
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide epitope-imprinted polymer microarrays for selective protein recognition. Application for SARS-CoV-2 RBD protein†
Zsófia Bognár a,
Eszter Supalaa,
Aysu Yarmanb,
Xiaorong Zhangb,
Frank F. Bierb,
Frieder W. Schellerb and
Róbert E. Gyurcsányi*a
a,
Eszter Supalaa,
Aysu Yarmanb,
Xiaorong Zhangb,
Frank F. Bierb,
Frieder W. Schellerb and
Róbert E. Gyurcsányi*a
aBME “Lendület” Chemical Nanosensors Research Group, Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, 1111 Budapest, Hungary. E-mail: gyurcsanyi.robert@vbk.bme.hu
bInstitute of Biochemistry and Biology, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam OT Golm, Germany
First published on 23rd November 2021
Abstract
We introduce a practically generic approach for the generation of epitope-imprinted polymer-based microarrays for protein recognition on surface plasmon resonance imaging (SPRi) chips. The SPRi platform allows the subsequent rapid screening of target binding kinetics in a multiplexed and label-free manner. The versatility of such microarrays, both as synthetic and screening platform, is demonstrated through developing highly affine molecularly imprinted polymers (MIPs) for the recognition of the receptor binding domain (RBD) of SARS-CoV-2 spike protein. A characteristic nonapeptide GFNCYFPLQ from the RBD and other control peptides were microspotted onto gold SPRi chips followed by the electrosynthesis of a polyscopoletin nanofilm to generate in one step MIP arrays. A single chip screening of essential synthesis parameters, including the surface density of the template peptide and its sequence led to MIPs with dissociation constants (KD) in the lower nanomolar range for RBD, which exceeds the affinity of RBD for its natural target, angiotensin-convertase 2 enzyme. Remarkably, the same MIPs bound SARS-CoV-2 virus like particles with even higher affinity along with excellent discrimination of influenza A (H3N2) virus. While MIPs prepared with a truncated heptapeptide template GFNCYFP showed only a slightly decreased affinity for RBD, a single mismatch in the amino acid sequence of the template, i.e. the substitution of the central cysteine with a serine, fully suppressed the RBD binding.
Introduction
Molecularly imprinted polymers (MIPs) are generally prepared by polymerizing functional monomers prearranged via non-covalent interactions around a template molecule. The template removal frees up recognizing sites in the polymer for the selective binding of the template that opens extensive prospects for the applicability of such synthetic sorbents.1–4 In this respect selective protein recognition within affinity assays by replacing antibodies5 contours as a natural application of MIPs. However, the generation of MIPs for macromolecular templates is considerably more complex than for small molecular weight templates.6 Owing to their large size the template proteins can be permanently entrapped in the polymeric matrix and their inherent conformational fragility needs mild polymerization conditions.7 Moreover, while for proteins, rich in functional groups, the cooperative contributions of multiple weak interactions with the functional monomers are generally expected to result in MIPs with high affinity they may also lead to cross-reactivity. Therefore, major enabling concepts had to be implemented to address these difficulties, which include surface imprinting,8–14 epitope imprinting,15–17 semicovalent imprinting,18,19 and various oriented immobilizations of the protein or peptide targets.20–22 Surface imprinting empowers the free exchange of the protein templates with the solution, which is the prerequisite of the recognition functionality of MIPs. Epitope imprinting, by using as templates characteristic short peptides of the target protein, is essential to foster selectivity by restricting the imprints to unique peptide sequences of the target protein.23 The use of peptide epitopes, amenable to routine peptide synthesis, instead of the target protein is also essential to decrease the cost of MIP fabrication. The oriented immobilization of the template epitope as compared with solution based target-monomer mixtures reportedly increased the capacity (density of recognition cavities) of the protein-selective MIPs.24 Finally, oriented epitope imprinting is also beneficial in terms of sensitive transduction of the binding events as well as homogeneity of imprints.21 The large molecular size of the protein targets makes computational approaches very demanding.25,26 Therefore, the rational design of MIPs is largely limited to target specific matching of the monomer functionality, i.e. using monomers with functionalities that are known to interact with parts of the template. Relevant examples are the use of boronate chemistry in case of glycoproteins,11,12 taking advantage of metal ion complexing effects27 for proteins rich in histidine or cysteine, and exploiting the charge and/or polarity of the protein targets. These are efficient strategies to enhance the success rate of generating highly affine MIPs, but depart from the original concept of a genuine molecular imprinting that would ideally imply the use of a generic monomer or monomer library that is made selective for different targets solely by imprinting. Additionally, even using template-tailored selection of monomers there is a considerable effort to adjust the polymerization conditions, template orientation, concentration or surface density of the target for optimal affinity. These largely empiric processes may be considered one of the major bottlenecks in developing high affinity protein MIPs. However, the development process may be alleviated by using high-throughput methodologies28,29 for both the synthesis of MIPs and characterization of their target binding properties. While MIP nanoparticles,30–32 are inherently compatible with conventional microplate screening assays, planar protein-MIP nanofilms with a few exceptions,33 largely devoid such possibilities. Especially, electrosynthesized MIPs which have clear advantages in terms of offering mild aqueous conditions for the synthesis and controlled deposition on sensor transducers.30 To address this demand, we introduced recently microelectrospotting34 for the electrosynthesis of surface-imprinted protein MIP-arrays. The procedure involves an electrochemical spotting pin35 for localized electropolymerization of monomer-template protein mixtures and enabled multiplexed screening of a large number of synthetic conditions for protein MIPs as well as their affinity. However, it is not suited for oriented epitope imprinting.Here we propose a practically universal strategy, that merges for the first time the essential advances in generating protein-selective MIPs: epitope imprinting, surface imprinting and oriented template immobilization; along with the control and mild conditions offered by the electrosynthesis, to generate MIP microarrays on gold surface plasmon resonance imaging (SPRi) chips (Scheme 1). The SPRi platform enables the fast post-synthetic screening of the target binding properties of MIPs. We show the proof of concept by generating MIPs for the selective recognition of the receptor binding domain (RBD) of the spike (S) protein of SARS-CoV-2 virus using for epitope imprinting a characteristic nonapeptide sequence, GFNCYFPLQ, of the RBD. Despite the actuality of this topic, MIP-based virus analytics largely lacks multiplexed development platforms for generating epitope-imprinted MIPs, i.e. the recognition has been solely attempted based on imprinting with large protein fragments of one of the virus proteins.36,37 Hence we present here the first epitope-based MIP and this concept may be adapted towards the recognition of different virus mutants and discriminating between closely homologue proteins.38
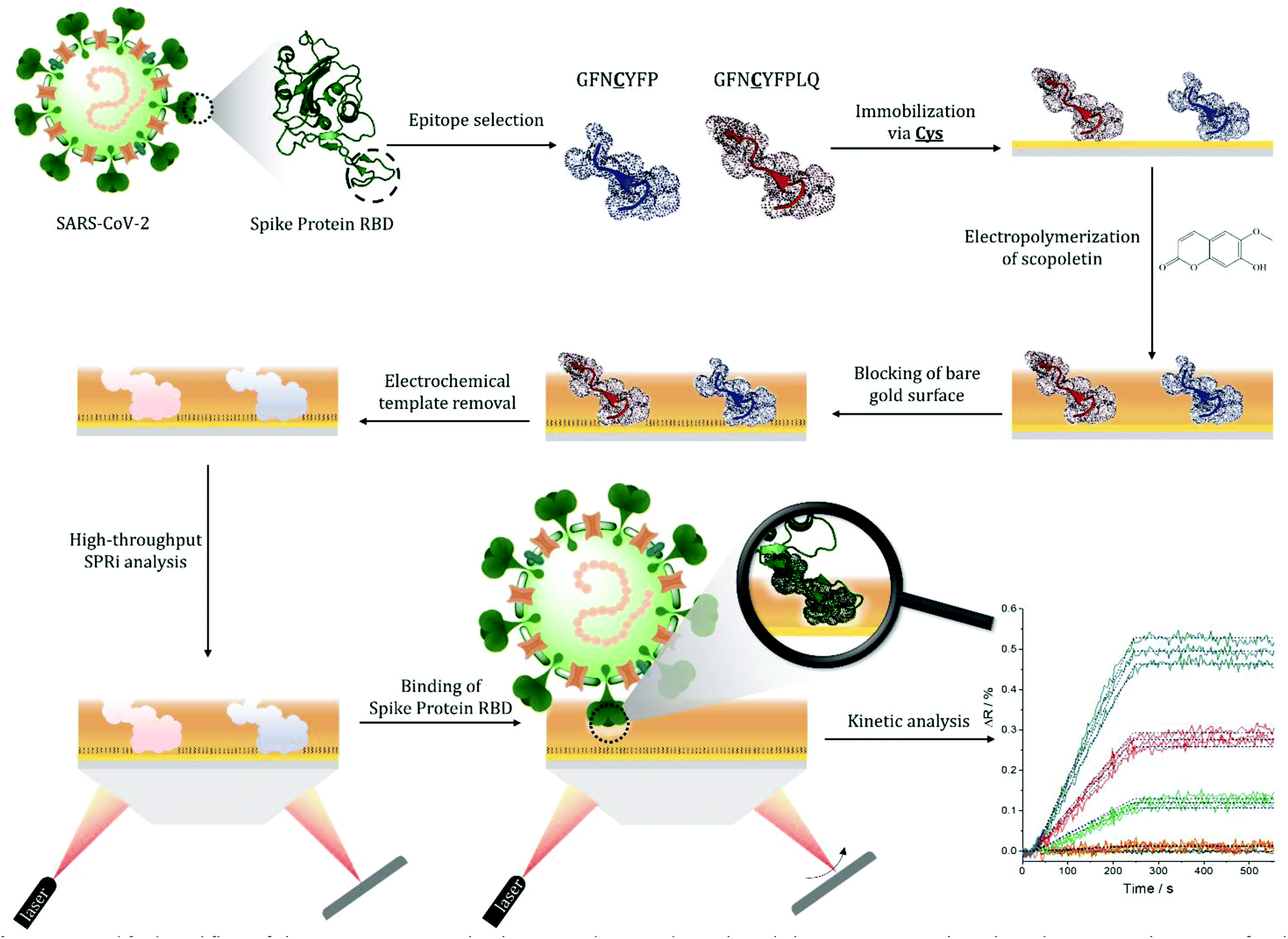 | ||
| Scheme 1 Simplified workflow of the epitope-imprinted polymer synthesis and SPRi-based characterization. The selected nonapeptide epitope for the RBD of S protein (GFNCYFPLQ) bearing a central cysteine (and other relevant peptides, e.g. the heptapeptide GFNCYFP) are contact microspotted onto the surface of a gold SPRi chip resulting in ca. 500 μm diameter spots of the surface-immobilized peptides. This is followed by electropolymerization of scopoletin to form the epitope-imprinted polymer on the peptide spots, and blocking the contingently exposed gold surfaces by HS-TEG. The peptide templates are removed electrochemically by oxidative stripping to liberate the RBD selective cavities in the nanofilm. The kinetics of the target binding is then determined by SPRi for all spots of the microarray in label-free and multiplexed manner (the inset shows SPRi measurements for 3 identical spots, the colours denote different RBD concentrations). | ||
Results and discussion
The S protein is a 150 kDa transmembrane protein on the surface of SARS-CoV-2. Its C-terminal 26 kDa RBD is the docking area of the virus to the angiotensin-convertase 2 enzyme (ACE2) for entering the host cell. This interaction has equilibrium dissociation constants (KD) in the range of 4.7 nM to ca. 15 nM.39 Despite some homologies with other corona viruses (the most related SARS-CoV-1) has a sequence identity of 73%),40 targeting RBD was reported to allow the selective identification of SARS-CoV-2 avoiding cross-reactivity with other corona viruses.41 This is especially true for the peptide sequence that has been chosen in this study. Docking simulations predicted that the peptide chain of RBD starting with L455 up to Y505 is the binding area of the RBD to the ACE2.42 This is in accordance with the structure of the RBD–ACE2 complex determined by X-ray crystallography, which shows that the residues F486, N487, Y489 and Q493 contact the binding area of ACE2.43 Therefore, we chose from this region as the epitope template the nonapeptide 485–493, because it contains four “interacting” amino acids of the RBD–ACE2 complex. The chip fabrication involves microspotting of cysteine (Cys) bearing peptides for epitope imprinting followed by the deposition of a polymer nanofilm by electropolymerization on the whole sensing surface of the chip (Fig. S1†) using scopoletin as monomer (Scheme S1†). This compound generated MIPs selective for a wide variety of peptide and protein targets through genuine imprinting.10,24,44–46 Polyscopoletin is electrically insulating; hence its growth by electropolymerization is self-limiting and results in highly conformal and uniform films with thicknesses up to ca. 10 nm (Fig. S2†). On the different peptide-covered spots, the deposition of the polyscopoletin film generated the respective epitope-imprinted polymer spots while on the bare gold (or PBS spots) the non-imprinted polymer (NIP) controls. To rule out the presence of exposed gold surfaces the chip was further treated with 0.5 mM (11-mercaptoundecyl)tetra(ethylene glycol) (HS-TEG) for 60 min. The binding sites were liberated by electrochemically stripping the template peptides using 1 V for 20 s, i.e. by oxidative desorption of the thiol-bearing peptides. No loss of HS-TEG was observed in the applied potential window (Fig. S3†). We have shown earlier by surface enhanced IR spectroscopy24 that unlike the commonly used chemical procedures the electrochemical template removal does not degrade the polymer film. After the template removal the target binding properties and selectivity of the MIP-based microarrays were investigated in real-time and multiplexed manner with a Horiba Plex II SPRi system. The contact microspotting of peptides enables the convenient adjustment of the surface density of the immobilized peptides by varying the peptide concentration of the microspotted solution.47,48 The effect of the epitope surface density on the amount of bound RBD (recombinant 2019-nCoV Spike RBD Protein) is shown in Fig. 1.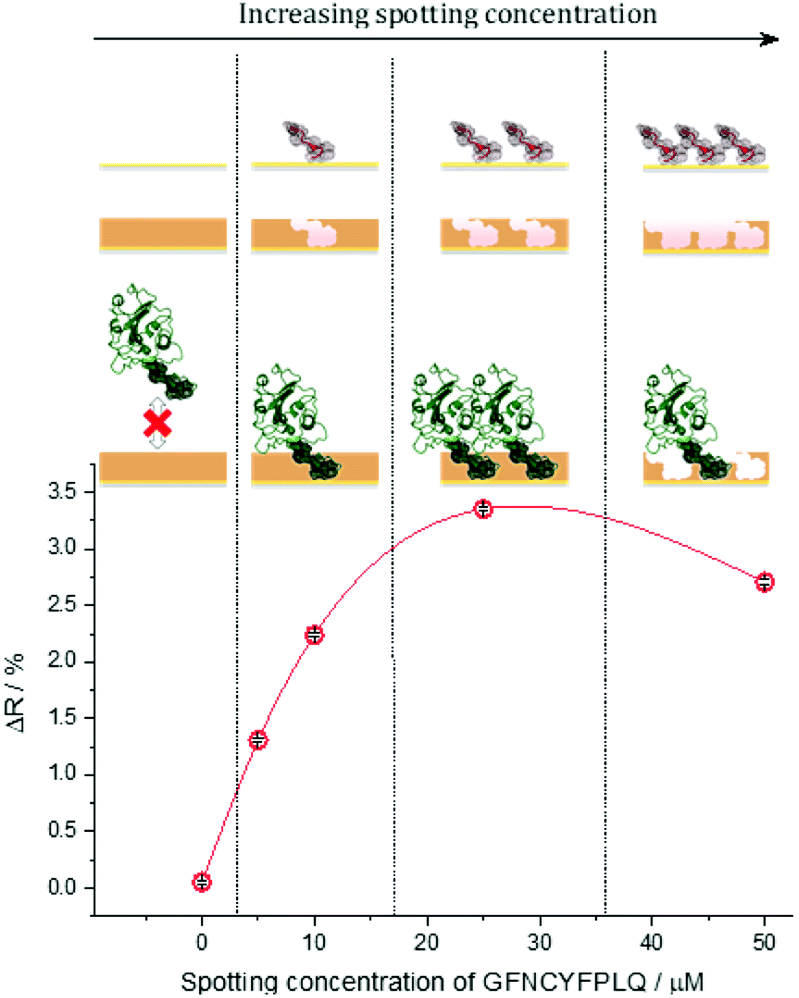 | ||
| Fig. 1 The effect of the microspotted GFNCYFPLQ peptide concentration on the subsequent binding of RBD at its saturation concentration (385 nM) to the respective epitope-imprinted spots in phosphate buffer saline with 0.05% Tween 20 (PBST). The reflectivity change (ΔR) is indicative of the amount of bound RBD, i.e. the binding capacity of the respective MIP spots. | ||
Increasing the epitope density on the gold surface increases the binding capacity of the epitope-imprinted polyscopoletin nanofilms till saturation (ca. 25 μM spotting concentration). Further increase in the surface concentration, however, slightly decreases the binding capacity since a “compact” peptide layer should be detrimental to the formation of a polymer film that fully surrounds the peptide template, i.e. to the formation of distinct recognition sites. Therefore, unless otherwise mentioned subsequent SPRi measurements are shown for 25 μM peptide concentration of the spotted solutions, which centers the broad maximum of the RBD binding curve.
To investigate in a microarray format the effect of the peptide template sequence on the RBD binding various peptides were microspotted onto the gold SPRi chip according to the layout shown in Fig. 2A.
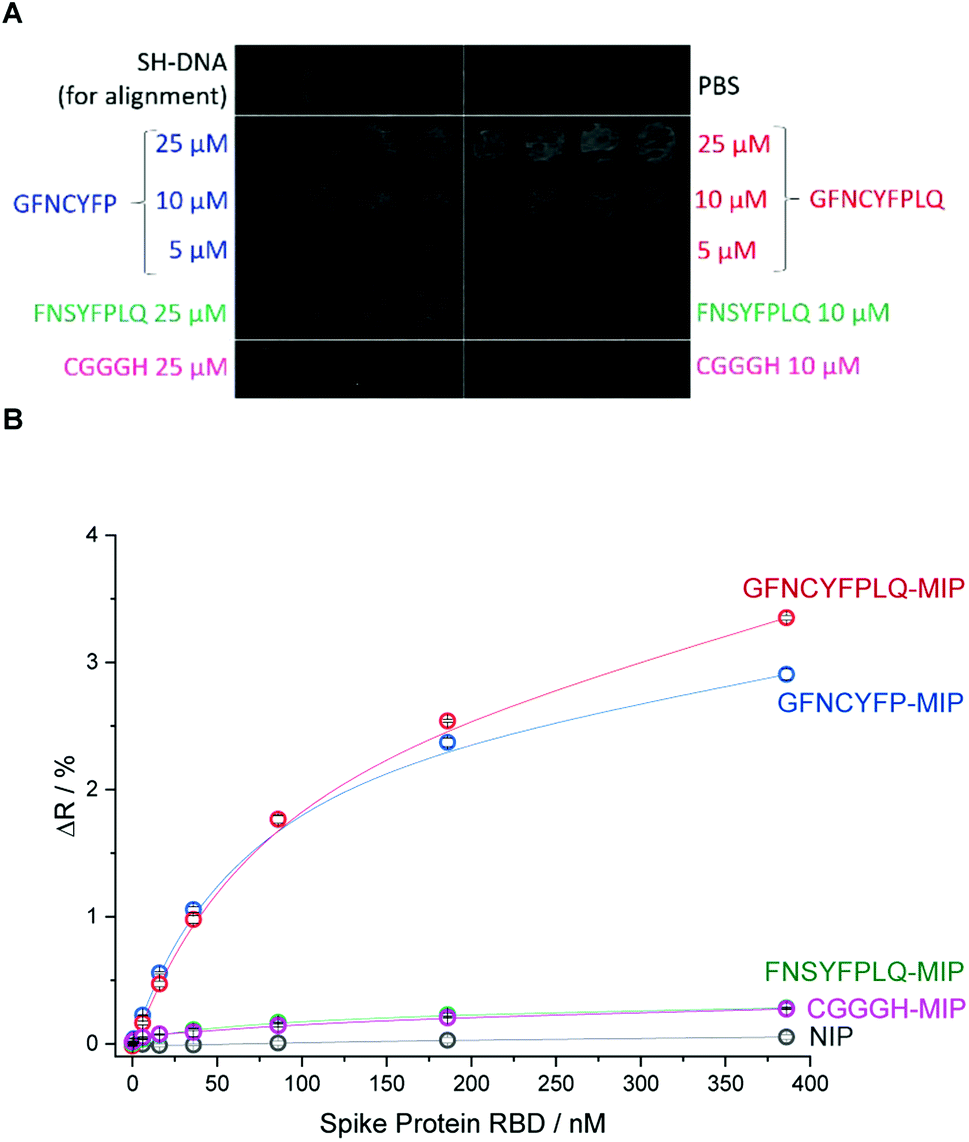 | ||
| Fig. 2 (A) The SPR image and microspotting layout of the epitope-imprinted polymer microarray. Each peptide concentration was spotted in triplicates. The diameter of the spots is ca. 500 μm with 420 μm spacing. (B) RBD binding to the different peptide-imprinted polyscopoletin spots. | ||
Beside the nonapeptide GFNCYFPLQ, we selected the shorter heptapeptide GFNCYFP (lacking terminal LQ) to explore the effect of the terminal Q493 hotspot. A similar octapeptide sequence with the cysteine substituted with serine (FNSYFPLQ) was selected to investigate the lack of the C residue on the immobilization and protein binding. The printing layout included also a random amino acid sequence peptide (CGGGH) as negative control. The binding of RBD to the different peptide-imprinted polymers was investigated in multiplexed manner by SPRi in the nanomolar range using kinetic titration49 (Fig. 2B). The nonapeptide GFNCYFPLQ and heptapeptide GFNCYFP showed an identical binding behavior in the low RBD concentration range. The lack of the Q493 residue manifested in a slightly decreased target binding solely at higher target concentrations (>200 nM). These results were fully consistent across parallel spots (Fig. S4†) and identically prepared microarrays on different SPRi chips (Fig. S5†). The chip-to-chip reproducibility is especially remarkable and supports the reliability of the fabrication approach. A single mismatch in the amino acid sequence by substitution of the central cysteine with serine resulted in a dramatic decrease in the RBD binding to the level of the random sequence CGGGH-based MIP. We have confirmed by voltammetric measurements (Fig. S6†) that the peptide adsorbs onto the gold despite the absence of the cysteine. Thus the lack of target binding suggests that the central C488 cysteine is essential for the formation of “open” cavities which can accommodate the epitope sequence of the ca. 26 kDa RBD.
The selectivity of the epitope-imprinted polymer nanofilm at optimal microspotting concentration of the GFNCYFPLQ peptide (25 μM) was studied by evaluating its cross-reactivity towards the human serum albumin (HSA) (Fig. 3). An excellent discrimination of the HSA was observed on the nonapeptide- imprinted spots, i.e. the amount of bound HSA was even lower than on NIP. Similar discrimination was found for MIPs prepared with different surface density of the RBD stemming peptides (Fig. 3A). This suggests that the recognition site density in the studied range influences primarily the binding capacity of the MIPs rather than their selectivity.
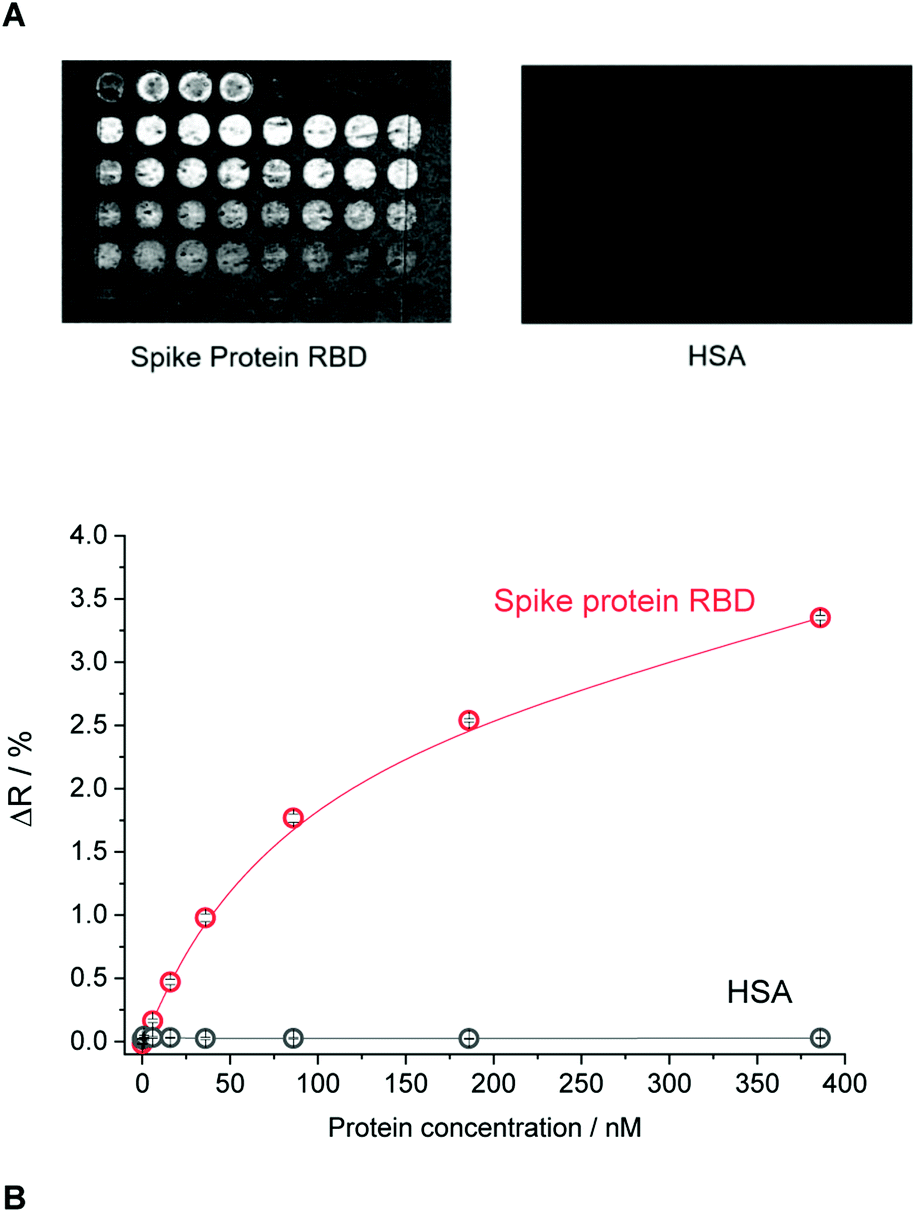 | ||
| Fig. 3 The selectivity of the epitope-imprinted polymer microarray. (A) The differential SPR images of the MIP chip for the layout shown in Fig. 2A exposed to RBD and HSA. (B) Binding isotherms of RBD and HSA to the nonapeptide GFNCYFPLQ-imprinted polyscopoletin spots recorded in PBST. | ||
A major advantage of the SPRi platform is that beside endpoint measurements (measuring the response at equilibrium) it allows the real-time monitoring of the binding events. To demonstrate the practical utility of such measurements we extended the kinetic analysis from PBST solution to 100-fold diluted artificial saliva and a commercial Covid-19 antigen test extraction buffer (Fig. S7†). The concentration of proteins in the diluted extraction buffer and the mucin concentration in the diluted artificial saliva were at least one order of magnitude higher than the RBD concentration. The kinetic analysis (Table 1) confirmed the slight superiority of the nonapeptide-imprinted polymers over the truncated heptapeptide sequence. However, for both MIPs the KD values revealed an even higher affinity to the RBD of the S protein than its natural target (ACE2). In the higher complexity sample matrices, the apparent affinities were significantly smaller, especially in case of the extraction buffer, but still in the applicable range. Remarkably, these experiments revealed the importance of using for imprinting the longer, nonapeptide sequence, with the relevant MIPs showing KD values lower than ca. 20 nM, compared to ca. 60 nM for the heptapeptide-imprinted polymers. This seems to be largely due to a better preservation of the association rate constants, ka, measured in PBST, which suggest better accessibility of the binding sites imprinted with the longer peptide in the presence of a high protein background.
| Solution | Imprinted peptide epitope | k a (M−1 s−1) | k d (s−1) | K D (nM) |
|---|---|---|---|---|
| PBST | GFNCYFP | 1.0 (±0.2) × 105 | 2.2 × 10−4 | 2.2 ± 0.4 |
| GFNCYFPLQ | 1.0 (±0.5) × 105 | 1.2 × 10−4 | 1.6 ± 0.9 | |
| Artificial saliva | GFNCYFP | 4.9 (±0.3) × 104 | 4.2 × 10−4 | 8.3 ± 0.5 |
| GFNCYFPLQ | 9.2 (±0.8) × 104 | 6.0 × 10−4 | 6.4 ± 0.5 | |
| Extraction buffer | GFNCYFP | 1.8 (±0.9) × 104 | 8.1 × 10−4 | 60 ± 32 |
| GFNCYFPLQ | 5.0 (±0.6) × 104 | 1.0 × 10−3 | 21 ± 2.5 |
The measurements in complex matrices confirmed the excellent resistance to non-specific adsorption of the NIPs and also the negative control peptide imprinted polymer spots (Fig. 4). However, in agreement with the decreased target affinity in the complex matrices as compared to PBST, the RBD-specific signal decreased with ca. 25 and 40% for the nonapeptide imprinted spots in the artificial saliva and extraction buffer, respectively (Fig. 4). We expected even higher affinities for the intact virus owing to cooperative binding of multiple RBD units on the surface of the virus to the epitope-imprinted polymers. This aspect was investigated with SARS-CoV-2 virus-like particles (VLPs) (Abnova) composed, beside the spike protein, of 3 other structural proteins (membrane protein (M), nucleocapsid protein (N) and envelope protein (E)). The concentration and integrity of the VLPs formulated in PBST was determined by nanoparticle tracking analysis (NTA). The results of the single particle detection method revealed a narrow distribution with an average particle diameter of 82 ± 2.8 nm (Fig. S8†) and 3.25 × 1011 ± 0.13 × 1011 particles per mL concentration for the stock solution. Furthermore, the measurements confirmed also the stability of the VLPs in PBST. The VLP binding to the GFNCYFPLQ peptide-imprinted polymer spots (see spotting layout in Fig. S1†) was detectable in the femtomolar range (the concentration units refer to the virus particles) in striking contrast with the RBD binding detected only at much higher concentrations (Fig. 5A). The binding of the VLPs to the nonapeptide-imprinted MIP was confirmed by atomic force microscopy (Fig. S9†) that revealed protuberances of ca. 80 nm height and diameter on the surface of the MIP in agreement with the VLP diameter.
 | ||
| Fig. 4 Comparison of the bound RBD (386 nM) to the various peptide-imprinted polymers in PBST, 100-fold diluted artificial saliva and Covid-19 antigen test extraction buffer. | ||
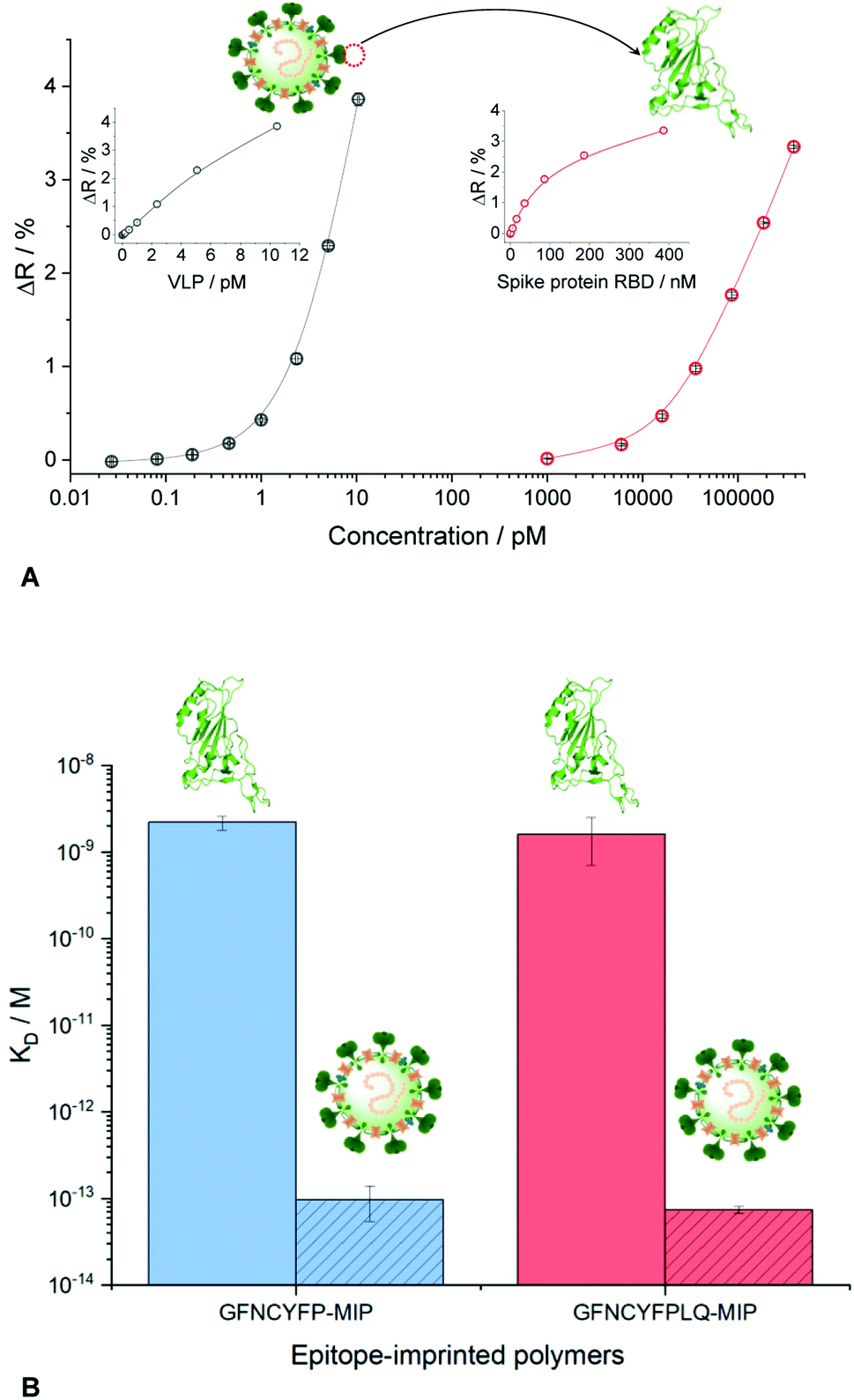 | ||
| Fig. 5 (A) Binding isotherms of SARS-CoV-2 VLP and RBD formulated in PBST to GFNCYFPLQ-imprinted polyscopoletin spots as revealed by SPRi. (B) Comparison of the KD values for the interaction of VLP and RBD with hepta-and nonapeptide imprinted polymer nanofilms. The KD values for the VLPs should be considered estimates given the limitation of the SPR technique to assess subpicomolar KD values. | ||
Comparing the KD values for the MIP–VLP and MIP–RBD interactions confirmed the expectations, i.e. much higher affinity of both the hepta- and nonapeptide imprinted MIPs for VLP with apparent KD values of ca. 100–500 fM (Fig. S10†). Of note, while these values are just estimates given the limitation of the SPR technique to assess subpicomolar KD values, they indicate a remarkable affinity increase with respect of the RBD protein. The increased affinity is reassuringly supported also by the binding curves as shown comparatively in Fig. 5B, i.e. while the RBD binding is detected in the nanomolar concentration range the VLP binding range is shifted to ca. 4 orders of magnitude lower concentrations.
The selectivity of the GFNCYFPLQ epitope-imprinted polymer nanofilm for SARS-CoV-2 VLPs was tested with β-propiolactone-inactivated influenza A (H3N2) virus particles of similar size (ca. 100 nm). As shown in Fig. 6 an excellent discrimination of the influenza A particles was obtained, i.e. less than 5% of the VLP binding signal.
 | ||
| Fig. 6 The selectivity of the GFNCYFPLQ epitope-imprinted polyscopoletin nanofilm to SARS-CoV-2 VLP against inactivated influenza A (H3N2). | ||
Conclusions
The combination of microcontact spotting of peptide epitopes with electropolymerization of scopoletin on SPRi chips offer a versatile platform for both the multiplexed synthesis and screening of epitope-imprinted polymers. The synthetic approach merges for the first time within a microarray format the essential advances of protein-imprinted polymers for protein recognition: epitope imprinting, surface imprinting and oriented template immobilization; along with the controlled surface confinement and mild conditions offered by electrosynthesis. The sequence of the peptide epitopes is essential for highly affine and selective epitope-imprinted polymers and this method can easily quantify, practically on a single chip, their effect on the target binding, along with the adjustment of the optimal surface density to maximize binding capacity. The platform allows also to evaluate the selectivity as well as the effect of the sample matrix on the affinity and kinetic parameters of the target binding. The efficiency of the approach is supported by the convenient generation of epitope-imprinted polyscopoletin ligands that bound the SARS-CoV-2 spike protein RBD with higher affinity than its natural target ACE2. Moreover, this translated into even higher affinity of SARS-CoV-2 VLP binding, along with excellent discrimination of influenza A (H3N2) virus. By extending the smaller format microarray used in this proof of concept study to hundreds of spots would allow the high-throughput screening of epitope-imprinted MIPs for (i) variants, e.g. virus mutants, (ii) homologous proteins and (iii) different protein targets on a single chip.Author contributions
Zs. Bognár: investigation, methodology, data curation, formal analysis, visualization, writing – original draft, writing – review & editing. E. Supala: investigation, methodology, data curation, formal analysis, writing – review & editing. A. Yarman: writing – review & editing. X. Zhang: methodology. F. F. Bier: conceptualization, F. W. Scheller: conceptualization, writing – review & editing, funding acquisition. R. E. Gyurcsányi: conceptualization, methodology, formal analysis, supervision, visualization, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research reported in this paper and carried out at the BME has been supported by the NRDI Fund (TKP2020 IES, BME-IE-NAT) based on the charter of bolster issued by the NRDI Office under the auspices of the Ministry for Innovation and Technology. F. W. S., and A. Y. received support from Germany’s Excellence Strategy–EXC 2008–390540038–UniSysCat. F. F. B. and X. Z. from the German Ministry of Education and Research (BMBF, 01DH20018). We thank Marc Eleveld and Dr Marien I. de Jonge for the production and providing the inactivated influenza A (H3N2) virus.References
- R. Arshady and K. Mosbach, Makromol. Chem., 1981, 182, 687–692 CrossRef CAS.
- G. Wulff and A. Sarhan, Angew. Chem., 1972, 84, 364 CrossRef.
- J. Xu, H. Miao, J. Wang and G. Pan, Small, 2020, 16, 1906644 CrossRef CAS.
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- M. Kempe, M. Glad and K. Mosbach, J. Mol. Recognit., 1995, 8, 35–39 CrossRef CAS PubMed.
- N. W. Turner, C. W. Jeans, K. R. Brain, C. J. Allender, V. Hlady and D. W. Britt, Biotechnol. Prog., 2006, 22, 1474–1489 CrossRef CAS PubMed.
- D. R. Kryscio, M. Q. Fleming and N. A. Peppas, Macromol. Biosci., 2012, 12, 1137–1144 CrossRef CAS.
- C. Sulitzky, B. Rückert, A. J. Hall, F. Lanza, K. Unger and B. Sellergren, Macromolecules, 2002, 35, 79–91 CrossRef CAS.
- A. Menaker, V. Syritski, J. Reut, A. Öpik, V. Horváth and R. E. Gyurcsányi, Adv. Mater., 2009, 21, 2271–2275 CrossRef CAS.
- D. Dechtrirat, K. J. Jetzschmann, W. F. M. Stöcklein, F. W. Scheller and N. Gajovic-Eichelmann, Adv. Funct. Mater., 2012, 22, 5231–5237 CrossRef CAS.
- J. Bognár, J. Szűcs, Z. Dorkó, V. Horváth and R. E. Gyurcsányi, Adv. Funct. Mater., 2013, 23, 4703–4709 CrossRef.
- M. Glad, O. Norrlöw, B. Sellergren, N. Siegbahn and K. Mosbach, J. Chromatogr. A, 1985, 347, 11–23 CrossRef CAS.
- H. Zhang, J. Jiang, H. Zhang, Y. Zhang and P. Sun, ACS Macro Lett., 2013, 2, 566–570 CrossRef CAS.
- A. Nematollahzadeh, W. Sun, C. S. A. Aureliano, D. Lütkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS.
- A. Rachkov and N. Minoura, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1544, 255–266 CrossRef CAS.
- H. Nishino, C.-S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45, 2392–2396 CrossRef CAS.
- K. Yang, S. Li, L. Liu, Y. Chen, W. Zhou, J. Pei, Z. Liang, L. Zhang and Y. Zhang, Adv. Mater., 2019, 31, 1902048 CrossRef CAS PubMed.
- M. Cieplak, K. Szwabinska, M. Sosnowska, B. K. C. Chandra, P. Borowicz, K. Noworyta, F. DSouza and W. Kutner, Biosens. Bioelectron., 2015, 74, 960–966 CrossRef CAS PubMed.
- J. U. Klein, M. J. Whitcombe, F. Mulholland and E. N. Vulfson, Angew. Chem., Int. Ed., 1999, 38, 2057–2060 CrossRef CAS.
- J. Kalecki, Z. Iskierko, M. Cieplak and P. S. Sharma, ACS Sens., 2020, 5, 3710–3720 CrossRef CAS PubMed.
- R. Xing, Y. Ma, Y. Wang, Y. Wen and Z. Liu, Chem. Sci., 2019, 10, 1831–1835 RSC.
- M. M. Titirici, A. J. Hall and B. Sellergren, Chem. Mater., 2003, 15, 822–824 CrossRef CAS.
- K. Yang, S. Li, J. Liu, L. Liu, L. Zhang and Y. Zhang, Anal. Chem., 2016, 88, 5621–5625 CrossRef CAS.
- G. Caserta, X. Zhang, A. Yarman, E. Supala, U. Wollenberger, R. E. Gyurcsányi, I. Zebger and F. W. Scheller, Electrochim. Acta, 2021, 381, 138236 CrossRef CAS.
- H. Cubuk, M. Ozbil and P. Cakir Hatir, Comput. Theor. Chem., 2021, 1199, 113215 CrossRef CAS.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- J. Liu, K. Yang, Q. Deng, Q. Li, L. Zhang, Z. Liang and Y. Zhang, Chem. Commun., 2011, 47, 3969–3971 RSC.
- T. Takeuchi, D. Fukuma and J. Matsui, Anal. Chem., 1999, 71, 285–290 CrossRef CAS.
- B. Dirion, Z. Cobb, E. Schillinger, L. I. Andersson and B. Sellergren, J. Am. Chem. Soc., 2003, 125, 15101–15109 CrossRef CAS.
- J. Erdőssy, V. Horváth, A. Yarman, F. W. Scheller and R. E. Gyurcsányi, TrAC, Trends Anal. Chem., 2016, 79, 179–190 CrossRef.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS.
- P. Çakir, A. Cutivet, M. Resmini, B. T. S. Bui and K. Haupt, Adv. Mater., 2013, 25, 1048–1051 CrossRef PubMed.
- A. Bossi, S. A. Piletsky, E. V. Piletska, P. G. Righetti and A. P. F. Turner, Anal. Chem., 2001, 73, 5281–5286 CrossRef CAS.
- M. Bosserdt, J. Erdőssy, G. Lautner, J. Witt, K. Köhler, N. Gajovic-Eichelmann, A. Yarman, G. Wittstock, F. W. Scheller and R. E. Gyurcsányi, Biosens. Bioelectron., 2015, 73, 123–129 CrossRef CAS.
- E. Supala, L. Tamás, J. Erdőssy and R. E. Gyurcsányi, Electrochem. Commun., 2020, 119, 106812 CrossRef CAS.
- A. Raziq, A. Kidakova, R. Boroznjak, J. Reut, A. Öpik and V. Syritski, Biosens. Bioelectron., 2021, 178, 113029 CrossRef CAS PubMed.
- N. Cennamo, G. Dagostino, C. Perri, F. Arcadio, G. Chiaretti, E. M. Parisio, G. Camarlinghi, C. Vettori, F. Di Marzo, R. Cennamo, G. Porto and L. Zeni, Sensors, 2021, 21, 1681 CrossRef CAS.
- P. Holenya, P. J. Lange, U. Reimer, W. Woltersdorf, T. Panterodt, M. Glas, M. Wasner, M. Eckey, M. Drosch, J.-M. Hollidt, M. Naumann, F. Kern, H. Wenschuh, R. Lange, K. Schnatbaum and F. F. Bier, Eur. J. Immunol., 2021, 51, 1839–1849 CrossRef CAS PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C.-L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- C. Corrêa Giron, A. Laaksonen and F. L. Barroso da Silva, Virus Res., 2020, 285, 198021 CrossRef.
- R. Kumavath, D. Barh, B. S. Andrade, M. Imchen, F. F. Aburjaile, A. Ch, D. L. N. Rodrigues, S. Tiwari, K. J. Alzahrani, A. Góes-Neto, M. E. Weener, P. Ghosh and V. Azevedo, Front. Immunol., 2021, 12, 663912 CrossRef CAS.
- Z. Liu, X. Xiao, X. Wei, J. Li, J. Yang, H. Tan, J. Zhu, Q. Zhang, J. Wu and L. Liu, J. Med. Virol., 2020, 92, 595–601 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Nature, 2020, 581, 215–220 CrossRef CAS.
- X. Zhang, A. Yarman, J. Erdossy, S. Katz, I. Zebger, K. J. Jetzschmann, Z. Altintas, U. Wollenberger, R. E. Gyurcsányi and F. W. Scheller, Biosens. Bioelectron., 2018, 105, 29–35 CrossRef CAS.
- K. J. Jetzschmann, G. Jágerszki, D. Dechtrirat, A. Yarman, N. Gajovic-Eichelmann, H.-D. Gilsing, B. Schulz, R. E. Gyurcsányi and F. W. Scheller, Adv. Funct. Mater., 2015, 25, 5178–5183 CrossRef CAS.
- Z. Stojanovic, J. Erdőssy, K. Keltai, F. W. Scheller and R. E. Gyurcsányi, Anal. Chim. Acta, 2017, 977, 1–9 CrossRef CAS PubMed.
- L. Simon, G. Lautner and R. E. Gyurcsányi, Anal. Methods, 2015, 7, 6077–6082 RSC.
- L. Simon and R. E. Gyurcsányi, Anal. Chim. Acta, 2019, 1047, 131–138 CrossRef CAS PubMed.
- R. Karlsson, P. S. Katsamba, H. Nordin, E. Pol and D. G. Myszka, Anal. Biochem., 2006, 349, 136–147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04502d |
| This journal is © The Royal Society of Chemistry 2022 |