
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific DNA functionalization through the tetrazene-forming reaction in ionic liquids†
Seiya
Ishizawa
 ,
Munkhtuya
Tumurkhuu
,
Elizabeth J.
Gross
and
Jun
Ohata
*
,
Munkhtuya
Tumurkhuu
,
Elizabeth J.
Gross
and
Jun
Ohata
*
Department of Chemistry, North Carolina State University, Raleigh, NC 27695, USA. E-mail: johata@ncsu.edu
First published on 20th January 2022
Abstract
Development of multiple chemical tools for deoxyribonucleic acid (DNA) labeling has facilitated wide use of their functionalized conjugates, but significant practical and methodological challenges remain to achievement of site-specific chemical modification of the biomacromolecule. As covalent labeling processes are more challenging in aqueous solution, use of nonaqueous, biomolecule-compatible solvents such as an ionic liquid consisting of a salt with organic molecule architecture, could be remarkably helpful in this connection. Herein, we demonstrate site-specific chemical modification of unprotected DNAs through a tetrazene-forming amine–azide coupling reaction using an ionic liquid. This ionic liquid-enhanced reaction process has good functional group tolerance and precise chemoselectivity, and enables incorporation of various useful functionalities such as biotin, cholesterol, and fluorophores. A site-specifically labeled oligonucleotide, or aptamer interacting with a growth factor receptor (Her2) was successfully used in the fluorescence imaging of breast cancer cell lines. The non-traditional medium-promoted labeling strategy described here provides an alternative design paradigm for future development of chemical tools for applications involving DNA functionalization.
Introduction
Chemically functionalized deoxyribonucleic acids (DNA) have become invaluable in various applications such as molecular beacons,1,2 polymerase chain reaction (PCR) technologies,3 deoxyribozymes (DNAzymes),4,5 asymmetric catalysis,6 nanotechnology,7 and therapeutics8 such as antisense sequences,9,10 transcription-factor decoys,11,12 and CpG motifs.13,14 The methods of chemical synthesis of DNA have significantly facilitated the introduction of desired functionalities onto a specific position,15 and post-synthetic modification methods are also powerful chemical tools,16–19 especially for labeling of DNA targets with large labeling groups incompatible during the chemical synthesis process.20–22 Bioinspired enzymatic post-synthetic labeling technologies have emerged as useful approaches through sequence- or structure-dependent substrate recognition,23 and chemical modification is an alternative means for site-specific DNA labeling by incorporation of a chemical handle that is inert toward endogenous chemical functional groups but reactive toward a labeling reagent.24,25 However, despite the remarkable advance of the bioconjugation technologies during the past decades, site-specific chemical modification remains a serious challenge due to its demanding criteria which include adequate reactivity of the labeling reagents toward the target site and high selectivity in biomolecule-compatible solvents such as aqueous buffer. Finding the appropriate balance between the reactivity and selectivity of a labeling reagent requires careful, time-consuming screening, and the need for an aqueous solution renders the realization of selective DNA bioconjugation even more problematic, as development of organic reactions in aqueous media is difficult.26Bioconjugation processes based on ionic liquids represent an alternative strategy to address the compatibility issue between DNAs and organic chemical reactions. Owing to the ionic nature and resulting solubility of DNAs, water or aqueous buffer is often a common choice for bioconjugation processes, although aqueous reaction conditions are not preferable from the perspective of the reaction because of their negative effects, which include the solubility and stability of labeling reagents and sluggish kinetics. As an alternative, ionic liquids, compounds completely composed of ions have organic frameworks with a constant charge state and have been successfully employed for dissolution and stabilization of various DNA molecules.27,28 However, ionic liquids have not been used in DNA bioconjugation processes even though their modular properties suggest the medium may be preferable for both the DNA and labeling process.
In this article, we report that a tetrazene forming reaction based on an ionic liquid enables site-specific labeling of the alkylamine tag on a desired position of DNA (Fig. 1). Our research program (Bioconjugation In Nonaqueous-Driven Reaction Solvent or BINDRS) to develop novel bioconjugation using nontraditional media, recently discovered that triphenylphosphine promotes a coupling reaction between an azide and an alkylamine in peptides or proteins, forming a tetrazene group with four consecutive nitrogen atoms.29 With no noticeable effect on the target peptides or proteins, this reaction, which takes place in an ionic liquid, displays excellent chemoselectivity for alkylamines in the presence of a variety of other NH groups in for example, guanidine or imidazole. The tetrazene formed in this reaction serves as a linker which can survive pH changes and redox mediators. We have found that the amine–azide coupling has no observable effect on native DNA functional groups such as DNA bases, phosphate backbone and ribose groups. This capability presents the possibility of site-specific modification at a desired location in DNA through incorporation of an exogenous alkylamine group. Reagent screening revealed the wide tolerance of the amine–azide coupling of various chemical functionalities, enabling preparation of a variety of functionalized DNA aptamers such as fluorophore, cholesterol, and biotin conjugates. The aptamer conjugates were successfully used in staining experiments of cancer cell lines, validating the compatibility of the ionic liquid-based modification processes with the biomolecule.
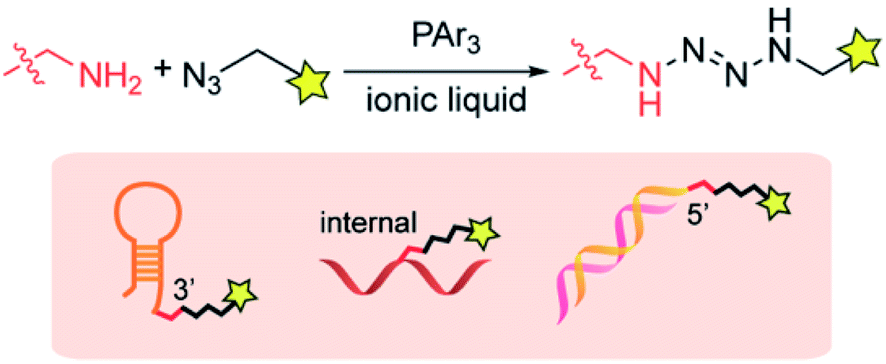 | ||
| Fig. 1 Site-specific tetrazene-forming reaction on DNA substrates. | ||
Results and discussion
Reactivity survey of the phosphine-mediated chemistry
Our initial survey of the reactivities of the adenine (A), thymine (T), cytosine (C), guanine (G), and uracil (U) nucleotides in a simple DNA substrate revealed their inertness toward the phosphine–azide coupling reaction (Fig. 2). In order to understand the applicability of the phosphine–azide coupling reaction, we first applied nonfunctional azide reagent 1a to a tetrathymidine containing an additional nucleotide at the 5′ terminus (Fig. 2A–C). Notwithstanding the presence of different types of NH2 groups in native DNAs, virtually no tetrazene-modified DNAs were observed in mass spectrometric analysis after the reaction and subsequent buffer exchange processes (Fig. 2D–H). On the other hand, introduction of alkylamine at the 5′-terminus of the backbone led to a significant increase in the signal from the peak corresponding to the formation of the tetrazene group (Fig. 2I). Those results suggest that the ionic liquid-based amine–azide coupling has a high specificity for alkylamines over other endogenous arylamine groups.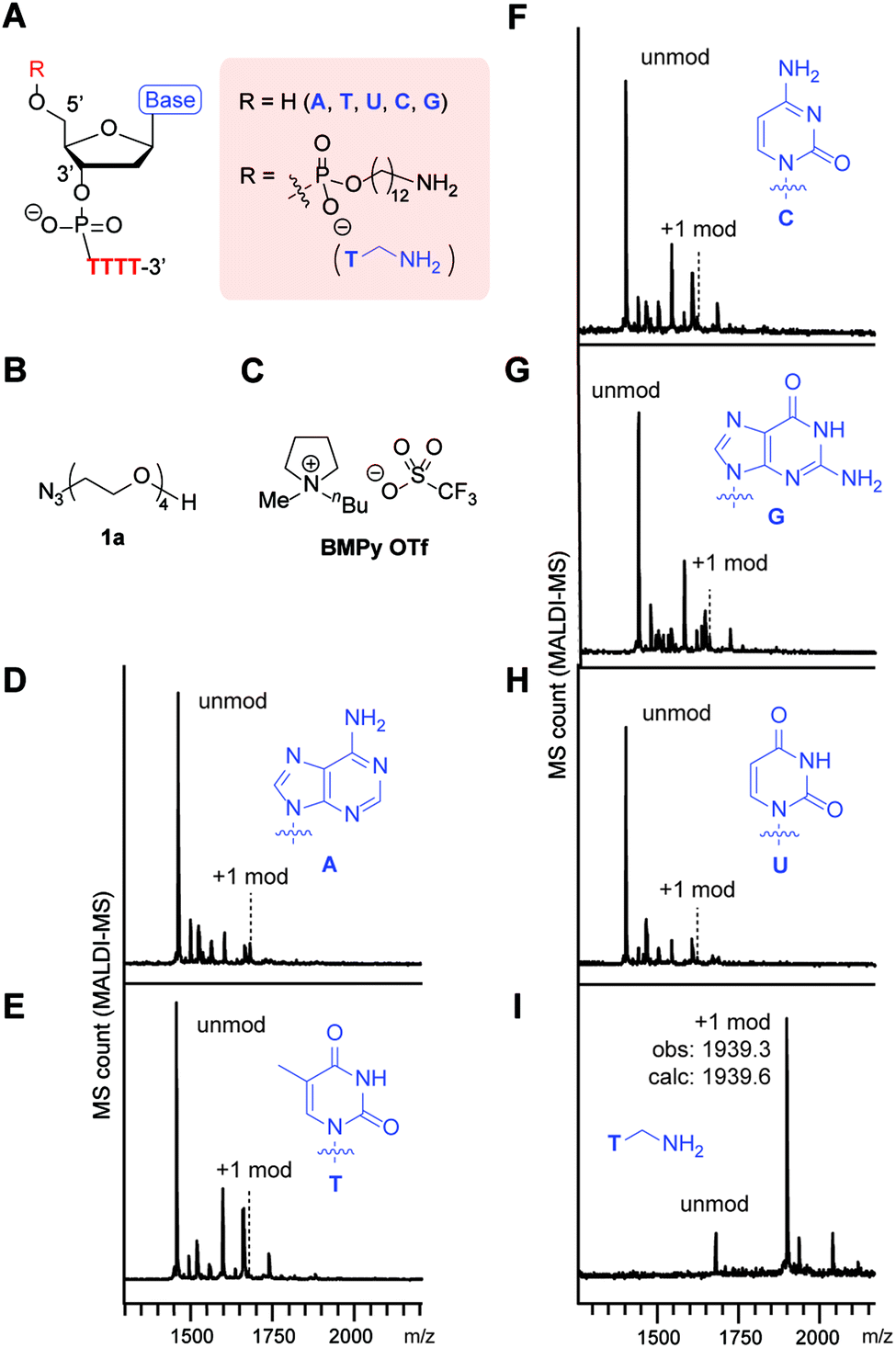 | ||
| Fig. 2 Alkylamine-selective tetrazene-forming reaction with pentanucleotides in an ionic liquid. Reaction conditions: KHCO3 (20 mM), XTTTT (0.2 mM), azide 1a (7.5 mM), and PPh3 (20 mM) in 1-butyl-1-methylpyrrolidinium trifluoromethanesulfonate (BMPy OTf) at 50 °C for 2 h. (A) Chemical structure of nucleotide backbone in an XTTTT sequence with or without an alkylamine. (B) Chemical structure of azide 1a. (C) Structure of the ionic liquid, BMPy OTf. (D–I) Matrix-assisted laser desorption/ionization (MALDI-MS) analysis of the reaction of XTTTT where X = adenosine (D), thymidine (E), cytidine (F), guanosine (G), deoxyuridine (H), and thymidine with alkylamine (I) containing a 12 carbon linker (5AmMC12). | ||
The high functional group tolerance of the modification process allowed incorporation of a variety of alkylazide reagents to the alkylamine-tagged DNA (Fig. 3, S1 and S2†). The trimethylsilyl (TMS) group has been used increasingly as a chemical reporter in structural biology research because of its characteristic chemical shift in nuclear magnetic resonance (NMR) spectra.30,31 Despite the relatively large steric bulk of the trimethylsilyl group, the TMS-methylazide (1b) could be used to introduce the TMS group to the DNA. Tertiary amine-containing reagents such as 1c or 1d did not decrease the efficiency of the modification, and could be useful for introduction of an additional positive charge to the biomolecule. Incorporation of the morpholine group (1d) is noteworthy as the resulting tag could be useful in endoplasmic reticulum-targeting applications.32 Aromatic rings and a fluorophore scaffold are also compatible with the modification, enabling preparation of a color-palette of DNA-fluorophore conjugates. Interestingly, despite the versatile reactivities of azides, phosphines, and their derivatives (e.g. phosphazide and iminophosphorane), a single product was observed with an alkyl bromide substituted azide (1g) without loss of the bromide group. As the incorporation of the dimethylalkyl bromide is common in radical polymerization processes,33–35 the successful attachment of the group assists further development of DNA–polymer conjugates. Thus, the phosphine azide reaction is not affected by a series of functional groups such as silyl, tertiary amino, ether, ester, aryl alcohol, tertiary alkyl bromide and alkene groups.
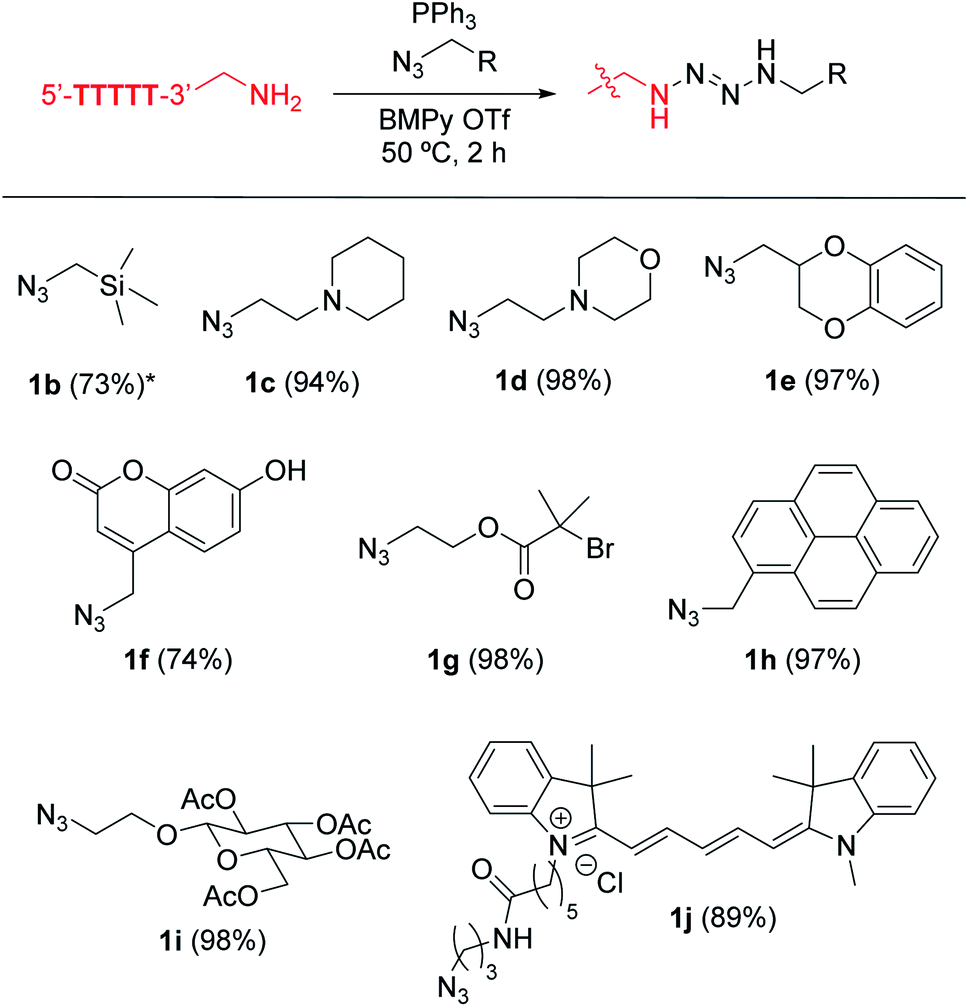 | ||
| Fig. 3 Tetrazene-forming DNA bioconjugation with a variety of alkylazides. Modification reaction conditions: KHCO3 (20 mM), 5′-TTTTT-3′-alkyl-NH2 (0.2 mM), azide derivatives 1b–1j (7.5 mM), and PPh3 (20 mM) in BMPy OTf at 50 °C for 2 h. *Reaction was incubated overnight. Conversion in the parentheses were calculated based on matrix-assisted laser desorption/ionization (MALDI-MS) analysis. Conversion obtained by liquid chromatography is available in Fig. S2.† | ||
In an effort to understand the compatibility of the functional groups and enhance the reaction efficiency, we tested different types of phosphine and phosphite reagents (Fig. 4). A DNA aptamer for human serum albumin was chosen as a model substrate for this study of phosphines.36 We focused only on reasonably air-stable phosphines including triaryl-phosphines with a range of substituents (2a–2g), alkylphosphine (JohnPhos, 2h), and arylphosphite (2i) and used triphenylphosphine oxide (3) as a negative control (Fig. 4A and C).37 The degree of the modification reaction was assessed with biotin-azide which provides an analysis handle for anti-biotin Southern blotting with a streptavidin–fluorophore conjugate. With the sole exception of tris(pentafluorophenyl)phosphines (2b), all the triarylphosphines (2a, 2c–2g) displayed a fluorescence signal at a similar level, indicating the minor effect of the subtle changes of the electronic properties of the aryl substituents (Fig. 4D and S3†). On the other hand, substantially weaker fluorescence signals were observed from reactions involving bisalkyl-monoarylphosphine (2h) or triphenylphosphite (2i), which suggests the necessity of triarylphosphine for the reaction. In order to visualize the total DNA amount on a blot membrane, Mayer's hemalum solution, which relies on coordination of aluminum with the phosphate backbone, was employed (Fig. S4†).38 In contrast to the Southern blot showing a varied fluorescence signal dependent on the biotin attachment on DNA, Mayer's hemalum stain showed similar intensity across all the conditions, confirming the validity of the experimental design (Fig. S3†). The triarylphosphine-dependent modification was also confirmed by mass spectrometry (Fig. S3†). We also found that different types of ionic liquid could be utilized for this chemical transformation (Fig. 4B and S5†). Furthermore, double-stranded DNAs with the alkylamine tag exhibited comparable reactivity to the single-stranded DNA system as well (Fig. S6†).
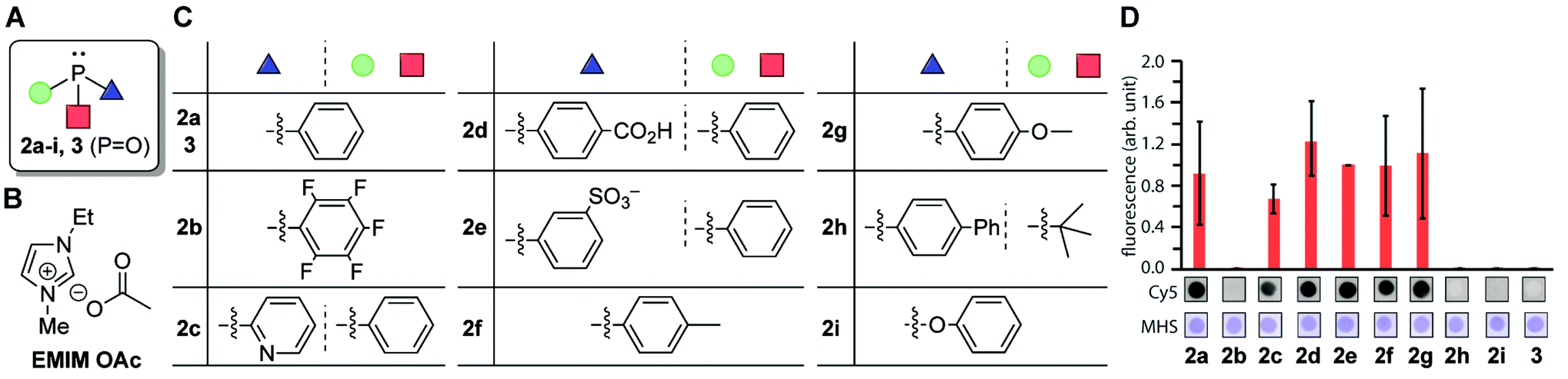 | ||
Fig. 4 Screening of phosphine reagents on tetrazene formation reaction on a DNA aptamer toward human serum albumin (HSA). Sequence of the HSA aptamer: 5′-TGCGGTTGTAGTACTCGTGGCCG-3′.36 Reaction conditions: HSA DNA aptamer (0.1 mM), KHCO3 (20 mM), biotin azide (7.5 mM), and phosphine (3.0 mM) in a mixture of 1-ethyl-3-methylimidazolium acetate (EMIM OAc)/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 50 °C for 2 h. (A) General chemical structure of phosphine. Green circle, red square, and blue triangle each represents the aryl or alkyl groups shown in Fig. 5C. (B) Chemical structure of ionic liquid EMIM OAc. (C) Chemical structures of the aryl or alkyl groups of different phosphine/phosphite reagents. (D) Bar graph showing the anti-biotin Southern blot after modification of the HSA DNA aptamer with biotin azide with different phosphines. Error bars represent standard deviation (n = 3). Representative blot membrane images for the anti-biotin Southern blot (Cy5) and total stain with Mayer's hemalum solution (MHS) are shown below the bar graphs. Full-width Southern blot membrane images are shown in Fig. S3.† :1) at 50 °C for 2 h. (A) General chemical structure of phosphine. Green circle, red square, and blue triangle each represents the aryl or alkyl groups shown in Fig. 5C. (B) Chemical structure of ionic liquid EMIM OAc. (C) Chemical structures of the aryl or alkyl groups of different phosphine/phosphite reagents. (D) Bar graph showing the anti-biotin Southern blot after modification of the HSA DNA aptamer with biotin azide with different phosphines. Error bars represent standard deviation (n = 3). Representative blot membrane images for the anti-biotin Southern blot (Cy5) and total stain with Mayer's hemalum solution (MHS) are shown below the bar graphs. Full-width Southern blot membrane images are shown in Fig. S3.† | ||
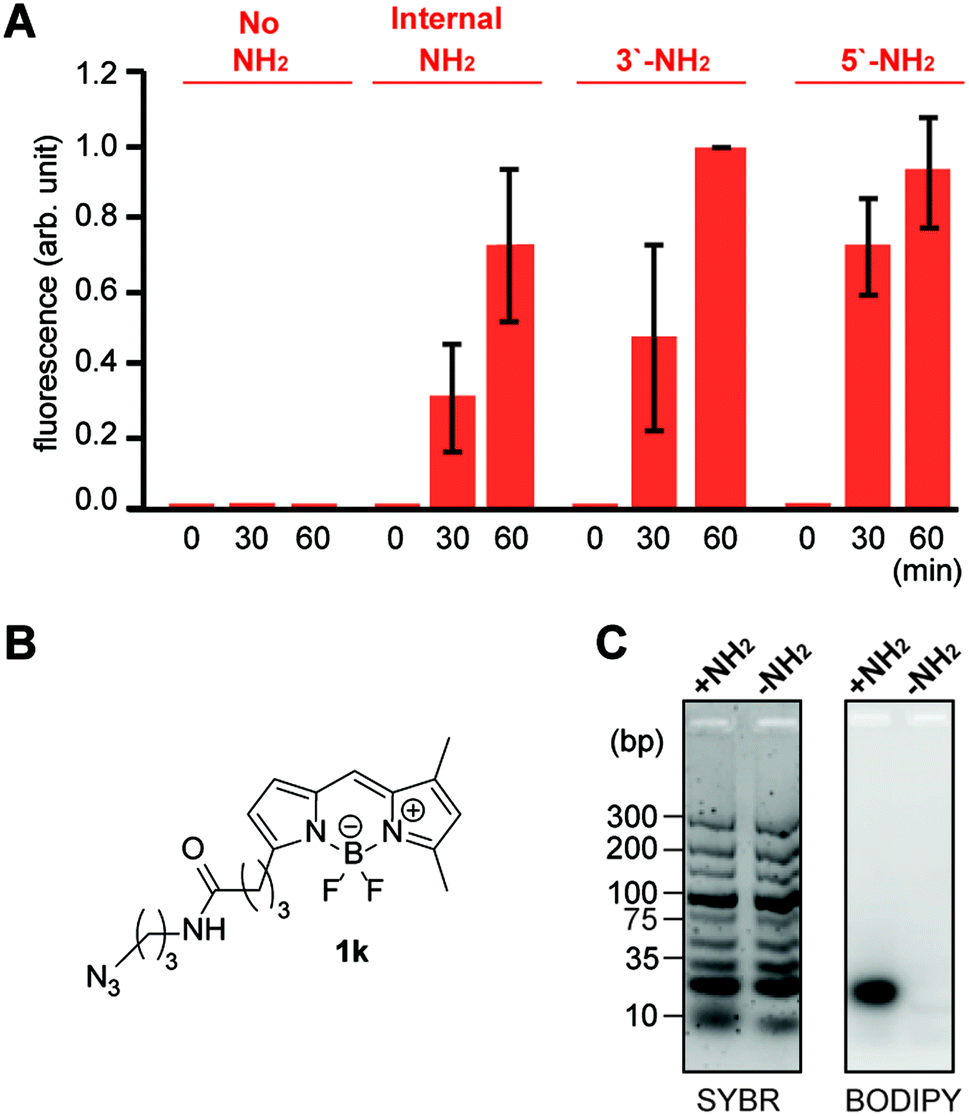 | ||
| Fig. 5 Reactivity and selectivity analysis of the tetrazene forming reaction on pentanucleotides with an alkylamine in different locations. (A) Bar graph showing fluorescence intensity of 5′-TTTTT-3′ with and without an alkylamine and treated with fluorophore azide 1k. Reaction conditions: KHCO3 (20 mM), 5′-TTTTT-3′ with and without amine group (0.2 mM), azide 1k (3 mM), and PPh3 (3 mM) in BMPy OTf at 50 °C for a specific time. Error bars represent standard deviation (n = 3). Statistical analysis of the data is available in Fig. S9.† (B) Chemical structure of alkylazide containing a boron dipyrromethene (BODIPY) group (1k). (C) Agarose gel images for the reaction of TTTTT-5′-NH2 with azide 1k in the presence of DNA ladder (10–300 bps). Total DNA samples were visualized by the fluorescence from SYBR Gold nucleic acid gel stain (Cy3 excitation and emission), while modified DNA samples were visualized by the fluorescence from BODIPY (Cy2 excitation and emission). Reaction conditions: KHCO3 (20 mM), TTTTT with or without NH2 tag at 5′ position (0.2 mM), DNA mixture (0.9 mg mL−1), azide 1k (3 mM), and PPh3 (3 mM) in BMPy OTf at 50 °C for 2 h. Chemical structures of the alkylamine groups on different positions (internal, 3′, and 5′) are available in Fig. S18.† | ||
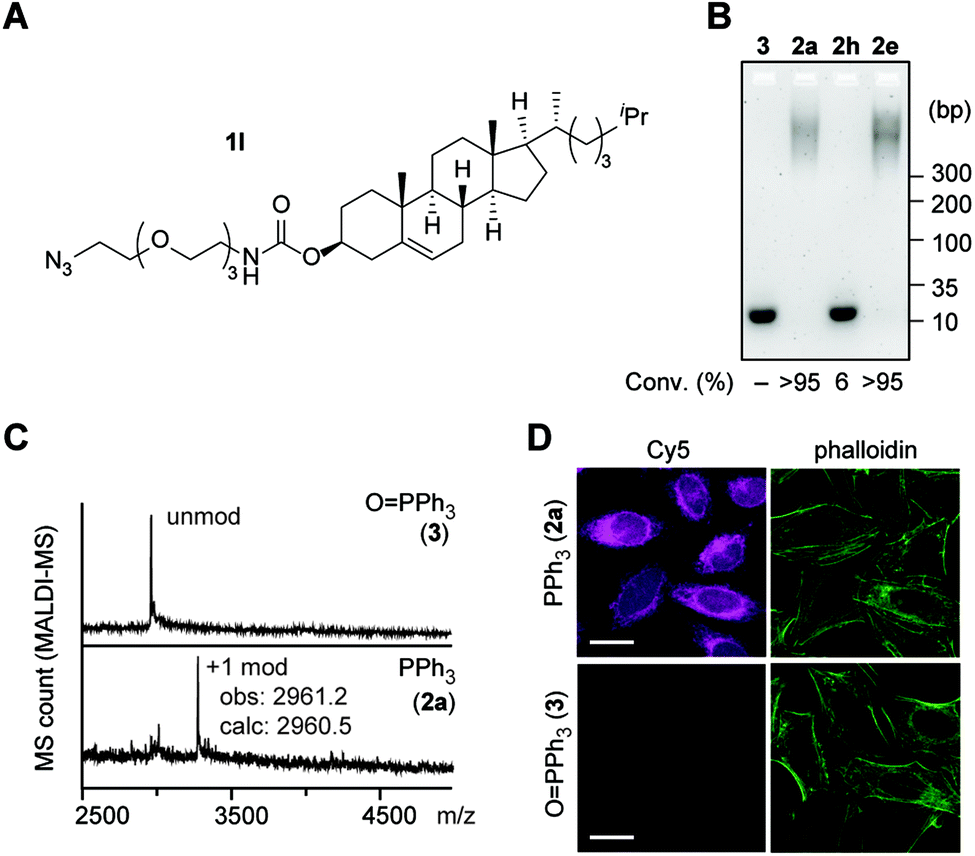 | ||
Fig. 6 Attachment of cholesterol to a DNA aptamer toward the RNA hairpin of human immunodeficiency virus (HIV)-1 transactivation-responsive (TAR) element through the tetrazene-forming reaction. Sequence of the HIV-1-TAR aptamer: 5′-CCCTAGTTAGCCATCTCCC-3′.41 Modification conditions: HIV-1-TAR-5′-NH2 aptamer (0.1 mM), KHCO3 (20 mM), azide 1l (7.5 mM), and PPh3 (2) or O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 (3) (20 mM) in EMIM OAc/DMF/DMSO (2:1:1) at 50 °C for 2 h. (A) Chemical structure of cholesterol azide (1l). (B) Agarose gel analysis of HIV-1-TAR-5′-NH2 aptamer modified with cholesterol azide (1l) in the presence of triphenylphosphine oxide (3), triphenylphosphine (2a) JohnPhos (2h), and sulfonate-substituted triphenylphosphine (2e). Degree of conversion was calculated by quantification of the unmodified DNA bands in comparison with the triphenylphosphine oxide condition as 100% of the starting material. (C) Matrix-assisted laser desorption/ionization (MALDI-MS) analysis of the modification of HIV-1-TAR-5′ NH2 aptamer with azide 1l and triphenylphosphine oxide (top, negative control) or triphenylphosphine (bottom). (D) Confocal microscopy images of HeLa cells stained with HIV-1-TAR aptamer hybridized with its complementary DNA-Cy5 conjugate (magenta). Top: cholesterol-modified aptamer (azide/PPh3-treated aptamer). Bottom: unmodified aptamer (azide/OPPh3-treated aptamer). Green: actin filament stain with phalloidin–CF488 conjugate. Scale bar: 20 μm. PPh3 (3) (20 mM) in EMIM OAc/DMF/DMSO (2:1:1) at 50 °C for 2 h. (A) Chemical structure of cholesterol azide (1l). (B) Agarose gel analysis of HIV-1-TAR-5′-NH2 aptamer modified with cholesterol azide (1l) in the presence of triphenylphosphine oxide (3), triphenylphosphine (2a) JohnPhos (2h), and sulfonate-substituted triphenylphosphine (2e). Degree of conversion was calculated by quantification of the unmodified DNA bands in comparison with the triphenylphosphine oxide condition as 100% of the starting material. (C) Matrix-assisted laser desorption/ionization (MALDI-MS) analysis of the modification of HIV-1-TAR-5′ NH2 aptamer with azide 1l and triphenylphosphine oxide (top, negative control) or triphenylphosphine (bottom). (D) Confocal microscopy images of HeLa cells stained with HIV-1-TAR aptamer hybridized with its complementary DNA-Cy5 conjugate (magenta). Top: cholesterol-modified aptamer (azide/PPh3-treated aptamer). Bottom: unmodified aptamer (azide/OPPh3-treated aptamer). Green: actin filament stain with phalloidin–CF488 conjugate. Scale bar: 20 μm. | ||
Finally, the ionic liquid-based amine–azide coupling was applied to labeling of a therapeutically important DNA aptamer (Fig. 7). The Her2 receptor is an epidermal growth factor related protein (ErbB2) family of receptor tyrosine kinases and emblematic example of overexpressed proteins in several types of cancer cells (Fig. 7A).44 We have adopted the sequence (42 nucleotides) of a DNA aptamer to the Her2 receptor originally reported by Yarden and co-workers.45 Modification of the Her2 aptamer with biotin-azide reagent proceeded smoothly in the same reaction conditions as those used with other aptamers, and was confirmed by MALDI-MS and anti-biotin Southern blotting (Fig. S11†). A gel shift assay using streptavidin also demonstrated the consumption of the unmodified aptamer (Fig. S11†). We used the biotinylated aptamer for cell imaging experiments on a Her2-overexpressing cell line, SK-BR-3. Treatment of SK-BR-3 cells with the biotinylated aptamer and streptavidin–fluorophore (Cy5) conjugate displayed strong fluorescence signals while negligible fluorescence was observed from the unconjugated aptamer (Fig. 7B). Importantly, cells treated with the biotinylated aptamer hybridized with its complementary sequence showed substantial decrease in the fluorescence signals, highlighting the importance of the single-strand aptamer structure for its binding with the target protein. In addition, treatment of the cells with the Her2 aptamer prior to the introduction of the biotinylated aptamer caused substantial decrease of fluorescence response (Fig. S12†), and this competition experiment shows the binding of the modified aptamer to the same antigen target. As such, we have confirmed the conservation of the aptamer's binding capability toward antigen targets even after treatment with ionic liquids and phosphine/azide reagents, demonstrating the practical utility of the ionic liquid-DNA bioconjugation approach.
 | ||
| Fig. 7 Tetrazene-forming reaction of DNA aptamer with the Her2 receptor. Sequence of the Her2 aptamer: 5′-GCAGCGGTGTGGGGGCAGCGGTGTGGGGGCAGCGGTGTGGGG-3′.45 Modification conditions: Her2 aptamer-5′-NH2 aptamer (0.1 mM), KHCO3 (20 mM), biotin azide (7.5 mM), and PPh3 (20 mM) in EMIM OAc/DMF/DMSO (2:1:1 ratio) at 50 °C for 2 h. (A) Partial crystal structure of Her2 receptor (PDB ID: 1N8Z). (B) Confocal microscopy images of Her2-overexpressing SK-BR-3 cells stained with biotin-modified Her2 aptamer (top), unmodified aptamer (middle), or biotin-modified Her2 aptamer hybridized with complementary DNA sequence (bottom). The bound aptamer was visualized by streptavidin–fluorophore (Cy5) conjugate (1:50 dilution) shown in magenta. Blue: nuclear stain with DAPI. DIC: differential interference contrast. Scale bar: 20 μm. | ||
Discussion: tetrazene formation by amine–azide coupling reaction
Nuclear magnetic resonance (NMR) and mass spectroscopic evidences support the formation of 1,4-disubstituted-2-trans-tetrazene by the phosphine-mediated chemistry. While NMR characterization of such disubstituted tetrazene has not been reported to date,46 we observed two broad peaks at 5.5 and 6.1 ppm for a model substrate in 1H NMR,29 which are akin to chemical shift of NH protons of a previously reported mono-substituted tetrazene compound (5.88 ppm).47 In order to further confirm the formation of disubstituted tetrazene, we have performed a deuterium exchange experiment by addition of D2O, and, indeed, disappearance of the two broad peaks was observed (Fig. S13†). 1H–1H COSY NMR demonstrated the correlation of broad peaks and methylene groups attached to the parent amine and azide reagents,29 and those observations also ascertained the formation of the disubstitued tetrazene. The observation of peaks at 75 (NH) and 300 ppm (NN) in 15N NMR in our previous report also support the formation of the disubstituted tetrazene29 as the tetrasubstituted tetrazene peaks are observed in the slightly more downfield region (NH: 100–180 ppm and NN: 360–400 ppm).48,49 With all the mass spectroscopic data in the current and previous report of peptide/protein/DNA substrates that shows the mass shift corresponding to the molecular weight of the azide reagents, the formation of 1,4-disubstituted tetrazene has been supported by our experimental data.
Tetrazene-forming reactions of azide species with amine derivatives are also documented and indicated in previous literature. Inorganic salt ammounium azide or [NH4][N3] is known to be interconverted to a constitutional isomer 2-tetrazene (H2N–NN–NH2) under high pressure,50,51 and this reaction represents the simplest example of the formal NH2–N3 coupling reaction (i.e. ammonia H–NH2 and hydrogen azide N3–H). Aryl- or sulfonyl–azide reagents are also known to form transient tetrazene intermediates through the reaction with anionic amine derivatives: Ar-NHLi52 or Ar-NHMgBr,53 respectively. Furthermore, reactions of amine compounds with sulfonylazide have been developed with the use of copper catalyst for the well-known azide transfer reaction through the formation of transient tetrazene species bound to the copper center.54,55 Thus, amine–azide coupling reactions have been accomplished through harsh reaction conditions or other reaction enhancement processes such as the deprotonation of amine or metal catalysis, and our system probably relies on the promotion of the azide reactivity through the aid of the phosphine reagent (vide infra).
The stability of tetrazene groups are dependent highly on their substitutions, and stable nature of alkyl-substituted tetrazene products are indicated in previous literature. The abovementioned aryl- and sulfonyl-tetrazene species generated from metal amide species undergoes rapid decomposition in a reaction mixture.52,53 Likewise, instability of aryl-substituted tetrazene generated from diazonium and hydrazine compounds are reported,56,57 which could be applied for radical coupling reactions.58 Consistent with those previous reports, the phosphine-mediated amine–azide coupling reaction of DNA with aryl and sulfonylazide did not provide the expected tetrazene products (Fig. S14†). On the other hand, alkyl-substituted tetrazene compounds could be substantially more stable; for instance, thermal decomposition temperature of a polymer form of tetraalkyl-substituted tetrazene occurs at 130 °C.59 Monoalkyl-substituted tetrazene's 1H NMR has been reported without special notes about the stability,47 which is also indicative of reasonable stability of the compound at ambient conditions. Our previous report indicated the product of amine–azide coupling reactions on proteins are stable toward heat denaturing condition (95 °C for 2 min) as well as biologically relevant molecules such as glutathione (thiol), hydrogen peroxide, and acidic/basic pH.29 The observation of the high stability also supports the formation of the dialkyl-substituted tetrazene by the amine–azide coupling reaction.
Discussion: roles of phosphine
The phosphine-mediated amine functionalization method relies on chemical reagents of Staudinger reduction converting the azide group into to amine group. The Staudinger reduction process initiates by the addition of phosphine reagents to the terminal nitrogen of azide group generating an intermediate called phosphazide, followed by its decomposition through the liberation of nitrogen gas to an iminophosphorane intermediate that is reactive toward water to produce the amine product and phosphine oxide.60 Though the rate-determining step of the Staudinger reactions are often the initial step (i.e. the addition of phosphine to azide),61 multiple factors could influence the reaction dynamics, allowing for various applications of the reactive phosphazide and iminophosphorane intermediates. Indeed, due to the chemoselectivity of those reagents to one another, numerous bioconjugation and bioorthogonal chemistry have been developed through different modification strategies which often takes advantage of the high nucleophilicity of the phosphazide and iminophosphorane intermediates.62–64On the other hand, the reactivity of phosphazide derivatives toward nucleophile has been recently reported as well. Formally, the phosphine-mediated amine–azide coupling can be considered the reaction of the phosphine- and azide-derived intermediate with nucleophile (amine) through the N–N bond formation. Such electrophilic behaviors of phosphazide derivatives were also observed in literature.65 In particular, a report by Zhang, Guo, and co-workers suggested that electrophilic reactivity of alkylated phosphazide intermediate toward an enolate nucleophile would produce 1,2,3-triazoline after the liberation of triphenylphosphine in an intramolecular manner (Fig. S15†).66 We propose that, for our tetrazene-forming process, such electrophilic/cationic phosphazide derivatives might be generated in situ by protonation of the nitrogen anion, and the nucleophilic attack of amine group as well as release of triphenylphosphine would afford the tetrazene product (Fig. S15†). The lifetime of the zwitterionic phosphazide species are dependent on their local environments (often controlled by bulkiness of the phosphine and azide substituents to destabilize the cis-phosphazide intermediate for the prevention of the iminophosphorane generation),64 and ionic liquid may be playing important roles in the stabilization of the zwitterionic species. Taking into account the higher basicity of alkyl-substituted nitrogen anion than aryl-substituted one, the protonation of the zwitterionic phosphazide would be more facile with the alkylazide-derived one than aryl- or sulfonyl ones, and the effective formation of the product with alkylazide rather than aryl and sulfonylazide might be a consequence of this basicity difference in addition to the aforementioned aryl- and sulfonyl-tetrazene instability (Fig. S14†).
Based on the proposed mechanism, only catalytic amount of triphenylphosphine would be necessary for the amine–azide coupling reaction; however, in practice, excess phosphine reagents are prerequisite to observe the tetrazene formation. We have performed the aptamer labeling using cholesterol-azide with varied amount of PPh3, and indeed, remarkable decrease of modification efficiency was observed with the lower PPh3 conditions (Fig. S16†), likely due to the competing Staudinger reduction pathway of the reaction between phosphine and azide reagents. Moreover, NMR study demonstrated that treatment of benzylamine (reactive for the amine–azide coupling) or aniline (unreactive) compounds with an alkylazide reagent with triphenylphosphine did not show meaningful difference in 31P NMR with regard to PPh3 and OPPh3 amounts (Fig. S17†), further supporting our mechanistic hypothesis that the coupling reaction does not generate triphenylphosphine oxide and that significant amount of the phosphine/azide reagents also would undergo the Staudinger reduction process. It would be reasonable to assume that the unique reaction conditions of bioconjugation with excess reagents enabled the development of the phosphine-mediated amine–azide coupling reaction.
Conclusion
The ionic liquid-based tetrazene-forming reaction has been successfully applied to the site-specific modification of unprotected DNA substrates. The high reaction efficiency at a desired location and high tolerance toward a variety of functional groups on azide and phosphine reagents could be of significant help in tailoring the technology to more specific applications. Thanks to the widespread use of azide–alkyne cycloaddition reactions in the chemistry and biology communities,67 there are numerous commercially available alkylazide reagents, and the current work can be readily adopted for diverse applications. The shelf-stable nature of the alkylazide and triarylphosphine reagents would also be practically helpful in this context. Persistent issues of common amine-targeting reagents originate from the reagent instability such as the hydrolytic decomposition of N-hydroxysuccinimide (NHS) ester reagents for the acylation reaction and the aerobic oxidation of aldehyde reagents used in the reductive alkylation reaction. Our initial success of ionic-liquid bioconjugation development for nucleotide substrates may serve to provide further access to untapped chemical labeling methodologies for preparation of nucleotide conjugates.Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Author contributions
J. O. designed research and wrote the manuscript. S. I., M. T., and E. J. G. performed experiments of chemical labeling of DNA; S. I., M. T., and E. J. G. prepared figures; S. I. and M. T. performed agarose gel experiments; S. I. performed mammalian cell staining experiments; S. I. and E. J. G. performed NMR experiments.Conflicts of interest
The authors declare the following competing financial interest(s): the authors have filed a patent application on the ionic-liquid-based tetrazene-forming bioconjugation method.Acknowledgements
This work was financially supported by North Carolina State University. E. J. G. was financially supported by an NC State Provost's Professional Experience Program (PEP). We thank Dr Alison Killilea and Wilmene Hercule of the UC Berkeley Cell Culture Facility for cell culture support, and Dr G. W. A. Milne for his editing of our manuscript. Confocal microscopy imaging was performed at the Cellular and Molecular Imaging Facility (CMIF), and MALDI-MS experiments were performed in Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University, which is supported by the State of North Carolina.References
- J. Zheng, R. Yang, M. Shi, C. Wu, X. Fang, Y. Li, J. Li and W. Tan, Chem. Soc. Rev., 2015, 44, 3036–3055 RSC.
- R. Monroy-Contreras and L. Vaca, J. Nucleic Acids, 2011, 2011, 741723 Search PubMed.
- C. J. Smith and A. M. Osborn, FEMS Microbiol. Ecol., 2009, 67, 6–20 CrossRef CAS PubMed.
- R. Ting, L. Lermer and D. M. Perrin, J. Am. Chem. Soc., 2004, 126, 12720–12721 CrossRef CAS PubMed.
- X. Wang, M. Feng, L. Xiao, A. Tong and Y. Xiang, ACS Chem. Biol., 2016, 11, 444–451 CrossRef CAS PubMed.
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083–2092 RSC.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CrossRef CAS PubMed.
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt, P. R. Cullis and R. van der Meel, Nat. Nanotechnol., 2021, 16, 630–643 CrossRef CAS PubMed.
- D. D. Young, M. O. Lively and A. Deiters, J. Am. Chem. Soc., 2010, 132, 6183–6193 CrossRef CAS PubMed.
- M. E. Østergaard, C. L. De Hoyos, W. B. Wan, W. Shen, A. Low, A. Berdeja, G. Vasquez, S. Murray, M. T. Migawa, X. Liang, E. E. Swayze, S. T. Crooke and P. P. Seth, Nucleic Acids Res., 2020, 48, 1691–1700 CrossRef PubMed.
- Y. Ma, X. Zhang, X. Xu, L. Shen, Y. Yao, Z. Yang and P. Liu, PLoS One, 2015, 10, e0124924 CrossRef PubMed.
- N. B. Struntz and D. A. Harki, ACS Chem. Biol., 2016, 11, 1631–1638 CrossRef CAS PubMed.
- C. Yu, M. An, M. Li and H. Liu, Mol. Pharmaceutics, 2017, 14, 2815–2823 CrossRef CAS PubMed.
- W. Meng, T. Yamazaki, Y. Nishida and N. Hanagata, BMC Biotechnol., 2011, 11, 88 CrossRef PubMed.
- R. A. Hughes and A. D. Ellington, Cold Spring Harbor Perspect. Biol., 2017, 9, a023812 CrossRef PubMed.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
- K. Tishinov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401–4406 RSC.
- S. N. Geigle, L. A. Wyss, S. J. Sturla and D. G. Gillingham, Chem. Sci., 2016, 8, 499–506 RSC.
- D. Gillingham and R. Shahid, Curr. Opin. Chem. Biol., 2015, 25, 110–114 CrossRef CAS PubMed.
- G. Li and R. E. Moellering, ChemBioChem, 2019, 20, 1599–1605 CAS.
- G. Li, M. A. Eckert, J. W. Chang, J. E. Montgomery, A. Chryplewicz, E. Lengyel and R. E. Moellering, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21493–21500 CrossRef CAS PubMed.
- P. V. Robinson, C. Tsai, A. E. de Groot, J. L. McKechnie and C. R. Bertozzi, J. Am. Chem. Soc., 2016, 138, 10722–10725 CrossRef CAS PubMed.
- E. K. Jang, R. G. Son and S. P. Pack, Nucleic Acids Res., 2019, 47, e102 CrossRef PubMed.
- D. T. Flood, K. W. Knouse, J. C. Vantourout, S. Kitamura, B. B. Sanchez, E. J. Sturgell, J. S. Chen, D. W. Wolan, P. S. Baran and P. E. Dawson, ACS Cent. Sci., 2020, 6, 1789–1799 CrossRef CAS PubMed.
- J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O’Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404–E6410 CrossRef CAS PubMed.
- D. K. Romney, F. H. Arnold, B. H. Lipshutz and C.-J. Li, J. Org. Chem., 2018, 83, 7319–7322 CrossRef CAS PubMed.
- S. K. Shukla and J.-P. Mikkola, Front. Chem., 2020, 8, 1219 Search PubMed.
- D. R. MacFarlane, A. L. Chong, M. Forsyth, M. Kar, R. Vijayaraghavan, A. Somers and J. M. Pringle, Faraday Discuss., 2017, 206, 9–28 RSC.
- H. M. El-Shaffey, E. J. Gross, Y. D. Hall and J. Ohata, J. Am. Chem. Soc., 2021, 143, 12974 CrossRef PubMed.
- Q. Liu, Q. He, X. Lyu, F. Yang, Z. Zhu, P. Xiao, Z. Yang, F. Zhang, Z. Yang, X. Wang, P. Sun, Q. Wang, C. Qu, Z. Gong, J. Lin, Z. Xu, S. Song, S. Huang, S. Guo, M. Han, K. Zhu, X. Chen, A. W. Kahsai, K.-H. Xiao, W. Kong, F. Li, K. Ruan, Z. Li, X. Yu, X. Niu, C. Jin, J. Wang and J. Sun, Nat. Commun., 2020, 11, 4857 CrossRef CAS PubMed.
- W. Hu, H. Wang, Y. Hou, Y. Hao and D. Liu, FEBS Lett., 2019, 593, 1113–1121 CrossRef CAS PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- X. Wu, X. He, L. Zhong, S. Lin, D. Wang, X. Zhu and D. Yan, J. Mater. Chem., 2011, 21, 13611–13620 RSC.
- S. Sun, Y. Cao, J. Feng and P. Wu, J. Mater. Chem., 2010, 20, 5605–5607 RSC.
- A. Nantalaksakul, A. Mueller, A. Klaikherd, C. J. Bardeen and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 2727–2738 CrossRef CAS PubMed.
- N. Kuntip, D. Japrung and P. Pongprayoon, Biopolymers, 2021, 112, e23421 CrossRef CAS PubMed.
- B. Stewart, A. Harriman and L. J. Higham, Organometallics, 2011, 30, 5338–5343 CrossRef CAS.
- J. Kiernan, Biotech. Histochem., 2018, 93, 133–148 CrossRef CAS PubMed.
- Z. Cao, R. Tong, A. Mishra, W. Xu, G. C. L. Wong, J. Cheng and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 6494–6498 CrossRef CAS PubMed.
- Y. Wan, L. Wang, C. Zhu, Q. Zheng, G. Wang, J. Tong, Y. Fang, Y. Xia, G. Cheng, X. He and S.-Y. Zheng, Cancer Res., 2018, 78, 798–808 CrossRef CAS PubMed.
- D. Sekkai, E. Dausse, C. Di Primo, F. Darfeuille, C. Boiziau and J.-J. Toulmé, Antisense Nucleic Acid Drug Dev., 2002, 12, 265–274 CrossRef CAS PubMed.
- A. Ohmann, K. Göpfrich, H. Joshi, R. F. Thompson, D. Sobota, N. A. Ranson, A. Aksimentiev and U. F. Keyser, Nucleic Acids Res., 2019, 47, 11441–11451 CrossRef CAS PubMed.
- S. F. Jones, H. Joshi, S. J. Terry, J. R. Burns, A. Aksimentiev, U. S. Eggert and S. Howorka, J. Am. Chem. Soc., 2021, 143, 8305–8313 CrossRef CAS PubMed.
- N. Iqbal and N. Iqbal, Mol. Biol. Int., 2014, 2014, e852748 Search PubMed.
- G. Mahlknecht, R. Maron, M. Mancini, B. Schechter, M. Sela and Y. Yarden, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8170–8175 CrossRef CAS PubMed.
- N. Wiberg, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1985, vol. 24, pp. 179–248 Search PubMed.
- D. R. Haines, N. J. Leonard and D. F. Wiemer, J. Org. Chem., 1982, 47, 474–482 CrossRef CAS.
- A. Dhenain, C. Darwich, C. M. Sabaté, D.-M. Le, A.-J. Bougrine, H. Delalu, E. Lacôte, L. Payen, J. Guitton, E. Labarthe and G. Jacob, Chem.–Eur. J., 2017, 23, 9897–9907 CrossRef CAS PubMed.
- C. M. Sebate and H. Delalu, Cent. Eur. J. Energ. Mater., 2014, 11, 515 Search PubMed.
- N. Yedukondalu, G. Vaitheeswaran, P. Anees and M. C. Valsakumar, Phys. Chem. Chem. Phys., 2015, 17, 29210–29225 RSC.
- M. Veith and G. Schlemmer, Z. Anorg. Allg. Chem., 1982, 494, 7–19 CrossRef CAS.
- S. W. Lee, G. A. Miller, C. F. Campana, M. L. Maciejewski and W. C. Trogler, J. Am. Chem. Soc., 1987, 109, 5050–5051 CrossRef CAS.
- W. Fischer and J. P. Anselme, J. Am. Chem. Soc., 1967, 89, 5284–5285 CrossRef CAS.
- A. K. Pandiakumar, S. P. Sarma and A. G. Samuelson, Tetrahedron Lett., 2014, 55, 2917–2920 CrossRef CAS.
- J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., Int. Ed., 2019, 58, 6176–6199 CrossRef CAS PubMed.
- Th. Curtius, Ber. Dtsch. Chem. Ges., 1893, 26, 1263–1271 Search PubMed.
- W. T. Evanochko and P. B. Shevlin, J. Am. Chem. Soc., 1979, 101, 4668–4672 CrossRef CAS.
- S. Shaaban, A. Jolit, D. Petkova and N. Maulide, Chem. Commun., 2015, 51, 13902–13905 RSC.
- J. Eymann, L. Joucla, G. Jacob, J. Raynaud, C. Darwich and E. Lacôte, Angew. Chem., Int. Ed., 2021, 60, 1578–1582 CrossRef CAS PubMed.
- T. Deb, J. Tu and R. M. Franzini, Chem. Rev., 2021, 121, 6850–6914 CrossRef CAS PubMed.
- W. Q. Tian and Y. A. Wang, J. Org. Chem., 2004, 69, 4299–4308 CrossRef CAS PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301 CrossRef CAS PubMed.
- M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117–12121 CrossRef CAS PubMed.
- T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126–4130 CrossRef CAS PubMed.
- A. A. Sediek, A. A. Shaddy and W. M. Abdou, J. Heterocycl. Chem., 2011, 48, 1258–1263 CrossRef CAS.
- H. Wang, L. Zhang, Y. Tu, R. Xiang, Y.-L. Guo and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 15787–15791 CrossRef CAS PubMed.
- N. Z. Fantoni, A. H. El-Sagheer and T. Brown, Chem. Rev., 2021, 121, 7122–7154 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05204g |
| This journal is © The Royal Society of Chemistry 2022 |