
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmyloid-β peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-β 42 aggregation†
Gabriel A.
Braun‡
 a,
Alexander J.
Dear
abcd,
Kalyani
Sanagavarapu§
a,
Henrik
Zetterberg
efgh and
Sara
Linse
*a
a,
Alexander J.
Dear
abcd,
Kalyani
Sanagavarapu§
a,
Henrik
Zetterberg
efgh and
Sara
Linse
*a
aBiochemistry and Structural Biology, Lund University, Lund, Sweden. E-mail: sara.linse@biochemistry.lu.se
bDepartment of Cell Biology, Harvard Medical School, Boston, MA, USA
cPaulson School of Engineering and Applied Science, Harvard University, Cambridge, MA, USA
dDepartment of Chemistry, University of Cambridge, Cambridge, UK
eDepartment of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, The Sahlgrenska Academy at the University of Gothenburg, Mölndal, Sweden
fClinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal, Sweden
gDepartment of Neurodegenerative Disease, UCL Institute of Neurology, Queen Square, London, UK
hUK Dementia Research Institute at UCL, London, UK
First published on 7th February 2022
Abstract
The pathology of Alzheimer's disease is connected to the aggregation of β-amyloid (Aβ) peptide, which in vivo exists as a number of length-variants. Truncations and extensions are found at both the N- and C-termini, relative to the most commonly studied 40- and 42-residue alloforms. Here, we investigate the aggregation of two physiologically abundant alloforms, Aβ37 and Aβ38, as pure peptides and in mixtures with Aβ40 and Aβ42. A variety of molar ratios were applied in quaternary mixtures to investigate whether a certain ratio is maximally inhibiting of the more toxic alloform Aβ42. Through kinetic analysis, we show that both Aβ37 and Aβ38 self-assemble through an autocatalytic secondary nucleation reaction to form fibrillar β-sheet-rich aggregates, albeit on a longer timescale than Aβ40 or Aβ42. Additionally, we show that the shorter alloforms co-aggregate with Aβ40, affecting both the kinetics of aggregation and the resulting fibrillar ultrastructure. In contrast, neither Aβ37 nor Aβ38 forms co-aggregates with Aβ42; however, both short alloforms reduce the rate of Aβ42 aggregation in a concentration-dependent manner. Finally, we show that the aggregation of Aβ42 is more significantly impeded by a combination of Aβ37, Aβ38, and Aβ40 than by any of these alloforms independently. These results demonstrate that the aggregation of any given Aβ alloform is significantly perturbed by the presence of other alloforms, particularly in heterogeneous mixtures, such as is found in the extracellular fluid of the brain.
Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disease, is the most common cause of dementia worldwide, afflicting nearly 50 million people.1 Although symptomatic treatments for AD exist, there are currently no available means of slowing or reversing the progression of the disease.2–4 Recently, the anti-amyloid β (Aβ) antibody aducanumab5–7 was approved for clinical use by the US Food and Drug Administration (FDA),8 and other promising antibody candidates are in the pipeline, but their clinical efficacy remains uncertain. This is due, in part, to the fact that the etiology of AD remains poorly understood. The most common theory of the progression of AD is the amyloid cascade hypothesis, which contends that aggregation of amyloid β peptide (Aβ) is the critical pathological event responsible for triggering the onset of the disease, with self-assembly of tau an important second step.9,10 In particular, dispersed Aβ oligomers, rather than the deposited plaques, are widely understood to be the primary cytotoxic forms of these aggregates,3,11 although fibrils may still play a significant role, directly12 or indirectly through generation of toxic oligomers through secondary nucleation.13,14 While the cascade model has been challenged in recent years,15–17 it is nevertheless commonly accepted that the aggregation of Aβ plays a critical part in the initiation and progression of AD.18–20 Understanding the Aβ aggregation process, its underlying microscopic steps, and their connection with toxicity, is essential for the development of effective treatments and better diagnostic tools.21Aβ is produced from the transmembrane amyloid precursor protein (APP). In the so-called amyloidogenic pathway for processing of APP, sequential cleavages of the protein by β- and γ-secretases result in the release of Aβ into the extracellular fluid.22,23 There is some variability in the cleavage sites by both secretases, leading to the presence of Aβ length-variants, or alloforms, that are generally between 37 and 43 residues long.24–27 Of these alloforms, the 40-residue length-variant APP 672-711, Aβ1–40, is the predominant form found in vivo, making up almost 60% the Aβ peptide present in cerebrospinal fluid (CSF).28,29 The next most abundant alloforms are Aβ1–38, Aβ1–42, and Aβ1–37, which make up approximately 15%, 10% and 8% of Aβ in CSF, respectively (although the concentration of Aβ1–42 in CSF decreases significantly in AD patients, which is proposed to be the result of peptide sequestration in plaques30,31). N-terminal length variants are observed both with extended25 and truncated32 N-termini, in which case extension retards33 and truncation accelerates34 aggregation.
Of the C-terminal Aβ length-variants found most commonly in vivo, the 40- and 42-residue alloforms are the most aggregation prone and are also found in the highest abundance in the characteristic AD plaques.35 As a result, the aggregation of these peptides has been extensively characterized, while the aggregation of shorter, less aggregation-prone alloforms has remained relatively less studied.36 Using a chemical kinetics approach, the microscopic steps underlying the in vitro aggregation of Aβ have previously been identified from global analyses of large sets of data over ranges of peptide concentration,37,38 showing that Aβ peptides, ending at residue 40 and 42, respectively, aggregate through a nucleation-and-growth pathway in which the formation of new aggregates occurs predominantly through the autocatalytic secondary nucleation on the surface of existing fibrils.14 However, the aggregation behavior of Aβ peptides is highly sensitive to solution conditions, including ionic strength39,40 and pH,41 and to the presence of foreign surfaces, such as phospholipid membranes42,43 and nanoparticles.44,45 Indeed, in the complex milieu of cerebrospinal fluid, it has been found that, while Aβ42 aggregates through a secondary-nucleation-dominated mechanism, the rate of this fibril-catalyzed nucleation is significantly reduced relative to that of the peptide in pure buffer.46,47 Additionally, the aggregation of Aβ peptides is perturbed by the co-existence of Aβ peptides of different lengths. For example, it has previously been shown that, while Aβ42 and Aβ40 aggregate to form homomolecular fibrils, monomeric Aβ42 strongly catalyzes the aggregation of Aβ40.48 Furthermore, Aβ42 has been shown to both co-aggregate with and cross-seed Aβ peptides with N-terminal extensions ranging from 5–40 residues in length.33 Consequently, it is important to account for the interactions between Aβ alloforms when considering the in vivo behavior of these peptides.
Although Aβ alloforms ending before residue 40 are less commonly found in plaques in AD patients and are not directly implicated in the onset of AD, these shorter alloforms are nevertheless important factors to consider in the disease-related aggregation of longer alloforms. Formation of oligomeric co-assemblies might decrease the rate of aggregation by peptides with a higher intrinsic nucleation rate, and acceleration the aggregation of peptides with a lower intrinsic nucleation rate. Indeed, there are indications that these shorter alloforms play an important role in vivo by modulating pathogenic Aβ1–42 aggregation. It has been shown these shorter alloforms are not inherently toxic, but are in fact neuroprotective in a dose-dependent manner and capable of reducing Aβ1–42 deposition.49,50 It has been proposed that this neuroprotective behavior could be caused by inhibition of Aβ42 oligomerization by the shorter alloforms51,52 or by impeded oligomer conversion to fibrillar structure,53 as has previously been observed for Aβ40.54,55 The interactions and cross-reactivity between Aβ alloforms other than Aβ1–42/Aβ1–40 have, to our knowledge, not been carefully studied.
In this study, we investigate the interactions and cross-reactivity between four Aβ naturally abundant alloforms, Aβ37, Aβ38, Aβ40, and Aβ42, with an aim to unravel any cross-catalysis or co-aggregation processes (Fig. 1). Working in vitro and using highly purified peptides to maximize reproducibility, we characterize their interactions primarily through kinetic aggregation assays using the fluorescent dye thioflavin T (ThT), the quantum yield of which reports quantitatively on aggregate mass concentration when properly calibrated.56 We investigate not only the effect that each alloform has on the aggregation of the others in binary mixtures, but also consider three-peptide and four-peptide mixtures in order to better understand how different ratios of Aβ alloforms affects the overall rate of conversion of monomer to amyloid fibril. In these experiments, we ask whether any particular ratio of the shorter length variants will maximally affect the aggregation of Aβ42.
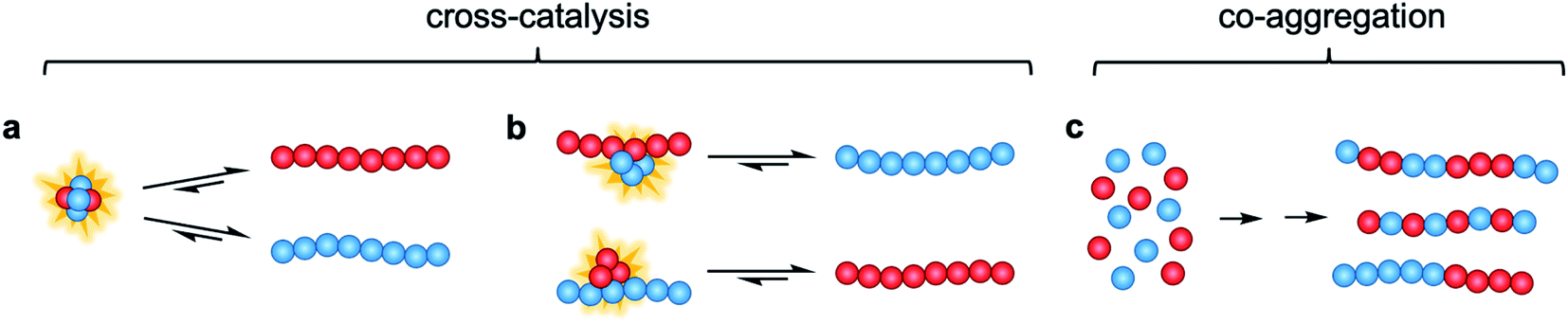 | ||
| Fig. 1 Possible modes of cross-catalysis through (a) heterogeneous primary nucleation in co-oligomers or (b) on fibril surfaces. (c) Co-aggregation is here defined as the process leading to any form of aggregates composed of more than one peptide alloform. | ||
Results
The aggregation of Aβ(M1-37), Aβ(M1-38), Aβ(M1-40), and Aβ(M1-42) (Fig. 2, referred to herein as Aβ37, Aβ38, Aβ40, and Aβ42, respectively) in binary, trinary, and quaternary mixtures was investigated using in vitro kinetic assays, analytical high performance liquid chromatography (HPLC), and cryo-transmission electron microscopy (cryo-TEM). First, the aggregation of Aβ37 or Aβ38 in the absence of other peptides was investigated. Next, the aggregation of two-peptide systems, including either Aβ37 or Aβ38 together with either Aβ40 or Aβ42, was investigated. Additionally, two-peptide cross-seeding experiments were also performed. Subsequently, three-peptide systems composed of Aβ40, Aβ42, and either Aβ37 or Aβ38 were studied. Finally, the aggregation behavior of four-peptide system, including all four alloforms, was investigated. All experiments were performed in 20 mM sodium phosphate buffer, 200 μM ethylenediaminetetraacetic acid (EDTA), 0.02% NaN3, at pH 7.4, as all alloforms aggregate fast enough to give relatively reproducible data under these conditions. The incorporation of an N-terminal Met residue has no observable effect on the ssNMR spectrum of Aβ42 fibrils and aggregation mechanism,57 and was chosen because it allows for expression “as is” with easy purification without the complication of tags and proteases.58 | ||
| Fig. 2 Amino acid sequences of the peptides investigated in this work. | ||
Independent aggregation of Aβ37 and Aβ38
Before studying the aggregation of Aβ37 and Aβ38 in heteromolecular mixtures, the aggregation of each peptide independent of the others was first investigated (Fig. 3). Aβ37 and Aβ38 displayed greatly reduced aggregation propensity relative to Aβ40 and Aβ42. In identical conditions and at the low end of the concentration range studied here, Aβ40 aggregates within two hours, while Aβ42 aggregates within a fraction of an hour.37,38,41 However, the aggregation propensity among these naturally occurring Aβ alloforms is evidently not directly correlated to peptide length, as Aβ38 was found here to aggregate much more slowly than Aβ37.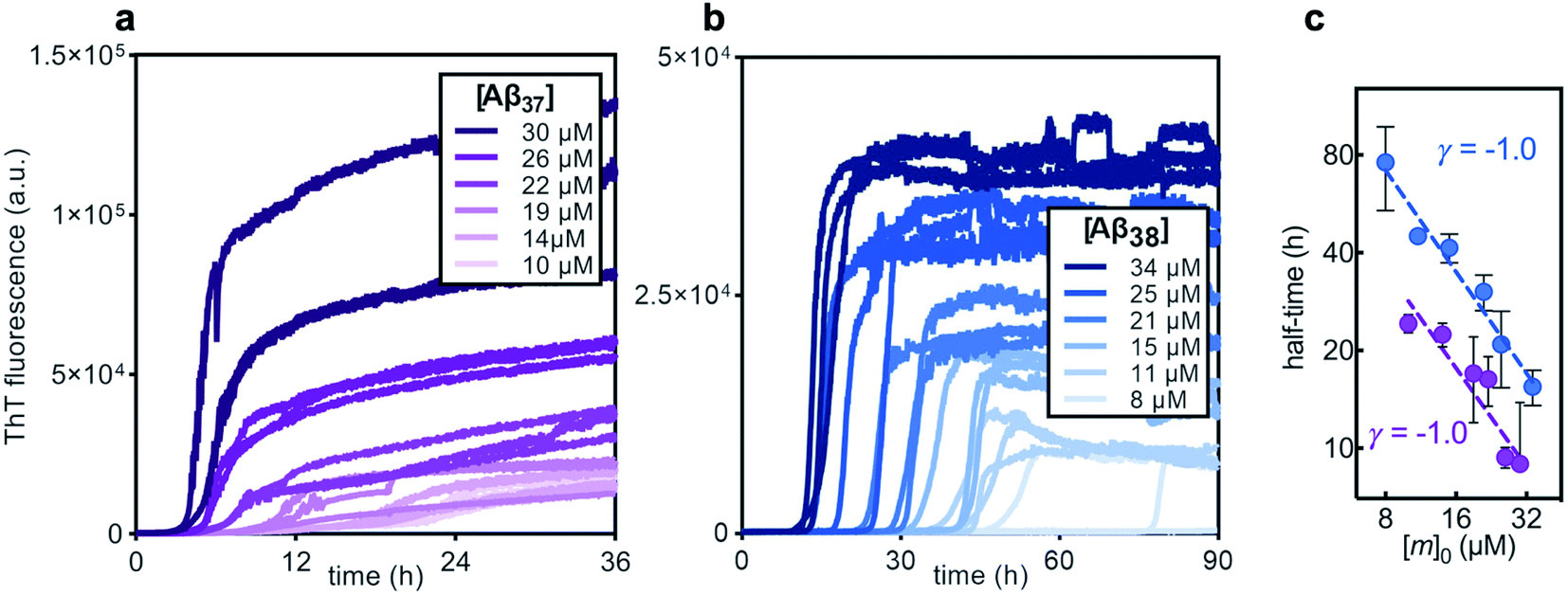 | ||
| Fig. 3 The aggregation kinetics of pure solutions of (a) Aβ37 and (b) Aβ38 in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. Three technical replicates are shown for each concentration. (c) Double-logarithmic plot of the aggregation half-time versus initial monomer concentration, [m]0, for Aβ37 (purple) and Aβ38 (blue), where the half-time is the time at which the fluorescence signal is half of its final plateau value. Each dashed line shows a fitted power function with scaling exponent, γ. Points show the mean of the three technical replicates, with error bars showing the standard deviation. | ||
The aggregation curve shapes for both Aβ37 and Aβ38 both displayed long lag phases, during which little aggregation was observable above the signal-to-noise ratio, followed by distinct growth phases, in which most aggregate growth happened rapidly. This distinctive curve shape, with a sharp transition between lag and growth phases, is characteristic of an aggregation mechanism driven by a self-replicating, fibril-catalyzed process. The concentration dependence of the rate of aggregation of each peptide was clearly shown by the half-time plots (Fig. 3c), in which the time at which half of the total peptide had aggregated was plotted versus starting monomer concentration. The scaling exponent, γ, of a power function fitted to the half-time data provides valuable insight into the underlying aggregation mechanism.59,60 Aggregation mechanisms driven by fragmentation and secondary nucleation, the two self-replicating processes observed in amyloid-related aggregation, display characteristic γ values: for fragmentation-dominated systems, γ = −0.5, while for secondary-nucleation-dominated systems γ ≤ −1.0, with the exact value depending on the reaction order of secondary nucleation and the balance between primary and secondary nucleation, and whether or not secondary nucleation saturates.38,61 For both Aβ37 and Aβ38, γ = −1.0, indicating that the aggregation of both peptides is driven by secondary nucleation. This is not surprising, as the aggregation of both Aβ40 and Aβ42 has been shown to be dominated by this same process.37,38 The high variability among technical repeats at low concentrations prevents data fitting to generate a quantitative description of the aggregation process for either peptide, as has previously been done for Aβ40 and Aβ42.37,38,62 However, such comprehensive characterization is not required for this study, for which the interactions between different Aβ alloforms, rather than the isolated behavior of any one peptide, is the primary focus.
Aggregation kinetics of mixtures of Aβ40 with Aβ37 or Aβ38
In kinetic assays of the aggregation process in binary mixtures of Aβ40 and either Aβ37 or Aβ38, only a single transition was observed (Fig. 4). This contrasts with the aggregation behavior of monomeric Aβ40/Aβ42 mixtures, which produced a double-sigmoidal profile that was found to correspond to the self-assembly of each peptide on distinct timescales into segregated, homomolecular fibrils.48 The effects of each peptide on the aggregation of the other were studied by holding the concentration of one peptide constant while varying the concentration of the other. The addition of Aβ40 to solutions of Aβ37 or Aβ38 decreased the lag time before the single observed fluorescence transition (Fig. 4a and b). Conversely, with a constant concentration of Aβ40, the addition of either Aβ37 or Aβ38 resulted in a delay in aggregation (Fig. 4c and d). In both cases, the addition of the shorter alloform to Aβ40 results in an increase in the intensity of ThT fluorescence, indicating that the shorter alloforms are aggregating concurrently with Aβ40, rather than at a later timepoint. The modulation of the kinetics of aggregation for both peptides in both Aβ37/Aβ40 and Aβ38/Aβ40 mixtures, as well as the fact that a monophasic aggregation profile is observed at all molar ratios tested despite the differing timescales of the independent aggregation of Aβ37 and Aβ38 compared to that of Aβ40, suggest that these peptides may co-aggregate to form some level of mixed aggregates. However, ThT fluorescence assays, which report only on total aggregate mass concentration, cannot distinguish co-aggregates from coexisting homomolecular fibrils.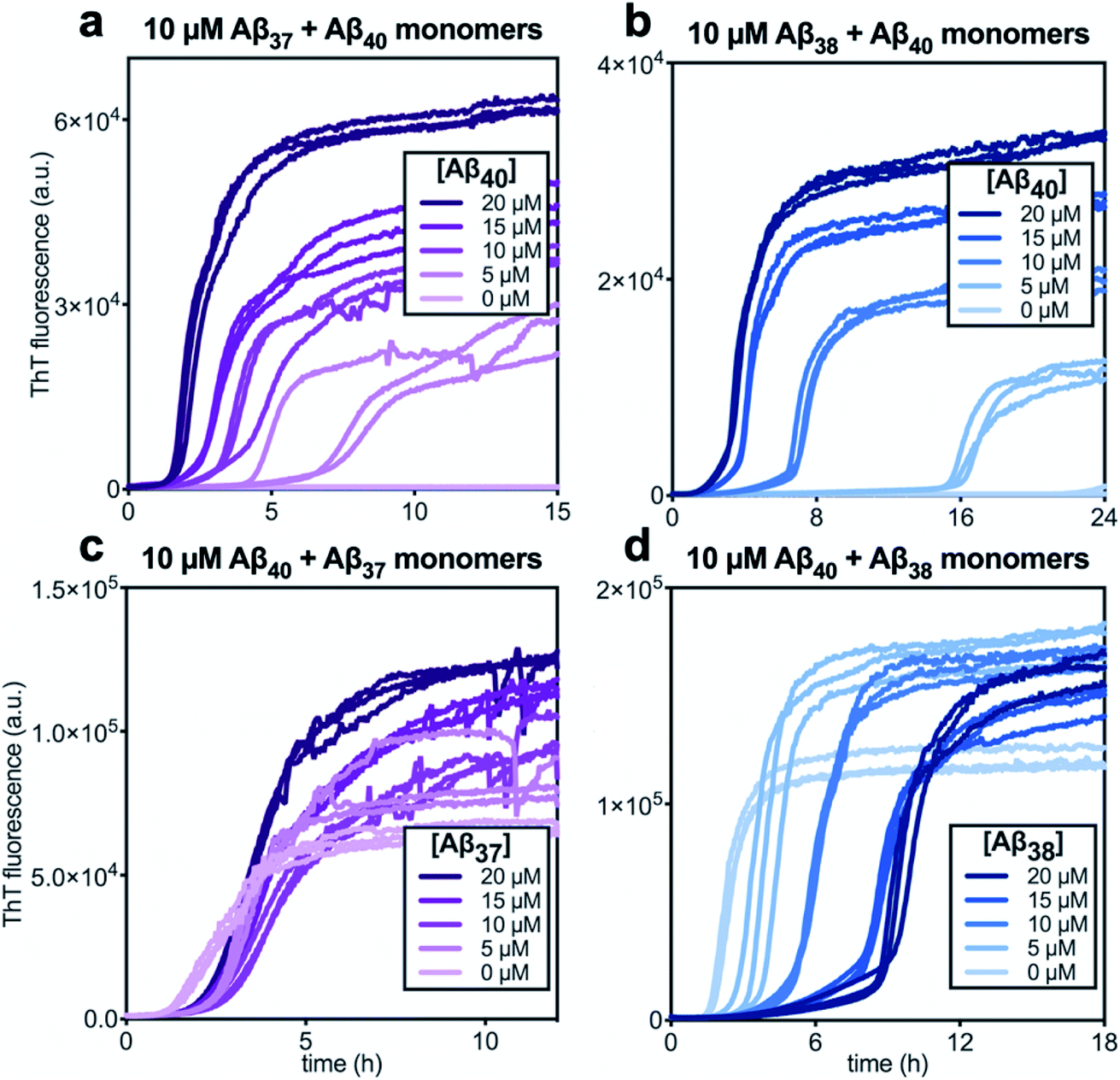 | ||
| Fig. 4 Aggregation kinetics of two-peptide systems composed of Aβ40 with (a & c) Aβ37 and (c & d) Aβ38 in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. The effect of Aβ40 at varying concentrations on the aggregation of (a) Aβ37 or (b) Aβ38. Aggregation kinetics for Aβ40 alone at the same concentrations are shown in Fig. S1.† The effect of varying concentrations of (c) Aβ37 or (d) Aβ38 on the aggregation of Aβ40. The extended time-courses for (c) and (d) are shown in Fig. S2.† | ||
The ultrastructure of the aggregates formed by these peptides, both alone and in mixtures, was investigated using cryo-TEM (Fig. 5). Images were collected of fibrils produced by each peptide individually, as well as by those formed by equimolar Aβ37/Aβ40 and Aβ38/Aβ40 mixtures. Samples were prepared in the same manner as for the kinetic assays, with fibril formation monitored by ThT fluorescence. The samples were frozen on grids once the final fluorescence plateau was reached. All of the fibrils formed from a pure peptide solution display relatively consistent morphology. Moreover, these homomolecular fibrils were morphologically similar between peptides: the fibrils formed by Aβ37, Aβ38, and Aβ40 had average helical half-pitch periods of 118 ± 30 nm, 131 ± 40 nm, and 125 ± 22 nm and average fibril diameters of 19 ± 6 nm, 15 ± 5 nm, and 18 ± 4, respectively. Notably, the morphology of the fibrils formed by each of these peptides was significantly different than that of the fibrils formed by Aβ42, which have a helical half pitch of 31 ± 17 nm.48 The fibrils formed from binary mixtures, on the other hand, displayed distinct morphology. For both Aβ37/Aβ40 and Aβ38/Aβ40 mixtures, fibrils were observed with helical half-pitch periodicities that did not correspond to the fibrils formed independently by either of the peptides present in the reaction mixture. In both cases, the average fibril half-pitch period (160 ± 90 nm for Aβ37/Aβ40 and 183 ± 106 for Aβ38/Aβ40) was both significantly longer and more variable than for any of the homomolecular fibrils (Fig. 5b). Additionally, the average width of the fibrils formed in each binary mixture (21 ± 10 nm for Aβ37/Aβ40 and 23 ± 12 for Aβ38/Aβ40) was both larger and more variable than for either of the constituent peptides, although this was statistically significant only for Aβ38/Aβ40 (Fig. 5c). The presence in both binary systems of fibrils with morphology distinct from those of homomolecular fibrils formed by either constituent peptide provided further evidence that some level of co-aggregation had occurred both between Aβ37 and Aβ40 and between Aβ38 and Aβ40.
 | ||
| Fig. 5 The morphology of fibrils formed by Aβ37, Aβ38, and Aβ40 in pure and equimolar binary solutions. (a) Representative cryo-TEM images of fibrils. Fibrils were formed in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. (b) Quantification of fibril node-to-node distance (half-pitch period). Representative grey scale profiles used for helical half-pitch period measurements are shown in Fig. S3.† (c) Quantification of fibril diameter. Plots represent >50 measurements, taken from at least 15 different fibrils. **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 by one-way ANOVA followed by Dunnett's post-hoc test. | ||
Aggregation kinetics of mixtures of Aβ42 with Aβ37 or Aβ38
Following the investigation of the aggregation behavior of Aβ40 with Aβ37 and Aβ38, similar experiments were performed on two-peptide systems with Aβ42 (Fig. 6). Starting from monomeric Aβ37/Aβ42 or Aβ38/Aβ42 mixtures, two distinct transitions were observed in the curves as monitored by ThT fluorescence. Such biphasic aggregation profiles were previously observed for the aggregation of a mixture of Aβ40 and Aβ42, which was found to correspond to aggregation into separate fibrils.48 This indicates that the nature of the interactions of these shorter alloforms with Aβ42 are quite different than with Aβ40. The biphasic profile suggests that two separate aggregation processes are occurring on discrete timescales, consistent with the formation of homomolecular fibrils.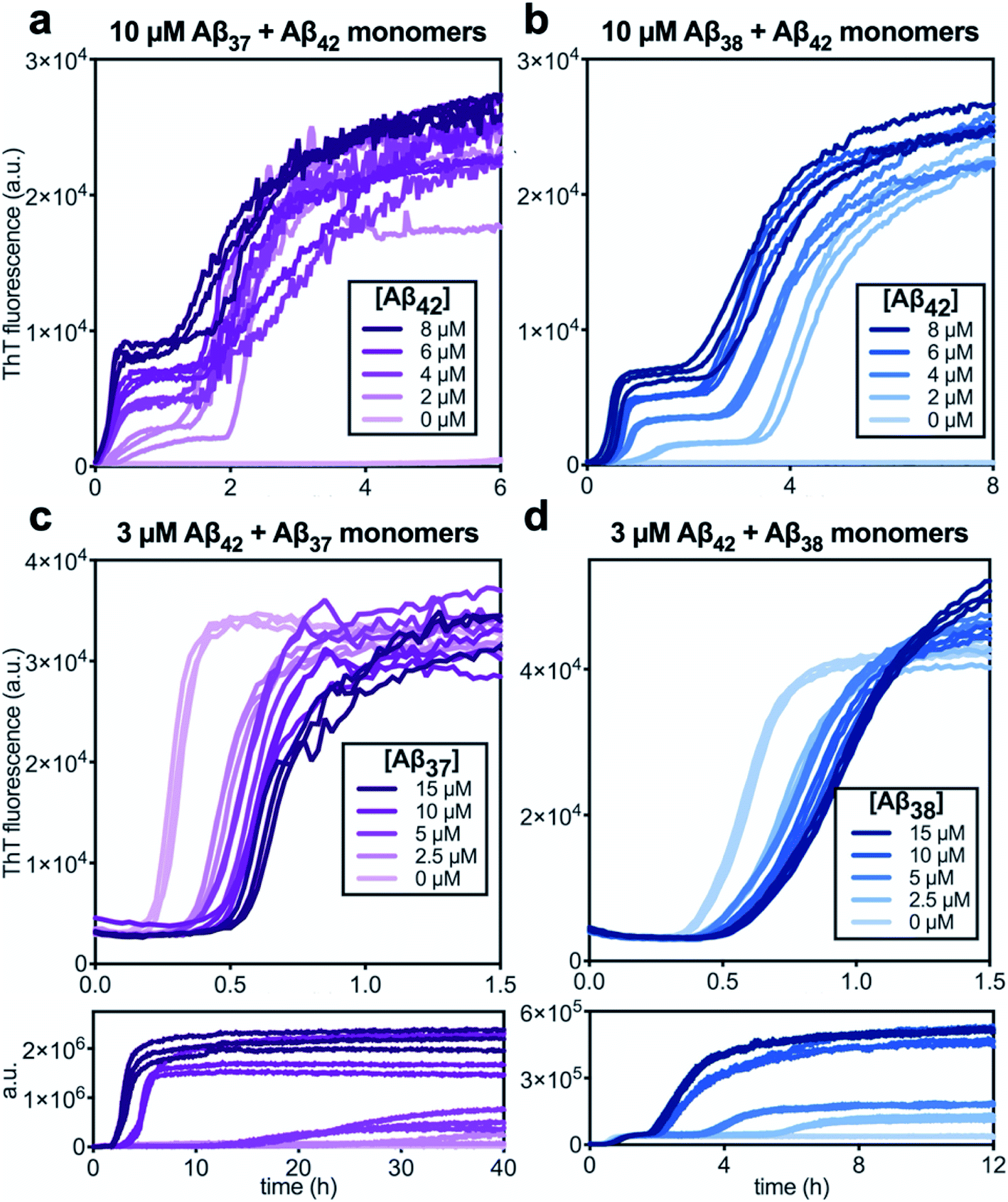 | ||
| Fig. 6 Aggregation kinetics of two-peptide systems composed of Aβ42 with (a & c) Aβ37 and (b & d) Aβ38 in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. (a and b) Increasing Aβ42 concentration leads to accelerated aggregation of Aβ37 and Aβ38, respectively. Aggregation kinetics for Aβ42 alone at the same concentrations are shown in Fig. S1.† (c and d) Increasing concentrations of Aβ37 and Aβ38, respectively, decrease the rate of Aβ42 aggregation; in both, the upper panel shows the first, low-intensity fluorescence transition and the lower panel shows the full biphasic aggregation profile. | ||
There were nevertheless significant interactions between the two peptides present in both Aβ37/Aβ42 and Aβ38/Aβ42 mixtures, as evidenced by the changes to the timescale of aggregation of both peptides in the mixture. The presence of Aβ42 strongly accelerated the aggregation of both Aβ37 and Aβ38 (Fig. 6a and b, respectively) by dramatically shortening their lag phases. Notably, each of Aβ37 and Aβ38 clearly inhibited the aggregation of Aβ42 (Fig. 6c and d, respectively), although this effect is less dramatic than the reciprocal acceleration of both Aβ37 and Aβ38 aggregation by Aβ42.
The aggregation of Aβ42 is known to proceed through three distinct processes: primary (non-fibril-catalyzed) nucleation, secondary (fibril-catalyzed) nucleation, and elongation.37 To inhibit aggregation, an effector must perturb one or more of these processes. Inhibition of each of the processes affects the kinetics of aggregation in a distinct manner,14,63 meaning that the change in the Aβ42 curve shape in the presence of each of the shorter alloforms can, in principle, be used to determine which process is being inhibited. Using the AmyloFit program,59 a model of secondary-nucleation-dominated aggregation was fit to the data shown in Fig. 6c and d; the parameters corresponding to the rate of each process were, one-by-one, allowed to vary while all other parameters were held constant (Fig. 7). This analysis shows that the observed changes to the curve shape of Aβ42 aggregation in the presence of both Aβ37 and Aβ38 is poorly modeled by inhibition of primary nucleation (Fig. 7a and d, respectively). However, the change to the Aβ42 aggregation curve shape, although significant, is too small for the kinetic analysis to distinguish between inhibition of secondary nucleation (Fig. 7b and e) and inhibition of elongation (Fig. 7c and f). Nevertheless, this fitting indicates that the inhibition of Aβ42 aggregation by both Aβ37 and Aβ38 involves interaction between the shorter peptide and the Aβ42 fibril, as both secondary nucleation and elongation are fibril-catalyzed processes.
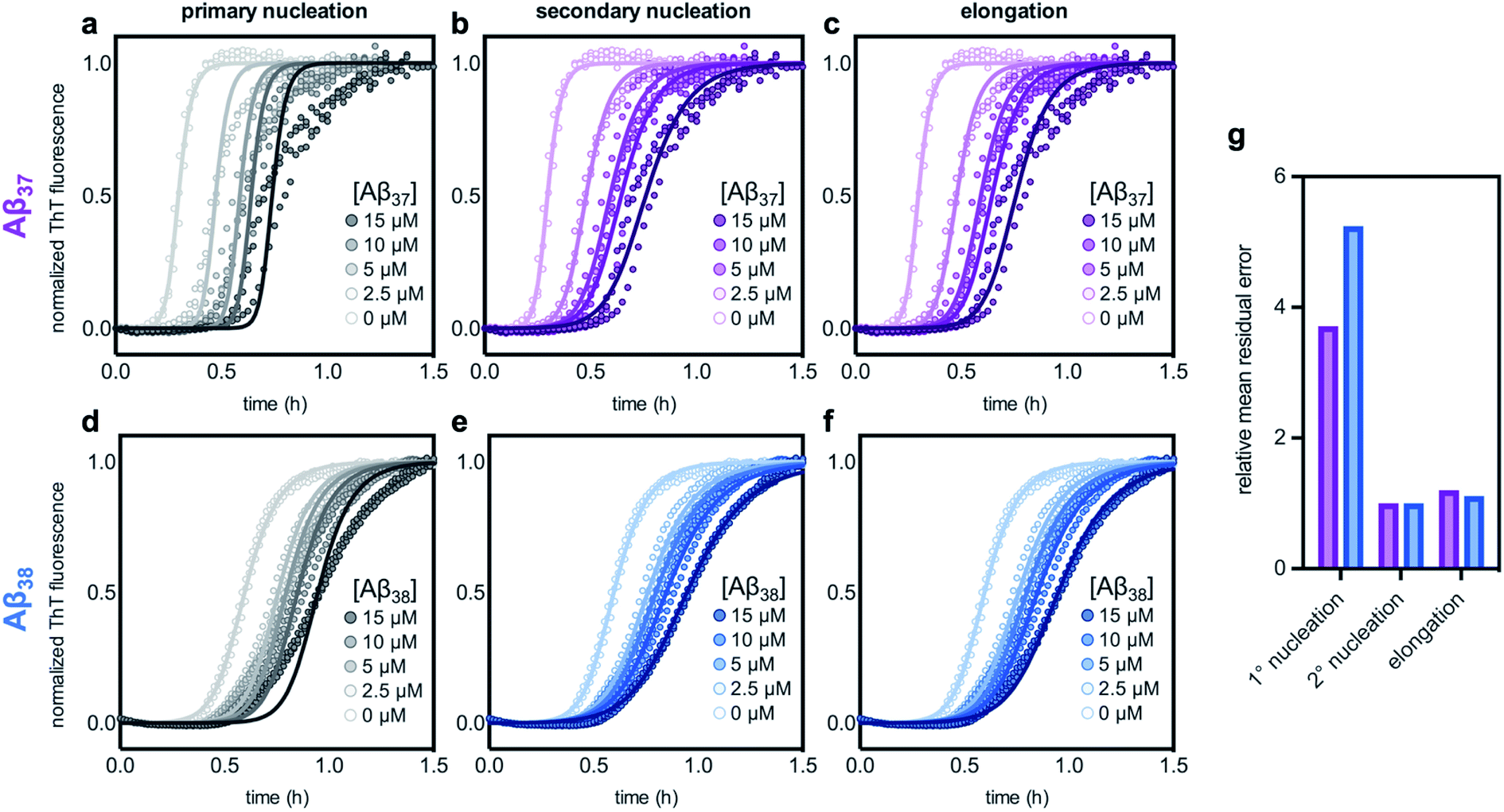 | ||
| Fig. 7 Fitting different models of inhibition to aggregation kinetics of 3 μM Aβ42 with varying concentrations of (a–c) Aβ37 and (d–f) Aβ38; the data are the same as are shown in Fig. 6c and d, respectively. The models used here correspond to inhibition of (a & d) primary nucleation, (b & e) secondary nucleation, and (c & f) elongation processes. (g) Mean residual errors for fitting shown in (a–f), normalized to the error for the best fit for each data set. Errors for Aβ37/Aβ42 fitting are shown in purple and errors for Aβ38/Aβ42 fitting are shown in blue. | ||
Cross-seeding of Aβ37 and Aβ38 with Aβ40 and Aβ42
Having studied the aggregation in systems composed of two Aβ alloforms, both starting in a monomeric state, we next investigated whether any of these peptide mixtures displayed cross-seeding behavior. In aggregating systems driven by a fibril-catalyzed secondary nucleation process, as is the case for all Aβ alloforms studied here, the addition of pre-aggregated seed fibrils to a solution of monomer can accelerate aggregation by providing a catalytic surface for nucleation from the start of the reaction process. The self-seeding of both Aβ42 and Aβ40 is well established.37,38,64 Although Aβ40 and Aβ42 have been shown to display some cross-seeding behavior, these effects are only evident at high seed concentrations (0.5 and 1 μM Aβ40 seed added to 2 μM Aβ42 monomer) and are much weaker than those observed for self-seeding.48In cross-seeding reactions, interactions between the monomeric peptide and the surface of the seed fibril can result in divergent kinetic outcomes.65 The seed fibril can serve as a surface for heterogenous primary nucleation, wherein monomer adsorption by the fibril increases local monomer concentration, thereby catalyzing aggregation.66,67 Conversely, higher-affinity monomer adsorption by the fibril surface leads to monomer sequestration, thus reducing the overall rate of aggregation, particularly during the fibril growth phase.48,67 In previous cross-seeding reactions between Aβ40 and Aβ42, Aβ42 seeds were shown to have no effect on Aβ40 aggregation; although high concentrations of Aβ40 seeds (25–50%) were shown to affect Aβ42 aggregation, these effects were much weaker than those of the self-seeding of Aβ42.48
Here, we found that the aggregation of Aβ37 is catalyzed by the presence of seeds of any of the four peptides (Fig. 8a). The effects were most dramatic for the self-seeding of Aβ37 (purple) and Aβ38 (blue), although they were nevertheless significant in all three cross-seeding cases. For the cross-seeding of Aβ37 with Aβ40 fibrils (green), the addition of seeds not only decreased the duration of the lag phase, but also reduced the slope of the curve during the aggregate growth phase. In contrast, either Aβ38 (blue) or Aβ42 (red) seed shortened the lag phase with minimal effects on the curve shape. This reduction in lag phase without a concomitant change in the curve shape suggests that the likely cause of the acceleration is the provision of a catalytic surface for the heterogeneous primary nucleation of Aβ37. However, at such long lag times, variability between experimental repeats is inevitable and prevents explicit verification of this proposed mechanism through fitting to kinetic models. Notably, the catalytic effect of Aβ42 fibrils on Aβ37 aggregation was smaller than that of monomeric Aβ42 at comparable concentrations (Fig. 6a), indicating that monomer–monomer interactions are a significant factor in the catalysis of Aβ37 aggregation by Aβ42. The fact that some cross-seeding behavior was observed, however, indicates that interactions between monomeric Aβ37 and fibrillar Aβ42 do also occur. This is consistent with the conclusions drawn from mechanistic fitting to the data of Aβ42 aggregation in the presence of monomeric Aβ37 (Fig. 7a–c).
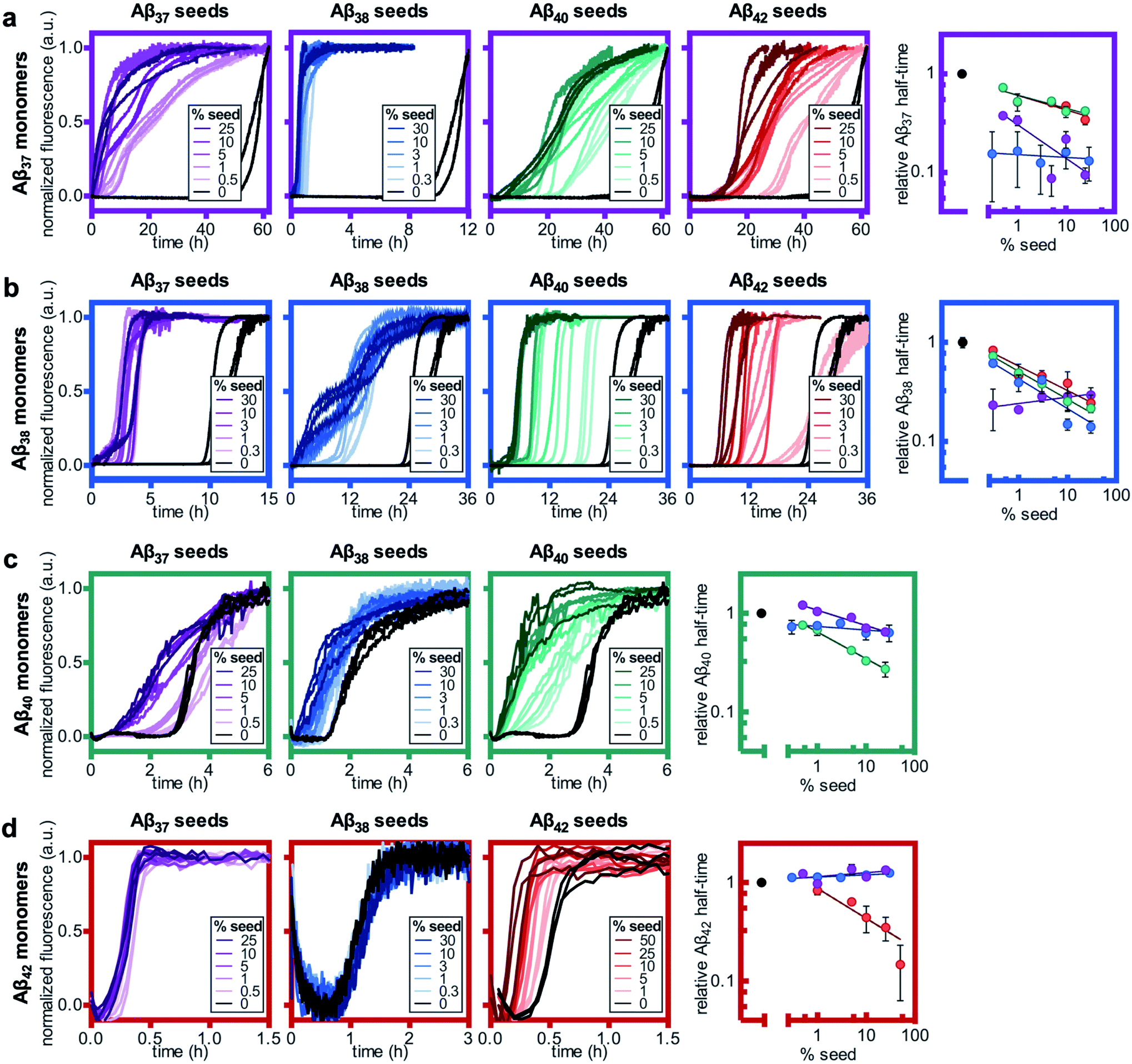 | ||
| Fig. 8 Self- and cross-seeding experiments for monomeric (a) Aβ37, (b) Aβ38, (c) Aβ40, and (d) Aβ42. In all panels, the color of the frame corresponds to the identity of the monomeric peptide and the color of the kinetic curves corresponds to the identity of the seed peptide, with Aβ37, Aβ38, Aβ40, and Aβ42 represented by purple, blue, green, and red, respectively. Seed concentrations are given as a molar percentage of the monomer concentration. The rightmost panel in each row shows the aggregation half-time as a function on seed percentage. Aβ37, Aβ38, and Aβ40 monomer solutions were prepared to a peptide concentration of 10 μM, and Aβ42 was prepared to a monomer concentration of 3 μM. All experiments were performed in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. | ||
As with Aβ37, the acceleration of Aβ38 aggregation was stronger in the self-seeded case than for cross-seeding (Fig. 8b). At high concentrations of Aβ38 seed, there was essentially no lag phase, with significant quantities of fibrils formed almost instantaneously. Having already found that Aβ38 fibrils can seed the aggregation of monomeric Aβ37 (Fig. 8a), the reciprocal effect is also observed, with the addition of Aβ37 seeds accelerating Aβ38 aggregation. For the cross-seeding of Aβ38 with both Aβ40 and Aβ42 seed fibrils, a clear concentration-dependent decrease in the lag time was observed; in both cases, there was no perturbation to the aggregation curve shape.
For Aβ40, clear self-seeding behavior was observed (Fig. 8c), consistent with previous findings.38,48 Additionally, clear reduction in the duration of the lag time was observed with the addition of both Aβ37 and Aβ38 seed fibrils, particularly at relatively high seed concentrations. As is the case for both Aβ37 and Aβ38, however, this cross-seeding effect was again weaker than the self-seeding effect. Similar to what was observed for the seeding of Aβ37 monomer with Aβ40 fibrils, the addition of Aβ37 fibrils to Aβ40 monomer affected not only the lag phase but also the slope of the curve during the aggregate growth phase. Indeed, at low concentrations of Aβ37 seed fibrils, this inhibitory effect was more significant than the reduction of the lag phase, resulting in an increase of the aggregation half-time relative to unseeded Aβ40.
Clear self-seeding behavior was observed for Aβ42, as has been reported previously;37,48 however, no cross-seeding catalysis was observed with either Aβ37 or Aβ38 seed fibrils (Fig. 8d). Even at high concentrations of Aβ37 and Aβ38 seed fibrils, both the timescale and curve shape of Aβ42 aggregation were completely unperturbed, indicating that these foreign aggregates are inert with respect to monomeric Aβ42, neither providing a surface for heterogeneous nucleation nor sequestering monomer. This suggests that the inhibition of Aβ42 aggregation observed in the presence of Aβ37 or Aβ38 (Fig. 6c and d, respectively) was caused by monomeric or oligomeric, rather than fibrillar, species. This is especially relevant given that, at the point in time at which Aβ42 aggregates, both Aβ37 and Aβ38 will be in a primarily monomeric state, due to the relatively slow aggregation of both peptides.
Aggregation kinetics of three-peptide mixtures
Having characterized the effects that the presence of either monomeric or fibrillar species of each Aβ alloform had on aggregation kinetics of the others, we next studied the behavior of Aβ37 or Aβ38 in three-peptide monomeric mixtures with Aβ40 and Aβ42. For both the Aβ37/Aβ40/Aβ42 and the Aβ38/Aβ40/Aβ42 mixtures, two discrete transitions in ThT fluorescence were observed, separated by an intermediate plateau phase (Fig. 9a and c, respectively). This is consistent with two aggregation processes happening on distinct timescales. The intensity of the second transition in ThT fluorescence increased with increasing concentration of Aβ37 or Aβ38, indicating that both peptides aggregate as part of the second, slower process. Based on the previous monomeric two-peptide aggregation kinetic assays (Fig. 4 and 6), it is most likely that the first fluorescence transition here represents the aggregation of Aβ42, with the second transition showing the co-aggregation of Aβ40 and Aβ37 or Aβ38.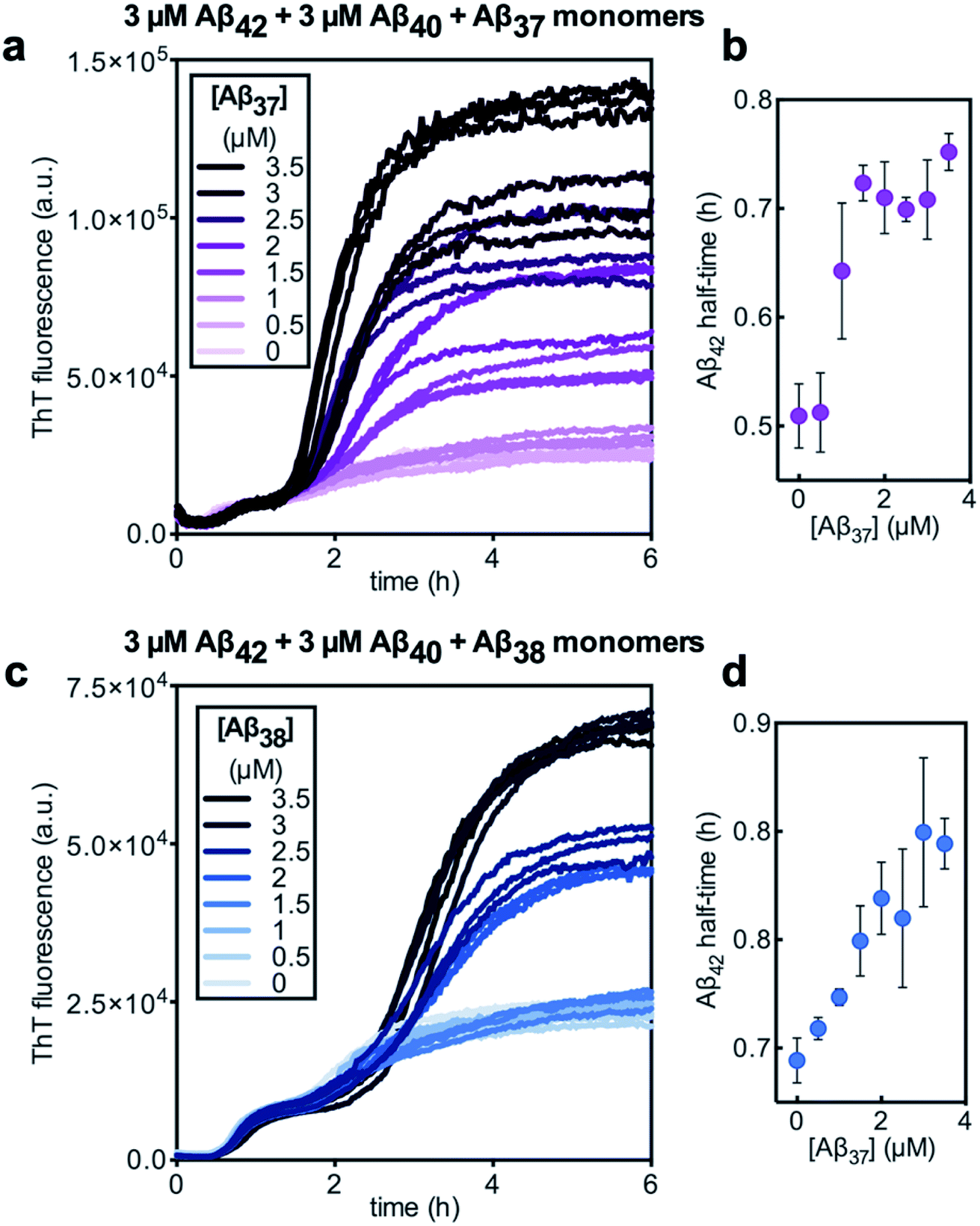 | ||
| Fig. 9 Aggregation of three-peptide systems composed of 3 μM each Aβ40 and Aβ42, with varying concentrations of (a and b) Aβ37 or (c and d) Aβ38 in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. Panels (a) and (c) show the aggregation kinetics curves. Panels (b) and (d) show the half-time of Aβ42 aggregation plotted as a function of Aβ37 and Aβ38 concentration, respectively. The points represent the average of the three technical triplicates, with the error bars showing the standard deviation. | ||
To analyze the identity and composition of the peptide aggregates that have formed during each transition, analytical HPLC was used to determine the concentration of each peptide over the time-course of the aggregation of an equimolar three-peptide mixture (Fig. 10). To characterize the composition of fibrillar species, aliquots were removed during the initial lag phase (tL) and each plateau phase (tP1 and tP2) (Fig. 10b and c for mixtures including Aβ37 and Aβ38, respectively). The fibrils present in these aliquots were isolated by centrifugation, washed to remove any oligomers associated with the fibril surface, then dissolved and injected on an HPLC instrument. In both three-peptide mixtures, it was found that the fibrils present in the intermediate plateau phase were primarily composed of Aβ42, while the fibrils in the final plateau phase were composed of all three peptides. These data are supported by the complementary analysis of the peptide composition of the supernatants (Fig. 10d and e), for which additional aliquots were taken during each aggregate growth phase (tG1 and tG2). For both Aβ37/Aβ40/Aβ42 and Aβ38/Aβ40/Aβ42 mixtures, it was found that the concentration of monomeric Aβ42 decreased during the first transition in ThT fluorescence and was undetectable by the intermediate plateau phase. During the second transition phase, the concentration of both Aβ40 and either Aβ37 or Aβ38 decreased. At the final plateau phase, the majority of peptides were converted to fibrils and the monomer concentration was below the detection limit of analytical HPLC. This indicates that, in these three-peptide mixtures, the behavior of each peptide is consistent with the behavior to in two-peptide systems, with Aβ42 forming fibrils during the first transition and Aβ40 co-aggregating with Aβ37 or Aβ38 during the second transition.
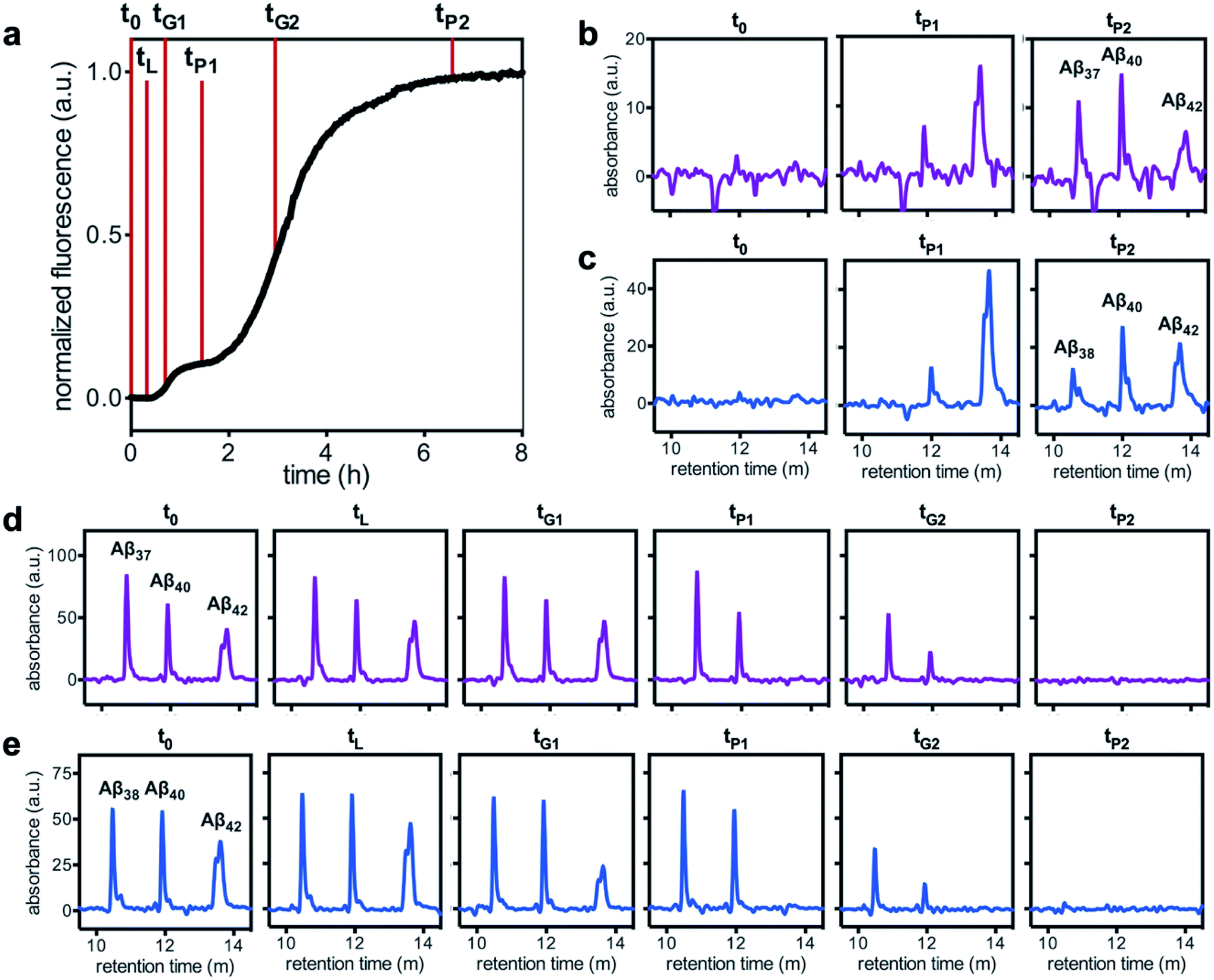 | ||
| Fig. 10 Alloform composition in fibrillar and monomeric species over the time-course of aggregation of trimolecular mixtures of 3 μM each Aβ40, Aβ42, and either (b & d) Aβ37 or (c & e) Aβ38, as analyzed using analytical HPLC. (a) A representative kinetic aggregation curve, with the time points at which aliquots were removed marked with red lines. (b & c) The composition of fibrillar species during the lag phase and each plateau phase. (d & e) The composition of monomeric species during the lag phase, both transition phases, and both plateau phases. Kinetic experiments were performed in 20 mM sodium phosphate, 200 μM EDTA, 0.02% NaN3, pH 7.4, with 20 μM ThT. | ||
Despite aggregating separately from Aβ42, the presence of either Aβ37 or Aβ38 perturbed the kinetics of Aβ42 aggregation, as was previously observed in two-peptide systems (Fig. 6c and d). The half-time of Aβ42 aggregation increased with increasing concentration of either Aβ37 or Aβ38 (Fig. 9b and d, respectively), with the half-time of Aβ42 aggregation defined here as the time at which the fluorescence intensity reached half-way between the initial baseline and the intermediate plateau. This shows that Aβ37/Aβ42, Aβ38/Aβ42, and Aβ40/Aβ42 (ref. 48) interactions observed in two-peptide systems persist in three-peptide systems.
Aggregation kinetics of four-peptide mixtures
Finally, the aggregation behavior in monomeric mixtures of all four alloforms was studied. First, the aggregation of 3 μM Aβ42 in mixtures with varying concentrations of Aβ37, Aβ38, and Aβ40 at an equimolar ratio was characterized (Fig. 11). For all four-peptide mixtures, two distinct transitions in ThT fluorescence intensity were observed. The intensity of the second transition (which was absent in a solution containing only Aβ42) scaled with increasing concentration of the Aβ37/Aβ38/Aβ40 mixture, while the intensity of the first transition did not. This indicates that all three of the shorter Aβ alloforms may form co-aggregates, while Aβ42 aggregates first, forming separate fibrils. As would be expected given the behavior observed in three-peptide mixtures, the aggregation of Aβ42 was perturbed by the presence of shorter Aβ alloforms despite the slower aggregation of these peptides, with increasing concentrations of the short-alloform mixture leading to longer Aβ42 aggregation half-times (Fig. 11b).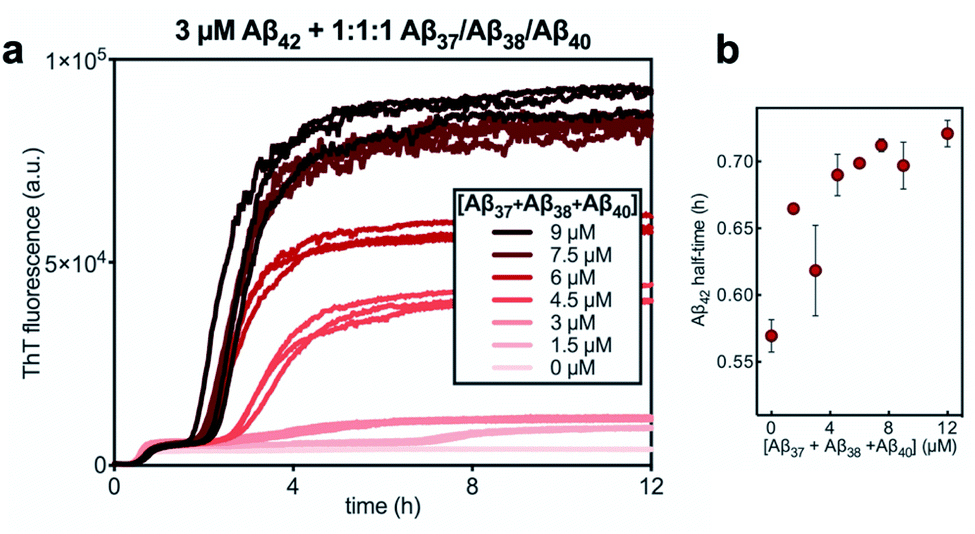 | ||
Fig. 11 (a) The aggregation of 3 μM Aβ42 with varying concentrations of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixture of Aβ37/Aβ38/Aβ40. (b) The half-time of the aggregation of Aβ42 shown in (a) versus the total concentration of the three shorter alloforms. Kinetic experiment was performed in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. :1:1 mixture of Aβ37/Aβ38/Aβ40. (b) The half-time of the aggregation of Aβ42 shown in (a) versus the total concentration of the three shorter alloforms. Kinetic experiment was performed in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. | ||
We next characterized the relative effects of each of the shorter Aβ alloforms on the aggregation of Aβ42, with an aim to determine whether specific ratios of these alloforms would maximally affect Aβ42 aggregation. For this, the aggregation of 3 μM Aβ42 was studied in the presence of a constant total concentration (6 μM) of mixtures of Aβ37, Aβ38, and Aβ40, with these three peptides in varying ratios (Fig. 12). For each ratio tested, only two fluorescence transitions were observed, providing further evidence that the three shortest alloforms co-aggregate irrespective of their ratio. Both the timescale and intensity of the second transition varied based on the ratio of Aβ37, Aβ38, and Aβ40, due to the differing aggregation propensities and ThT quantum yielded of each peptide. The aggregation of Aβ42 was found to be inhibited by all solutions tested, irrespective of the peptide ratios, although to varying extents. Included in this study were 6 μM each Aβ37, Aβ38, and Aβ40 alone, to allow for side-by-side comparison of the degree to which each alloform inhibits Aβ42 aggregation. Of the three, Aβ38 inhibited the aggregation of Aβ42 to the greatest degree, followed by Aβ37 and Aβ40. This was found to be reproducible between repeats of the whole experiment. Notably, mixtures of all three peptides produced a stronger inhibitory effect than any single peptide alone. Although the relative strength of inhibition of these different mixtures was not found to be highly reproducible between repeats of the experiment, mixtures with higher concentrations of Aβ38 generally exhibited stronger inhibitory effects than did mixtures with low concentrations of Aβ38. Still, Aβ38 alone was not the most effective, with the strongest inhibition observed at the 2:3:1 and 1:4:1 ratios of Aβ37:Aβ38:Aβ40.
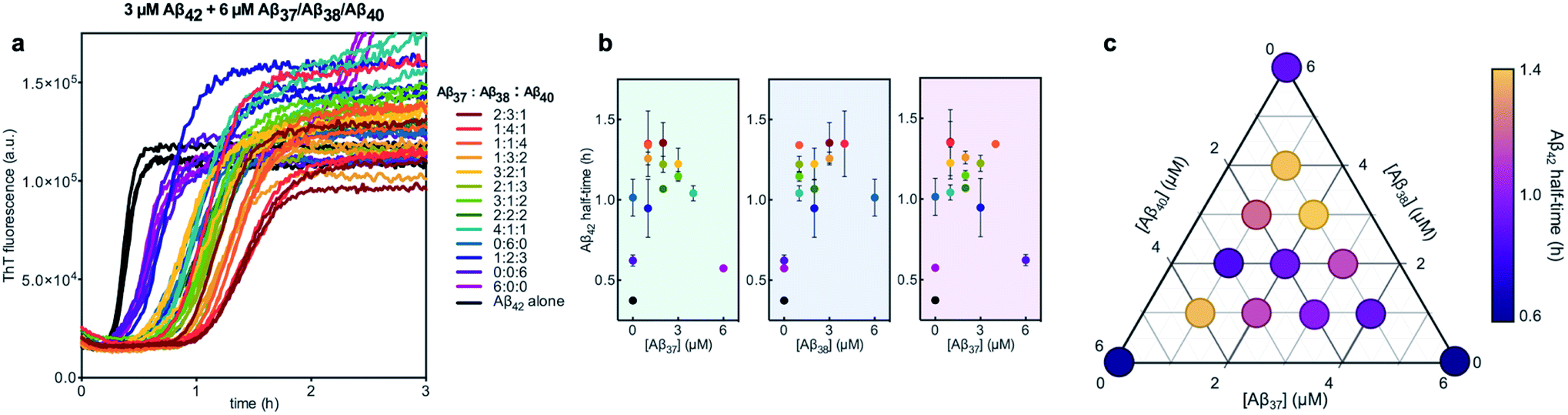 | ||
| Fig. 12 Aggregation kinetics of 3 μM Aβ42 with 6 μM of Aβ37/Aβ38/Aβ40 mixtures in varying molar ratios. (a) The first ThT transition, corresponding to the aggregation of Aβ42 is shown; the full aggregation profiles are shown in Fig. S4.† (b) The half-times of Aβ42 aggregation in (a), versus the concentration of Aβ37 (green panel), Aβ38 (blue panel), and Aβ40 (red panel). The color of each point corresponds to the curves of the same color in (a). (c) Triangular heat-map representation of the half-time of Aβ42 aggregation depending on the Aβ37/Aβ38/Aβ40 concentrations. Experiments were performed in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT. | ||
Discussion
The results of this study points to significant cross-reactivity in quaternary mixtures of Aβ42 and shorter alloforms, beyond those observed in binary mixtures.48 Notably, both monomeric Aβ37 and Aβ38 appear to interact with Aβ42 in much the same manner as does monomeric Aβ40. Most fundamentally, all three of the shortest alloforms aggregate independently of Aβ42, giving rise to two distinct fluorescence transitions. Furthermore, similar reciprocal kinetic effects are observed in binary monomeric mixtures of Aβ42 with each of the three shorter alloforms. As was found here for both Aβ37 and Aβ38, the presence of monomeric Aβ42 accelerates the aggregation of Aβ40 by significantly shortening the lag phase,48 although this effect is significantly more pronounced with Aβ37 and Aβ38, which both aggregate much more slowly than Aβ40. Additionally, the presence of any of the three shortest alloforms serves to inhibit the aggregation of Aβ42. In each case, this effect is weaker than the reciprocal Aβ42-induced acceleration of aggregation of the shorter alloform.The cross-seeding between Aβ42 and each of the shorter alloforms showed a stronger dependence on alloform length. As reported by Cukalevski et al., the addition of Aβ42 seeds to Aβ40 monomers results in minimal change to the kinetics of Aβ40 aggregation.48 This contrasts with the cross-seeding interactions between Aβ42 seeds and both Aβ37 and Aβ38 monomers, for which a clear Aβ42-concentration-dependent decrease in the aggregation half-time of the shorter alloform was observed in both cases. Furthermore, Cukalevski et al. reported that the addition of high concentrations of Aβ40 seeds to Aβ42 monomers leads to a moderate reduction of both the lag time and the rate of Aβ42 aggregation during the fibril growth phase (these converse effects largely cancel out, leaving the half-time of Aβ42 essentially unchanged).48 This behavior differs from the interactions observed here for the cross-seeding of Aβ42 monomers with either Aβ37 or Aβ38, for which even high seed concentrations had no effect on the kinetics of Aβ42 aggregation.
The results of this study serve to extend our understanding of interactions between Aβ alloforms by investigating the aggregation behavior of tri- and tetra-molecular mixtures. The fact that Aβ37, Aβ38, and Aβ40 each affected Aβ42 aggregation to a different extent indicates that inhibition of Aβ42 aggregation by these shorter alloforms is not simply due to non-specific interactions with Aβ42, but rather includes some level of sequence specificity. That Aβ38 is a more potent inhibitor of Aβ42 aggregation than are either Aβ37 or Aβ40 indicates that this effect is not directly correlated to alloform length. Likewise, this inhibition does not appear to be correlated with aggregation propensity, as Aβ37 and Aβ40 inhibited Aβ42 aggregation to a similar extent, despite Aβ40 displaying a much higher intrinsic aggregation rate than Aβ37. Determination of the various factors, including peptide length and sequence, that affect the relative inhibitory potency of these shorter alloforms towards Aβ42 aggregation would provide greater physical insight into the interactions that underlie this inhibition.
Notably, the results of this study indicate that, in addition to being alloform-specific, the effect of short Aβ alloforms on the aggregation of Aβ42 is inherently cooperative. Were this inhibition non-cooperative, it would be expected that the decrease in Aβ42 aggregation would simply scale with the relative concentrations of the short alloforms. Instead, a mixture of these shorter alloforms affected the rate of Aβ42 aggregation more significantly than any single alloform independently, with the strongest inhibition seen at the 2:3:1 and 1:4:1 Aβ37:Aβ38:Aβ40 ratios. While the underlying molecular interactions that give rise to this cooperative inhibition are not readily evident, these results further buttress the conclusion that inhibition of Aβ42 aggregation by shorter Aβ alloforms is sequence specific. One possible explanation for inhibition observed in certain quaternary mixtures may be the formation of mixed oligomers that cannot readily convert to fibrils and that compete for catalytic sites on the fibril surface with more conversion-competent homomolecular oligomers.68 Although Aβ42 in heteromolecular mixtures aggregates at reduced rate compared to pure Aβ42, it seems to promote nucleation of Aβ37, Aβ38 and Aβ40 in these mixed oligomers, and after Aβ42 has formed homomolecular fibrils, these three peptides seem form joint fibrils with different morphology than any of the pure fibrils of Aβ37, Aβ38 and Aβ40.
When studied alone as single peptides at a range of concentrations, Aβ38 appears to aggregate much more slowly than Aβ37. The origin of this difference is not clear but may be a consequence of primary or secondary nucleation being less effective with the extra Gly residue at the C-terminus of Aβ38. In general, longer sequences aggregate more slowly if the additional length difference lies in a non-amyloidogenic segment.33,69,70
These results suggest that in the brain extracellular fluids, where a number of different short Aβ alloforms are present, the disease-associated aggregation of Aβ1–42 may in fact be modulated by these short alloforms and, furthermore, that the rate of Aβ1–42 aggregation may be sensitive to specific ratios of these alloforms. The relevance of these findings is further underscored by the disease-promoting effect of familial AD-causing presenilin mutations, which lead to a loss of Aβ1–37 and Aβ1–38 whilst affecting neither Aβ1–40 nor Aβ1–42 production.71
Conclusions
In this study, we show that the Aβ alloforms Aβ37 and Aβ38 both aggregate through a secondary nucleation driven process to form β-sheet rich fibrils. The aggregation of both of these peptides is comparable to the well-characterized aggregation processes of Aβ40 and Aβ42, although the aggregation of the shorter two alloforms is significantly slower, which is consistent with their reduced aggregation propensity in vivo. Furthermore, we find that the aggregation of all four Aβ alloforms is highly sensitive to the presence of other alloforms. Both Aβ37 and Aβ38 co-aggregate with Aβ40, forming fibrils that are ultrastructurally distinct from the fibrils formed by either of the constituent peptides alone. Furthermore, the kinetics of aggregation in these binary mixtures is highly dependent on the ratio of Aβ37/Aβ40 or Aβ38/Aβ40 present in solution. In contrast to their co-aggregation with Aβ40, we find that both Aβ37 and Aβ38 aggregate independently from Aβ42. There is nevertheless significant kinetic modulation between Aβ42 and both Aβ37 and Aβ38, with the shorter alloforms impeding the aggregation of Aβ42 in a concentration-dependent manner and Aβ42 reciprocally accelerating the aggregation of the shorter alloforms. Finally, we find that the aggregation of Aβ42 is sensitive to the specific ratio of shorter alloforms present, indicating a level of sequence specificity in these interactions; we furthermore demonstrate that a mixture of Aβ37, Aβ38, and Aβ40 more potently affects Aβ42 aggregation than any one of the shorter alloforms alone, indicating that this inhibition is inherently cooperative.These results demonstrate that interactions between Aβ alloforms significantly modulate the aggregation behavior of these peptides in vitro. As the presence of Aβ38 has been shown to attenuate Aβ42 fibril formation and deposition in vivo,49,50 it is likely that the interactions described here are pertinent to the biological behavior of these peptides. Indeed, short Aβ alloforms, which make up as much as 90% of the Aβ peptide present in the extracellular fluid of the brain, may play a pathologically relevant role in moderating the aggregation of Aβ42. These results are relevant not only to understanding the pathogenesis of AD, but also to the development of AD therapeutics. As modulation of γ-secretase activity is increasingly being proposed as a promising route to AD therapeutics,72–74 an understanding of how different Aβ alloforms—and how different combinations of these alloforms—affect pathological Aβ42 aggregation is becoming increasingly important.
Materials and methods
Expression and purification of Aβ peptides
The genes encoding wild-type Aβ(M1-42), Aβ(M1-40), Aβ(M1-38), and Aβ(M1-37) (referred to herein as Aβ42, Aβ40, Aβ38, and Aβ37, respectively) were produced by overlapping PCR and cloned into the PetSac vector.58 The peptides were expressed in E. coli strains BL21 Star (DE3) pLysS (for Aβ42, Aβ40, and Aβ37) or BL21-Gold (DE3) pLysS (for Aβ38). Cells were cultured in LB medium with 50 mg L−1 ampicillin and (for BL21 Star cells) 30 mg L−1 chloramphenicol. Well-isolated bacterial colonies were used to inoculate 50 mL cultures grown in 250 mL baffled flasks at 37 °C with 130 rpm shaking for ca. 7 h. Once an OD of 0.7–1.0 was reached, 500 μL of these cultures were then added to 500 mL LB medium (containing the relevant antibiotics, as detailed above) in 2 L baffled flasks, which were grown at 37 °C with 125 rpm shaking for ca. 15 h. Cells were then harvested by centrifugation at 6000 × g for 10 min at 4 °C.The peptides were isolated from inclusion bodies after iterative sonication and centrifugation. The inclusion bodies were dissolved in 8 M urea in 10 mM Tris–HCL, pH 8.5, 1 mM EDTA, and the peptide purified by ion exchange chromatography on a DEAE cellulose resin, as previously described.58 The purity of the eluted fractions was assessed by SDS-PAGE. The purest fractions were pooled. Monomer isolation and removal of inclusion-body-bound proteins was achieved by filtration using 30 kDa MWCO filters, and size exclusion chromatography on a 26 × 600 mm Superdex 75 column. The peptide was then aliquoted, frozen and lyophilized, and stored at −20 °C.
Monomer isolation for kinetic assays
To isolate pure monomer, Aβ peptides were subjected to size-exclusion chromatography (SEC) immediately before preparing aggregation kinetic assays. Aliquots of purified, lyophilized Aβ were dissolved in 1.0 mL 6 M GuHCl, injected on a Superdex 75 10/300 GL column, and eluted with an isocratic 0.7 mL min−1 flow of 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA and 0.02% NaN3. Typical retention times were between 19 and 21 minutes for all peptides. The center of the monomer peak was collected on ice using low-binding Eppendorf tubes. The concentration of purified peptide was determined by integrating the absorbance at 280 nm of the collected peak, using ε280 = 1400 L mol−1 cm−1 (based on the presence of a single tyrosine residue in each alloform). Concentration determination in this manner is accurate within ±20%.Preparation of unseeded kinetic assays for pure peptides
A dilution series was prepared from the SEC-purified peptide. First, thioflavin T (ThT, Calbiochem) was added to the sample from a concentrated stock solution (filtered through 0.2 μm filter) to a final concentration of 20 μM. The dilution series was then prepared from this peptide solution, using 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT (at this concentration, ThT fluorescence was found to scale linearly with aggregate concentration for all alloforms, Fig. S5†). All samples were prepared in low-binding tubes on ice (Axygen). The samples were loaded from low to high peptide concentration, with five wells per sample and 90 μL per well, into a 96-well, half-area, clear bottomed, PEG-coated, black polystyrene plate (Corning 3881), which was subsequently sealed. The whole setup was repeated at least three times for each peptide.Preparation of multimolecular unseeded kinetic assays
All four peptides were purified separately by SEC, as described above. ThT was added from a concentrated stock solution to each of the purified peptides to a final concentration of 20 μM. Additionally, a dilution solution of 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA, 0.02% NaN3, and 20 μM ThT was prepared. The purified peptide solutions and the dilution solution were combined in varying quantities to give the desired molar ratios and total concentrations. The samples were then loaded from low to high concentration of the most aggregation-prone peptide in the mixture, with four to five 90 μL wells per sample, into a 96-well plate (Corning 3881), which was subsequently sealed. The whole setup was repeated at least three times for each peptide combination.Preparation of self- and cross-seeded kinetic assays
Seed fibrils were made from SEC-purified peptide, with fibril formation monitored by ThT fluorescence, as described above. Seed fibrils were removed once the fluorescence plateau was reached, approximately 20 hours for Aβ37, 48 hours for Aβ38, 5 hours for Aβ40, and 1 hour for Aβ42. These seed fibrils were then added in varying concentrations to aliquots of a solution of SEC-purified monomeric peptide, to which ThT had been added to a final concentration of 20 μM. For all seeded experiments, the aggregation of a set of unseeded control samples was run in parallel to the seeded reactions. Four technical replicates of each sample were then plated into a 96-well plate (Corning 3881). The whole setup was repeated at least twice for each peptide combination.Kinetic assays
ThT fluorescence was measured using a FLUOstar Omega or FLUOstar Optima plate reader (BMG Labtech) at 37 °C under quiescent conditions. Fluorescence was measured through the bottom of the plate using an excitation filter of 440 nm and an emission filter of 480 nm, with measurements taken every 60–120 s (depending on the aggregation propensity of the alloform being studied).Kinetic analysis
All kinetic analyses were performed using the free online program AmyloFit (www.amylofit.ch.cam.ac.uk).59 To facilitate half-time determination and fitting of various mechanistic models, fluorescence data were normalized, using both a zero-point offset (in case of seeded experiments) and end-point normalization. For data sets with multiple fluorescence transitions, only the first transition, corresponding to Aβ42 aggregation, was normalized; for this, the intermediate plateau phase was used as the aggregation endpoint.Aggregation half-times were determined by performing a linear fit to the normalized ThT fluorescence data for the period in which the signal intensity was between 0.4 and 0.6. The half-time was taken to be the time at which the linear fit equals 0.5. When applicable, the scaling exponent was determined by plotting the half-time versus the relevant starting monomer concentration and fitting to this data a power function
| t1/2 = α[m]γ0 |
The fitting of mechanistic models was used to assess which of the microscopic Aβ42 aggregation steps are perturbed in Aβ42/Aβ38 and Aβ42/Aβ37 binary mixtures. For this, the data from the sample containing only Aβ42 was fitted first, using the integrated rate law
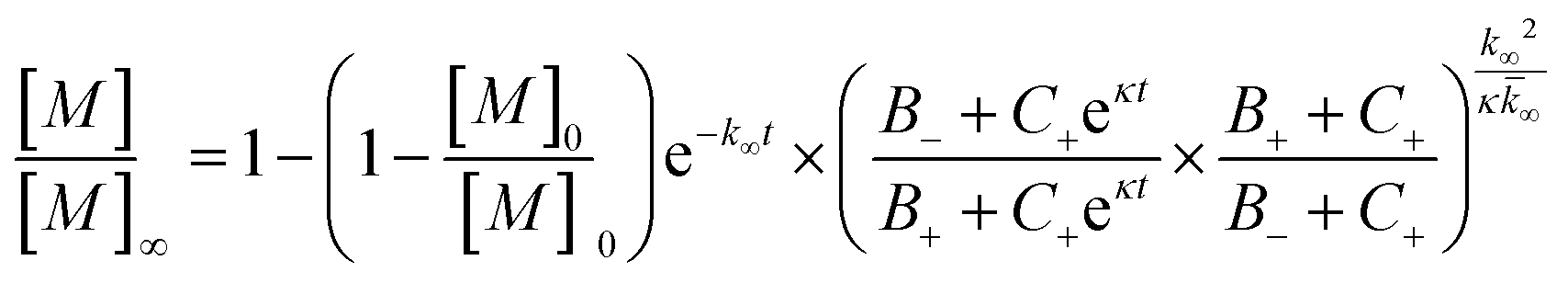
for which the parameters are defined
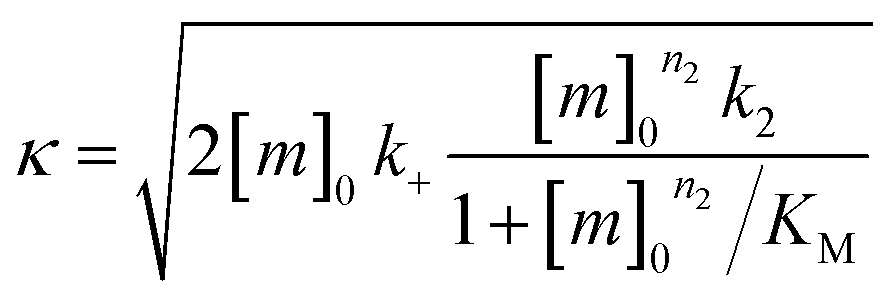
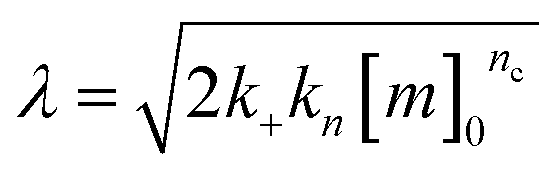
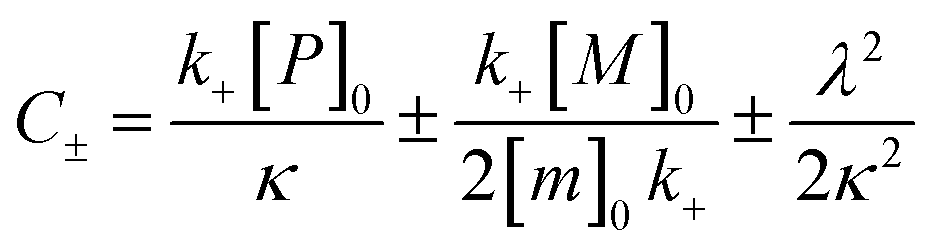
| k∞ = 2k+[P]∞ |
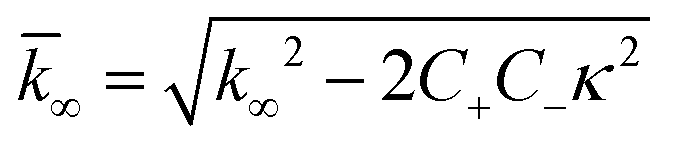
were [m]0 is the initial monomer concentration; [P]0 is the fibril number at the start of the reaction; [P]∞ is the fibril number at equilibrium, when the reaction has reached completion (see ref. 38 for the detailed expression of [P]∞); [M]0 is the fibril mass concentration at the start of the reaction; [M]∞ is the fibril mass concentration at equilibrium; kn, k2, and k+ are the rate constants for primary nucleation, secondary nucleation, and elongation, respectively; KM is the saturation constant for secondary nucleation; and nc and n2 are the reaction orders of primary and secondary nucleation, respectively.
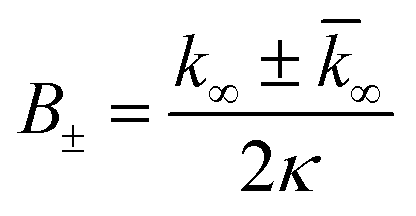
For this fitting, the primary and secondary nucleation reaction orders (nc and n2, respectively) both set to 2, based on previously reported data for Aβ42 under similar conditions,37,41 and the initial seed concentration was set to 0. The rate constants k+, kn, and k2 (corresponding to elongation, primary nucleation, and secondary nucleation, respectively) were simultaneously fitted to the data, thus accounting for any minor differences in the starting monomer concentration. For the analysis of each binary aggregation data set, three different fits were performed with one rate constant as a variable parameter and the other two rate constants fixed to the values obtained for Ab42 alone. The comparison of the three fits reveals whether selective perturbation of a given process could accurately model the observed inhibition of Aβ42 aggregation.
Monomer depletion by HPLC
The samples used for monomer depletion studies were prepared and incubated as described above, with aggregation monitored by ThT fluorescence. All samples were prepared to a concentration of 3 μM each Aβ42, Aβ40, and either Aβ38 or Aβ37 and plated in 100 μL aliquots. One sample was removed into a low-binding tube (Axygen) at the start of incubation, with additional samples removed during the initial lag phase, the first fluorescence transition, the intermediate plateau phase, the second fluorescence transition, and the final plateau phase. In addition to the removed samples, two additional samples were left in the plate to monitor the entire aggregation process.Upon removal, each sample was centrifugated at 20000 × g for 5 min at room temperature to sediment the aggregated peptide. Following centrifugation, the upper 50 μL of the supernatant was removed and injected on an analytical HPLC instrument (Agilent 1100), equipped with a C8 column (Agilent ZORBAX 300SB, 200 × 4.6 mm), run at a flow rate of 1 mL min−1 at 70 °C. The mobile phases used were (solvent A) 0.1% formic acid in water and (solvent B) 0.1% formic acid in acetonitrile. A linear gradient of 15–30% B over 10 min, followed by an isocratic flow of 30% B for an additional five min was used. The approximate retention times for each peptide were as follows: Aβ42 = 13.7 min, Aβ40 = 12.0 min, Aβ38 = 10.6 min, Aβ37 = 10.8 min.
Fibril composition by HPLC
Samples were prepared and incubated as described above, with aggregation monitored by ThT fluorescence. All samples were prepared to a concentration of 3 μM each of Aβ42, Aβ40, and either Aβ38 or Aβ37, and plated in 125 μL aliquots. Samples were removed at the start of incubation, during the intermediate plateau phase, and the final plateau phase. At each time point, the contents of two wells were removed and combined to give a total sample volume of 250 μL. In addition to the removed samples, two additional samples were left in the plate to monitor the aggregation process.Upon removal, samples were filtrated through 0.2 μm spin filters (VIVASPIN 500) at 15000 × g for 5 min at room temperature to trap the fibrils. The filter-trapped fibrils were then washed five times with 500 μL ultrapure water (to a total wash volume of 10x the sample volume) to remove any monomeric or oligomeric peptide on the fibril and filter surface. After each wash, the sample was centrifugated at 15000 × g for 5 min at room temperature, leaving a retentate volume of ca. 25 μL. After the final wash, the retentate was mixed in a 1:3 ratio with 6 M GuHCl to dissolve the fibrils for 15 min before being injected on an analytical HPLC instrument. HPLC was performed as described above.
Cryo-EM
For Cryo-EM samples, monomer isolation for all peptides was performed as described above. Solutions were prepared with a total monomeric peptide concentration of 10 μM and incubated in the same manner as for the kinetic assays, with aggregation monitored by ThT fluorescence. Samples were taken once the final fluorescence plateau was reached. Specimens were prepared for imaging in a controlled environment vitrification system (CEVS) to maintain stable temperature and to minimize solution loss. The sample was prepared as a thin liquid film, <300 nm thick, on lacey carbon filmed copper grids. This was plunged into liquid ethane at −180 °C to vitrify the sample; this minimizes water crystallization as well as component segmentation and rearrangement, thus maintaining original microstructures. The vitrified sample was stored under liquid nitrogen until imaged. The grid was transferred into the electron microscope (JEM 2200FS) using a Fischione Model 2550 cryo transfer tomography holder. The microscope was equipped with an in-column energy filter (Omega filter), thus allowing for zero-loss imaging. The acceleration voltage was 200 kV and zero-loss images were recorded digitally with a TVIPS F416 camera using SerialEM under low dose conditions and with a 30 eV energy selecting slit in place.To quantify fibril ultrastructure, the node-to-node distance and fibril diameter were measured using ImageJ (version 2.1.0/1.53c, NIH). For each sample, at least 50 measurements were performed, measuring at least fifteen total fibrils from five different images, each taken from different parts of the sample grid. Measurements are presented in boxplots with Tukey whiskers, with the median represented by the center line, the box containing the 25th–75th percentiles, and individually plotted points representing statistical outliers. Statistical significance was assessed using a one-way ANOVA followed by Dunnett's post-hoc test, with p < 0.05 considered significant. All statistical analysis was performed in GraphPad Prism (version 9.0.2).
Data availability
All data presented in this article will be made available upon reasonable request.Author contributions
H. Z. and S. L. designed the study. G. A. B., K. S. and S. L. performed the experiments. G. A. B. and A. J. D. analyzed data. G. A. B. wrote the paper with input from all co-authors.Conflicts of interest
H. Z. has served at scientific advisory boards for Eisai, Denali, Roche Diagnostics, Wave, Samumed, Siemens Healthineers, Pinteon Therapeutics, Nervgen, AZTherapies and CogRx, has given lectures in symposia sponsored by Cellectricon, Fujirebio, Alzecure and Biogen, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work).Acknowledgements
Cryo-EM images were obtained with expert assistance from Anna Carnerup (Lund University). This work was funded by the Swedish Research Council (grants #2015-00143 to S. L. and #2018-02532 to H. Z.), the European Research Council (#681712 to H. Z.), Swedish State Support for Clinical Research (#ALFGBG-720931 to H. Z.), the Lindemann Trust Fellowship (A. J. D.), the Novo Nordisk Foundation (#NNF19OC0054635 to S. L.), and the Fulbright U.S. Student Program (G. A. B). H. Z. is a Wallenberg Scholar.References
- C. A. Lane, J. Hardy and J. M. Schott, Eur. J. Neurol., 2018, 25, 59–70 CrossRef CAS PubMed.
- Developing therapeutics for Alzheimer's disease: progress and challenges, ed., M. S. Wolfe, Elsevier/AP, Academic Press is an imprint of Elsevier, Amsterdam; Boston, 2016 Search PubMed.
- A. B. Reiss, H. A. Arain, M. M. Stecker, N. M. Siegart and L. J. Kasselman, Rev. Neurosci., 2018, 29, 613–627 CAS.
- J. L. Cummings, G. Tong and C. Ballard, J. Alzheimer's Dis., 2019, 67, 779–794 Search PubMed.
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50–56 CrossRef CAS PubMed.
- S. B. Haeberlein, Presented in part at the 12th Clinical Trials on Alzheimer's Disease (CTAD) Congress, San Diego, CA, 2019 Search PubMed.
- S. Linse, T. Scheidt, K. Bernfur, M. Vendruscolo, C. M. Dobson, S. I. A. Cohen, E. Sileikis, M. Lundqvist, F. Qian, T. O’Malley, T. Bussiere, P. H. Weinreb, C. K. Xu, G. Meisl, S. R. A. Devenish, T. P. J. Knowles and O. Hansson, Nat. Struct. Mol. Biol., 2020, 27, 1125–1133 CrossRef CAS PubMed.
- “FDA Grants Accelerated Approval for Alzheimer's Drug.” June 2021. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug. Accessed October 2021 Search PubMed.
- J. Hardy and D. Allsop, Trends Pharmacol. Sci., 1991, 12, 383–388 CrossRef CAS PubMed.
- J. Hardy and G. Higgins, Science, 1992, 256, 184–185 CrossRef CAS PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, J. Alzheimer's Dis., 2018, 64, S567–S610 CAS.
- M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson and S. E. Radford, Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773 CrossRef CAS PubMed.
- S. Linse, Biophys. Rev., 2017, 9, 329–338 CrossRef CAS PubMed.
- M. Törnquist, T. C. T. Michaels, K. Sanagavarapu, X. Yang, G. Meisl, S. I. A. Cohen, T. P. J. Knowles and S. Linse, Chem. Commun., 2018, 54, 8667–8684 RSC.
- R. M. Ransohoff, Science, 2016, 353, 777–783 CrossRef CAS PubMed.
- V. Calsolaro and P. Edison, Alzheimer's Dementia, 2016, 12, 719–732 CrossRef PubMed.
- F. Kametani and M. Hasegawa, Front. Neurosci., 2018, 12, 25 CrossRef PubMed.
- G. S. Bloom, JAMA Neurol., 2014, 71, 505 CrossRef PubMed.
- G. K. Gouras, T. T. Olsson and O. Hansson, Neurotherapeutics, 2015, 12, 3–11 CrossRef CAS PubMed.
- D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595–608 CrossRef CAS PubMed.
- P. Arosio, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Trends Pharmacol. Sci., 2014, 35, 127–135 CrossRef CAS PubMed.
- Y. Zhang, R. Thompson, H. Zhang and H. Xu, Mol. Brain, 2011, 4, 3 CrossRef CAS PubMed.
- C. Haass, C. Kaether, G. Thinakaran and S. Sisodia, Cold Spring Harbor Perspect. Med., 2012, 2, a006270 Search PubMed.
- K. G. Mawuenyega, T. Kasten, W. Sigurdson and R. J. Bateman, Anal. Biochem., 2013, 440, 56–62 CrossRef CAS PubMed.
- N. Kaneko, R. Yamamoto, T.-A. Sato and K. Tanaka, Proc. Jpn. Acad., Ser. B, 2014, 90, 104–117 CrossRef CAS PubMed.
- J. Reinert, B. C. Richard, H. W. Klafki, B. Friedrich, T. A. Bayer, J. Wiltfang, G. G. Kovacs, M. Ingelsson, L. Lannfelt, A. Paetau, J. Bergquist and O. Wirths, Acta Neuropathol. Commun., 2016, 4, 24 CrossRef PubMed.
- G. Brinkmalm, W. Hong, Z. Wang, W. Liu, T. T. O'Malley, X. Sun, M. P. Frosch, D. J. Selkoe, E. Portelius, H. Zetterberg, K. Blennow and D. M. Walsh, Brain, 2019, 142, 1441–1457 CrossRef PubMed.
- M. Bibl, B. Mollenhauer, H. Esselmann, P. Lewczuk, H.-W. Klafki, K. Sparbier, A. Smirnov, L. Cepek, C. Trenkwalder, E. Rüther, J. Kornhuber, M. Otto and J. Wiltfang, Brain, 2006, 129, 1177–1187 CrossRef PubMed.
- J. Wiltfang, H. Esselmann, M. Bibl, A. Smirnov, M. Otto, S. Paul, B. Schmidt, H.-W. Klafki, M. Maler, T. Dyrks, M. Bienert, M. Beyermann, E. Rüther and J. Kornhuber, J. Neurochem., 2002, 81, 481–496 CrossRef CAS PubMed.
- D. Galasko, L. Chang, R. Motter, C. M. Clark, J. Kaye, D. Knopman, R. Thomas, D. Kholodenko, D. Schenk, I. Lieberburg, B. Miller, R. Green, R. Basherad, L. Kertiles, M. A. Boss and P. Seubert, Arch. Neurol., 1998, 55, 937 CrossRef CAS PubMed.
- P. E. Spies, M. M. Verbeek, T. van Groen and J. A. H. R. Claassen, Front. Biosci., Landmark Ed., 2012, 17, 2024–2034 CrossRef PubMed.
- S. Zampar, H. W. Klafki, K. Sritharen, T. A. Bayer, J. Wiltfang, A. Rostagno, J. Ghiso, L. A. Miles and O. Wirths, Neuropathol. Appl. Neurobiol., 2020, 46, 673–685 CrossRef CAS PubMed.
- O. Szczepankiewicz, B. Linse, G. Meisl, E. Thulin, B. Frohm, C. Sala Frigerio, M. T. Colvin, A. C. Jacavone, R. G. Griffin, T. Knowles, D. M. Walsh and S. Linse, J. Am. Chem. Soc., 2015, 137, 14673–14685 CrossRef CAS PubMed.
- T. Weiffert, G. Meisl, P. Flagmeier, S. De, C. J. R. Dunning, B. Frohm, H. Zetterberg, K. Blennow, E. Portelius, D. Klenerman, C. M. Dobson, T. P. J. Knowles and S. Linse, ACS Chem. Neurosci., 2019, 10, 2374–2384 CrossRef CAS PubMed.
- M. P. Murphy and H. LeVine, J. Alzheimer's Dis., 2010, 19, 311–323 Search PubMed.
- A. Vandersteen, E. Hubin, R. Sarroukh, G. De Baets, J. Schymkowitz, F. Rousseau, V. Subramaniam, V. Raussens, H. Wenschuh, D. Wildemann and K. Broersen, FEBS Lett., 2012, 586, 4088–4093 CrossRef CAS PubMed.
- S. I. A. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. A. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef CAS PubMed.
- A. Abelein, J. Jarvet, A. Barth, A. Gräslund and J. Danielsson, J. Am. Chem. Soc., 2016, 138, 6893–6902 CrossRef CAS PubMed.
- G. Meisl, X. Yang, C. M. Dobson, S. Linse and T. P. J. Knowles, Chem. Sci., 2017, 8, 4352–4362 RSC.
- G. Meisl, X. Yang, B. Frohm, T. P. J. Knowles and S. Linse, Sci. Rep., 2016, 6, 18728 CrossRef CAS PubMed.
- R. Sabaté, A. Espargaró, L. Barbosa-Barros, S. Ventura and J. Estelrich, Biochimie, 2012, 94, 1730–1738 CrossRef PubMed.
- A. K. Srivastava, J. M. Pittman, J. Zerweck, B. S. Venkata, P. C. Moore, J. R. Sachleben and S. C. Meredith, Protein Sci., 2019, 28, 1567–1581 CAS.
- C. Cabaleiro-Lago, F. Quinlan-Pluck, I. Lynch, S. Lindman, A. M. Minogue, E. Thulin, D. M. Walsh, K. A. Dawson and S. Linse, J. Am. Chem. Soc., 2008, 130, 15437–15443 CrossRef CAS PubMed.
- K. Ezzat, M. Pernemalm, S. Pålsson, T. C. Roberts, P. Järver, A. Dondalska, B. Bestas, M. J. Sobkowiak, B. Levänen, M. Sköld, E. A. Thompson, O. Saher, O. K. Kari, T. Lajunen, E. Sverremark Ekström, C. Nilsson, Y. Ishchenko, T. Malm, M. J. A. Wood, U. F. Power, S. Masich, A. Lindén, J. K. Sandberg, J. Lehtiö, A.-L. Spetz and S. EL Andaloussi, Nat. Commun., 2019, 10, 2331 CrossRef PubMed.
- E. R. Padayachee, H. Zetterberg, E. Portelius, J. Borén, J. L. Molinuevo, N. Andreasen, R. Cukalevski, S. Linse, K. Blennow and U. Andreasson, Brain Res., 2016, 1651, 11–16 CrossRef CAS PubMed.
- R. Frankel, M. Törnquist, G. Meisl, O. Hansson, U. Andreasson, H. Zetterberg, K. Blennow, B. Frohm, T. Cedervall, T. P. J. Knowles, T. Leiding and S. Linse, Commun. Biol., 2019, 2, 365 CrossRef PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215–4233 RSC.
- B. D. Moore, J. Martin, L. de Mena, J. Sanchez, P. E. Cruz, C. Ceballos-Diaz, T. B. Ladd, Y. Ran, Y. Levites, T. L. Kukar, J. J. Kurian, R. McKenna, E. H. Koo, D. R. Borchelt, C. Janus, D. Rincon-Limas, P. Fernandez-Funez and T. E. Golde, J. Exp. Med., 2018, 215, 283–301 CrossRef CAS PubMed.
- M. O. Quartey, J. N. K. Nyarko, J. M. Maley, J. R. Barnes, M. A. C. Bolanos, R. M. Heistad, K. J. Knudsen, P. R. Pennington, J. Buttigieg, C. E. De Carvalho, S. C. Leary, M. P. Parsons and D. D. Mousseau, Sci. Rep., 2021, 11, 431 CrossRef CAS PubMed.
- E. Portelius, U. Andreasson, J. M. Ringman, K. Buerger, J. Daborg, P. Buchhave, O. Hansson, A. Harmsen, M. K. Gustavsson, E. Hanse, D. Galasko, H. Hampel, K. Blennow and H. Zetterberg, Mol. Neurodegener., 2010, 5, 2 CrossRef PubMed.
- E. Portelius, B. Van Broeck, U. Andreasson, M. K. Gustavsson, M. Mercken, H. Zetterberg, H. Borghys and K. Blennow, J. Alzheimer's Dis., 2010, 21, 1005–1012 CAS.
- T. C. T. Michaels, A. Šarić, S. Curk, K. Bernfur, P. Arosio, G. Meisl, A. J. Dear, S. I. A. Cohen, C. M. Dobson, M. Vendruscolo, S. Linse and T. P. J. Knowles, Nat. Chem., 2020, 12, 445–451 CrossRef CAS PubMed.
- A. Jan, O. Gokce, R. Luthi-Carter and H. A. Lashuel, J. Biol. Chem., 2008, 283, 28176–28189 CrossRef CAS PubMed.
- M. M. Murray, S. L. Bernstein, V. Nyugen, M. M. Condron, D. B. Teplow and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 6316–6317 CrossRef CAS PubMed.
- S. G. Bolder, L. M. C. Sagis, P. Venema and E. van der Linden, Langmuir, 2007, 23, 4144–4147 CrossRef CAS PubMed.
- R. Silvers, M. T. Colvin, K. K. Frederick, A. C. Jacavone, S. Lindquist, S. Linse and R. G. Griffin, Biochemistry, 2017, 56, 4850–4859 CrossRef CAS PubMed.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266–1281 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. T. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef CAS PubMed.
- G. Meisl, T. C. T. Michaels, S. Linse and T. P. J. Knowles, in Amyloid Proteins, ed. E. M. Sigurdsson, M. Calero and M. Gasset, Springer New York, New York, NY, 2018, vol. 1779, pp. 181–196 Search PubMed.
- G. Meisl, L. Rajah, S. A. I. Cohen, M. Pfammatter, A. Šarić, E. Hellstrand, A. K. Buell, A. Aguzzi, S. Linse, M. Vendruscolo, C. M. Dobson and T. P. J. Knowles, Chem. Sci., 2017, 8, 7087–7097 RSC.
- A. J. Dear, G. Meisl, T. C. T. Michaels, M. R. Zimmermann, S. Linse and T. P. J. Knowles, J. Chem. Phys., 2020, 152, 045101 CrossRef CAS PubMed.
- A. Munke, J. Persson, T. Weiffert, E. De Genst, G. Meisl, P. Arosio, A. Carnerup, C. M. Dobson, M. Vendruscolo, T. P. J. Knowles and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6444–6449 CrossRef CAS PubMed.
- C. J. Sarell, P. G. Stockley and S. E. Radford, Prion, 2013, 7, 359–368 CrossRef CAS PubMed.
- M. I. Ivanova, Y. Lin, Y.-H. Lee, J. Zheng and A. Ramamoorthy, Biophys. Chem., 2021, 269, 106507 CrossRef CAS PubMed.
- S. Chia, P. Flagmeier, J. Habchi, V. Lattanzi, S. Linse, C. M. Dobson, T. P. J. Knowles and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8005–8010 CrossRef CAS PubMed.
- S. Bondarev, K. Antonets, A. Kajava, A. Nizhnikov and G. Zhouravleva, Int. J. Mol. Sci., 2018, 19, 2292 CrossRef PubMed.
- F. Hasecke, C. Niyangoda, G. Borjas, J. Pan, G. Matthews, M. Muschol and W. Hoyer, Angew. Chem., Int. Ed., 2021, 60, 3016–3021 CrossRef CAS PubMed.
- J. C. Kessler, J.-C. Rochet and P. T. Lansbury, Biochemistry, 2003, 42, 672–678 CrossRef CAS PubMed.
- S. Abeln and D. Frenkel, PLoS Comput. Biol., 2008, 4, e1000241 CrossRef PubMed.
- C. Arber, J. Toombs, C. Lovejoy, N. S. Ryan, R. W. Paterson, N. Willumsen, E. Gkanatsiou, E. Portelius, K. Blennow, A. Heslegrave, J. M. Schott, J. Hardy, T. Lashley, N. C. Fox, H. Zetterberg and S. Wray, Mol. Psychiatry, 2020, 25, 2919–2931 CrossRef PubMed.
- S. Mekala, G. Nelson and Y.-M. Li, RSC Med. Chem., 2020, 11, 1003–1022 RSC.
- M. S. Wolfe, Molecules, 2021, 26, 388 CrossRef CAS PubMed.
- G. Yang, R. Zhou, X. Guo, C. Yan, J. Lei and Y. Shi, Cell, 2021, 184, 521–533.e14 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02990h |
| ‡ Current address: University of California, San Francisco, Program in Chemistry and Chemical Biology, San Francisco, CA, United States. |
| § Current address: Wren Therapeutics Sweden AB, Lund, Sweden. |
| This journal is © The Royal Society of Chemistry 2022 |