-poly-1100. Therefore, the iodine capture efficiency was 91%, 77%, 55%, and 42%, for C-poly-120, C-poly-150, C-poly-180, and C-poly-1100, respectively. Further analyses revealed that the iodine uptake process exhibited the characteristics of a pseudo-second-order mechanism, and the rate constant (kobs) values were 8.4 × 10−4, 6.2 × 10−4, 3.9 × 10−4, 3.5 × 10−4, and 3.4 × 10−4 g (mg min−1)−1, respectively, for C-poly-15, C-poly-120, C-poly-150, C-poly-180, and C-poly-1100 (Fig. S21, ESI†).58 Evidence thus suggests that the rate and capacity of the iodine uptake decreased as the pore size of the polymers increased. Probably, C-poly-15 has an appropriate pore size for iodine uptake, so this polymer exhibited the highest efficiency and largest adsorption capacity among the tested samples (Fig. S22, ESI†). The excellent iodine adsorption by the porous polymers was probably due to the interactions between iodine molecules and the phenyl rings and nitrogen atoms of the polymers; it was also assumed to depend on the pore size. Since these polymers had relatively low surface areas, the helical structure of the polyisocyanide backbone also played a vital role in the efficient iodine capture. The iodine adsorption kinetics of C-poly-15 in water was then investigated by estimating the variation of maximum adsorption of I-3 (KI–I2).59 It was found that more than 98% of I-3 was captured within 6 minutes, as disclosed by UV-vis absorption spectroscopy (Fig. S23, ESI†). The equilibrium values of I-3 adsorption were fitted well by the Langmuir isotherm model (Fig. S24, ESI†).58 Based on the simulation results, the maximum adsorption capacity of C-poly-15 for I-3 was determined to be as high as 3.2 g g−1.
 | ||
| Fig. 4 Time-dependent UV-visible spectra of a saturated aqueous iodine solution (1.18 mM) in the presence of porous networks C-poly-15 (a), C-poly-120 (b), C-poly-150 (c), C-poly-180 (d) and C-poly-1100 (e) at 25 °C (polymer loading: 0.25 mg mL−1; insets: photographs of the solutions). (f) Calculated iodine capture efficiency of C-poly-1ms at different time points. | ||
Since C-poly-15 exhibited the highest iodine capture performance in water, its ability to capture iodine in organic solvent was also investigated. As can be evidenced from the photos shown in Fig. 5a, the dark red color of iodine-dissolved n-hexane solution gradually faded, and turned nearly colorless within 30 minutes, following the addition of C-poly-15 with only 0.5 mg mL−1 loading (Fig. 5a). Time-dependent UV-vis spectroscopy data suggested that the concentration of iodine dropped rapidly to 68.2 ppm within 30 minutes and to 28.8 ppm after 1 h (Fig. 5b). The iodine capture efficiency was thus 98% within 1 h, confirming the highly efficient iodine capture by C-poly-15 in n-hexane. According to the Langmuir isotherm model, the adsorption capacity of C-poly-15 for iodine/n-hexane was 2.1 g g−1 (Fig. S25 and S26, ESI†).59 Other porous polymers such as C-poly-120, C-poly-150, and C-poly-1100 also exhibited excellent iodine adsorption performance in n-hexane, although their efficiencies and capacities were lower than the corresponding parameters for C-poly-15 (Fig. S25 and S27, ESI†).59 The different iodine adsorption behaviours of C-poly-15 in water and n-hexane were ascribed to different equilibrium concentrations of iodine in water and n-hexane and also due to the different solvation effects of these solvents.60
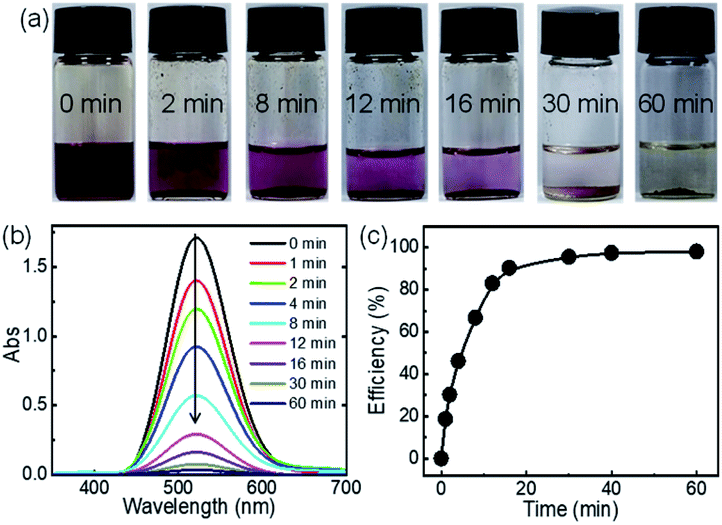 | ||
| Fig. 5 Color change (a) and UV-visible spectra (b) of iodine/n-hexane (3.94 mM, 4 mL) recorded at different time points after the addition of crosslinked polymer C-poly-15 (2.1 mg). (c) Efficiency of the adsorption of iodine from iodine/n-hexane by C-poly-15. | ||
Iodine capture in the vapor phase was performed by exposing the powdered polymers to iodine vapor according to the procedure reported in the literature (Fig. 6a).53 Briefly, the polymer samples (10 mg) were placed into pre-weighted small glass vials, which were then transferred into larger vials containing iodine (500 mg). The vials were then sealed and heated to 80 °C in an oven at ambient pressure (Fig. S28, ESI†). Following exposure to iodine vapor, the yellow-brown polymers gradually became darker. The values for the iodine capture capacity were calculated by weighing the samples every 20 minutes before and after the adsorption experiment. As can be evidenced from the data reported in Fig. 6b–e, it took just about 4 hours to reach the absorption maximum; moreover, the adsorption capacity was 5.74 g g−1 for C-poly-15, 4.31 g g−1 for C-poly-120, 3.75 g g−1 for C-poly-150, and 3.02 g g−1 for C-poly-1100. The iodine adsorption capacity of C-poly-15 in particular was extremely high at 574 wt%, which is a higher value than those reported for most materials for which iodine adsorption has been investigated.10,53–55 Probably, the small pore size of C-poly-15 and the abundant phenyl rings and nitrogen atoms in the said helical polymer synergistically promoted iodine adsorption. For comparison, the iodine adsorption was also performed using uncrosslinked 4-poly-15 and linear analogue poly-15. The adsorption capacities are much lower than that of the porous polymers. For example, the uptake capacity of 4-poly-15 is just 0.28 g g−1 in water and 0.42 g g−1 in iodine vapor, respectively, while the iodine adsorption capacity of the linear poly-15 is only 0.18 g g−1 in iodine vapor (Fig. S22 and S29†).
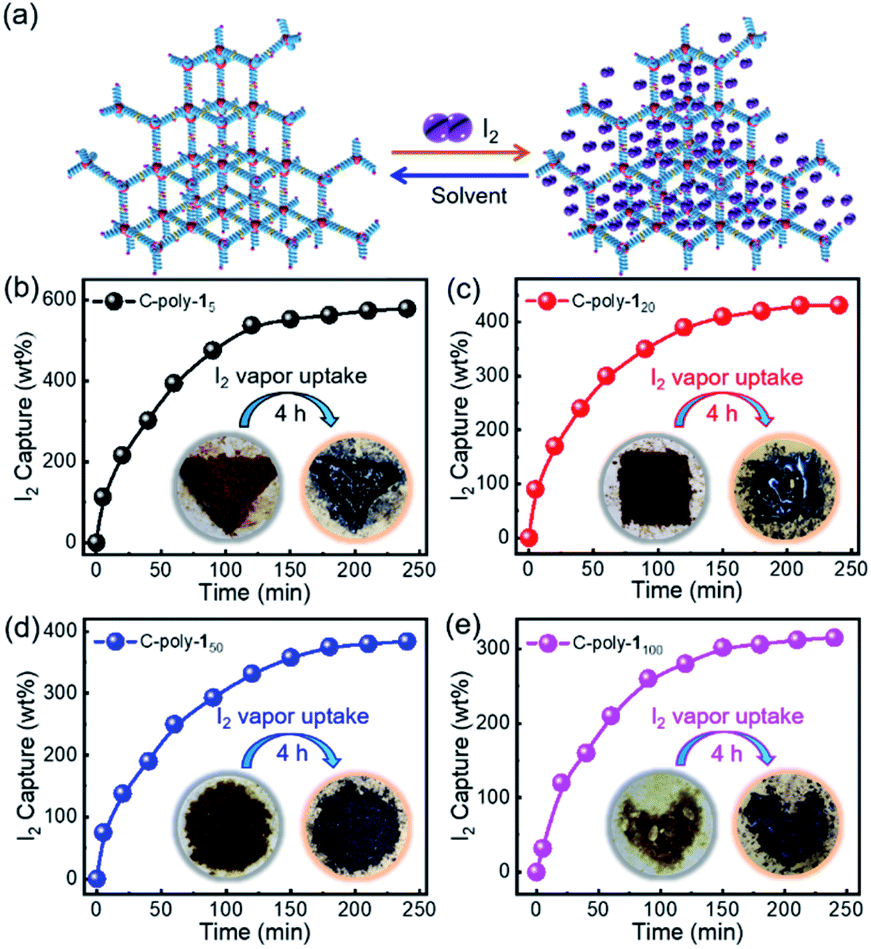 | ||
| Fig. 6 (a) Process of iodine (I2) capture and release by C-poly-1m polymers. Time-dependent iodine capture plots of C-poly-15 (b), C-poly-120 (c), C-poly-150 (d), and C-poly-1100 (e) at 80 °C. Panels (b–e) include photographs of C-poly-1m polymers taken before and after their exposure to iodine vapor. | ||
PXRD patterns of the C-poly-1m polymers recorded after their capture of iodine in the vapor phase to form a series of species dubbed I2@C-poly-1ms included no obvious diffraction signals in the 2θ range from 3° to 25°, probably because the samples were covered with the adsorbed iodine molecules (Fig. S30a, ESI†). SEM images of I2@C-poly-1ms obtained similar to the reported literature were indicative of the occurrence of morphological changes stemming from iodine adsorption.53 The pores of the polymers were occupied by iodine and became rougher (Fig. S31, ESI†). Moreover, the results of energy-dispersive spectroscopy experiments indicated that a large amount of iodine was present on the surface of I2@C-poly-1ms. TGA conducted on I2@C-poly-15, I2@C-poly-120, I2@C-poly-150, and I2@C-poly-1100 revealed that the mass losses were 85.3%, 80.5%, 77.5%, and 74.5%, respectively (Fig. S32, ESI†). These results are consistent with the I2 capture capacity data for each polymer determined as described above. Based on the TGA results, the captured iodine was released from the polymers below 200 °C in all cases, indicating that iodine had been physically adsorbed. FT-IR spectroscopy measurements further corroborated the idea that iodine had been physically, rather than chemically, adsorbed onto the polymers, given that no appreciable changes were observed in the spectra of the polymers recorded before and after iodine adsorption (Fig. S33, ESI†).
Lastly, release of the captured iodine and the reuse of the polymers for iodine adsorption were investigated. The release of iodine from I2@C-poly-15 was conducted in methanol and monitored by UV-vis absorption spectroscopy (Fig. 7a). The initially colorless methanol solution first became yellow and eventually turned deep brown (see the inset in Fig. 7a), a process attributed to iodine release. The concentration of iodine in methanol increased as the experiment progressed, reaching an equilibrium level within 20 minutes; at this point, >95% of iodine had been released from I2@C-poly-15, based on the analyses of UV-vis absorption data (Fig. 7b). Since iodine was physically adsorbed and can be readily released, the porous polymer was recycled to be used in a new round of iodine capture. Since C-poly-15 proved to be insoluble in methanol, after iodine release, the polymer was recovered via filtration and washed with methanol several times. Indeed, the same C-poly-15 sample was re-used for capturing iodine from the vapor phase via the described recycling process for a total of six times, and it was determined to maintain high efficiency and large capacity (Fig. 7c). The recovered C-poly-15 could also recycle captured iodine from aqueous solution with maintaining good performance (Fig. S34, ESI†). Note that the iodine release in water is much slower than that in methanol (Fig. S35, ESI†), probably because methanol has a high solvation effect and also due to the poor solubility of iodine in water.48 Thus, the iodine release is not only a diffusion-controlled mechanism but also rely on the solvation effect of the used solvents.60
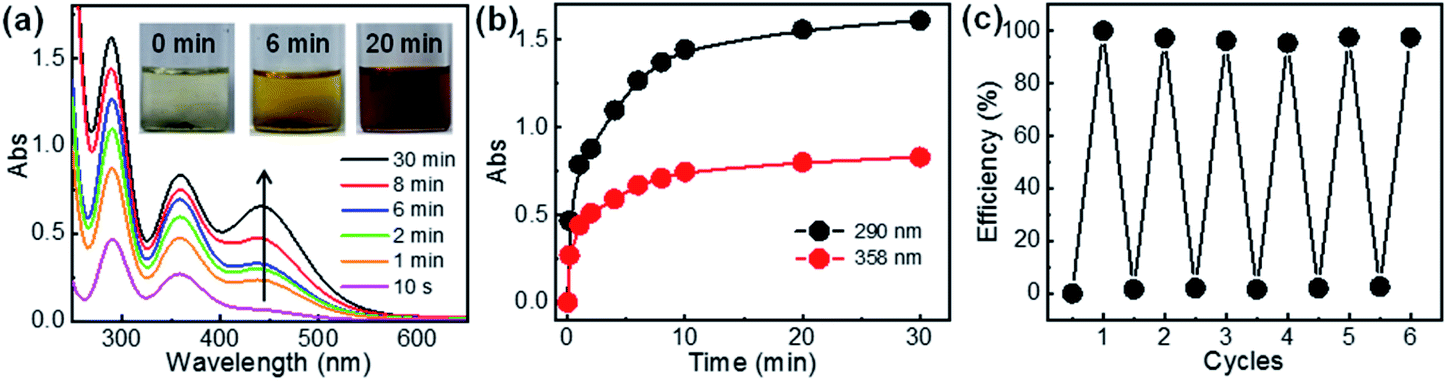 | ||
| Fig. 7 (a) Time-dependent UV-visible spectra of the methanol solution kept in contact with C-poly-15 that had adsorbed iodine from the vapor phase (species dubbed I2@C-poly-15). In the inset are the reported photographs of the said methanol solution. (b) Changes over time of the absorbance of UV-visible spectroscopy peaks at 290 and 358 nm associated with iodine release from I2@C-poly-15 into the methanol solution. (c) Efficiency of cycles of iodine desorption from and adsorption onto the C-poly-15 polymer. | ||
Conclusions
A series of tetrahedral four-arm star helical polyisocyanides with controlled Mn and low Mw/Mn were prepared for the first time. The chain-ends of these polyisocyanides were then crosslinked through the insertion of a bifunctional isocyanide linker; as a result, porous polymer frameworks with defined pore sizes and narrow distributions were generated. The porosity of these polymers could be facilely regulated by tuning the length of the helical polyisocyanide blocks. Remarkably, members of this novel polymer family characterized by the appropriate pore size were observed to efficiently capture iodine from aqueous solution and in vapor as well. More than 98% of iodine can be removed from a saturated iodine aqueous solution within 1 minute, with an adsorption capacity of up to 3.2 g g−1 measured for the herein developed porous polymer frameworks. Furthermore, the most efficient iodine-adsorbing polymers synthesized in the present study could capture iodine from the vapor phase, exhibiting a capacity of 5.74 g g−1 over 4 hours; these data indicate that the said novel polymer frameworks exhibit relatively fast action and large iodine capacity with respect to previously reported materials.This study provides a new approach for the controlled synthesis of novel polymer frameworks with tunable porosity that relies on rigid polymers as building blocks; by the approach described herein, a family of porous materials has been generated that can be used for the efficient capture of iodine. In view of the feasibility of modifications of Pd(II)-based catalysts and isocyanide monomers, the development of a diverse range of distinct polymer frameworks with great application potential can be anticipated.
Data availability
The detailed experimental data associated with this work is available in the ESI.†Author contributions
X.-H. X. and Y.-X. L. performed the experiments and analyses. X.-H. X. and Z.-Q. W. wrote and edited the paper. L. Z. analysed the data and conducted the calculations. N. L. and Z.-Q. W. conceived the idea and supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China for financial support (NSFC, No. 22071041, 21971052, 51903072, and 21871073). L. Zhou and Z.-Q. Wu appreciate the Fundamental Research Funds for the Central Universities of China (Grant No. PA2019GDPK0057, and PA2020GDJQ0028).Notes and references
- J. Zhu, C. Yang, C. Lu, F. Zhang, Z. Yuan and X. Zhuang, Acc. Chem. Res., 2018, 51, 3191–3202 CrossRef CAS PubMed.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611 CrossRef CAS.
- Y. Gu, J.-J. Zheng, K.-I. Otake, M. Shivanna, S. Sakaki, H. Yoshino, M. Ohba, S. Kawaguchi, Y. Wang, F. Li and S. Kitagawa, Angew. Chem., Int. Ed., 2021, 60, 11688–11694 CrossRef CAS PubMed.
- Y.-B. Huang, Q. Wang, J. Liang, X. Wang and R. Cao, J. Am. Chem. Soc., 2016, 138, 10104–10107 CrossRef CAS PubMed.
- A. Mähringer, M. Hennemann, T. Clark, T. Bein and D. D. Medina, Angew. Chem., Int. Ed., 2021, 60, 5519–5526 CrossRef PubMed.
- J. Lv, Y.-X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M.-D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- Y. Xie, W. Huang, B. Zheng, S. Li, Q. Liu, Z. Chen, W. Mai, R. Fu and D. Wu, Adv. Mater., 2019, 31, 1900104 CrossRef PubMed.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- Z.-J. Yin, S.-Q. Xu, T.-G. Zhan, Q.-Y. Qi, Z.-Q. Wu and X. Zhao, Chem. Commun., 2017, 53, 7266–7269 RSC.
- S. Sharma, A. V. Desai, B. Joarder and S. K. Ghosh, Angew. Chem., Int. Ed., 2020, 59, 7788–7792 CrossRef CAS PubMed.
- H.-S. Xu, Y. Luo, P. Z. See, X. Li, Z. Chen, Y. Zhou, X. Zhao, K. Leng, I.-H. Park, R. Li, C. Liu, F. Chen, S. Xi, J. Sun and K. P. Loh, Angew. Chem., Int. Ed., 2020, 59, 11527–11532 CrossRef CAS PubMed.
- A. Chaix, G. Mouchaham, A. Shkurenko, P. Hoang, B. Moosa, P. M. Bhatt, K. Adil, K. N. Salama, M. Eddaoudi and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 14571–14575 CrossRef CAS PubMed.
- T.-N. Gao, T. Wang, W. Wu, Y. Liu, Q. Huo, Z.-A. Qiao and S. Dai, Adv. Mater., 2019, 31, 1806254 CrossRef PubMed.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- L. Tana and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- A. Comotti, S. Bracco and P. Sozzani, Acc. Chem. Res., 2016, 49, 1701–1710 CrossRef CAS PubMed.
- Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS PubMed.
- J. Liu, W. Duan, J. Song, X. Guo, Z. Wang, X. Shi, J. Liang, J. Wang, P. Cheng, Y. Chen, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 12064–12070 CrossRef CAS PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- S.-Y. Dinga and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- X. Han, J. Huang, C. Yuan, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 892–895 CrossRef CAS PubMed.
- W. Xu, B. Tu, Q. Liu, Y. Shu, C.-C. Liang, C. S. Diercks, O. M. Yaghi, Y.-B. Zhang, H. Deng and Q. Li, Nat. Rev. Mater., 2020, 5, 764–779 CrossRef CAS.
- H. Li, F. Chen, X. Guan, J. Li, C. Li, B. Tang, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2021, 143, 2654–2659 CrossRef CAS PubMed.
- S. You, M. Ma, W. Wang, D. Qi, X. Chen, J. Qu and N. Ren, Adv. Energy Mater., 2017, 7, 1601364 CrossRef.
- M.-Q. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef PubMed.
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS PubMed.
- M.-Q. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Chem. Soc. Rev., 2006, 35, 622–629 RSC.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Noltea and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- Y.-X. Xue, Y.-Y. Zhu, L.-M. Gao, X.-Y. He, N. Liu, W.-Y. Zhang, J. Yin, Y. Ding, H. Zhou and Z.-Q. Wu, J. Am. Chem. Soc., 2014, 136, 4706–4713 CrossRef CAS PubMed.
- L. Zhou, X.-H. Xu, Z.-Q. Jiang, L. Xu, B.-F. Chu, N. Liu and Z.-Q. Wu, Angew. Chem., Int. Ed., 2021, 60, 806–812 CrossRef CAS PubMed.
- W.-B. Liu, X.-H. Xu, S.-M. Kang, X. Song, L. Zhou, N. Liu and Z.-Q. Wu, Macromolecules, 2021, 54, 3158–3168 CrossRef CAS.
- E. Elacqua, K. B. Manning, D. S. Lye, S. K. Pomarico, F. Morgia and M. Weck, J. Am. Chem. Soc., 2017, 139, 12240–12250 CrossRef CAS PubMed.
- Q. Wang, B.-F. Chu, J.-H. Chu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2018, 7, 127–131 CrossRef CAS.
- E. Elacqua, A. Croom, K. B. Manning, S. K. Pomarico, D. Lye, L. Young and M. Weck, Angew. Chem., Int. Ed., 2016, 55, 15873–15878 CrossRef CAS PubMed.
- Z.-Q. Jiang, S.-Q. Zhao, Y.-X. Su, N. Liu and Z.-Q. Wu, Macromolecules, 2018, 51, 737–745 CrossRef CAS.
- R. C. Ewing and F. N. von Hippel, Science, 2009, 325, 151–152 CrossRef CAS PubMed.
- A. M. Leung and L. E. Braverman, Nat. Rev. Endocrinol., 2014, 10, 136–142 CrossRef CAS PubMed.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther III, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598–11620 CrossRef PubMed.
- A. Saiz-Lopez, J. M. C. Plane, A. R. Baker, L. J. Carpenter, R. V. Glasow, J. C. G. Martín, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773–1804 CrossRef CAS PubMed.
- K. Jie, Y. Zhou, E. Li, Z. Li, R. Zhao and F. Huang, J. Am. Chem. Soc., 2017, 139, 15320–15323 CrossRef CAS PubMed.
- D. F. Sava, M. A. Rodriguez, K. W. Chapman, P. J. Chupas, J. A. Greathouse, P. S. Crozier and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 12398–12401 CrossRef CAS PubMed.
- B. Li, B. Wang, X. Huang, L. Dai, L. Cui, J. Li, X. Jia and C. Li, Angew. Chem., Int. Ed., 2019, 58, 3885–3889 CrossRef CAS PubMed.
- X. Guo, Y. Li, M. Zhang, K. Cao, Y. Tian, Y. Qi, S. Li, K. Li, X. Yu and L. Ma, Angew. Chem., Int. Ed., 2020, 59, 22697–22705 CrossRef CAS PubMed.
- P. Wang, Q. Xu, Z. Li, W. Jiang, Q. Jiang and D. Jiang, Adv. Mater., 2018, 30, 1801991 CrossRef PubMed.
- X. Zhang, I. d. Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitórica-Yrezábal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudić, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289–16296 CrossRef CAS PubMed.
- T. C. T. Pham, S. Docao, I. C. Hwang, M. K. Song, D. Y. Choi, D. Moon, P. Oleynikovc and K. B. Yoon, Energy Environ. Sci., 2016, 9, 1050–1062 RSC.
- Y.-D. Yang, X.-L. Chen, J. L. Sessler and H.-Y. Gong, J. Am. Chem. Soc., 2021, 143, 2315–2324 CrossRef CAS PubMed.
- Z. Yan, Y. Yuan, Y. Tian, D. Zhang and G. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12733–12737 CrossRef CAS PubMed.
- D. Dai, J. Yang, Y.-C. Zou, J.-R. Wu, L.-L. Tan, Y. Wang, B. Li, T. Lu, B. Wang and Y.-W. Yang, Angew. Chem., Int. Ed., 2021, 60, 8967–8975 CrossRef CAS PubMed.
- L. Xie, Z. Zheng, Q. Lin, H. Zhou, X. Ji, J. L. Sessler and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e202113724 CrossRef CAS PubMed.
- D. Gambhir, M. Venkateswarulu, T. Verma and R. R. Koner, ACS Appl. Polym. Mater., 2020, 2, 152–158 CrossRef CAS.
- J. Zhu, L. Wu, Z. Bu, S. Jie and B.-G. Li, Ind. Eng. Chem. Res., 2017, 56, 10155–10163 CrossRef CAS.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed.
- S. Karak, K. Dey, A. Torris, A. Halder, S. Bera, F. Kanheerampockil and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 7572–7581 CrossRef CAS PubMed.
- H. Ma, J.-J. Chen, L. Tan, J.-H. Bu, Y. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther III, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598–11620 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05361b |
| This journal is © The Royal Society of Chemistry 2022 |