
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcentration of stimulant regulates initial exocytotic molecular plasticity at single cells†
Xiulan
He
and
Andrew G.
Ewing
 *
*
Department of Chemistry and Molecular Biology, University of Gothenburg, 412 96 Gothenburg, Sweden. E-mail: andrewe@chem.gu.se
First published on 20th January 2022
Abstract
Activity-induced synaptic plasticity has been intensively studied, but is not yet well understood. We examined the temporal and concentration effects of exocytotic molecular plasticity during and immediately after chemical stimulation (30 s K+ stimulation) via single cell amperometry. Here the first and the second 15 s event periods from individual event traces were compared. Remarkably, we found that the amount of catecholamine release and release dynamics depend on the stimulant concentration. No changes were observed at 10 mM K+ stimulation, but changes observed at 30 and 50 mM (i.e., potentiation, increased number of molecules) were opposite to those at 100 mM (i.e., depression, decreased number of events), revealing changes in exocytotic plasticity based on the concentration of the stimulant solution. These results show that molecular changes initiating exocytotic plasticity can be regulated by the concentration strength of the stimulant solution. These different effects on early plasticity offer a possible link between stimulation intensity and synaptic (or adrenal) plasticity.
Introduction
Synaptic plasticity provides the basis for most modes of learning, memory and development in neural circuits.1–4 The variation of weight of individual synaptic connections is an indicator of synaptic plasticity, which also can be regulated both postsynaptically and presynaptically in each case giving plasticity.5,6 Postsynaptic plasticity can be achieved by adjusting the number or properties of neurotransmitter receptors on the post synaptic cell, whereas presynaptic plasticity can be achieved by regulating the efficacy of transmitter release at the presynapse or the molecular structure (e.g., Munc13) which is related to this efficacy.7–9 This efficacy is determined by two quantal parameters: the number of neurotransmitter release sites and the probability of neurotransmitter release, which can be scaled by the number of exocytotic events or amount (or fraction) of release during exocytosis.8 Therefore, exocytosis from single cells (e.g., pheochromocytoma (PC12) cells and chromaffin cells) evoked by repetitive chemical stimuli or electrical pulse stimuli has been used to study factors that affect synaptic plasticity.10–12Chromaffin cells have been studied for about a century and are widely used as a model cell to study neural differentiation and neurosecretion. They synthesize and store catecholamines in vesicles that can be triggered by stimulations (e.g., ions, molecules, electrical pulses).13,14 Moreover, their exocytotic fusion mode can be regulated from “kiss-and-run” to “open and closed” (or even full release) by adjusting the intracellular concentration of calcium, which is related to the stimulus conditions and extracellular concentration of calcium.15 As the main producers of stress hormones, adrenal chromaffin cells play a major role in the response to physiological stress and adaptation to the stress.16 Stress can induce various human diseases including depression, obesity, and Parkinson's disease without adrenal adaptation (i.e., adrenal plasticity).17,18 Thus, in addition to modeling synaptic plasticity, it is fundamentally important to understand exocytotic dynamics and changes of chromaffin cells to further our understanding of stress. Plasticity during a single continuous chemical stimulation (e.g., 30 s K+ stimulation) is critical to the initiation of cellular and molecular changes. This has not been investigated to date, primarily owing to a lack of mature measurement techniques and protocols, although several papers have reported that exocytosis at chromaffin cells is affected by prolonged K+-treatment (e.g., 40 min)19 and K+ concentration in stimulation solution.15 In addition, the effects of stimulant solution K+ concentration on plasticity, revealed by exocytosis during a short continuous K+ stimulation without treatment, have not been reported.
In this paper, we used single cell amperometry (SCA) to monitor chemical secretion by exocytosis from single chromaffin cells which were triggered by stimulation with a continuous 30 s (e.g., 10, 30, 50, 100 mM) K+ solution. This 30 s evoked SCA trace was then divided into two equal parts: 15 s K+ triggered time periods, and the amount of release and dynamics of exocytosis obtained from these two phases of release were compared to revealing and astoundingly rapid and concentration-dependent plasticity during the early time period of chemical stimulation. We found that both the amount of release and dynamics of exocytosis from early and late parts of the stimulus remain consistent at a stimulation of 10 mM K+, whereas, they were significantly different upon stimulation with a higher concentration of K+ (30, 50, 100 mM), additionally revealing contrasting plasticity at 30 (or 50) mM versus 100 mM K+. A larger amount of catecholamine release was observed in the second 15 s time period following a 30 or 50 mM K+ stimulus, which was attributed to the duration of the open fusion pore being longer. However, a lower amount was obtained during the second 15 s time period following a 100 mM stimulation, owing to a lower number of exocytosis events and a smaller release flux, although the exocytosis events are themselves also slower. We used different concentration of K+ stimulation solution to mimic different strength of stimuli or stress, to study the effect of different concentrations of K+ on exocytotic plasticity, which providing an understanding of exocytotic plasticity during cellular communication and stress.
Results and discussion
Number of exocytotic events induced by 100 mM K+ causes depression plasticity
Single cell amperometry (SCA) was used to measure exocytosis. Detailed information about exocytosis was gained by analyzing the transient current spikes, such as the amount of catecholamine release and the dynamics of exocytosis.20 A carbon-fiber disk microelectrode was placed on top of a chromaffin cell to record exocytosis, which was triggered by a 30 s delivery of 10, 30, 50, or 100 mM K+. These exocytotic release events were observed as current transients for the oxidation of catecholamine released from the vesicle via the fusion pore formed. Typical SCA amperometric traces obtained are shown in Fig. 1. Each trace represents a train of current transients following each stimulation, in which each current transient corresponds to a single vesicle release event. We then divided these 30 s evoked SCA traces into two equal parts: the initial 15 s and the second 15 s of the K+-triggered event periods.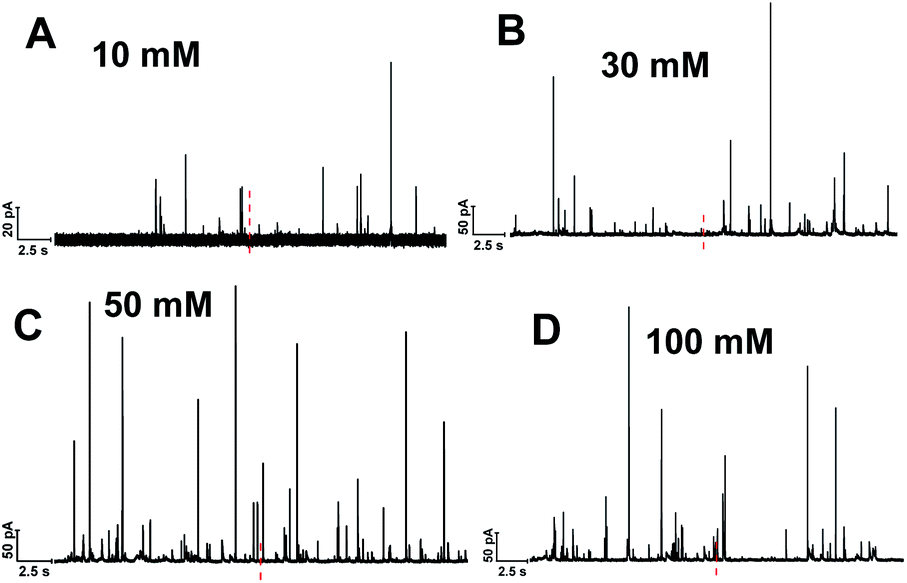 | ||
| Fig. 1 Representative SCA amperometric traces of exocytosis from single chromaffin cell stimulated with different concentration of K+: (A) 10, (B) 30, (C) 50, (D) 100 mM. The black line underneath each trace indicates stimulus period (30 s). The red dashed line highlighted the central position to divide the trace to two 15 s time periods. | ||
This allowed us to study the effects of stimulation time and K+ concentration on neurotransmitter release from single chromaffin cells, and to reveal plasticity during the 30 s continuous chemical stimulation (Fig. 2). Upon stimulation with 100 mM K+, the number of exocytotic events (Nevents) significantly decreased from the first to second 15 s interval. However, there was no significant difference in the number of exocytotic release events per cell between uninterrupted consecutive 15 s time periods during K+ stimulation with lower concentrations (e.g., 10, 30, 50 mM) (p values are listed in ESI Appendix, Table S1†). At 100 mM K+, the number of spikes detected gradually decreased in the second 15 s time period, indicating that the number of exocytotic release events per cell significantly decreased over the stimulation time, suggesting a form of depression plasticity.
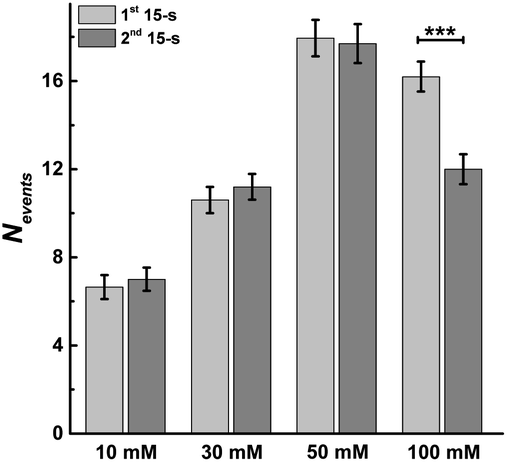 | ||
| Fig. 2 Average number of exocytotic events obtained from 1st and 2nd 15 s time period of stimulation for single chromaffin cells which were stimulated by different concentrations of K+. The number of cells was 20. Data represent means ± SEM. Datasets were compared with a 2-tailed Mann–Whitney rank-sum test, p < 0.05, *; p < 0.01, **; p < 0.001, ***. All of the p values were calculated and are included in ESI Appendix, Table S1.† | ||
Amount of catecholamine release per event causes potentiation plasticity
As longer stimulus time doesn't change the number of vesicular release events under most stimulation conditions (10, 30 and 50 mM K+), we investigated the effect on the number of molecules released from individual vesicles. Dynamic information about exocytosis also can be obtained by analyzing SCA traces, including the opening, duration and closure of the fusion pore, revealed by the time from 25% to 75% of the maximum amplitude at the rising part of the SCA amperometric peak, trise, the width of the peak, t1/2, and the time from 75% to 25% of maximum amplitude at the falling part of the peak, tfall, as shown in Fig. S1.† The peak amplitude (Imax) indicates the flux of catecholamine through the open fusion pore and the number of catecholamine molecules (Nmolecules) released from single vesicles can be quantified from the integrated current under individual spikes by use of the Faraday equation. The mean values for amount of release for chromaffin cells over the first and second 15 s time periods during K+ stimulation and at different K+ concentration are shown in Fig. 3. The number of molecules released per individual exocytotic event increased significantly from the first and second 15 s time period following stimulation with either 30 or 50 mM K+ solution, but not at 10 or 100 mM (p values are listed in ESI Appendix, Table S2†). This suggests plasticity where the response was potentiated for the middle stimulation concentrations, whereas, this was not observed at the low and high concentrations (10 and 100 mM K+).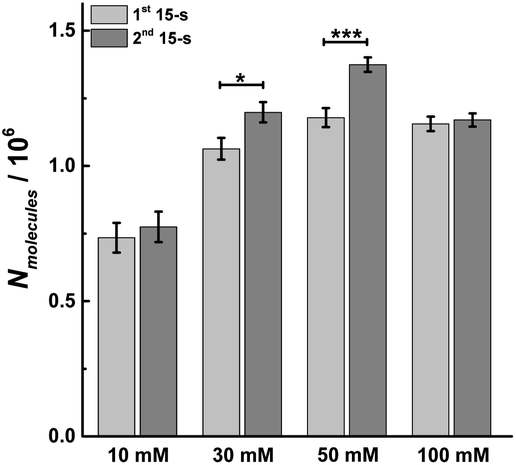 | ||
| Fig. 3 Average amount of exocytotic release obtained from 1st and 2nd 15 s time period following stimulation of single chromaffin cells which were stimulated by different concentrations of K+. The number of cells was 20. Data represent means ± SEM. Datasets were compared with a 2-tailed Mann–Whitney rank-sum test, p < 0.05, *; p < 0.01, **; p < 0.001, ***. All of the p values were calculated and are included in ESI Appendix, Table S2.† | ||
Slower exocytotic events cause plasticity in the second 15 s after stimulation
We evaluated total release as the amount per release event, Nmolecules, times the Nevents. When these were compared for 30 and 50 mM stimulation, the lack of a significant change in the Nevents indicated that total exocytotic catecholamine release (Nmolecules × Nevents) was increased in the second 15 s time period suggesting an overall positive plasticity. In contrast, owing to smaller Nevents, for 100 mM stimulation the total catecholamine release was reduced in the second 15 s time period, showing negative plasticity. There is no plasticity observed during the stimulation of 30 s 10 mM K+, the lowest stimulant concentration.Considering single cell heterogeneity, we calculated the values of the increasing ratio of Nmolecules and Nevents across 20 cells to compare the parameters obtained from the paired 1st and 2nd 15 s of each event period. As shown in Fig. S2,† similar results were obtained which suggests the number of molecules released per individual exocytotic event increased from the first and second 15 s time period following stimulation with either 30 or 50 mM K+ solution, while the number of events decreased in the second 15 s time period of 100 mM K+ stimulation. In addition, as shown in Fig. S3,† we analyzed the relationship between the value of Nevents and the concentration of stimulant K+, and found the turning-point concentration of stimulant K+ for a single cell from potentiation to depression plasticity is around 80 mM.
To better understand the mechanism behind these different effects, we performed peak analysis for the current spikes from SCA to obtain the dynamic parameters of exocytosis, i.e., Imax, t1/2, trise, and tfall. As shown in Fig. 4, these parameters, which are regulated most by the protocol used, are the times defining the release event (t1/2, trise, and tfall) rather than the flux of catecholamines through the fusion pore (Imax). In Fig. 4A, the peak current, Imax, did not significantly change between the first and second 15 s time periods following 10, 30 or 50 mM K+ stimulation, which might indicate that the size of the fusion pore and the vesicle content remained almost the same (p values are listed in ESI Appendix, Table S3†). However, Imax significantly decreased in the second 15 s time period during 100 mM K+, suggesting a smaller fusion pore or a smaller vesicle content. The dynamics are interesting as the duration of the open fusion pore (Fig. 4B), t1/2, was not significantly different between the two 15 s time periods during a stimulation of 10 mM K+. However, an obvious increase in t1/2 was observed during the second 15 s period for stimulation with 30, 50, and 100 mM K+ (p values are listed in ESI Appendix, Table S4†). This suggests that a longer lasting and possibly more stable fusion pore was formed during this stimulation time of 30 s. The values of trise and tfall correspond to the time of opening and closing of the fusion pore, respectively. The data in Fig. 4C and D show that the opening time became longer for the second 15 s time period following stimulation with 50 and 100 mM K+ solution, and the pore closing time increased at higher K+ concentration for the second time period for 30, 50, and 100 mM K+ stimulations (p values are listed in ESI Appendix, Tables S5 and S6†). We interpret this to mean that a higher K+ concentration and a longer stimulus time lead to a slowing down of the exocytotic release event by increasing the pore opening and stabilizing the closing time, which results in an increase of vesicular transmitter load being released during the second 15 s time period following 30 or 50 mM K+. For 100 mM K+ stimulation, similar catecholamine release was observed owing to a smaller release flux that is matched by the longer pore opening time.
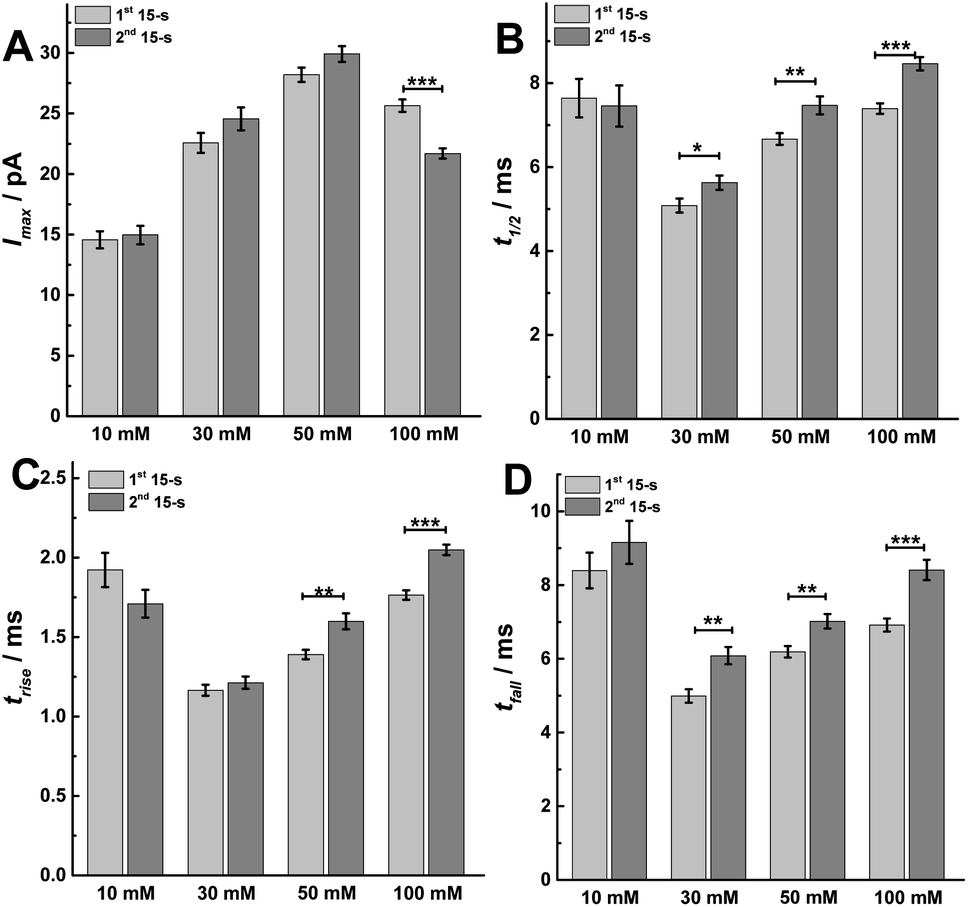 | ||
| Fig. 4 Parameters including (A) peak current: Imax, (B) half peak width: t1/2, (C) rise time: trise, and (D) fall time: tfall from SCA were calculated to compare. The number of cells used was 20. Data are mean of medians ± SEM (ESI Appendix, Methods†). Datasets were compared with a 2-tailed Mann–Whitney rank-sum test, p < 0.05, *; p < 0.01, **; p < 0.001, ***. All of the p values were calculated and are included in ESI Appendix, Tables S3–S6.† | ||
Intracellular calcium changes are consistent with observed rapid plasticity
Vesicular release can be regulated by intracellular Ca2+ concentration, which is evoked by different concentration K+ stimulation solution.15 To better understand the changes in the exocytosis process during single continuous 30 s K+ stimulation, we carried out calcium imaging experiments with fura-2 to monitor the real-time change of intracellular calcium level after stimulation with 10, 30, 50, or 100 mM K+ solution.21 A higher intracellular calcium concentration was observed as the concentration of K+ was increased, as depicted in ESI, Fig. S4A.† As shown in Fig. S4B,† the ratio of fluorescence decreased linearly after a rapid rise during the stimulation for a continuous 30 s K+ solution, which is similar with previous observation.22 The slope of the best-fit line suggests the rate of decline, which negatively increased following the concentration of K+ in stimulation solution (i.e., −7.87, −25.96, −33.04, and −43.06 at 10, 30, 50, and 100 mM, respectively). To further understand the difference of intracellular Ca2+ concentration between the first and second 15 s periods during the 30 s stimulation, the relative intensity of fluorescence at 25 and 40 s was compared, which was presented as the ratio of fluorescence at the end of each 15 s stimulation period, respectively. As shown in Fig. 5, upon stimulation with 30, 50 and 100 mM K+, the ratio of fluorescence decreased significantly during the second 15 s event period (Fig. 5A, p values are listed in ESI Appendix, Table S7†). Meanwhile, there was a linear relationship between the ratio of fluorescence and log10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CK+ (Fig. 5B), which is consistent with a previous report.23 This monotonically increased intracellular Ca2+ concentration induces the increased Nmolecules and Nevents from 10 mM to 50 mM stimulant concentration, but decreased at 100 mM, which is consistent with previous observations24,25 and suggests that the release fraction (or fusion mode) and the endocytotic mode might be changed following increased K+ concentration in the stimulation solution. The duration of the fusion pore dynamics (trise, t1/2, and tfall) decreased when the concentration of K+ was changed from 10 mM to 30 mM, then increased when the concentration of K+ increases from 30 mM to 100 mM. This might be explained by intracellular Ca2+-dominated actin changes (i.e., actin polymerization and F-actin disruption).26
CK+ (Fig. 5B), which is consistent with a previous report.23 This monotonically increased intracellular Ca2+ concentration induces the increased Nmolecules and Nevents from 10 mM to 50 mM stimulant concentration, but decreased at 100 mM, which is consistent with previous observations24,25 and suggests that the release fraction (or fusion mode) and the endocytotic mode might be changed following increased K+ concentration in the stimulation solution. The duration of the fusion pore dynamics (trise, t1/2, and tfall) decreased when the concentration of K+ was changed from 10 mM to 30 mM, then increased when the concentration of K+ increases from 30 mM to 100 mM. This might be explained by intracellular Ca2+-dominated actin changes (i.e., actin polymerization and F-actin disruption).26
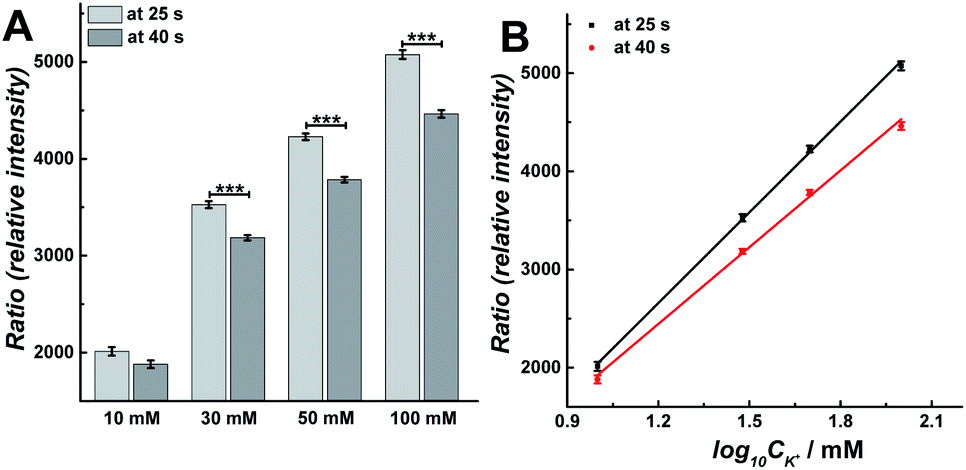 | ||
| Fig. 5 (A) Compared calcium imaging results at 25 s and 40 s to compare the difference between 10, 30, 50, and 100 mM K+ stimuli. The number of cells analyzed for all stimuli was 170 and the error bars represent SEM. Datasets were compared with a 2-tailed Mann–Whitney rank-sum test, p < 0.05, *; p < 0.01, **; p < 0.001, ***. The p values were calculated and are included in ESI Appendix, Table S7.† (B) Plot of calcium imaging results versus log10CK+: black curve (at 25 s): y = 3077x − 1040.6, R2 = 0.999; red curve (at 40 s): y = 2602.8x − 691.06, R2 = 0.998. The data was obtained from Fig. S4.† | ||
Fraction of catecholamine released during individual exocytosis events drives plasticity
In order to confirm the fusion mode and the fraction of release, we performed intracellular vesicle impact electrochemical cytometry (IVIEC).27 By piercing a nano-tip conical carbon-fiber microelectrode through the cell membrane, we were able to monitor and quantify the electroactive content of vesicles inside the cytoplasm when they adsorbed onto the electrode surface and subsequently ruptured. An IVIEC amperometric trace is shown in Fig. S5A,† and the average vesicular content of the vesicles in these cells was (26.02 ± 1.26) × 105 molecules (mean of the median ± SEM, n = 20). Knowing the amount of release and the total vesicular content, the fraction of release was calculated by comparison of release to vesicle content as shown in Fig. S5B.† The fraction of release was similar in both 15 s time periods for a 30 s stimulus with 10 or 100 mM K+. When compared to the first 15 s time period, a slight increase of 2.4% and 3.5% is observed during the second 15 s time period for 10 or 100 mM K+, respectively. However, when we compare the second to the first 15 s time period for 30 or 50 mM K+ stimulation, an increase in release fraction of 14.8% and 16.7% was observed, respectively. This suggests a potentiation effect at 30 and 50 mM stimulation concentration, and where the main fusion mode is partial release (i.e., “open and closed exocytosis”).28We also investigated the parameters of prespike feet obtained from SCA, as shown in Fig. S1.† The prespike foot is recorded as a small current transient immediately before the main exocytotic peak, and it represents the initial fusion pore opening with a small amount of neurotransmitter release before the pore continues to expand.20 The parameters for analysis include Ifoot, the peak current of the foot, tfoot, the duration of the foot, and Qfoot, the charge of the prespike foot which is correlated with the number of catecholamines being released through the foot, and the results obtained are shown in Fig. 6. Examining data for two 15 s time periods during a 30 s K+ stimulation, Ifoot remains same in these two 15 s periods for 10, 50, and 100 mM K+, but increases during the second 15 s time period for 30 mM K+ (Fig. 6A, p values are listed in ESI Appendix, Table S8†). Moreover, tfoot increases at 30 mM, but decreases for 50 and 100 mM K+ (Fig. 6B, p values are listed in ESI Appendix, Table S9†). This consequently resulted in a larger amount of neurotransmitter released (Nmolecules in foot = Ifoot × tfoot) via the foot during the second 15 s time period with a 30 mM K+ stimulation, and less neurotransmitter released via the foot during the second 15 s time period for 100 mM K+ stimulation (Fig. 6C, p values are listed in ESI Appendix, Table S10†). Both values are consistent with the results obtained for the main exocytosis peaks. The proportion of feet (Nfoot/Nevents) also shows a similar trend as the Nmolecules and increases in the second 15 s time period after 30 mM K+ stimulation, but decreases during the second 15 s time period with 100 mM K+ (Fig. 6D, p values are listed in ESI Appendix, Table S11†). The variations of Nfoot/Nevents and Nmolecules in foot also indicate a potentiation effect following 30 mM K+ and a depression effect at 100 mM K+, which is consistent with the conclusions for Nevents and Nmolecules for the main peaks.
 | ||
| Fig. 6 Different parameters for prespike foot analysis, including (A) Ifoot, (B) tfoot, (C) Nmolecules in foot, and (D) the proportion of foot in total events (Nfoot/Nevents) obtained from 1st and 2nd 15 s time period of SCA to compare. Data represent means ± SEM (ESI Appendix, Methods†). The number of cells used was 20. Datasets were compared with a 2-tailed Mann–Whitney rank-sum test, p < 0.05, *; p < 0.01, **; p < 0.001, ***. All of the p values were calculated and are included in ESI Appendix, Tables S8–S11.† | ||
Discussion
The time of 15 s is long enough to distinguish the rapid (from millisecond to second) and slow endocytosis (tens of second)29–31 and to arouse the exocytosis from the chromaffin cells at low concentration of K+ stimulation solution, and matched with the time scale of the short-term plasticity.32–34 The data presented show that different K+ concentrations (e.g., 10, 30, 50, and 100 mM) in the stimulation solution induce three variations of plasticity. These differences are revealed by use of a slightly prolonged 30 s K+ stimulated exocytosis from chromaffin cells and examining exocytosis during the first and second 15 s of the stimulation.At low stimulation concentration (10 mM K+), both the amount of release and the dynamics are similar in the first and second 15 s exocytosis periods, indicating no plasticity. At high stimulation concentration (100 mM K+), the number of exocytotic events significantly decreases in the second 15 s time period compared to the first, implying depression plasticity. Third, intermediate stimulation concentrations (30 and 50 mM K+) result in a larger number of catecholamine molecules released during the second 15 s time period versus the first 15 s, owing to a slower exocytotic process, with trise, t1/2 and tfall all increased, suggesting potentiation plasticity. To our knowledge, exocytotic plasticity changes evoked by a short continuous chemical stimulation with different concentration K+ have not, until now, been demonstrated at the cellular level. This short time period is interesting as it is the molecular initiation of plasticity. This reveals a fundamental molecular process to understand spiking-rate-dependent plasticity in which the plasticity depends on the frequency and intensity of stimulation.
Exocytosis from chromaffin cells has been shown to be regulated by the intracellular Ca2+ concentration.15 In our work, imaging of Ca2+ with fura-2 shows a low intracellular change in concentration of Ca2+ following 10 mM K+ stimulation. In this case, vesicles fuse with the plasma membrane for a longer duration through a narrow fusion pore, resulting in amperometric spikes with small amplitude and wide duration. Additionally, low Ca2+ concentration is observed to induce fewer exocytotic events and a lower fraction released via partial release. Due to the rapid endocytosis,29–31 the size of the readily releasable pool doesn't change during 30 s stimulation, showing no plasticity at 10 mM.
In contrast to the observations with 10 mM K+ stimulation, a higher intracellular Ca2+ concentration is observed when chromaffin cells are triggered by 100 mM K+ stimulation, and this induces a higher fraction of release by partial release. Rapid endocytosis dependent on dynamin-1 is blocked when intracellular Ca2+ is high, whereas slow endocytosis depended on dynamin-2 is activated.29–31 In those instances where full expansion of the vesicular pore occurs with collapse of the vesicle into the plasma membrane, the vesicles are retrieved by slow endocytosis which depends on clathrin and dynamin-2, and are then recycled. Rapid endocytosis or partial release during exocytosis allows vesicles to be retrieved in the millisecond to second time frame. Hence, depletion of the content of readily releasable pool vesicles occurs during partial release leading to fewer exocytotic events in the second 15 s for 100 mM K+-triggered events, indicating exocytotic depression. Furthermore, as many proteins have been implicated in the calcium-dependent exocytotic process,35–37 such as protein kinase C (PKC), and their activities can be regulated by intracellular Ca2+ concentration to induce plasticity.38 We speculate that this activity-dependent depression arises from a deactivation of PKC, a decreased size of the readily releasable pool, and the decreased number of fused vesicles, by a lowered cytosolic Ca2+ level during the second 15 s time period for 100 mM K+ stimulation.
The fusion process during exocytosis is mediated by the SNARE complex which includes synaptobrevin (syb), vesicle associated membrane protein (VAMP), syntaxin and synaptosomal-associated protein (25 kDa, SNAP-25).39–41 PKC can phosphorylate proteins in the vesicle fusion machinery, such as munc-18 which is a syntaxin-interacting protein.42,43 The core complex of SNARE is the minimum release capable structure that organizes into a rod-like coiled-coil structure of four α-helices.41 SNARE complex-enhanced motion is reversed by elevation of the intracellular calcium level.44 Fewer and slower vesicle fusion occurs in the highly mobile vesicles present in SNARE complex-enhanced motion. Thus, the SNARE complex also appears to be negatively affected by a lower intracellular Ca2+ concentration in the second 15 s time period to reduce the frequency of exocytotic release as observed in our data.
A moderate concentration of intracellular Ca2+ is obtained upon 30 s stimulation with 30 or 50 mM K+, which can disrupt the actin network and induce the formation of new actin filaments facilitating pore closure.26 This appears to cause transiently fusing vesicles to fuse for a shorter duration but with larger release than that observed at 10 mM. Rapid endocytosis31 ensures the content of vesicles in the readily releasable pool remain unchanged, resulting in almost the same number of exocytotic events during both 15 s intervals during stimulation for 30 s. Decreased intracellular Ca2+ in the second 15 s time period might work against the reorganization of the F-actin network to slow down the rates of both dilation and constriction of the fusion pore, resulting in an increased amount of release (Nmolecules) and longer duration time for the pore opening (trise, t1/2, and tfall). In addition to actin,45–47 dynamin also has been indicated previously to regulate exocytotic dynamics and plasticity, and it is involved in the duration and kinetics of exocytotic release.48–52 This suggests that the interplay between dynamin and actin, as regulators, could offer another explanation for the enhanced fusion pore stability during 30 s (30, 50 or 100 mM K+) stimulation.
In addition to proteins, lipids are major components of cellular and cystic membranes. Lipids also play an important role in exocytosis, plasticity, and memory.53–56 Both exocytotic amount and dynamics can be regulated by regulating the species (e.g., PC (phosphatidylcholine), PE (phosphatidylethanolamine), PI (phosphatidylinositol), and cholesterol) or amount of lipids.53–55 Different lipids have different intrinsic geometries such as cylindrical (e.g., PC), conical (e.g., PE), inverse-conical (e.g., PI), leading to favoring structures with low curvature, negative curvature, or positive curvature, respectively. Cholesterol also has an intrinsic negative curvature and thus promotes high-curvature pore formation during membrane fusion.57 In contrast, PC prefers to distribute in the low curvature membrane areas, such as the outer leaflet of the cell membrane.58–61 The conical PE preferentially localizes to the inner leaflet of the membrane, especially the inner leaflet of the cystic membrane because of higher curvature of vesicles with smaller diameter than cells.58–61 PI prefers to locate in the outer leaflet of the cystic membrane.58–61 This leads us to speculate that another possible explanation during stimulation with intermediate to high concentration of (30, 50 and 100 mM) K+, is that in the exocytotic active zones the low curvature lipids (PC) on the cell membrane are exchanged to provide a higher ratio of high curvature lipid species (PE, PI and cholesterol) owing to exchange with vesicular lipids during pore opening.54,55 This altered cell membrane with higher curvature would then stabilize the high curvature fusion pore formed during exocytosis, leading to the increased duration of the open pore and a larger amount of neurotransmitter release during the second 15 s time period after 30 or 50 mM K+ stimulation.
Conclusions
Short-term activity-dependent plasticity has been studied in chromaffin cells through exocytosis of LDCVs following repetitive stimuli, which was shown to be independent of Ca2+ accumulation, but instead attributed to other biochemical modifications including protein kinase-mediated phosphorylation.11 In our study, during 30 s stimulation with 10, 30, 50, and 100 mM K+, the intracellular Ca2+ concentration in chromaffin cells is shown to increase with larger K+ concentrations in the stimulation solution. However, the Nmolecules for release is biphasic increasing first (10, 30, 50 mM), and then decreasing (100 mM), which is consistent with previous observations.24,25 Interestingly, during a 30 s K+ stimulation the intracellular Ca2+ concentration increases rapidly to reach a maximum amplitude in few seconds, then decreases linearly vs. time. However, compared to Ca2+, the different trends are observed for exocytosis during consecutive 15 s K+ time periods by regulating K+ concentration. This plasticity in exocytosis is closely related to the fraction of chemical release during exocytosis events, which appears to be dominated by the concentration of intracellular Ca2+. A large decrease of intracellular Ca2+ might be attributed to the depression plasticity revealed by a decrease in Nevents at 100 mM stimulation by a depletion of the content of the readily releasable pool of vesicles, a deactivation of PKC, and restraint of the SNARE complex. While the potentiation plasticity revealed by a larger release, which can be attributed to a sufficient content of vesicles located in the readily releasable pool, an inhibition of actin cytoskeleton, and a high proportion of high-curvature lipids might be involved in this response during the second time period in the 30 s stimulation triggered by 30 or 50 mM K+ solution. Our work reveals a link between K+ concentration in the stimulation solution and activity-dependent plasticity during a single continuous chemical stimulation, and helps to better understand the adrenal plasticity or different molecular and cellular plasticity in response to stress.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
All authors have approved the final version of the manuscript. X. H. and A. E. conceived the idea. X. H. performed the experiments, analysed and interpreted the data, and wrote and edited the manuscript. A. E. was involved in funding acquisition, supervising, discussions of data interpretation, and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The European Research Council (ERC Advanced Grant Project No. 787534 NanoBioNext), Knut and Alice Wallenberg Foundation, and the Swedish Research Council (VR Grant No. 2017-04366) are acknowledged for financial support. We would like to thank Dalsjöfors Kött AB, Sweden, and their employees for their kind help with providing the adrenal glands.Notes and references
- L. F. Abbott and S. B. Nelson, Nat. Neurosci., 2000, 3, 1178–1183 CrossRef CAS PubMed.
- D. Fioravante and W. G. Regehr, Curr. Opin. Neurobiol., 2011, 21, 269–274 CrossRef CAS PubMed.
- A. Critri and R. C. Malenka, Neuropsychopharmacology, 2008, 33, 18–41 CrossRef PubMed.
- S. J. Martin, P. D. Grimwood and R. G. M. Morris, Annu. Rev. Neurosci., 2000, 23, 649–711 CrossRef CAS PubMed.
- S. Royer and D. Paré, Nature, 2003, 422, 518–522 CrossRef CAS PubMed.
- B. Barbour, N. Brunel, V. Hakim and J.-P. Nadal, Trends Neurosci., 2007, 30, 622–629 CrossRef CAS PubMed.
- C. Lüscher, R. A. Nicoll, R. C. Malenka and D. Muller, Nat. Neurosci., 2000, 3, 545–550 CrossRef PubMed.
- S. S. Zakharenko, L. Zablow and S. A. Siegelbaum, Nat. Neurosci., 2001, 4, 711–717 CrossRef CAS PubMed.
- J. Xu, M. Camacho, Y. Xu, V. Esser, X. Liu, T. Trimbuch, Y.-Z. Pan, C. Ma and D. R. Tomchick, eLife, 2017, 6, e22567 CrossRef PubMed.
- C. Gu, A. Larsson and A. G. Ewing, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21409–21415 CrossRef CAS PubMed.
- Y. Park and K.-T. Kim, Cell. Signalling, 2009, 21, 1465–1470 CrossRef CAS PubMed.
- C. Smith, J. Neurosci., 1999, 19, 589–598 CrossRef CAS PubMed.
- T. Fulop, S. Radabaugh and C. Smith, J. Neurosci., 2005, 25, 7324–7332 CrossRef CAS PubMed.
- A. M. G. De Diego, L. Gandia and A. G. Garcia, Acta Physiol., 2008, 192, 287–301 CrossRef CAS PubMed.
- A. Elhamdani, F. Azizi and C. R. Artalejo, J. Neurosci., 2006, 26, 3030–3036 CrossRef CAS PubMed.
- D. S. Goldstein, Cell. Mol. Neurobiol., 2010, 30, 1433–1440 CrossRef CAS PubMed.
- G. P. Chrousos, Nat. Rev. Endocrinol., 2009, 5, 374–381 CrossRef CAS PubMed.
- I. Berger, M. Werdermann, S. R. Bornstein and C. Steenblock, J. Steroid Biochem., 2019, 190, 198–206 CrossRef CAS PubMed.
- E. N. Pothos, E. Mosharov, K.-P. Liu, W. Selik, M. Haburcak, G. Baldini, M. D. Gershon, H. Tamir and D. Sulzer, J. Physiol., 2002, 542, 453–476 CrossRef CAS PubMed.
- C. Amatore, S. Arbault, I. Bonifas, Y. Bouret, M. Erard, A. G. Ewing and L. A. Sombers, Biophys. J., 2005, 88, 4411–4420 CrossRef CAS PubMed.
- E. Neher, Neuropharmacology, 1995, 34, 1423–1442 CrossRef CAS PubMed.
- T. R. Bagalkot, N. Leblanc and G. L. Craviso, Sci. Rep., 2019, 9, 11545 CrossRef PubMed.
- J. M. Finnegan and R. M. Wightman, J. Biol. Chem., 1995, 270, 5353–5359 CrossRef CAS PubMed.
- S. M. Ali, M. J. Geisow and R. D. Burgoyne, Nature, 1989, 340, 313–315 CrossRef CAS PubMed.
- A. Morgan and R. D. Burgoyne, Nature, 1992, 355, 833–836 CrossRef CAS PubMed.
- A. M. Cárdenas and F. D. Marengo, J. Neurochem., 2016, 137, 867–879 CrossRef PubMed.
- X. Li, S. Majdi, J. Dunevall, H. Fathali and A. G. Ewing, Angew. Chem., Int. Ed., 2015, 54, 11978–11982 CrossRef CAS PubMed.
- L. Ren, L. J. Mellander, J. Keighron, A.-S. Cans, M. E. Kurczy, I. Svir, A. Oleinick, C. Amatore and A. G. Ewing, Q. Rev. Biophys., 2016, 49, e12 CrossRef PubMed.
- L.-G. Wu, E. Hamid, W. Shin and H.-C. Chiang, Annu. Rev. Physiol., 2014, 76, 301–331 CrossRef CAS PubMed.
- K. Liang, L. Wei and L. Chen, Front. Mol. Neurosci., 2017, 10, 109 Search PubMed.
- C. R. Artalejo, A. Elhamdani and H. C. Palfrey, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6358–6363 CrossRef CAS PubMed.
- E. Iezzi, A. Suppa, A. Conte, P. Li Voti, M. Bologna and A. Berardelli, Eur. J. Neurosci., 2011, 33, 1908–1915 CrossRef PubMed.
- C. E. Young and C. R. Yang, Eur. J. Neurosci., 2005, 21, 3310–3320 CrossRef PubMed.
- K. Gerrow and A. Triller, Curr. Opin. Neurobiol., 2010, 20, 631–639 CrossRef CAS PubMed.
- A. G. García, A. M. García-de-Diego, L. Gandía, R. Borges and J. García-Sancho, Physiol. Rev., 2006, 86, 1093–1131 CrossRef PubMed.
- G. Alvarez de Toledo, M. Angeles Montes, P. Montenegro and R. Borges, FEBS Lett., 2018, 592, 3532–3541 CrossRef CAS PubMed.
- T. Fulop and C. Smith, Biochem. J., 2006, 399, 111–119 CrossRef CAS PubMed.
- K. D. B. Wierda, R. F. G. Toonen, H. de Wit, A. B. Brussaard and M. Verhage, Neuron, 2007, 54, 275–290 CrossRef CAS PubMed.
- J. B. Sørensen, U. Matti, S.-H. Wei, R. B. Nehring, T. Voets, U. Ashery, T. Binz, E. Neher and J. Retting, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1627–1632 CrossRef PubMed.
- Y. Zhao, Q. Fang, A. D. Herbst, K. N. Berberian, W. Almers and M. Lindau, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14249–14254 CrossRef CAS PubMed.
- R. B. Sutton, D. Fasshauer, R. Jahn and A. T. Brunger, Nature, 1998, 395, 347–353 CrossRef CAS PubMed.
- N. Katayama, S. Yamamori, M. Fukaya, S. Kobayashi, M. Watanabe, M. Takahashi and T. Manabe, Sci. Rep., 2017, 7, 7996 CrossRef PubMed.
- U. Nili, H. De Wit, A. Gulyas-Kovacs, R. F. Toonen, J. B. Sørensen, M. Verhage and U. Ashery, Neuroscience, 2006, 143, 487–500 CrossRef CAS PubMed.
- I. López, J. A. Ortiz, J. Villanueva, V. Torres, C. J. Torregrosa-Herland, M. D. M. Francés, S. Viniegra and L. M. Gutiérrez, Traffic, 2009, 10, 172–185 CrossRef PubMed.
- K. W. Eyring and R. W. Tsien, Cell, 2018, 173, 819–821 CrossRef CAS PubMed.
- R. Trouillon and A. G. Ewing, ACS Chem. Biol., 2014, 9, 812–820 CrossRef CAS PubMed.
- J. Jędrzejewska-Szmek and K. T. Blackwell, Semin. Cell Dev. Biol., 2019, 95, 120–129 CrossRef PubMed.
- J. E. Hinshaw, Annu. Rev. Cell Dev. Biol., 2000, 16, 483–519 CrossRef CAS PubMed.
- B. Antonny, C. Burd, P. De Camilli, E. Chen, O. Daumke, K. Faelber, M. Ford, V. A. Frolov, A. Frost, J. E. Hinshaw, T. Kirchhausen, M. M. Kozlov, M. Lenz, H. H. Low, H. McMahon, C. Merrifield, T. D. Pollard, P. J. Robinson, A. Roux and S. Schmid, EMBO J., 2016, 35, 2270–2284 CrossRef CAS PubMed.
- A. M. González-Jamett, X. Báez-Matus, M. A. Hevia, M. J. Guerra, M. J. Olivares, A. D. Martínez, A. Neely and A. M. Cárdenas, J. Neurosci., 2010, 30, 10683–10691 CrossRef PubMed.
- Q. Wu, Q. Zhang, B. Liu, Y. Li, X. Wu, S. Kuo, L. Zheng, C. Wang, F. Zhu and Z. Zhou, J. Neurosci., 2019, 39, 199–211 CrossRef CAS PubMed.
- R. Trouillon and A. G. Ewing, ChemPhysChem, 2013, 14, 2295–2301 CrossRef CAS PubMed.
- M. R. Ammar, N. Kassas, S. Chasserot-Golaz, M.-F. Bader and N. Vitale, Front. Endocrinol., 2013, 14, 125 CrossRef PubMed.
- M. Aref, E. Ranjbari, A. Romiani and A. G. Ewing, Chem. Sci., 2020, 11, 11869 RSC.
- C. Gu, M. H. Philipsen and A. G. Ewing, Int. J. Mol. Sci., 2020, 21, 9519 CrossRef CAS PubMed.
- Y. Uchiyama, M. M. Maxson, T. Sawada, A. Nakano and A. G. Ewing, Brain Res., 2007, 1151, 46–54 CrossRef CAS PubMed.
- M. A. Churchward, T. Rogasevskaia, J. Höfgen, J. Bau and J. R. Coorssen, J. Cell Sci., 2005, 118, 4833–4848 CrossRef CAS PubMed.
- M. Da Prada, A. Pletscher and J. P. Tranzer, Biochem. J., 1972, 127, 681–683 CrossRef CAS PubMed.
- H. Blaschko, H. Firemark, A. D. Smith and H. Winkler, Biochem. J., 1967, 104, 545 CrossRef CAS PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- J. E. Vance, Traffic, 2015, 16, 1–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemicals and solutions, isolation of adrenal chromaffin cells, fabrication of disk carbon fiber microelectrodes, fabrication of nano-tip conical carbon fiber microelectrodes, electrochemical measurements, data acquisition and analysis, calcium imaging experiments, Fig. S1–S5 and Tables S1–S11. See DOI: 10.1039/d1sc05278k |
| This journal is © The Royal Society of Chemistry 2022 |