
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaterials by design at high pressures
Meiling
Xu
 a,
Yinwei
Li
*a and
Yanming
Ma
*bc
a,
Yinwei
Li
*a and
Yanming
Ma
*bc
aLaboratory of Quantum Functional Materials Design and Application, School of Physics and Electronic Engineering, Jiangsu Normal University, Xuzhou 221116, China. E-mail: yinwei_li@jsnu.edu.cn
bState Key Laboratory of Superhard Materials & International Center for Computational Method and Software, College of Physics, Jilin University, Changchun 130012, China. E-mail: mym@jlu.edu.cn
cInternational Center of Future Science, Jilin University, Changchun 130012, China
First published on 9th December 2021
Abstract
Pressure, a fundamental thermodynamic variable, can generate two essential effects on materials. First, pressure can create new high-pressure phases via modification of the potential energy surface. Second, pressure can produce new compounds with unconventional stoichiometries via modification of the compositional landscape. These new phases or compounds often exhibit exotic physical and chemical properties that are inaccessible at ambient pressure. Recent studies have established a broad scope for developing materials with specific desired properties under high pressure. Crystal structure prediction methods and first-principles calculations can be used to design materials and thus guide subsequent synthesis plans prior to any experimental work. A key example is the recent theory-initiated discovery of the record-breaking high-temperature superhydride superconductors H3S and LaH10 with critical temperatures of 200 K and 260 K, respectively. This work summarizes and discusses recent progress in the theory-oriented discovery of new materials under high pressure, including hydrogen-rich superconductors, high-energy-density materials, inorganic electrides, and noble gas compounds. The discovery of the considered compounds involved substantial theoretical contributions. We address future challenges facing the design of materials at high pressure and provide perspectives on research directions with significant potential for future discoveries.
1 Introduction
Pressure, a fundamental thermodynamic variable, can dramatically reduce interatomic distances and alter chemical bonds to induce structural transformations and new high-pressure phases.1,2 In general, each material is expected to experience several structural transformations when compressed up to a pressure of a million atmospheres. Additionally, new oxidation states appearing under high pressure can stabilize new compounds with unconventional stoichiometries.3–7 As an example, we can consider the unconventional Na3Cl and NaCl3 stoichiometries of Na and Cl that form under pressure rather than the known chemical species NaCl.8 These new high-pressure phases and compounds often exhibit exotic physical and chemical properties and are thus potentially functional materials. Pressure is therefore an efficient tool in developing new materials that are inaccessible at ambient pressure.Since the first achievement of lab pressure at ∼10 GPa in a so-called large-volume apparatus obtained by Percy Bridgeman in 1905, many high-pressure generating techniques have been developed.9–12 This includes the static-pressure generating equipment such as diamond anvil cells (DAC) and large-volume presses (multiple-anvil system, piston-cylinder devices and sealed vessel systems), and large-scale shock-wave facilities that generate dynamic compression. DAC is the most widely used device to generate steady-state pressure with a record upper pressure of above 1000 GPa (ref. 12) achieved by using nanodiamonds as second-stage anvils in double-stage DAC, much higher than that (∼30 GPa (ref. 13)) generated by large-volume presses. Dynamic-compression facilities could generate pressures well above terapascal by using shock waves produced by gas guns, laser-driven compression or hemispherically converging explosives, capable of reaching 5000 GPa on a ∼1 mm3 sample for a fraction of a second.13 The fairly high upper frontiers of pressure expanded tremendously the searching space for new functional materials.
Exploration of new materials often involves experimental trial and error, with a range of possible compounds being synthesized and compared to identify the best. This is a time-consuming and expensive process. A classic example is Edison's testing of approximately 8000 materials over two years to find a material durable enough to provide electric lighting for 1000 hours. An experimental search for target functional materials among the large number of new phases and unconventional compounds available under high pressure would be challenging. Considering hydrides as an example, little experimental progress in these potential high-temperature superconductors was made before 2014 due to the great variety of hydrides at high pressure. Only when theoretical work explicitly identified highly compressed H2S as a superconductor,14 the H3S superconductor with a critical temperature (Tc) of 200 K was subsequently developed.15 This demonstrates the successes possible when theoretical and experimental efforts are applied together to the discovery of new materials at high pressure.
Developments in crystal structure prediction (CSP) and first-principles calculations have improved the accuracy of the prediction of the structures and properties of new phases and unconventional compounds formed at high pressure, thus prompting the discovery of new materials by design.15–20 Unbiased CSP methods without any experimental input can predict the energetically most stable or metastable structures at a given pressure, leading to the construction of the phase diagram of a considered system. First-principles calculations can simulate the mechanical, thermal, optical, electronic, and magnetic properties of these high-pressure unconventional compounds and the pressure and temperature conditions for their synthesis, thus allowing theoretical findings to guide the choice of experiments attempted. In 2011, the U.S.A., Japan, and China successively announced the Materials Genome Projects, aiming to combine theory and experiment to accelerate the development of new materials.
Theoretical design for high-pressure synthesis has aided several major experimental discoveries of new materials. A milestone example is the theory-initiated discovery of the record-breaking high-temperature superconductors H3S14,15,21 and LaH10 (ref. 6, 7, 22 and 23) with Tc values of 200 K and 260 K, respectively. Other discoveries include high-energy-density materials (e.g., cg-N,24,25 layered Pba2 polymeric nitrogen,26 and LiN5 (ref. 27)), inorganic electrides (e.g., Na,16 Ca2N,28 and Sr5P3 (ref. 29)), and noble gas compounds (e.g., XeFe3,4 XeNi3,4 Na2He,5 and Xe3O2 (ref. 30)).
This review surveys these materials, with particular emphasis on their theory-oriented discovery at high pressure. First, we highlight the effect of pressure on materials and discuss the stabilization of new materials. Second, the theoretical methods for material design, especially CSP methods, are discussed. Third, we summarize and discuss recent discoveries of materials at high pressure initiated by theoretical design. We particularly focus on hydrogen-rich superconductors, high-energy-density materials, inorganic electrides, and noble gas compounds. Our work concludes with descriptions of the main drawbacks of high-pressure materials and a discussion of the possible solutions and future research directions. Actually, these available reviews1,2,31–33 either focus on new materials with a given function such as superconductivity, or summarize all designed materials at high pressure. Here, our review intends to summarize those materials discovery initiated by theoretical design at high pressure, with a purpose to highlight the leading role of structure prediction in material discovery.
2 Effect of pressure on materials
High pressure provides two main routes to stabilize new materials. First, the decrease in the interatomic distance under compression redistributes valence electrons and alters bonding patterns, thereby effectively modulating the relative energetic stability of possible structures on the potential energy surface and generating new high-pressure phases via structural transformations. A typical example is the pressure-induced phase transformation of sp2-hybrid graphite to sp3-hybrid diamond.34 Pressure helps overcome the energy barrier for the conversion between the two carbon phases, making diamond the global energy minimum. Each element or compound undergoes several structural transformations at high pressure. For example, at least six phase transformations have been observed or predicted for the alkali metal Na (bcc → fcc (65 GPa) → cI16 (103 GPa) → tI19 (156 GPa) → hP4 (200 GPa) → oP8 (1.75 TPa) → cI24 (>15.5 TPa)),16,35 among which the transformation into the transparent hP4 phase is particularly interesting as it characterizes a so called anti-Wilson transition, i.e., transformation of a good metal into an insulator, that violates the traditional wisdom of Wilson transition.16 Many high-pressure phases with exotic properties have been discovered with potential applications, including superconducting phases (e.g., H2S14 and AlH3 (ref. 36)), polymeric nitrogen (e.g., cg-N,24,25 LP-Pba2 (ref. 26) and LP-N37), and electrides (e.g., Li38 and Na16,35).Pressure can also stabilize compounds and phases by reordering the energies of outer atomic orbitals, causing charge transfer between different orbitals, thus modifying the chemical identity of atoms via the appearance of new oxidation states and leading to the formation of unconventional compounds that are inaccessible at ambient pressure. For example, the alkali metal Cs shows oxidation states of +3 and +5 at high pressure rather than the +1 state observed at ambient pressure, as evidenced by the unconventional CsF3 and CsF5 compounds predicted at high pressure in addition to the typical CsF39 known at ambient pressure. Another example is the formation of compounds of the inert element Xe. Its fully occupied 5p valence states can be partially excited to unoccupied orbitals at high pressure, activating the Xe 5p electrons to form compounds such as XeFe3.4 Advanced structure prediction methods have predicted various unconventional compounds formed at high pressure that have since been experimentally confirmed, including high-Tc superconducting hydrides (e.g., H3S,14,15,21 LaH10,6,7 YH6,40 and C–S–H41,42), high-energy-density nitrogen-rich compounds (e.g., LiN5 (ref. 27) and CsN5 (ref. 43)), electrides (e.g., Sr5P3 (ref. 29) and Na2He5), and noble gas compounds (e.g., XeFe3,4 XeFe5,4 and Na2He5).
3 Theoretical methods for material design
The crystal structure is one of the most fundamental pieces of information needed to characterize a material. Its prediction for a given compound at high pressures without any prior information is therefore a primary task in material design. This is now possible due to the development of CSP methods such as Crystal structure AnaLYsis by Particle Swarm Optimization (CALYPSO) (based on particle swarm optimization44,45), ab initio random structure searching (AIRSS) (based on random sampling46), Universal Structure Predictor: Evolutionary Xtallography (USPEX) (based on a genetic algorithm47), Xtallography Optimization (XtalOpt),48 Global Space-Group Optimization (GSGO),49 and Evolutionary Algorithm (EVO).50These methods have been proven efficient in identifying the global energy minimum among the vast number of local minima on the potential energy surface, making them powerful tools for materials by design.51–61 Consider, for example, a binary system formed by elements A and B; material-by-design research requires construction of a convex hull map of composition vs. formation enthalpy to screen the thermodynamically most stable stoichiometries. This can be achieved via systematic structure predictions of candidate AxBy stoichiometries at selected pressures through the combination of CSP and first-principles calculations. Once the most stable structure of each AxBy is obtained, a formation enthalpy vs. composition convex hull can be constructed to screen out the thermodynamically stable compounds. The convex hull presents also the possible precursors to synthesize the candidate stable compounds, providing essential information to guide the subsequent experiments. Finally, the useful properties (e.g., electronic band structure, phonons, and electron-phonon coupling parameters, etc.) of the stable compound can be simulated by performing first-principle calculations.
4 Theory-oriented discovery of pressure-induced materials
Here, we summarize and discuss recent accomplishments in material discovery at high pressure initiated by theoretical design. These materials include hydrogen-rich superconductors, high-energy-density materials, inorganic electrides, and noble gas compounds.4.1 Hydrogen-rich superconductors
Room-temperature superconductivity is the holy grail of condensed-matter physics. Before 2014, there is no report on the Tc values that have ever exceeded 40 K for conventional superconductors. Hydrogen-rich compounds at high pressure have long been sought as high Tc superconductors.62 However, the large amount of unknown hydrogen-containing compounds that can possibly exist under high pressure poses a great challenge to experimentalists seeking candidate hydrides to test. Little progress was initially made experimentally, but theoretical designs of hydrogen-rich superconductors by the aid of CSP methods have led to a series of experimental breakthroughs. The two main categories of superconducting hydrides discovered thus far are the covalent H3S and the ionic clathrate structured, the superconductivity of which arises mainly through S–H covalent bonds and caged H sublattices with encaged metals, respectively (Table 1).| Materials | Brief description | Year | References |
|---|---|---|---|
| H–S | The first theoretical prediction of H2S with 80 K superconductivity at 160 GPa | 2014 (September) | 14 |
| Theoretical prediction of H3S with ∼200 K superconductivity at 200 GPa | 2014 (November) | 21 | |
| Experimental confirmation of the superconductivity in compressed H2S and the observation of the record 203 K superconductivity | 2015 | 15 | |
| Experimental confirmation of the pressure-driven disproportionation of H2S to H3S, contributing the 203 K superconductivity | 2016 | 66 and 69 | |
| C–S–H | Theoretical prediction of a metastable CSH7 with 190 K superconductivity at 150 GPa | 2021 (April) | 42 |
| Theoretical prediction of a metastable CSH7 with 181 K superconductivity at 100 GPa | 2021 (May) | 41 | |
| Experimental synthesis of CSHx with 288 K superconductivity at 267 GPa | 2021 (October) | 20 | |
| LaH10 | Theoretical prediction of LaH10 with 255–288 K superconductivity at 250 GPa | 2017 | 6 and 7 |
| Experimental observation of superconductivity at 260 K in LaH10 by compressing La and hydrogen at 200 GPa | 2019 (January) | 22 | |
| Experimental observation of superconductivity at 250 K in LaH10 by compressing NH3BH3 and La at 170 GPa | 2019 (May) | 23 | |
| CaH6 | The first predicted clathrate hydride with superconductivity of 235 K at 150 GPa | 2012 | 79 |
| Experimental synthesis of CaH6 with 215 K superconductivity at 172 GPa | 2021 | 81 |
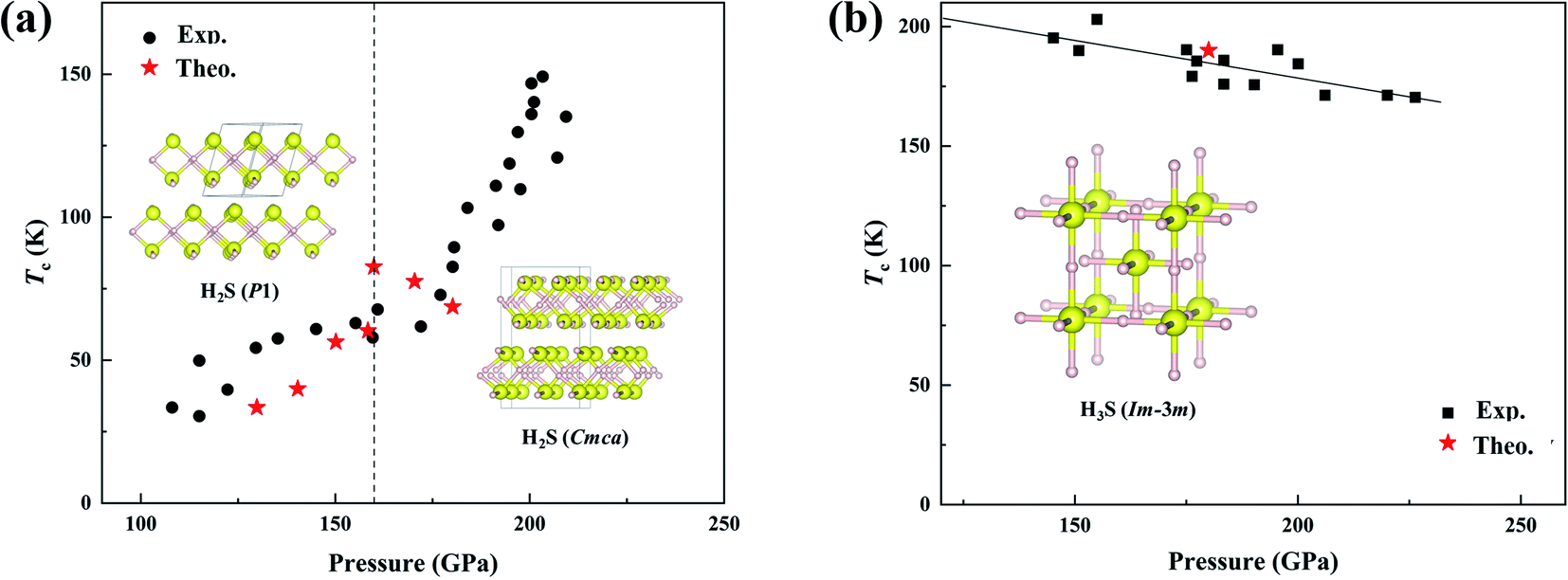 | ||
| Fig. 1 (a) Comparison of the calculated14 and measured Tc for H2S pressurized at low temperature.15 Insets show two predicted structures.14 (b) Comparison of the calculated21 and measured Tc for H2S pressurized at high temperature.15 The inset shows the predicted structure.21 | ||
Theoretical studies have demonstrated that strong covalent bonding is a key determinant of the high superconductivity in H3S, thus providing a new route to design superconducting hydrides. Many attempts have been made to design covalent superconducting hydrides, including H3Se,71 PH2,72 PH3,73,74 TeH4,75 H2Cl/Br/I,76,77 H6SSe,78 and C–S–H,20,41,42 as summarized in Fig. 2. Two independent theoretical studies in early 2020 proposed that the intercalation of methane (CH4) into the H3S framework forms a new candidate superconductor, CSH7, with an estimated Tc of 100–190 K.41,42 This proposal stimulated subsequent experimental synthesis of CSHx by compressing elemental C, S, and H2 above 4 GPa, where a maximum Tc of ∼288 K at ∼267 GPa (ref. 20) was claimed. However, the undetermined value of x and lack of structural information prevent elucidation of the origin of the superconductivity. We expect future experimental confirmation from an independent group for the record high superconductivity observed in CSHx and theoretical studies, especially those using CSP, may help provide further details on the structures.
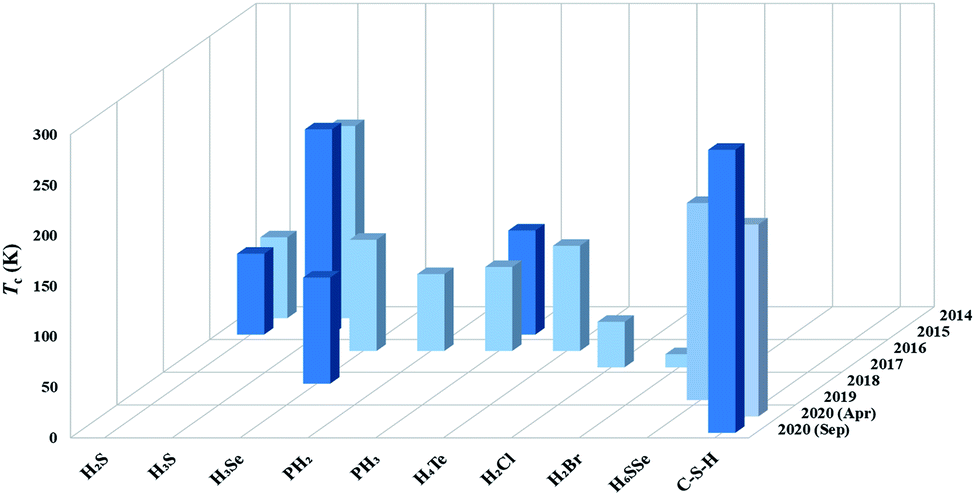 | ||
| Fig. 2 Computed (light blue) and measured (dark blue) Tc of covalent hydrides since 2014. | ||
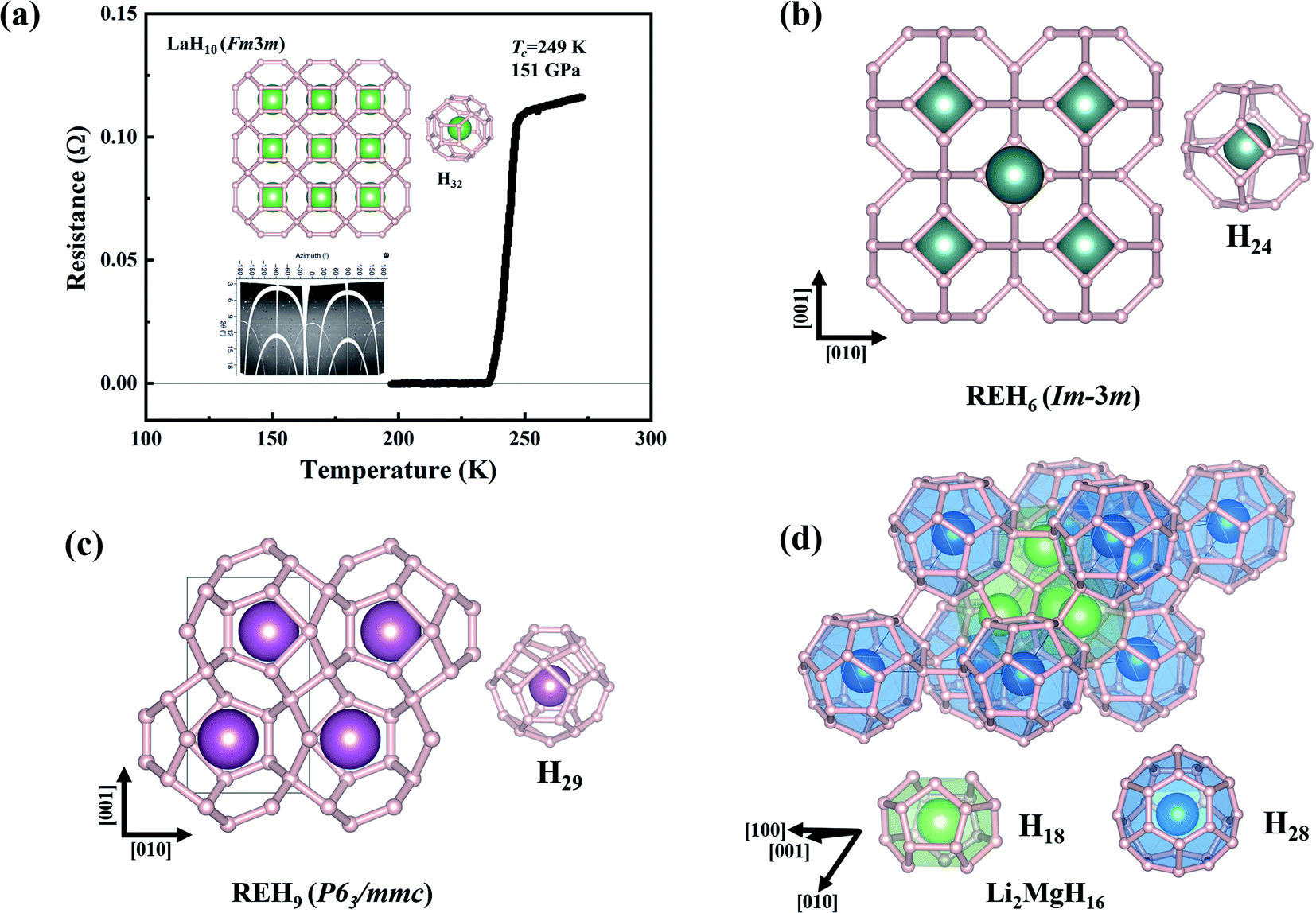 | ||
| Fig. 3 (a) Measured resistance as a function of temperature of LaH10 at 151 GPa.23 The inset presents the predicted clathrate structure. (b–d) Three typical clathrate structures of superhydrides formed at high pressures. | ||
Theoretical studies have established the crucial contribution of hydrogen cages to the high superconductivity of the above clathrate hydrides. The cages represent another pathway to the design of high-Tc superconductors, namely, pressure-stabilized clathrate superhydrides. Ref. 11 proposed that there is common rule for the formation of clathrate structures in rare-earth (RE) superhydrides with appearing three different H24, H29, and H32 cages in stoichiometric superhydrides of REH6, REH9, and REH10, respectively. Subsequent theoretical attempts have further confirmed hydrogen cages as common species at high pressure.6,7,82–94Fig. 4a summarizes the cage-structured hydrides predicted at high pressure, most of which show high superconductivity, such as Tc at 473 K in H18 + H28 cage-structured Li2MgH16,88Tc at ∼146–243 K in H29 cage-structured Y(Ce, Th)H9,6,86,94Tc at 160–303 K in H32 cage-structured Y(Th, Ac)H10,6,7,86,93 and Tc at 173 K in H40 cage-structured AcH12.93 Besides the above mentioned experimental confirmation on the theoretical prediction of high superconductivity in CaH6 and YH6, subsequent experimental studies have also confirmed high superconductivities in, for example, YH9,95 CeH9,91 ThH9,92 and ThH10 (ref. 92) with measured Tc values at ∼262, 110, 146 and 160 K, respectively. These successful examples on the theory-orientated finding of high Tc superconductors among pressure-stabilized superhydrides demonstrate that the leading role of theoretical methods.
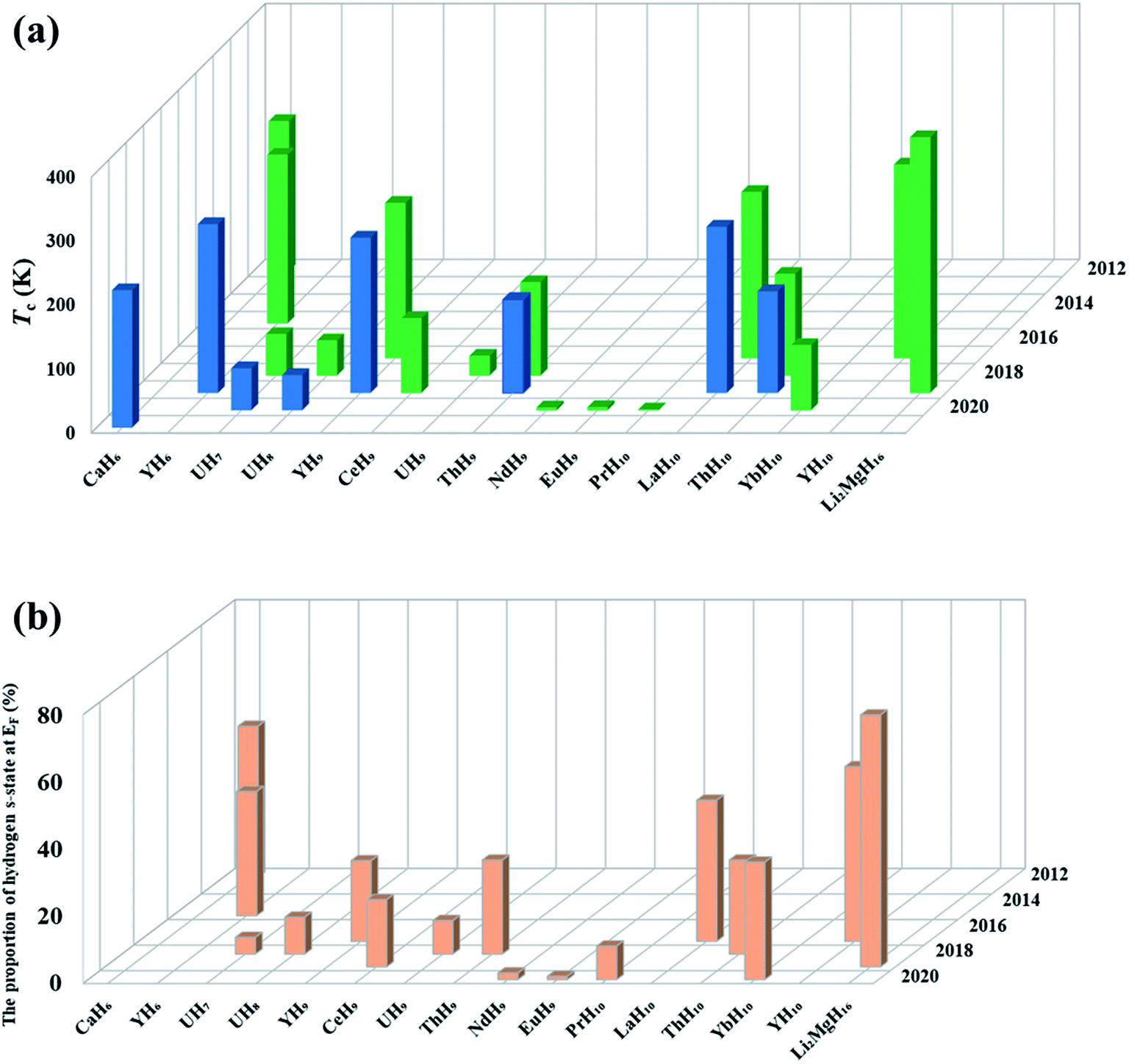 | ||
| Fig. 4 (a) Summary of the computed (green) and measured (blue) Tc of several typical superconducting superhydrides. (b) Proportion of H-s electron states at the Fermi level for selected superconducting superhydrides. | ||
The superconducting Tc of hydrides can be estimated by using the Allen–Dynes modified McMillan equation, which is suitable for those compounds with electron-phonon coupling parameter (λ) less than 1.5. However, the recent studies have demonstrated that certain superhydrides have very strong electron-phonon coupling parameters λ (e.g., 2.19 for H3S, 2.69 for CaH6, and ∼4 for Li2MgH16), posing a challenge to use Allen–Dynes modified McMillan equation for the calculation of the Tc values for the predicted hydrides. An alternative method is to estimate Tc directly from the spectral function (α2F(ω)) by numerically solving the Eliashberg equations,103 which gives a better description of systems with λ larger than 1.5.
A limitation of high-Tc hydrides is that their stability is maintained only under extremely high pressures, which precludes any immediate practical application. A research aim is therefore to search for superhydrides that can be synthesized at moderate pressures and are quenchable to ambient conditions as metastable phases. Currently, ternary hydrides display high potential as high-Tc superconductors, as exemplified by CSHx (288 K)20 and Li2MgH16 (473 K).88 We expect that the relatively more complex interactions in ternary systems could form hydrides at moderate pressures. Theoretical predictions, especially those employing CSP methods, will undoubtedly play a key role in achieving this goal.
4.2 Polymeric nitrogen
Solid nitrogen at ambient pressure adopts a diatomic molecular form, characterized by strong triple N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds within each N2 molecule (Fig. 5a). High pressure can efficiently break the triple bonds to form single N–N bonds, allowing the formation of polymeric phases. The large energy difference between the single (∼160 kJ mol−1) and triple (∼954 kJ mol−1) bonds means that the transformation from single to triple bonds releases much energy, making polymeric nitrogen or nitrides containing N–N bonds ideal high-energy-density materials. Theoretical studies have guided experimental efforts to synthesize the proposed high-energy-density materials. Here, we provide an overview of recent discoveries initiated by theoretical design of high-energy-density materials at high pressure, specifically polymeric nitrogen crystals and N-rich compounds (Table 2).
N bonds within each N2 molecule (Fig. 5a). High pressure can efficiently break the triple bonds to form single N–N bonds, allowing the formation of polymeric phases. The large energy difference between the single (∼160 kJ mol−1) and triple (∼954 kJ mol−1) bonds means that the transformation from single to triple bonds releases much energy, making polymeric nitrogen or nitrides containing N–N bonds ideal high-energy-density materials. Theoretical studies have guided experimental efforts to synthesize the proposed high-energy-density materials. Here, we provide an overview of recent discoveries initiated by theoretical design of high-energy-density materials at high pressure, specifically polymeric nitrogen crystals and N-rich compounds (Table 2).
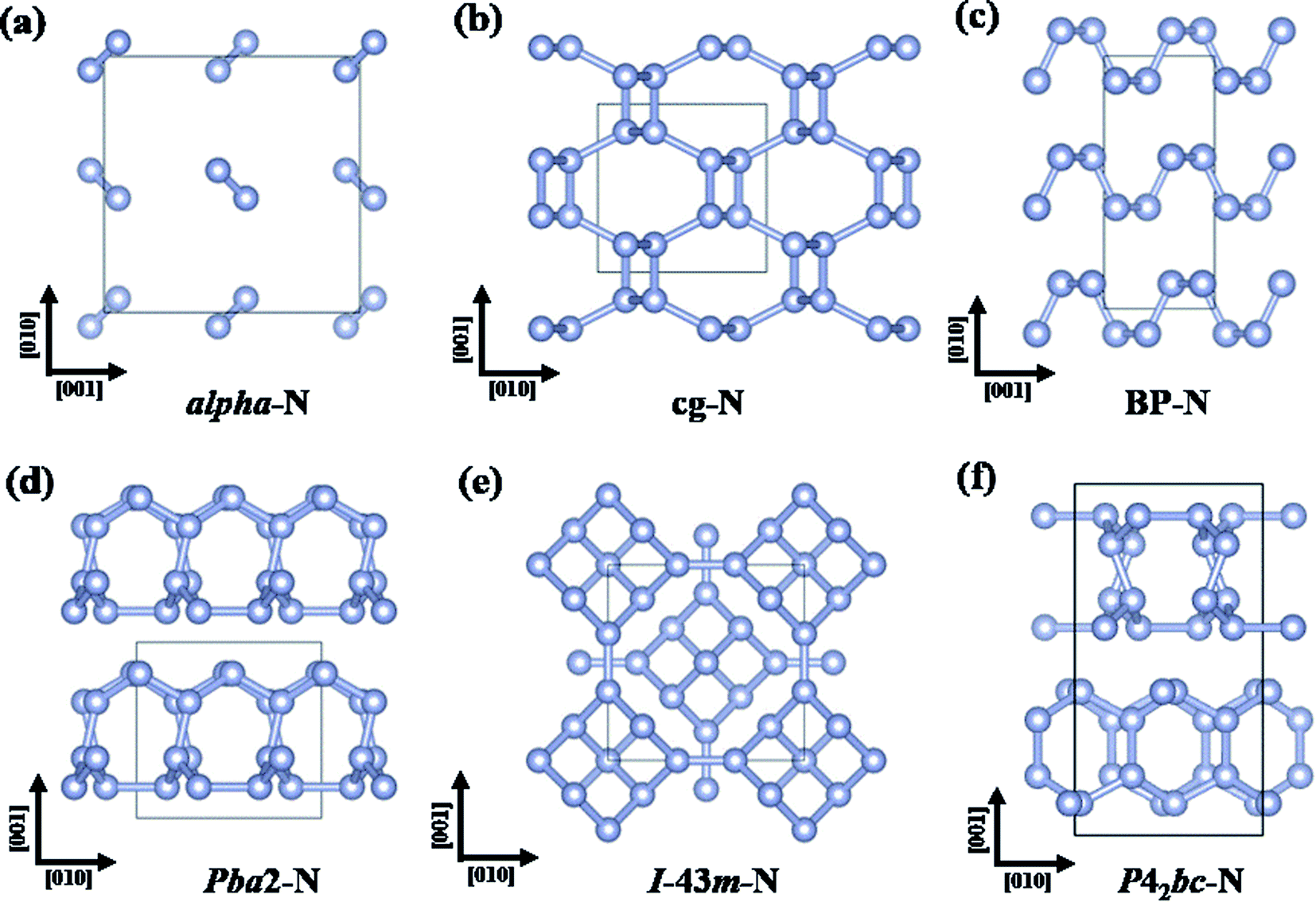 | ||
| Fig. 5 Crystal structures of (a) alpha-N, (b) cg-N, (c) BP-N, (d) Pba2-N, (e) I-43m-N, and (f) P42bc-N. | ||
| Materials | Brief description | Year | References |
|---|---|---|---|
| cg-N | The first theoretical predicted polymetric nitrogen at ∼50 GPa | 1992 | 104 |
| Experimental synthesis of cg-N at 110 GPa and 2000 K | 2004 | 24 and 25 | |
| Pba2-N | Theoretical prediction of stable layered polymeric N at 188–263 GPa | 2009 | 26 |
| Experimental synthesis of Pba2-N at 125–180 GPa | 2014 | 37 | |
| P42bc-N | Theoretical prediction of a metastable polymeric N | 2012 | 111 |
| Experimental synthesis of P42bc-N at 250 GPa | 2019 | 115 | |
| LiN5 | Theoretical prediction of stable LiN5 containing a polymeric N5 framework above 9.9 GPa | 2015 | 27 |
| Experimental synthesis of LiN5 by compressing LiN3 and N2 at ∼40 GPa | 2020 | 136 | |
| Experimental synthesis of LiN5 by compressing pure lithium and nitrogen at ∼45 GPa | 2018 | 137 and 138 | |
| WN6 | Theoretical predictions of stable WN6 containing N6 rings at 16 GPa | 2017 | 139 |
| Theoretical predictions of stable WN6 containing N6 rings at 65 GPa | 2018 | 140 | |
| Experimental synthesis of WN6 at 126 GPa | 2021 | 141 |
Many other stable or metastable polymeric nitrogen phases at high pressures have also been predicted by CSP methods.26,107–114 Among the most-energetically stable phases, a phase sequence of cg-N → orthorhombic LP-Pba2 (188 GPa)26 → bcc I-43m (263 GPa)111 → orthorhombic Cmca (2200 GPa)112 → tetragonal P4/nbm (2500 GPa)112 was suggested (Fig. 5b–d). Besides, a tetragonal P42bc structure (Fig. 5f) was predicted to have energy indistinguishable from that of the Pba2 structure.111 Among these structures, LP-Pba2 and P42bc have been experimentally synthesized at 125–180 (ref. 37) and ∼250 GPa,115 respectively.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonded linear N3− anions initially attracted attention, as double NN bonds with lower bonding energy (418 kJ mol−1) than triple NN bonds could transform to single bonds much more easily than pure N2. Indeed, theoretical studies have shown that azides can polymerize at low pressures of ∼50 GPa, with the formation of pseudobenzene N6 rings containing both double NN and single N–N bonds (Fig. 6). However, the pressures needed to form three-dimensional polymerized nitrogen networks are unexpectedly high. For example, NaN3 requires 150 GPa, and other azides require pressures exceeding 200 GPa (Fig. 6).116–122 Extensive experiments have been performed to investigate the polymerization of nitrogen in azides, and the dissociation of double NN bonds was only observed in NaN3 considering the disappearance of molecular vibrations above 120 GPa.123 However, polymerization of nitrogen has not been observed in other azides, as the experimental pressures considered for other azides are lower than 50 GPa.124–129 We expect that future high-pressure experiments might observe polymerization of nitrogen in other azides.
N bonded linear N3− anions initially attracted attention, as double NN bonds with lower bonding energy (418 kJ mol−1) than triple NN bonds could transform to single bonds much more easily than pure N2. Indeed, theoretical studies have shown that azides can polymerize at low pressures of ∼50 GPa, with the formation of pseudobenzene N6 rings containing both double NN and single N–N bonds (Fig. 6). However, the pressures needed to form three-dimensional polymerized nitrogen networks are unexpectedly high. For example, NaN3 requires 150 GPa, and other azides require pressures exceeding 200 GPa (Fig. 6).116–122 Extensive experiments have been performed to investigate the polymerization of nitrogen in azides, and the dissociation of double NN bonds was only observed in NaN3 considering the disappearance of molecular vibrations above 120 GPa.123 However, polymerization of nitrogen has not been observed in other azides, as the experimental pressures considered for other azides are lower than 50 GPa.124–129 We expect that future high-pressure experiments might observe polymerization of nitrogen in other azides.
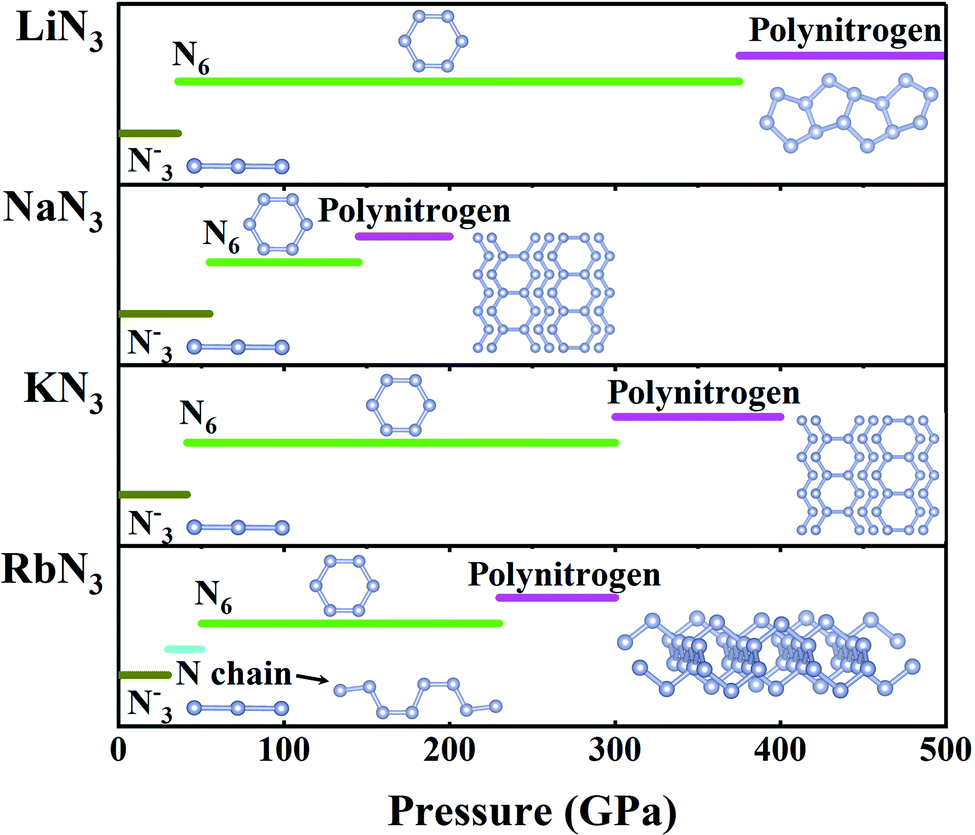 | ||
| Fig. 6 Polymerization of nitrogen in alkali metal azides at high pressure. | ||
Studies have demonstrated that high pressure can stabilize unconventional nitrogen-rich compounds with high ratios of nitrogen, providing another route to polymerization of nitrogen. Theoretical calculations have predicted that many unconventional nitrides, MN4–10 (M = Li, Be, Na, Mg, Ca, Cr, Ru, or OS), stabilize at high pressure.3,130–135 In 2015, unconventional LiN5 containing a polymeric N5−1 framework that becomes energetically stable at pressures as low as 9.9 GPa was predicted in addition to conventional LiN3.27 The study also proposed compressing solid LiN3 and N2 as a feasible way to synthesize LiN5; subsequent experimental work successfully synthesized LiN5 by compressing LiN3 and N2 at pressures up to ∼40 GPa.136 Two independent experiments also synthesized LiN5 by compressing pure lithium embedded in a much greater quantity of molecular nitrogen at 45 GPa.137,138 The discovery of CsN5 was similar. It was first predicted to be energetically stable at 9.1 GPa, and the first synthesis was achieved by compressing a mixture of CsN3 and N2 to 60 GPa.43 Another example is WN6, which was hypothesized to contain single-bonded armchair-like N6 rings and was first proposed to be stable at 16 GPa (ref. 139) or 65 GPa,140 and it was recently successfully synthesized at a much higher pressure of 126 GPa.141
N bond is the strongest bond in nature, making a need for an extra pressure or temperature to overcome the energy barrier for the transformation of a triple bond into a single bond. For example, in actual experiments, an extra pressure of ∼60 GPa evidences as compared to the theoretical one for the synthesis of cg-N and WN6. However, it is still a challenge to evaluate the exact extra pressure needed to synthesize the predicted nitrides. One might calculate energy barrier by using methods such as climbing image nudged elastic band for a good estimation of the aforementioned extra pressure. Besides the above mentioned theoretical examples that received the subsequent experimental confirmation, many other proposed nitrogen-rich compounds3,130–135,142–146 are to be verified, representing significant challenges to experimentalists. Furthermore, although nitrogen-rich compounds could be synthesized at low pressure and even recovered under ambient conditions, their energy densities are lower than that (9.7 kJ g−1) of pure cg-N due to the introduction of other elements, for example, 3.48 kJ g−1 in MgN10 (ref. 147) and 5.39 kJ g−1 in BeN10.147 Theoretical studies have identified two candidate routes to design new materials with high energy density. One is to search for nitrides with extremely high nitrogen contents, such as HeN22,148 whose energy density reaches 10.44 kJ g−1. The other is to design pure polymeric nitrogen by removing the non-N elements from compounds that can be obtained at high pressure; for example, polymeric t-N has been predicted to be achievable by removing He from HeN4 (ref. 149) and to have a high energy density of 11.3 kJ g−1. We expect that further study using CSP methods can help design alternative N-containing high-energy-density materials for their future synthesis.
4.3 Inorganic electrides
Electrides represent a distinct class of ionic compounds, in which electrons distributed in lattice cavities or channels can occupy non-nuclear orbitals and serve as anions individually rather than being attached to atoms. They are potentially useful as catalysts, electron donors, and reducing agents due to their unique physicochemical properties. At the pressures currently achievable, the hybridized valence electrons could be repulsed by core electrons into lattice interstices when compression is sufficiently strong that atomic cores start to overlap, leading to the formation of electrides. Therefore, pressure application is an efficient approach to the design of electrides. Their formation under high pressure is often accompanied by changes in electronic properties, for example, the metal–insulator transition in alkali metals or the emergence of superconductivity.16,38 Little progress was made in the experimental synthesis of electrides until the development of computational approaches for screening electride materials.58,150–152 Here, we introduce some important progress in the theory-guided discovery of electrides, including elemental Na and Li and the binary systems of Ca2N and Sr5P3 (Table 3).| Materials | Brief description | Year | References |
|---|---|---|---|
| Na | Na is predicted and observed to form an insulating electride at 200 GPa | 2019 | 16 |
| Li | Li is predicted to form a semiconducting electride at 60–80 GPa | 2011 | 38 |
| Experimental observation of Li electride above 60 GPa | 2011 | 158 and 159 | |
| Ca2N | Ca2N is predicted to transform into a zero-dimensional insulating electride at 9.7 GPa | 2017 | 28 |
| Experimental confirmation of the insulating electride Ca2N at 11 GPa | 2018 | 161 |
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2d structure (Fig. 7c) at 9.7 GPa, accompanied by a metal-to-insulator transformation.28 The predicted structure was subsequently synthesized at 11 GPa, proving the possibility of Ca2N forming a 0D electride at high pressure.161 CSP methods have helped in predicting many electrides that can be formed at ambient or high pressures, such as Y2C,131 Sr2N,52 and Ba2N52 at ambient pressure and Na2He,5 Li6P,162 Na3S,163 Ti2O,164 Mg2Xe,165 and Mg3O2 (ref. 166) at high pressures. Among the predicted electride structures, 2D Y2C,167 1D Sr5P3,29 and 0D Na2He5 have been experimentally confirmed.
2d structure (Fig. 7c) at 9.7 GPa, accompanied by a metal-to-insulator transformation.28 The predicted structure was subsequently synthesized at 11 GPa, proving the possibility of Ca2N forming a 0D electride at high pressure.161 CSP methods have helped in predicting many electrides that can be formed at ambient or high pressures, such as Y2C,131 Sr2N,52 and Ba2N52 at ambient pressure and Na2He,5 Li6P,162 Na3S,163 Ti2O,164 Mg2Xe,165 and Mg3O2 (ref. 166) at high pressures. Among the predicted electride structures, 2D Y2C,167 1D Sr5P3,29 and 0D Na2He5 have been experimentally confirmed.
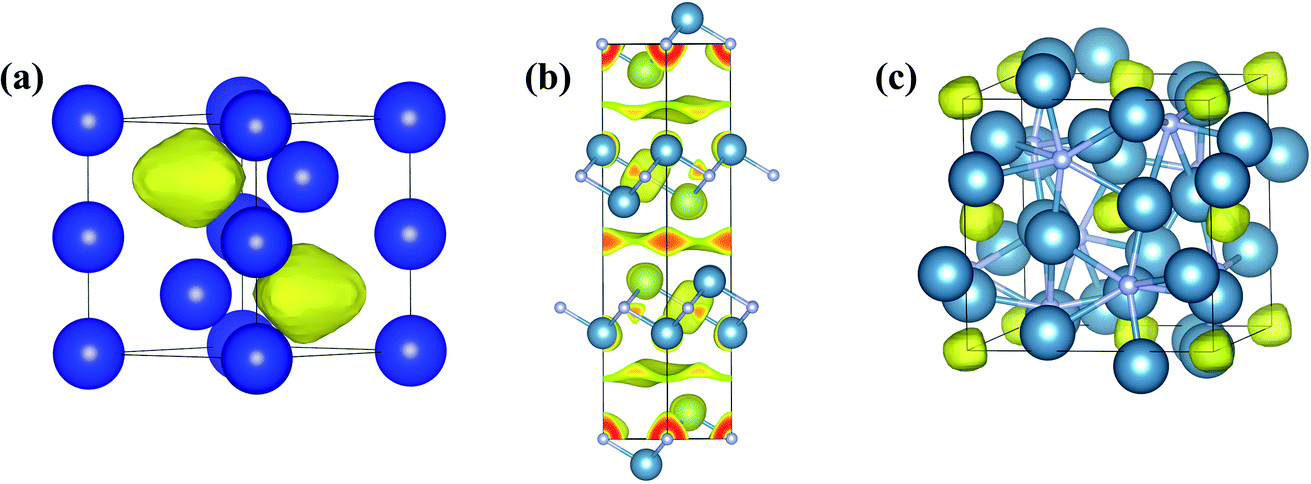 | ||
| Fig. 7 Three-dimensional electron localization function maps of (a) hP4-Na at 200 GPa, (b) anti-CdCl2-type Ca2N at ambient pressure, and (c) I2d Ca2N at 20 GPa to show electron localization in the lattice interstices. | ||
4.4 Chemistry of noble elements
Noble elements are characterized by their full electron shells and chemical inertness. The long-held belief that an element cannot react further if it has full valence orbitals was questioned with the first theoretical proposal of the chemical reaction of Xe by Pauling in 1933 (ref. 168) and disproved with the first synthesis of XePtF6 in 1962 by Neil Bartlett.169 Noble gases are common in the universe and are generally considered to exist only in planetary atmospheres rather than in their interiors due to their chemical inertness. Recent studies using CSP methods have predicted a large number of noble gas compounds that are stable at high pressures and temperatures corresponding to the conditions inside the earth or large icy planets. These compounds are therefore essential to the understanding of the interior structure of planets.4,170–177 Here, we summarize the theory-guided synthesis of noble gas compounds, paying particular attention to Xe and He compounds (Table 4).| Materials | Brief description | Year | References |
|---|---|---|---|
| XeFe3 (XeNi3) | Theoretical predictions of stable XeFe3 and XeNi3 above 200 and 150 GPa, respectively | 2014 | 4 |
| Experimental synthesis of XeNi3 at ∼150 GPa | 2017 | 18 | |
| Experimental synthesis of XeFe3 and XeNi3 at 210 and 155 GPa, respectively | 2018 | 19 | |
| Xe3O2 | Theoretical prediction of stable Xe3O2 above 75 GPa | 2014 | 30 |
| Experimental synthesis of Xe3O2 at 97 GPa | 2016 | 17 | |
| Na2He | Theoretical prediction and experimental confirmation of Na2He at high pressure | 2017 | 5 |
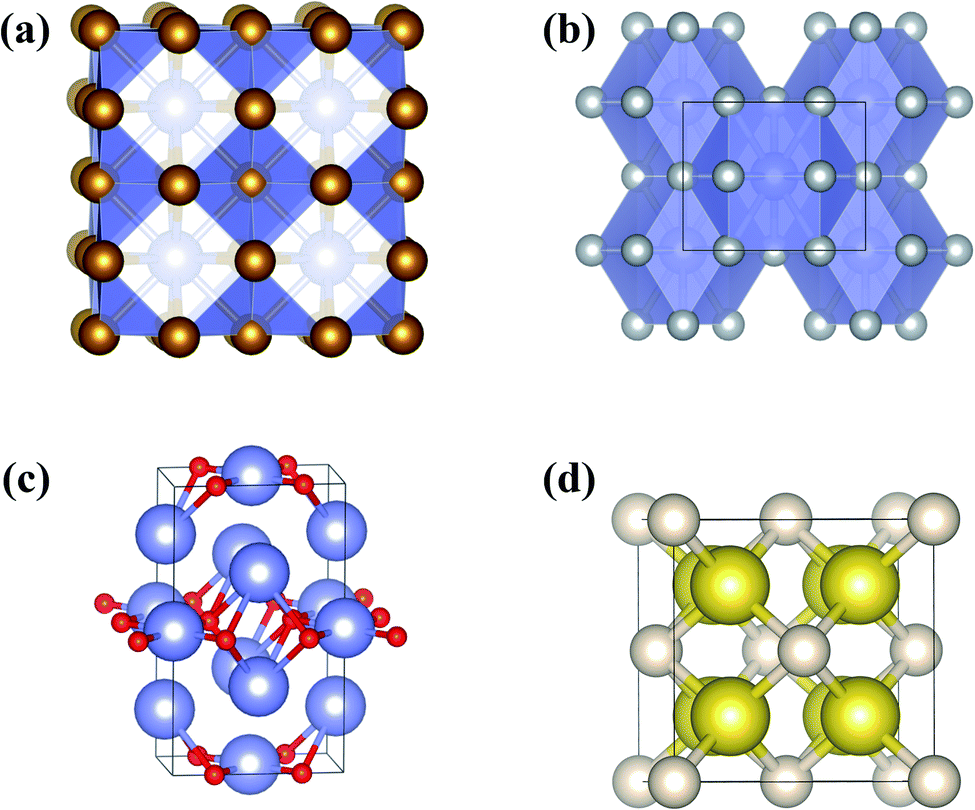 | ||
| Fig. 8 Predicted and confirmed crystal structures of (a) XeFe3, (b) XeNi3, (c) Xe3O2, and (d) Na2He. | ||
Theoretical calculations also uncovered the reason for the emergence of chemical activity in Xe at high pressure. The full 5p shell of Xe opens at high pressure, making Xe a 5p-like element with the ability to transfer electrons to Fe or Ni. This form of Xe has been predicted to react with many other elements or molecules at high pressure to produce, for example, Xe3O2,30 XeN6,178 LiXe,179 MgXe,165 Xe2F,180 Xe4O12H12,181 Xe2FeO2,170 CsXe,182 and Xe–H2.183–185 Among these, Xe3O2 and XeN6 were subsequently synthesized. The reactivity of Xe with O was previously confirmed to occur at pressures as low as 3 GPa, with the formation of the van der Waals compound Xe(O2)2.186 Later, CSP methods predicted the formation of a new Xe3O2 (Fig. 8c) compound above 75 GPa.30 Recent experiments successfully synthesized this compound (at 97 GPa) as well as the unpredicted compound Xe2O5 (88 GPa).17 A prediction of Xe–N was made at pressures of 100–300 GPa, with the finding of stable XeN6 above 146 GPa, indicating the reactivity of Xe with N2.178 These elements were later experimentally reacted at much lower pressure (5 GPa) to form the van der Waals compound Xe(N2)2.187 Much higher pressures are needed to synthesize the predicted stoichiometric XeN6.
A recent theoretical study demonstrated that helium could also be trapped in Earth's lower mantle by the prediction of stable FeO2He.171 Helium was even predicted to react with a broad range of ionic A2B- and AB2-type compounds, such as Li2O, CaF2, and MgF2,188 and react with nitrogen to form stable HeN4,149 HeN6, HeN10, and HeN22 (ref. 148) at high pressures. A common feature of these compounds is the formation of host–guest structures with guest helium atoms intercalated into the lattice interstices, with almost no charge transfer between helium and the other elements. These predicted He compounds have yet to be experimentally confirmed.
5 Outlook
In addition to the abovementioned examples, other types of functional materials have been frequently discovered at high pressure, such as superhard, ferromagnetic, ferroelectric, optoelectronic, catalytic, negative compressibility, and thermoelectric materials190 where theory can also play a vital role in aiding the discovery. Though CSP-based theory is quite successful for the design of materials, challenges remain, in particular for large simulation systems, which contain usually a large number of atoms in the simulation cells, reaching several hundreds and thousands of atoms. The number of the candidate structures increases exponentially with the increasing number of atoms in the simulation system, leading to an extremely complex potential energy landscape with a tremendous number of energy minima for a large system.191 Generally, one has to optimize all candidate structures to find the global energy minima, however, a practical problem is that the much time-consuming geometry optimizations makes it is apparently unaffordable for the available first-principles methods. Also, for ternary or quaternary systems, the existence of huge candidate compositions results in unbearable complicated composition landscape, posing a big challenge for the current first-principle methods to calculate the countlessly possible structures. One strategy might be considered on the development of alternative algorithms that guarantee fast and reliable calculations on total energy and geometry optimization. Force field methods, machine learning potentials,192 and linear scaling methods (e.g., orbital free density functional theory193) might be relied on, but particular caution must be taken for the transferability of the method in dealing with entirely different structures. Besides the use of machine learning potentials, machine learning technique (e.g., deep learning, etc.) can offer an alternative opportunity to aid the material discovery. As a data-driven method, machine learning uses large amounts of data to continuously optimize models and to make reasonable predictions under the guidance of algorithms.194–196 Generally, high-quality data can be selected as training and testing sets from the available material databases, such as Open Quantum Material Database, Material Project, Computational Materials Repository, Inorganic Crystal Structure Database, AFLOWLIB, and CALYPSO structure database, as well as those obtained by local structure optimizations of plenty of structures generated by CSP methods.197,198The products via high-pressure experiments are largely influenced by many factors, such as the precursors, the grain size of sample, the rate of pressure rise, as well as the temperature used. The current theoretical methods are ready to predict the static crystal structures at a given pressure and zero temperature, however, the lack of the study on the dynamic process of material formation provides no information about the synthetic pathway. A reaction pathway describes the successive steps at the molecular level that takes place in a chemical reaction, while a deep understanding of all reaction pathways is essential to explore the evolution of a system over time given a set of initial conditions such as precursors, pressure and temperature. This provides insight into the choices of reaction conditions that can affect the overall reaction outcome, purity, and reaction rates.199,200 The use of CSP simulations in combination with the method for calculations of kinetic energy barriers (KEB) for the chemical reaction or phase transition may help to screen out the possible synthetic routes, thus narrowing down the scope of the experiment trials. To obtain the KEB, one has to find out the minimum energy path and transition state from the astronomically large number of possible solutions. The calculation of KEB can be achieved by a recent reported evolution strategy combining a matrix particle swarm optimization algorithm and an improved nudged elastic band method with the knowledge of the initial state and the final state.61 However, there is lack of a generally applicable technique on how to account for the dynamic environment of a reacting system with quantum accuracy, posing a challenge for the investigation of the KEB. It is believed that the development of useful strategy to probe the mechanism of chemical reaction is one of the essential issues to accelerate the material discovery initialized by theoretical design.
Data availability
Figures, tables and detailed crystallographic information are available from the corresponding author by request.Author contributions
All authors contributed to the preparation of the manuscript. M. X. and Y. L. participated in original draft writing and graphic visualization. Y. M. contributed to the substantial revision of the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12074154, 11904142, and 11722433), the Major Program of the National Natural Science Foundation of China (Grant No. 52090024), and the National Key R&D Program of China (Grant No. 2018YFA0305900).References
- L. Zhang, Y. Wang, J. Lv and Y. Ma, Nat. Rev. Mater., 2017, 2, 17005 CrossRef CAS.
- M. Miao, Y. Sun, E. Zurek and H. Lin, Nat. Rev. Chem., 2020, 4, 508–527 CrossRef CAS.
- Y. Li, J. Hao, H. Liu, S. Lu and J. S. Tse, Phys. Rev. Lett., 2015, 115, 105502 CrossRef PubMed.
- L. Zhu, H. Liu, C. J. Pickard, G. Zou and Y. Ma, Nat. Chem., 2014, 6, 644–648 CrossRef CAS PubMed.
- X. Dong, A. R. Oganov, A. F. Goncharov, E. Stavrou, S. Lobanov, G. Saleh, G.-R. Qian, Q. Zhu, C. Gatti, V. L. Deringer, R. Dronskowski, X.-F. Zhou, V. B. Prakapenka, Z. Konôpková, I. A. Popov, A. I. Boldyrev and H.-T. Wang, Nat. Chem., 2017, 9, 440–445 CrossRef CAS PubMed.
- H. Liu, I. I. Naumov, R. Hoffmann, N. W. Ashcroft and R. J. Hemley, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6990–6995 CrossRef CAS PubMed.
- F. Peng, Y. Sun, C. J. Pickard, R. J. Needs, Q. Wu and Y. Ma, Phys. Rev. Lett., 2017, 119, 107001 CrossRef PubMed.
- W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel, A. O. Lyakhov, E. Stavrou, M. Somayazulu, V. B. Prakapenka and Z. Konopkova, Science, 2013, 342, 1502–1505 CrossRef CAS PubMed.
- R. J. Hemley, High Press. Res., 2010, 30, 581–619 CrossRef CAS.
- Z. Jenei, E. F. O'Bannon, S. T. Weir, H. Cynn, M. J. Lipp and W. J. Evans, Nat. Commun., 2018, 9, 3563 CrossRef PubMed.
- L. Dubrovinsky, N. Dubrovinskaia, E. Bykova, M. Bykov, V. Prakapenka, C. Prescher, K. Glazyrin, H. P. Liermann, M. Hanfland, M. Ekholm, Q. Feng, L. V. Pourovskii, M. I. Katsnelson, J. M. Wills and I. A. Abrikosov, Nature, 2015, 525, 226–229 CrossRef CAS PubMed.
- N. Dubrovinskaia, L. Dubrovinsky, N. A. Solopova, A. Abakumov, S. Turner, M. Hanfland, E. Bykova, M. Bykov, C. Prescher, V. B. Prakapenka, S. Petitgirard, I. Chuvashova, B. Gasharova, Y. Mathis, P. Ershov, I. Snigireva and A. Snigirev, Sci. Adv., 2016, 2, 1–12 Search PubMed.
- W. A. Phelan, J. Zahn, Z. Kennedy and T. M. McQueen, J. Solid State Chem., 2019, 270, 705–709 CrossRef CAS.
- Y. Li, J. Hao, H. Liu, Y. Li and Y. Ma, J. Chem. Phys., 2014, 140, 174712 CrossRef PubMed.
- A. P. Drozdov, M. I. Eremets, I. A. Troyan, V. Ksenofontov and S. I. Shylin, Nature, 2015, 525, 73–76 CrossRef CAS PubMed.
- Y. Ma, M. Eremets, A. R. Oganov, Y. Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle and V. Prakapenka, Nature, 2009, 458, 182–185 CrossRef CAS PubMed.
- A. Dewaele, N. Worth, C. J. Pickard, R. J. Needs, S. Pascarelli, O. Mathon, M. Mezouar and T. Irifune, Nat. Chem., 2016, 8, 784–790 CrossRef CAS PubMed.
- A. Dewaele, C. M. Pépin, G. Geneste and G. Garbarino, High Press. Res., 2017, 37, 137–146 CrossRef CAS.
- E. Stavrou, Y. Yao, A. F. Goncharov, S. S. Lobanov, J. M. Zaug, H. Liu, E. Greenberg and V. B. Prakapenka, Phys. Rev. Lett., 2018, 120, 096001 CrossRef CAS PubMed.
- E. Snider, N. Dasenbrock-Gammon, R. McBride, M. Debessai, H. Vindana, K. Vencatasamy, K. V. Lawler, A. Salamat and R. P. Dias, Nature, 2020, 586, 373–377 CrossRef CAS PubMed.
- D. Duan, Y. Liu, F. Tian, D. Li, X. Huang, Z. Zhao, H. Yu, B. Liu, W. Tian and T. Cui, Sci. Rep., 2015, 4, 6968 CrossRef PubMed.
- M. Somayazulu, M. Ahart, A. K. Mishra, Z. M. Geballe, M. Baldini, Y. Meng, V. V. Struzhkin and R. J. Hemley, Phys. Rev. Lett., 2019, 122, 027001 CrossRef CAS PubMed.
- A. P. Drozdov, P. P. Kong, V. S. Minkov, S. P. Besedin, M. A. Kuzovnikov, S. Mozaffari, L. Balicas, F. F. Balakirev, D. E. Graf, V. B. Prakapenka, E. Greenberg, D. A. Knyazev, M. Tkacz and M. I. Eremets, Nature, 2019, 569, 528–531 CrossRef CAS PubMed.
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed.
- E. Gregoryanz, A. F. Goncharov, C. Sanloup, M. Somayazulu, H. Mao and R. J. Hemley, J. Chem. Phys., 2007, 126, 184505 CrossRef PubMed.
- Y. Ma, A. R. Oganov, Z. Li, Y. Xie and J. Kotakoski, Phys. Rev. Lett., 2009, 102, 065501 CrossRef PubMed.
- F. Peng, Y. Yao, H. Liu and Y. Ma, J. Phys. Chem. Lett., 2015, 6, 2363–2366 CrossRef CAS PubMed.
- Y. Zhang, W. Wu, Y. Wang, S. A. Yang and Y. Ma, J. Am. Chem. Soc., 2017, 139, 13798–13803 CrossRef CAS PubMed.
- J. Wang, K. Hanzawa, H. Hiramatsu, J. Kim, N. Umezawa, K. Iwanaka, T. Tada and H. Hosono, J. Am. Chem. Soc., 2017, 139, 15668–15680 CrossRef CAS PubMed.
- A. Hermann and P. Schwerdtfeger, J. Phys. Chem. Lett., 2014, 5, 4336–4342 CrossRef CAS PubMed.
- E. Zurek and T. Bi, J. Chem. Phys., 2019, 150, 050901 CrossRef PubMed.
- C. J. Pickard, I. Errea and M. I. Eremets, Annu. Rev. Condens. Matter Phys., 2020, 11, 57–76 CrossRef CAS.
- J. A. Flores-Livas, L. Boeri, A. Sanna, G. Profeta, R. Arita and M. Eremets, Phys. Rep., 2020, 856, 1–78 CrossRef CAS.
- F. P. Bundy, J. Chem. Phys., 1963, 38, 631–643 CrossRef CAS.
- Y. Li, Y. Wang, C. J. Pickard, R. J. Needs, Y. Wang and Y. Ma, Phys. Rev. Lett., 2015, 114, 125501 CrossRef PubMed.
- W. Feng, S. Cui and M. Feng, J. Phys. Chem. Solids, 2014, 75, 803–807 CrossRef CAS.
- D. Tomasino, M. Kim, J. Smith and C.-S. Yoo, Phys. Rev. Lett., 2014, 113, 205502 CrossRef PubMed.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Phys. Rev. Lett., 2011, 106, 015503 CrossRef PubMed.
- M. Miao, Nat. Chem., 2013, 5, 846–852 CrossRef CAS PubMed.
- Y. Li, J. Hao, H. Liu, J. S. Tse, Y. Wang and Y. Ma, Sci. Rep., 2015, 5, 9948 CrossRef CAS PubMed.
- Y. Sun, Y. Tian, B. Jiang, X. Li, H. Li, T. Iitaka, X. Zhong and Y. Xie, Phys. Rev. B, 2020, 101, 174102 CrossRef CAS.
- W. Cui, T. Bi, J. Shi, Y. Li, H. Liu, E. Zurek and R. J. Hemley, Phys. Rev. B, 2020, 101, 134504 CrossRef CAS.
- B. A. Steele, E. Stavrou, J. C. Crowhurst, J. M. Zaug, V. B. Prakapenka and I. I. Oleynik, Chem. Mater., 2017, 29, 735–741 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- C. J. Pickard and R. J. Needs, J. Phys. Condens. Matter, 2011, 23, 053201 CrossRef PubMed.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- D. C. Lonie and E. Zurek, Comput. Phys. Commun., 2011, 182, 372–387 CrossRef CAS.
- G. Trimarchi and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 104113 CrossRef.
- S. Bahmann and J. Kortus, Comput. Phys. Commun., 2013, 184, 1618–1625 CrossRef CAS.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, J. Chem. Phys., 2012, 137, 084104 CrossRef PubMed.
- Y. Wang, M. Miao, J. Lv, L. Zhu, K. Yin, H. Liu and Y. Ma, J. Chem. Phys., 2012, 137, 224108 CrossRef PubMed.
- M. Xu, C. Huang, Y. Li, S. Liu, X. Zhong, P. Jena, E. Kan and Y. Wang, Phys. Rev. Lett., 2020, 124, 067602 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, J. Lv, C. Zhu, Q. Li, M. Zhang, Q. Li and Y. Ma, J. Chem. Phys., 2013, 138, 114101 CrossRef PubMed.
- S. Lu, Y. Wang, H. Liu, M. Miao and Y. Ma, Nat. Commun., 2014, 5, 3666 CrossRef CAS PubMed.
- B. Gao, X. Shao, J. Lv, Y. Wang and Y. Ma, J. Phys. Chem. C, 2015, 119, 20111–20118 CrossRef CAS.
- P. Gao, Q. Tong, J. Lv, Y. Wang and Y. Ma, Comput. Phys. Commun., 2017, 213, 40–45 CrossRef CAS.
- Y. Zhang, H. Wang, Y. Wang, L. Zhang and Y. Ma, Phys. Rev. X, 2017, 7, 011017 Search PubMed.
- P. Gao, S. Wang, J. Lv, Y. Wang and Y. Ma, RSC Adv., 2017, 7, 39869–39876 RSC.
- B. Gao, P. Gao, S. Lu, J. Lv, Y. Wang and Y. Ma, Sci. Bull., 2019, 64, 301–309 CrossRef CAS.
- K. Yin, P. Gao, X. Shao, B. Gao, H. Liu, J. Lv, J. S. Tse, Y. Wang and Y. Ma, npj Comput. Mater., 2020, 6, 16 CrossRef.
- N. W. Ashcroft, Phys. Rev. Lett., 2004, 92, 187002 CrossRef CAS PubMed.
- M. Sakashita, H. Yamawaki, H. Fujihisa, K. Aoki, S. Sasaki and H. Shimizu, Phys. Rev. Lett., 1997, 79, 1082–1085 CrossRef CAS.
- R. Rousseau, M. Boero, M. Bernasconi, M. Parrinello and K. Terakura, Phys. Rev. Lett., 2000, 85, 1254–1257 CrossRef CAS PubMed.
- N. Bernstein, C. S. Hellberg, M. D. Johannes, I. I. Mazin and M. J. Mehl, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 060511 CrossRef.
- Y. Li, L. Wang, H. Liu, Y. Zhang, J. Hao, C. J. Pickard, J. R. Nelson, R. J. Needs, W. Li, Y. Huang, I. Errea, M. Calandra, F. Mauri and Y. Ma, Phys. Rev. B, 2016, 93, 020103 CrossRef.
- I. Errea, M. Calandra, C. J. Pickard, J. Nelson, R. J. Needs, Y. Li, H. Liu, Y. Zhang, Y. Ma and F. Mauri, Phys. Rev. Lett., 2015, 114, 157004 CrossRef PubMed.
- D. Duan, X. Huang, F. Tian, D. Li, H. Yu, Y. Liu, Y. Ma, B. Liu and T. Cui, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 180502 CrossRef.
- M. Einaga, M. Sakata, T. Ishikawa, K. Shimizu, M. I. Eremets, A. P. Drozdov, I. A. Troyan, N. Hirao and Y. Ohishi, Nat. Phys., 2016, 12, 835–838 Search PubMed.
- T. A. Strobel, P. Ganesh, M. Somayazulu, P. R. C. Kent and R. J. Hemley, Phys. Rev. Lett., 2011, 107, 255503 CrossRef PubMed.
- S. Zhang, Y. Wang, J. Zhang, H. Liu, X. Zhong, H.-F. Song, G. Yang, L. Zhang and Y. Ma, Sci. Rep., 2015, 5, 15433 CrossRef CAS PubMed.
- A. Shamp, T. Terpstra, T. Bi, Z. Falls, P. Avery and E. Zurek, J. Am. Chem. Soc., 2016, 138, 1884–1892 CrossRef CAS PubMed.
- Y. Yuan, Y. Li, G. Fang, G. Liu, C. Pei, X. Li, H. Zheng, K. Yang and L. Wang, Natl. Sci. Rev., 2019, 6, 524–531 CrossRef CAS PubMed.
- H. Liu, Y. Li, G. Gao, J. S. Tse and I. I. Naumov, J. Phys. Chem. C, 2016, 120, 3458–3461 CrossRef CAS.
- X. Zhong, H. Wang, J. Zhang, H. Liu, S. Zhang, H.-F. Song, G. Yang, L. Zhang and Y. Ma, Phys. Rev. Lett., 2016, 116, 057002 CrossRef PubMed.
- D. Duan, F. Tian, Y. Liu, X. Huang, D. Li, H. Yu, Y. Ma, B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2015, 17, 32335–32340 RSC.
- Q. Zeng, S. Yu, D. Li, A. R. Oganov and G. Frapper, Phys. Chem. Chem. Phys., 2017, 19, 8236–8242 RSC.
- B. Liu, W. Cui, J. Shi, L. Zhu, J. Chen, S. Lin, R. Su, J. Ma, K. Yang, M. Xu, J. Hao, A. P. Durajski, J. Qi, Y. Li and Y. Li, Phys. Rev. B, 2018, 98, 174101 CrossRef CAS.
- H. Wang, J. S. Tse, K. Tanaka, T. Iitaka and Y. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6463–6466 CrossRef CAS PubMed.
- X. Feng, J. Zhang, G. Gao, H. Liu and H. Wang, RSC Adv., 2015, 5, 59292–59296 RSC.
- L. Ma, K. Wang, Y. Xie, X. Yang, Y. Wang, M. Zhou, H. Liu, G. Liu, H. Wang and Y. Ma, 2021, arXiv:2103.16282v1.
- I. A. Troyan, D. V. Semenok, A. G. Kvashnin, A. V. Sadakov, O. A. Sobolevskiy, V. M. Pudalov, A. G. Ivanova, V. B. Prakapenka, E. Greenberg, A. G. Gavriliuk, I. S. Lyubutin, V. V. Struzhkin, A. Bergara, I. Errea, R. Bianco, M. Calandra, F. Mauri, L. Monacelli, R. Akashi and A. R. Oganov, Adv. Mater., 2021, 2006832 CrossRef CAS PubMed.
- P. P. Kong, V. S. Minkov, M. A. Kuzovnikov, A. P. Drozdov, S. P. Besedin, S. Mozaffari, L. Balicas, F. F. Balakirev, V. B. Prakapenka, S. Chariton, D. A. Knyazev, E. Greenberg and M. I. Eremets, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- D. Zhou, D. V. Semenok, H. Xie, X. Huang, D. Duan, A. Aperis, P. M. Oppeneer, M. Galasso, A. I. Kartsev, A. G. Kvashnin, A. R. Oganov and T. Cui, J. Am. Chem. Soc., 2020, 142, 2803–2811 CrossRef CAS PubMed.
- L. Ma, M. Zhou, Y. Wang, S. Kawaguchi, Y. Ohishi, F. Peng, H. Liu, G. Liu, H. Wang and Y. Ma, Phys. Rev. Res., 2021, 3, 043107 CrossRef.
- A. G. Kvashnin, D. V. Semenok, I. A. Kruglov, I. A. Wrona and A. R. Oganov, ACS Appl. Mater. Interfaces, 2018, 10, 43809–43816 CrossRef CAS PubMed.
- Y. Li, J. Hao, H. Liu, J. S. Tse, Y. Wang and Y. Ma, Sci. Rep., 2015, 5, 9948 CrossRef CAS PubMed.
- Y. Sun, J. Lv, Y. Xie, H. Liu and Y. Ma, Phys. Rev. Lett., 2019, 123, 097001 CrossRef CAS PubMed.
- W. Sun, X. Kuang, H. D. J. Keen, C. Lu and A. Hermann, Phys. Rev. B, 2020, 102, 144524 CrossRef CAS.
- M. Peña-Alvarez, J. Binns, A. Hermann, L. C. Kelsall, P. Dalladay-Simpson, E. Gregoryanz and R. T. Howie, Phys. Rev. B, 2019, 100, 184109 CrossRef.
- N. P. Salke, M. M. Davari Esfahani, Y. Zhang, I. A. Kruglov, J. Zhou, Y. Wang, E. Greenberg, V. B. Prakapenka, J. Liu, A. R. Oganov and J.-F. Lin, Nat. Commun., 2019, 10, 4453 CrossRef PubMed.
- D. V. Semenok, A. G. Kvashnin, A. G. Ivanova, V. Svitlyk, V. Y. Fominski, A. V. Sadakov, O. A. Sobolevskiy, V. M. Pudalov, I. A. Troyan and A. R. Oganov, Mater. Today, 2020, 33, 36–44 CrossRef CAS.
- D. V. Semenok, A. G. Kvashnin, I. A. Kruglov and A. R. Oganov, J. Phys. Chem. Lett., 2018, 9, 1920–1926 CrossRef CAS PubMed.
- X. Li, X. Huang, D. Duan, C. J. Pickard, D. Zhou, H. Xie, Q. Zhuang, Y. Huang, Q. Zhou, B. Liu and T. Cui, Nat. Commun., 2019, 10, 3461 CrossRef PubMed.
- E. Snider, N. Dasenbrock-Gammon, R. McBride, X. Wang, N. Meyers, K. V Lawler, E. Zurek, A. Salamat and R. P. Dias, Phys. Rev. Lett., 2021, 126, 117003 CrossRef CAS PubMed.
- H. Xie, Y. Yao, X. Feng, D. Duan, H. Song, Z. Zhang, S. Jiang, S. A. T. Redfern, V. Z. Kresin, C. J. Pickard and T. Cui, Phys. Rev. Lett., 2020, 125, 217001 CrossRef CAS PubMed.
- D. Zhou, X. Jin, X. Meng, G. Bao, Y. Ma, B. Liu and T. Cui, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014118 CrossRef.
- J. Hooper, B. Altintas, A. Shamp and E. Zurek, J. Phys. Chem. C, 2013, 117, 2982–2992 CrossRef CAS.
- Y. Liu, D. Duan, F. Tian, H. Liu, C. Wang, X. Huang, D. Li, Y. Ma, B. Liu and T. Cui, Inorg. Chem., 2015, 54, 9924–9928 CrossRef CAS PubMed.
- Z. Wang, H. Wang, J. S. Tse, T. Iitaka and Y. Ma, Chem. Sci., 2015, 6, 522–526 RSC.
- Y. Wang, H. Wang, J. S. Tse, T. Iitaka and Y. Ma, Phys. Chem. Chem. Phys., 2015, 17, 19379–19385 RSC.
- X. Ye, N. Zarifi, E. Zurek, R. Hoffmann and N. W. Ashcroft, J. Phys. Chem. C, 2018, 122, 6298–6309 CrossRef CAS.
- G. M. Eliashberg, Sov. Phys. JETP, 1960, 11, 696 Search PubMed.
- A. K. McMahan and R. LeSar, Phys. Rev. Lett., 1985, 54, 1929–1932 CrossRef CAS PubMed.
- C. Mailhiot, L. H. Yang and A. K. McMahan, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 14419–14435 CrossRef CAS PubMed.
- D. Laniel, B. Winkler, T. Fedotenko, A. Pakhomova, S. Chariton, V. Milman, V. Prakapenka, L. Dubrovinsky and N. Dubrovinskaia, Phys. Rev. Lett., 2020, 124, 216001 CrossRef CAS PubMed.
- M. M. G. Alemany and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 024110 CrossRef.
- W. D. Mattson, D. Sanchez-Portal, S. Chiesa and R. M. Martin, Phys. Rev. Lett., 2004, 93, 125501 CrossRef PubMed.
- F. Zahariev, A. Hu, J. Hooper, F. Zhang and T. Woo, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 214108 CrossRef.
- C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2009, 102, 125702 CrossRef PubMed.
- X. Wang, Y. Wang, M. Miao, X. Zhong, J. Lv, T. Cui, J. Li, L. Chen, C. J. Pickard and Y. Ma, Phys. Rev. Lett., 2012, 109, 175502 CrossRef PubMed.
- J. Sun, M. Martinez-Canales, D. D. Klug, C. J. Pickard and R. J. Needs, Phys. Rev. Lett., 2013, 111, 175502 CrossRef PubMed.
- M. J. Greschner, M. Zhang, A. Majumdar, H. Liu, F. Peng, J. S. Tse and Y. Yao, J. Phys. Chem. A, 2016, 120, 2920–2925 CrossRef CAS PubMed.
- S. Liu, L. Zhao, M. Yao, M. Miao and B. Liu, Adv. Sci., 2020, 7, 1902320 CrossRef CAS PubMed.
- D. Laniel, G. Geneste, G. Weck, M. Mezouar and P. Loubeyre, Phys. Rev. Lett., 2019, 122, 066001 CrossRef CAS PubMed.
- J. Zhang, Z. Zeng, H.-Q. Lin and Y.-L. Li, Sci. Rep., 2015, 4, 4358 CrossRef PubMed.
- X. Wang, J. Li, N. Xu, H. Zhu, Z. Hu and L. Chen, Sci. Rep., 2015, 5, 16677 CrossRef PubMed.
- J. Li, X. Wang, N. Xu, D. Li, D. Wang and L. Chen, Europhys. Lett., 2013, 104, 16005 CrossRef.
- X. Wang, J. Li, J. Botana, M. Zhang, H. Zhu, L. Chen, H. Liu, T. Cui and M. Miao, J. Chem. Phys., 2013, 139, 164710 CrossRef PubMed.
- M. Zhang, H. Yan, Q. Wei, H. Wang and Z. Wu, Europhys. Lett., 2013, 101, 26004 CrossRef.
- X. Huang, D. Li, F. Li, X. Jin, S. Jiang, W. Li, X. Yang, Q. Zhou, B. Zou, Q. Cui, B. Liu and T. Cui, J. Phys. Chem. C, 2012, 116, 9744–9749 CrossRef CAS.
- S. A. Medvedev, I. A. Trojan, M. I. Eremets, T. Palasyuk, T. M. Klapötke and J. Evers, J. Phys. Condens. Matter, 2009, 21, 195404 CrossRef CAS PubMed.
- M. I. Eremets, M. Y. Popov, I. A. Trojan, V. N. Denisov, R. Boehler and R. J. Hemley, J. Chem. Phys., 2004, 120, 10618–10623 CrossRef CAS PubMed.
- C. Ji, F. Zhang, D. Hou, H. Zhu, J. Wu, M.-C. Chyu, V. I. Levitas and Y. Ma, J. Phys. Chem. Solids, 2011, 72, 736–739 CrossRef CAS.
- D. Hou, F. Zhang, C. Ji, T. Hannon, H. Zhu, J. Wu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 064127 CrossRef.
- C. Ji, R. Zheng, D. Hou, H. Zhu, J. Wu, M.-C. Chyu and Y. Ma, J. Appl. Phys., 2012, 111, 112613 CrossRef.
- D. Li, X. Wu, J. Jiang, X. Wang, J. Zhang, Q. Cui and H. Zhu, Appl. Phys. Lett., 2014, 105, 071903 CrossRef.
- D. Li, F. Li, Y. Li, X. Wu, G. Fu, Z. Liu, X. Wang, Q. Cui and H. Zhu, J. Phys. Chem. C, 2015, 119, 16870–16878 CrossRef CAS.
- H. Zhu, F. Zhang, C. Ji, D. Hou, J. Wu, T. Hannon and Y. Ma, J. Appl. Phys., 2013, 113, 033511 CrossRef.
- Z. Zhao, K. Bao, D. Li, D. Duan, F. Tian, X. Jin, C. Chen, X. Huang, B. Liu and T. Cui, Sci. Rep., 2015, 4, 4797 CrossRef PubMed.
- S. Zhu, F. Peng, H. Liu, A. Majumdar, T. Gao and Y. Yao, Inorg. Chem., 2016, 55, 7550–7555 CrossRef CAS PubMed.
- Y. Zhang, L. Wu, B. Wan, Y. Lin, Q. Hu, Y. Zhao, R. Gao, Z. Li, J. Zhang and H. Gou, Sci. Rep., 2016, 6, 33506 CrossRef CAS PubMed.
- S. Zhang, Z. Zhao, L. Liu and G. Yang, J. Power Sources, 2017, 365, 155–161 CrossRef CAS.
- S. Yu, B. Huang, Q. Zeng, A. R. Oganov, L. Zhang and G. Frapper, J. Phys. Chem. C, 2017, 121, 11037–11046 CrossRef CAS.
- B. A. Steele and I. I. Oleynik, J. Phys. Chem. A, 2017, 121, 8955–8961 CrossRef CAS PubMed.
- M. Zhou, M. Sui, X. Shi, Z. Zhao, L. Guo, B. Liu, R. Liu, P. Wang and B. Liu, J. Phys. Chem. C, 2020, 124, 11825–11830 CrossRef CAS.
- D. Laniel, G. Weck, G. Gaiffe, G. Garbarino and P. Loubeyre, J. Phys. Chem. Lett., 2018, 9, 1600–1604 CrossRef CAS PubMed.
- D. Laniel, G. Weck and P. Loubeyre, Inorg. Chem., 2018, 57, 10685–10693 CrossRef CAS PubMed.
- Q. Li, L. Sha, C. Zhu and Y. Yao, Europhys. Lett., 2017, 118, 46001 CrossRef.
- K. Xia, H. Gao, C. Liu, J. Yuan, J. Sun, H.-T. Wang and D. Xing, Sci. Bull., 2018, 63, 817–824 CrossRef CAS.
- N. P. Salke, K. Xia, S. Fu, Y. Zhang, E. Greenberg, V. B. Prakapenka, J. Liu, J. Sun and J.-F. Lin, Phys. Rev. Lett., 2021, 126, 065702 CrossRef CAS PubMed.
- F. Peng, Y. Ma, A. Hermann and M. Miao, Phys. Rev. Mater., 2020, 4, 103610 CrossRef CAS.
- Z. Raza, C. J. Pickard, C. Pinilla and A. M. Saitta, Phys. Rev. Lett., 2013, 111, 235501 CrossRef PubMed.
- Z. Liu, D. Li, S. Wei, Y. Liu, F. Tian, D. Duan and T. Cui, Phys. Lett. A, 2019, 383, 125859 CrossRef CAS.
- K. Yin, Y. Wang, H. Liu, F. Peng and L. Zhang, J. Mater. Chem. A, 2015, 3, 4188–4194 RSC.
- W. Wang, H. Wang, Y. Liu, D. Li, F. Tian, D. Duan, H. Yu and T. Cui, Inorg. Chem., 2019, 58, 2397–2402 CrossRef CAS PubMed.
- K. Xia, X. Zheng, J. Yuan, C. Liu, H. Gao, Q. Wu and J. Sun, J. Phys. Chem. C, 2019, 123, 10205–10211 CrossRef CAS.
- J. Hou, X.-J. Weng, A. R. Oganov, X. Shao, G. Gao, X. Dong, H.-T. Wang, Y. Tian and X.-F. Zhou, Phys. Rev. B, 2021, 103, L060102 CrossRef CAS.
- Y. Li, X. Feng, H. Liu, J. Hao, S. A. T. Redfern, W. Lei, D. Liu and Y. Ma, Nat. Commun., 2018, 9, 722 CrossRef PubMed.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Nat. Rev. Mater., 2019, 4, 331–348 CrossRef.
- A. R. Oganov, Modern Methods of Crystal Structure Prediction, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- T. Inoshita, S. Jeong, N. Hamada and H. Hosono, Phys. Rev. X, 2014, 4, 031023 CAS.
- Y. Ma, A. R. Oganov and Y. Xie, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 014102 CrossRef.
- K. Takemura, N. E. Christensen, D. L. Novikov, K. Syassen, U. Schwarz and M. Hanfland, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14399–14404 CrossRef CAS.
- P. Li, G. Gao, Y. Wang and Y. Ma, J. Phys. Chem. C, 2010, 114, 21745–21749 CrossRef CAS.
- H. L. Skriver, Phys. Rev. Lett., 1982, 49, 1768–1772 CrossRef CAS.
- A. H. Wilson, Proc. R. Soc. Lond. - Ser. A Contain. Pap. a Math. Phys. Character, 1932, 138, 594–606 CAS.
- C. L. Guillaume, E. Gregoryanz, O. Degtyareva, M. I. McMahon, M. Hanfland, S. Evans, M. Guthrie, S. V. Sinogeikin and H.-K. Mao, Nat. Phys., 2011, 7, 211–214 Search PubMed.
- M. Marqués, M. I. McMahon, E. Gregoryanz, M. Hanfland, C. L. Guillaume, C. J. Pickard, G. J. Ackland and R. J. Nelmes, Phys. Rev. Lett., 2011, 106, 095502 CrossRef PubMed.
- M.-S. Miao and R. Hoffmann, Acc. Chem. Res., 2014, 47, 1311–1317 CrossRef CAS PubMed.
- H. Tang, B. Wan, B. Gao, Y. Muraba, Q. Qin, B. Yan, P. Chen, Q. Hu, D. Zhang, L. Wu, M. Wang, H. Xiao, H. Gou, F. Gao, H. Mao and H. Hosono, Adv. Sci., 2018, 5, 1800666 CrossRef PubMed.
- Z. Zhao, S. Zhang, T. Yu, H. Xu, A. Bergara and G. Yang, Phys. Rev. Lett., 2019, 122, 097002 CrossRef CAS PubMed.
- B. Wan, S. Xu, X. Yuan, H. Tang, D. Huang, W. Zhou, L. Wu, J. Zhang and H. Gou, J. Mater. Chem. A, 2019, 7, 16472–16478 RSC.
- X. Zhong, M. Xu, L. Yang, X. Qu, L. Yang, M. Zhang, H. Liu and Y. Ma, npj Comput. Mater., 2018, 4, 70 CrossRef CAS.
- M. Miao, X. Wang, J. Brgoch, F. Spera, M. G. Jackson, G. Kresse and H. Lin, J. Am. Chem. Soc., 2015, 137, 14122–14128 CrossRef CAS PubMed.
- Q. Zhu, A. R. Oganov and A. O. Lyakhov, Phys. Chem. Chem. Phys., 2013, 15, 7696 RSC.
- K. Horiba, R. Yukawa, T. Mitsuhashi, M. Kitamura, T. Inoshita, N. Hamada, S. Otani, N. Ohashi, S. Maki, J. Yamaura, H. Hosono, Y. Murakami and H. Kumigashira, Phys. Rev. B, 2017, 96, 045101 CrossRef.
- L. Pauling, J. Am. Chem. Soc., 1933, 55, 1895–1900 CrossRef CAS.
- N. Bartlett, Proc. Chem. Soc. Lond., 1962, 6, 197–236 Search PubMed.
- F. Peng, X. Song, C. Liu, Q. Li, M. Miao, C. Chen and Y. Ma, Nat. Commun., 2020, 11, 1–7 Search PubMed.
- J. Zhang, J. Lv, H. Li, X. Feng, C. Lu, S. A. T. Redfern, H. Liu, C. Chen and Y. Ma, Phys. Rev. Lett., 2018, 121, 255703 CrossRef PubMed.
- J. Shi, W. Cui, J. Hao, M. Xu, X. Wang and Y. Li, Nat. Commun., 2020, 11, 3164 CrossRef CAS PubMed.
- Y. Bai, Z. Liu, J. Botana, D. Yan, H.-Q. Lin, J. Sun, C. J. Pickard, R. J. Needs and M.-S. Miao, Commun. Chem., 2019, 2, 102 CrossRef.
- C. Liu, H. Gao, Y. Wang, R. J. Needs, C. J. Pickard, J. Sun, H.-T. Wang and D. Xing, Nat. Phys., 2019, 15, 1065–1070 Search PubMed.
- B. Monserrat, M. Martinez-Canales, R. J. Needs and C. J. Pickard, Phys. Rev. Lett., 2018, 121, 015301 CrossRef CAS PubMed.
- H. Gao, C. Liu, A. Hermann, R. J. Needs, C. J. Pickard, H.-T. Wang, D. Xing and J. Sun, Natl. Sci. Rev., 2020, 7, 1540–1547 CrossRef CAS PubMed.
- C. Liu, H. Gao, A. Hermann, Y. Wang, M. Miao, C. J. Pickard, R. J. Needs, H.-T. Wang, D. Xing and J. Sun, Phys. Rev. X, 2020, 10, 021007 CAS.
- F. Peng, Y. Wang, H. Wang, Y. Zhang and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 094104 CrossRef.
- Z. Liu, J. Botana, M. Miao and D. Yan, Europhys. Lett., 2017, 117, 26002 CrossRef.
- F. Peng, J. Botana, Y. Wang, Y. Ma and M. Miao, J. Phys. Chem. Lett., 2016, 7, 4562–4567 CrossRef CAS PubMed.
- C. Sanloup, S. A. Bonev, M. Hochlaf and H. E. Maynard-Casely, Phys. Rev. Lett., 2013, 110, 265501 CrossRef PubMed.
- S. Zhang, H. Bi, S. Wei, J. Wang, Q. Li and Y. Ma, J. Phys. Chem. C, 2015, 119, 24996–25002 CrossRef CAS.
- M. Somayazulu, P. Dera, A. F. Goncharov, S. A. Gramsch, P. Liermann, W. Yang, Z. Liu, H. Mao and R. J. Hemley, Nat. Chem., 2010, 2, 50–53 CrossRef CAS PubMed.
- M. Somayazulu, P. Dera, J. Smith and R. J. Hemley, J. Chem. Phys., 2015, 142, 104503 CrossRef PubMed.
- X. Yan, Y. Chen, X. Kuang and S. Xiang, J. Chem. Phys., 2015, 143, 124310 CrossRef PubMed.
- A. Dewaele, P. Loubeyre, P. Dumas and M. Mezouar, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014103 CrossRef.
- D. Laniel, G. Weck and P. Loubeyre, Phys. Rev. B, 2016, 94, 174109 CrossRef.
- Z. Liu, J. Botana, A. Hermann, S. Valdez, E. Zurek, D. Yan, H. Lin and M. Miao, Nat. Commun., 2018, 9, 951 CrossRef PubMed.
- T. Plisson, G. Weck and P. Loubeyre, Phys. Rev. Lett., 2014, 113, 025702 CrossRef CAS PubMed.
- G. Xiao, T. Geng and B. Zou, ACS Mater. Lett., 2020, 2, 1233–1239 CrossRef CAS.
- Y. Wang and Y. Ma, J. Chem. Phys., 2014, 140, 040901 CrossRef PubMed.
- V. Vassilev-Galindo, G. Fonseca, I. Poltavsky and A. Tkatchenko, J. Chem. Phys., 2021, 154, 094119 CrossRef CAS PubMed.
- Y. A. Wang and E. A. Carter, in Theoretical Methods in Condensed Phase Chemistry, Kluwer Academic Publishers, Dordrecht, 2005, pp. 117–184 Search PubMed.
- R. Lopez de Mantaras and E. Armengol, Data Knowl. Eng., 1998, 25, 99–123 CrossRef.
- W. WU and Q. SUN, Sci. Sin. Phys. Mech. Astron., 2018, 48, 107001 CrossRef.
- J. Wei, X. Chu, X. Sun, K. Xu, H. Deng, J. Chen, Z. Wei and M. Lei, InfoMat, 2019, 1, 338–358 CrossRef CAS.
- Q. Tong, L. Xue, J. Lv, Y. Wang and Y. Ma, Faraday Discuss., 2018, 211, 31–43 RSC.
- Q. Tong, P. Gao, H. Liu, Y. Xie, J. Lv, Y. Wang and J. Zhao, J. Phys. Chem. Lett., 2020, 11, 8710–8720 CrossRef CAS PubMed.
- G. N. Simm, A. C. Vaucher and M. Reiher, J. Phys. Chem. A, 2019, 123, 385–399 CrossRef CAS PubMed.
- M. B. Smith, March's advanced organic chemistry: reactions, mechanisms, and structure. John Wiley & Sons, 2020 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |