
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing a sustainable manufacturing route to lapatinib†
Roderick T.
Stark
a,
Dominic R.
Pye
a,
Wenyi
Chen
a,
Oliver J.
Newton
 a,
Benjamin J.
Deadman
b,
Philip W.
Miller
a,
Jenny-Lee
Panayides
c,
Darren L.
Riley
d,
Klaus
Hellgardt
e and
King Kuok (Mimi)
Hii
*ab
a,
Benjamin J.
Deadman
b,
Philip W.
Miller
a,
Jenny-Lee
Panayides
c,
Darren L.
Riley
d,
Klaus
Hellgardt
e and
King Kuok (Mimi)
Hii
*ab
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 82, Wood Lane, London W12 0BZ, UK. E-mail: mimi.hii@imperial.ac.uk
bCentre for Rapid Online Analysis of Reactions (ROAR), Molecular Sciences Research Hub, 82, Wood Lane, London W12 0BZ, UK
cPharmaceutical Technologies, Future Production: Chemicals, Council for Scientific & Industrial Research (CSIR), Meiring Naude Rd, Brummeria, Pretoria, 0184, South Africa
dDepartment of Chemistry, Faculty of Natural and Agricultural Sciences, University of Pretoria, Lynnwood Rd, Hatfield, Pretoria, 0002, South Africa
eDepartment of Chemical Engineering, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK
First published on 9th August 2022
Abstract
A synthetic route to an anti-cancer drug, lapatinib, was devised to support the development of a sustainable manufacturing process in South Africa. Quantitative metrics were employed to evaluate the sustainability of the key steps of the reaction.
Introduction
Breast cancer is the fifth leading cause of cancer mortality worldwide, recognized by the World Health Organization (WHO) to be an urgent health priority. In 2020, there were an estimated 2.3 million new cases of breast cancer diagnosed globally with 685![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths.1 Risk factors for breast cancer include genetic mutations (BRCA1 and BRCA2 genes), reproductive and hormonal risk factors (central role of oestrogen), and lifestyle factors including alcohol intake, excess body weight and lack of physical activity.2 Whilst the incidence of breast cancer is higher in developed countries (in part due to better screening), it is also rising in developing nations.3 This partly reflects changes to women's lifestyles; including a shift into working environments that have resulted in increased risk factors (e.g. giving birth to children later in life, having fewer children, increased body weight and less physical activity). In the past two decades, sub-Saharan Africa has seen an increased incidence of breast cancer and some of the world's highest mortality rates.4 Contributing factors to these poor outcomes include under-resourced healthcare provision, lack of screening, poor infrastructure, and limited access to treatments.5
000 deaths.1 Risk factors for breast cancer include genetic mutations (BRCA1 and BRCA2 genes), reproductive and hormonal risk factors (central role of oestrogen), and lifestyle factors including alcohol intake, excess body weight and lack of physical activity.2 Whilst the incidence of breast cancer is higher in developed countries (in part due to better screening), it is also rising in developing nations.3 This partly reflects changes to women's lifestyles; including a shift into working environments that have resulted in increased risk factors (e.g. giving birth to children later in life, having fewer children, increased body weight and less physical activity). In the past two decades, sub-Saharan Africa has seen an increased incidence of breast cancer and some of the world's highest mortality rates.4 Contributing factors to these poor outcomes include under-resourced healthcare provision, lack of screening, poor infrastructure, and limited access to treatments.5
Increased cancer prevalence presents a significant burden for low and middle-income countries (LMICs), who cannot afford expensive therapies.6 Driven by this emerging global healthcare challenge, we initiated an ambitious project to enable the development of local pharmaceutical manufacturing capabilities in South Africa. Herein, we describe our efforts to deliver a cost-effective and sustainable synthesis route for the chemotherapy agent lapatinib (Fig. 1).
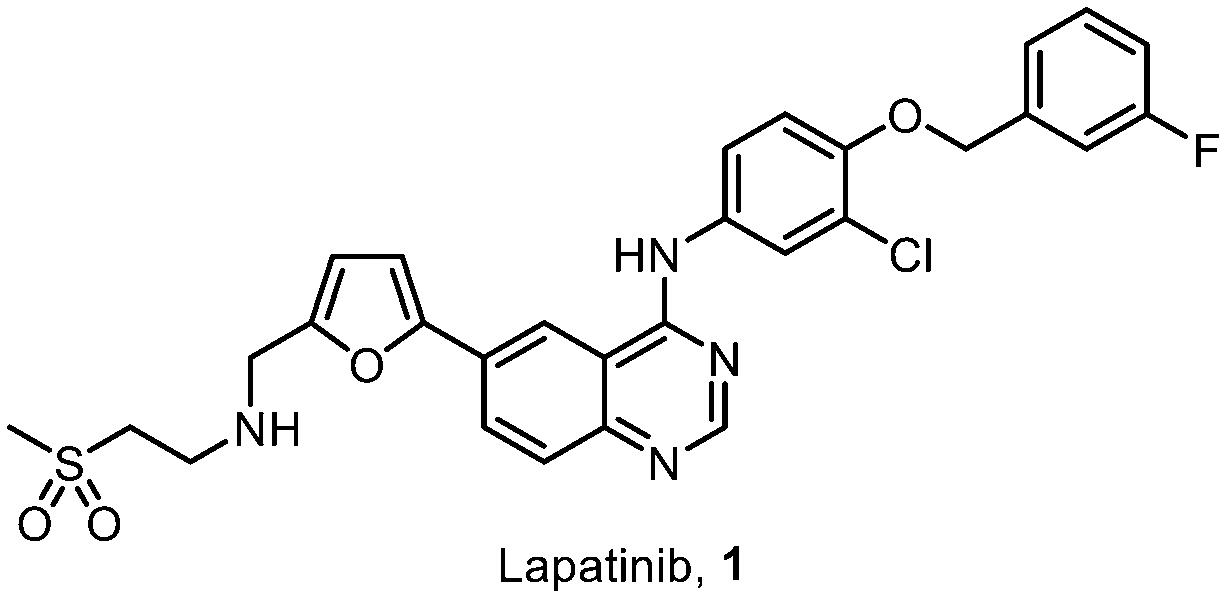 | ||
| Fig. 1 Structure of lapatinib, 1. | ||
Lapatinib (1) is a small molecule active pharmaceutical ingredient (API) patented in 1999,7 as a chemotherapeutic drug for the treatment of breast cancer, acting as a tyrosine kinase inhibitor (TKI) of oncogenes ErbB1 and HER2.8 The observed bioactivity is attributed to the in vivo binding of the 4-arylaminoquinazoline core – also found in other TKI's9 – in conjunction with the hydrophilic amino-sulfone chain providing hydrogen-bonding sites. Additionally, the 4-(3-fluorobenzyloxy) motif interacts strongly within enzymatic binding pockets, providing hydrophobic contacts.10–12 The combination of lapatinib and capecitabine has been found to be one of the most cost-effective treatments for HER2+ metastatic breast cancer.13
The original synthesis route for lapatinib was disclosed in a patent published in 2002 (Scheme 1);7 comprising of the following steps: (i) construction of the quinazoline heterocyclic ring by a Niementowski-type reaction, followed by chlorination (2 → 4); (ii) Nucleophilic substitution to form a 4-aminoquinazoline ring (9a); (iii) biaryl cross-coupling to attach a 5-formyl furan moiety; and finally (iv) the attachment of the amino sulfone side chain by reductive amination (11 → 1). During the last two decades or so, modifications to each stage of the synthetic route had been reported in patent and medicinal chemistry literature for lapatinib, as well as closely related derivatives. However, critical details, such as the amount of catalyst and reaction times, are often omitted from these publications, which hampered the techno-economical evaluation of a potential localised production process in South Africa. With this in mind, we set out to re-evaluate and modify the reported synthetic procedures into a sustainable manufacturing route. Combining qualitative (12 Principles of Green Chemistry)15 and quantitative metrics, the work aims to optimize each step of the synthesis, while also minimising both material and energy requirements of the process. The key approaches are:
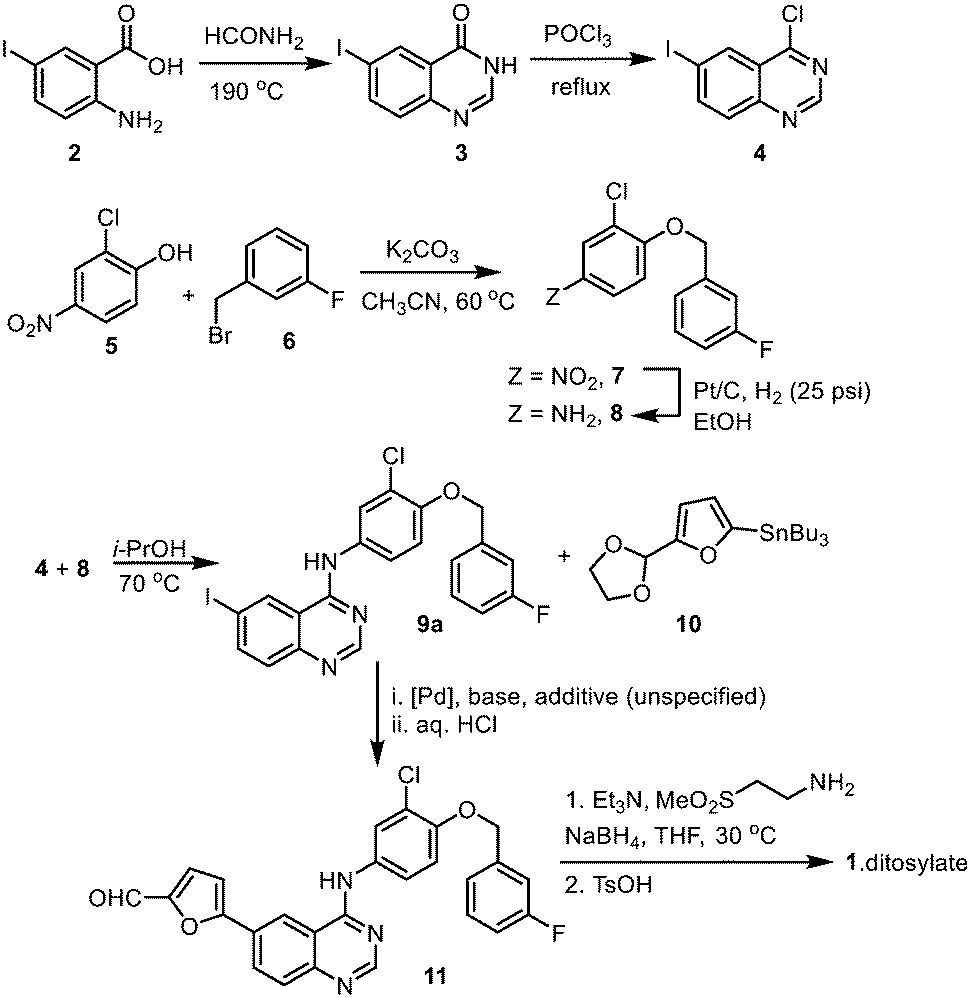 | ||
| Scheme 1 Original reported synthesis of lapatinib.14 | ||
(i) Minimising waste: by choosing atom-efficient reactions;
(ii) Working closely to solubility limits and precise reaction stoichiometry;
(iii) Telescope reactions where possible;
(iv) Select less toxic and ‘greener’ reagents and solvents wherever possible;
(v) No column chromatography: using only filtration and (re)crystallization as means of purification.
Green metrics will be used throughout the work to assess the environmental impact of the procedures.
Results and discussion
Construction of the 4-aminoquinazoline, 9 (Scheme 2)
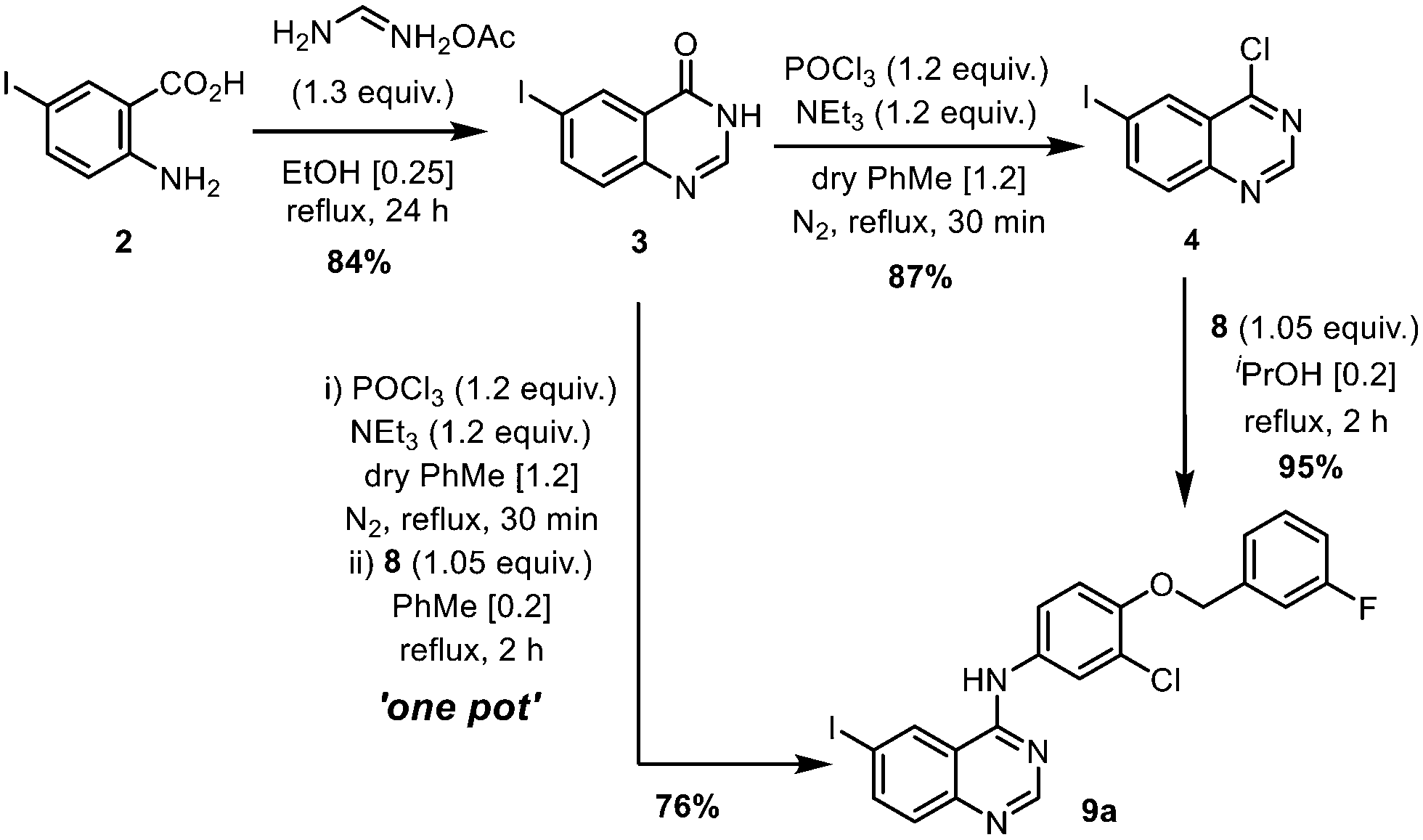 | ||
| Scheme 2 Optimised conditions for the preparation of 4-aminoquinazoline 9. | ||
Chlorination of 3 can be achieved using phosphorus oxychloride (POCl3),16,17 thionyl chloride (SOCl2)22 or oxalyl chloride [(COCl)2].19 In this case, the phosphorus reagent was preferred as it does not emit toxic gaseous by-products (such as SO2 and CO). The chlorination of similar quinazolones by POCl3 had been extensively studied by a process chemistry team,23 who reported the importance of controlling the reaction conditions, particularly pH and the reaction temperature, to suppress side product formation. Guided by this, we were able to optimize the reaction relatively quickly (Table S2, ESI†). Deploying only slight excesses of POCl3 and triethylamine (1.2 equivalents) in toluene, 4 was attained as a light brown solid in 87% yield, following a basic workup.
The introduction of the 4-(3-fluorobenzyloxy) motif via a nucleophilic substitution (SNAr) reaction was found to proceed in different solvents (Table S3, ESI†), such as in refluxing propan-2-ol, to afford 9a in 95% yield. Using toluene as a common solvent, it is possible to telescope the chlorination and substitution steps, to convert compound 3 to 9avia a one-pot process, albeit in a lower yield of 76% (compared to 82% over two steps). The limited solubility of compound 9a enables convenient isolation and purification by filtration and washing without the need for further purification.
Using the CHEM21 toolkit,24 the sustainability metrices of the two procedures were calculated (Table 1). The comparison revealed that while the one-pot procedure may be practically more convenient, it is, in fact, less efficient in terms of mass and overall efficiencies (RME, OE) than the two-step process. This is partly due to the lower yield of the one-pot process, but also largely because the two-step procedure is already highly efficient, as we were able to reduce excess amounts of the reagents (POCl3 and NEt3), as well as working closely to the solubility limits of the reactants (thereby reducing the amount of solvent).
| Procedure | Yieldb/% | AEc/% | RMEd/% | OEe% | PMIf |
|---|---|---|---|---|---|
| a Calculated using CHEM21 Metrics Toolkit. b Isolated yield. c Atom economy, = (Mw of product/total Mw of reactants) × 100. d Reaction mass efficiency = (mass of isolated product/total mass of reactants) × 100. e Optimum efficiency = (RME/AE) × 100. f Process mass intensity = total mass in a process/mass of product. | |||||
| 2 steps, via4 | 81.9 | 96.6 | 82.4 | 85.3 | 64.6 |
| ‘One-pot’ | 75.5 | 96.6 | 73.0 | 75.6 | 74.4 |
Biaryl coupling between quinazoline and furan rings
The Stille cross coupling reaction employed in the original synthetic route (Scheme 1, 9a + 10 → 11) has since been superseded by the Suzuki–Miyaura (SM) reaction (Scheme 3), where the toxic organostannane (10) is replaced by 5-formylfurylboronic acid (12). Many SM protocols are reported in the patent literature, where different palladium catalyst precursors were employed under a variety of reaction conditions: from Pd(OAc)225 or Pd/C26 under ‘ligandless’ conditions, as well as discreet palladium complexes such as Pd(PPh3)2Cl217 and (dppf)PdCl2.27
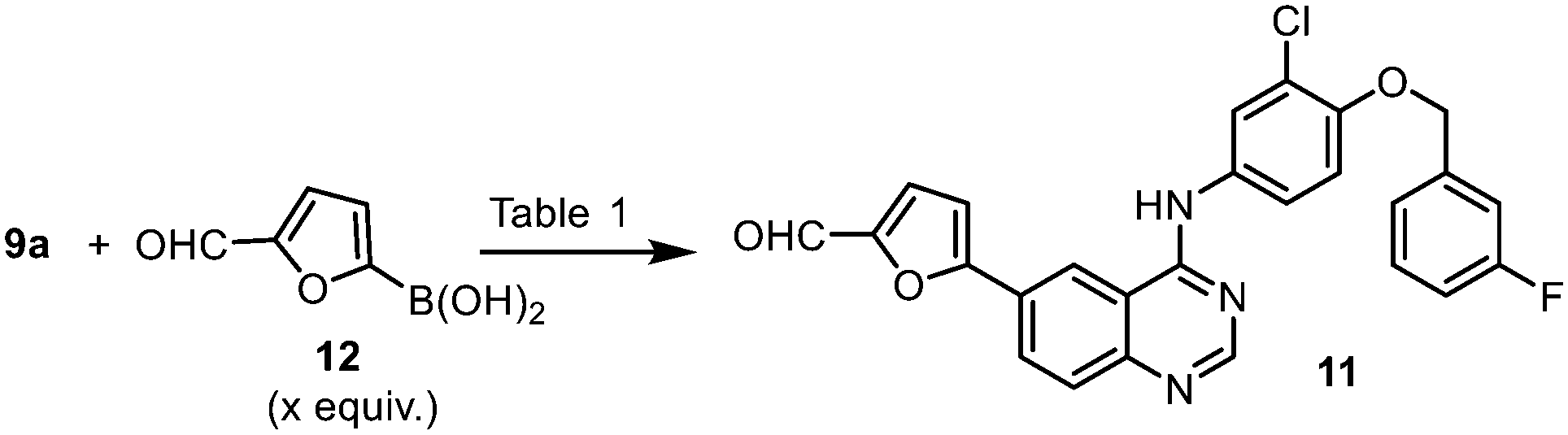 | ||
| Scheme 3 Suzuki–Miyaura cross coupling between 4 and 12. | ||
Given that the aryl iodides are considered as highly activated substrates in cross-coupling reactions, we predicted that the use of air-sensitive or expensive ligands should not be necessary. With this in mind, the SM reaction was optimised using Pd(OAc)2 as the catalyst precursor without extraneous ligands (Table 2). As expected, the reaction proceeded well, even with a very low catalyst loading of 0.01 mol% (entry 10 and Fig. S1, ESI†), even without rigorous drying of the solvent or the need to purge the mixture with inert gas (entries 5–11). The ability to reduce the amount of the precious metal catalyst not only has economic benefits, but also reduces the amount of metal residue in the product mixture and greatly simplifying the workup process.
| Entry | [Pd] (mol%) | [9]/M | x/equiv. | T/h | Yieldb/% |
|---|---|---|---|---|---|
|
a General reaction conditions: 4a (1 mmol), NEt3 (4 equiv.), degassed DME–MeOH (2:1 v/v), 50 °C (see ESI†).
b Isolated yield.
c Solvents were not degassed prior to use.
d Performed at 40 mmol scale.
e Reaction run in MeOH–propan-2-ol (2:1 v/v).
|
|||||
| 1 | Pd(OAc)2 (5) | 0.05 | 1.5 | 1 | 68 |
| 2 | Pd(OAc)2 (5) | 0.05 | 1.5 | 2 | 82 |
| 3 | Pd(OAc)2 (5) | 0.05 | 1.3 | 2 | 71 |
| 4 | Pd(OAc)2 (5) | 0.05 | 1.1 | 2 | 77 |
| 5c | Pd(OAc)2 (1) | 0.05 | 1.1 | 2 | 84 |
| 6c | Pd(OAc)2 (1) | 0.1 | 1.1 | 2 | 84 |
| 7c | Pd(OAc)2 (1) | 0.2 | 1.1 | 2 | 82 |
| 8c | Pd(OAc)2 (1) | 0.05 | 1.1 | 4 | 90 |
| 9c,d | Pd(OAc)2 (0.1) | 0.05 | 1.1 | 24 | 90 |
| 10c | Pd(OAc)2 (0.01) | 0.05 | 1.1 | 24 | 84 |
| 11c | 5% Pd/C (1) | 0.05 | 1.1 | 4 | 90 |
| 12c,e | Pd(OAc)2 (1) | 0.05 | 1.1 | 4 | 42 |
The reaction was subsequently replicated successfully on a larger scale (40 mmol) using 0.1 mol% of catalyst (entry 9). Finally, we showed that Pd(OAc)2 can be replaced by the heterogeneous Pd/C catalyst without any deleterious effect (entry 11).
An alternate synthesis of compound 11 is to couple the aryl halide with furfural directly, without pre-activation of the furan ring by a boronic acid. This reaction was investigated by a GSK team in 2014,16 where the bromide derivative 9b was coupled with furfural without the need for solvents (Scheme 4). Given that furfural can be derived from renewable biomass sources, this is an attractive approach. However, the reaction required the presence of potassium acetate (2 equiv.), pivalic acid (0.5 equiv.), an air-sensitive phosphine ligand, elevated reaction temperature (>110 °C) and a large excess of the furfural to proceed. More critically, the reaction was found to be highly dependent on careful monitoring of the reaction conditions to avoid the competitive reaction occurring at the aryl chloride.
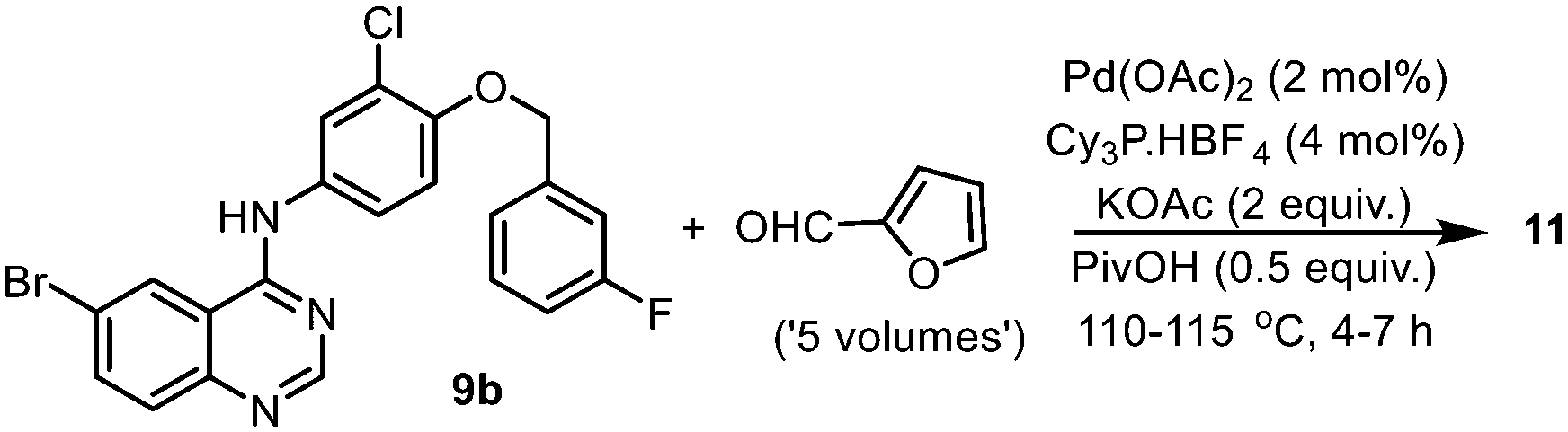 | ||
| Scheme 4 Reported synthesis of compound 11via a direct arylation of furfural (optimized conditions). | ||
Again, using the CHEM21 toolkit,24 the sustainability metrics for the two different catalytic methodologies were evaluated (Table 3). The comparison revealed that while the direct coupling methodology has better atom economy and lower PMI, it was, in fact, not as efficient (RME, OE) as the SM reaction due to its lower reaction yield. Furthermore, the toxicity and potential carcinogenicity of furfural (used in excess), and the need to deploy a higher catalyst loading and an air-sensitive phosphine ligand (compared to 0.1 mol% utilized in the ligandless SM reaction), counteracted the potential atom economy. Thus, while the direct coupling reaction does not require pre-activation of the furan ring and might be considered as more ‘elegant’ than the SM reaction, it is unlikely that it will be implemented on a larger scale without substantial improvement in its selectivity and robustness.
| Reaction | Yieldb/% | AEc/% | RMEd/% | OEe% | PMIf |
|---|---|---|---|---|---|
| a See Table 1. b See Table 1. c See Table 1. d See Table 1. e See Table 1. f See Table 1. g Health and safety of substances which triggers amber or red flags (H phrases). | |||||
| Suzuki–Miyauraa | 90 | 73.4 | 64.5 | 87.8 | 107.5 |
| H&S:g MeOH (amber: H301, 311, 331), DME (red: H360FD) | |||||
| Direct arylation16 | 63 | 85.4 | 12.2 | 14.3 | 35.3 |
| H&S:g furfural (amber: H301, 331, 351) | |||||
However, while the SM reaction may be more process efficient, an important issue to be addressed is the solvents required: both methanol and dimethoxyethane (DME) are toxic;28 the latter is listed as a ‘substance of very high concern’ (SVHC) by the European Chemicals Agency as it may damage fertility and the unborn child.29 Nevertheless, the use of DME was found to be essential to ensure solubility of 9a in the reaction mixture: an attempt to substitute the solvent mixture with propan-2-ol led to the dramatic erosion in yield of 11 from 90 to 42% (Table 2, entries 8 and 12). The formation of Pd black was particularly noticeable in the alcoholic solvent. Presumably, the presence of the glycol ether (DME) is necessary to stabilize the active Pd catalyst.
Given that the use of DME–MeOH is unavoidable for the C–C coupling reaction, it was decided that the remaining steps of the synthetic sequence should also be investigated using the same solvent mixture, with the intention that sequential steps can potentially be telescoped, effectively reducing the amounts of these solvents in the overall process.30
Reductive amination
The last chemical step of the lapatinib synthesis involves the installation of the sulfone side chain by a reductive amination (Scheme 5). This was typically achieved with the reaction of 11 with 2-aminoethylmethylsulfone and a hydride reducing agent such as NaBH4 or NaBH(OAc)3. Clearly, it will be desirable to replace these hazardous stoichiometric reductants with a catalytic method that utilises H2 as the more atom-efficient reductant. Although the catalytic hydrogenation of the imine intermediate 13 over Pt/C and Pd/C had been previously disclosed in the patent literature,31 the described procedure employed dichloromethane as a solvent – a restricted substance under current REACH regulations32 due to its potential carcinogenicity and volatility.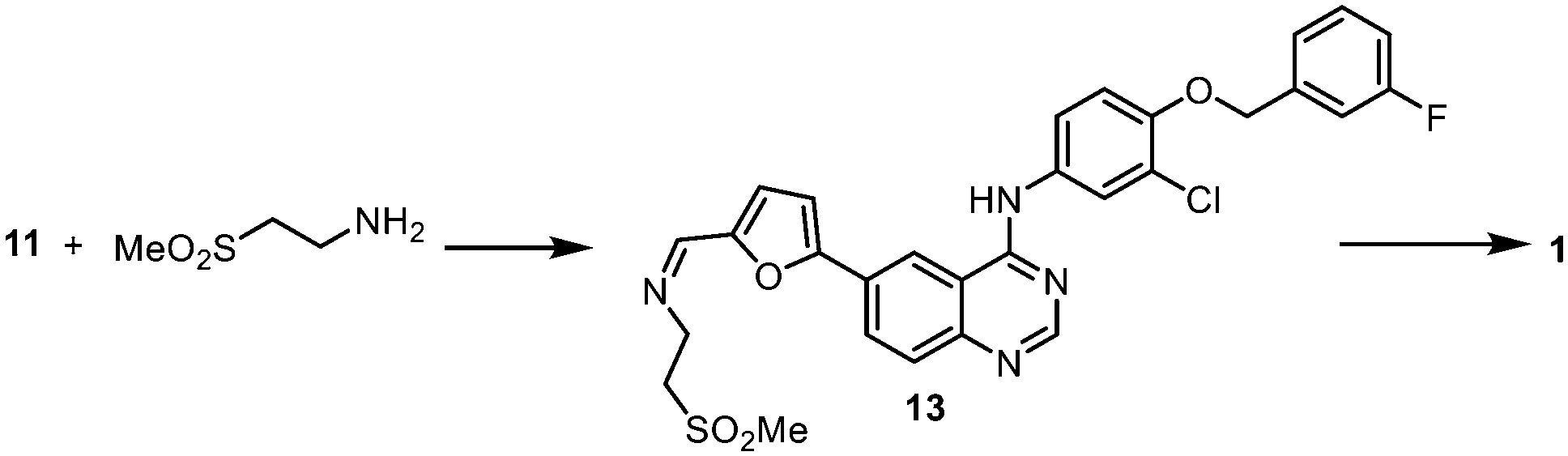 | ||
| Scheme 5 Introduction of the sulfone sidechain vs. reductive amination. | ||
In the presence of triethylamine, the reaction of 2-aminoethylmethyl sulfone hydrochloride and carboxaldehyde 11 in refluxing methanol afforded the imine 13 as a stable off-white solid, which can be isolated in 87% yield (ESI†), and may be kept at room temperature for several months, without any noticeable decomposition. The reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond was initially studied using the isolated imine as a precursor, before we attempted to integrate the condensation and the reduction steps. In this work, two catalytic hydrogenation strategies were evaluated in the same solvent mixture and reaction temperature deployed for the SM reaction.
N bond was initially studied using the isolated imine as a precursor, before we attempted to integrate the condensation and the reduction steps. In this work, two catalytic hydrogenation strategies were evaluated in the same solvent mixture and reaction temperature deployed for the SM reaction.
The first reduction method involves the use of ammonium formate or amine–formic acid adducts as H-surrogates (transfer hydrogenation protocol). While this may not be as atom-economical as using H2, it bypasses the need for high-pressure equipment, or when H2 is not available at the production facility. The results of a small initial screening of a selection of homogeneous and heterogeneous Pd catalysts, as well as hydrogen surrogates (ammonium formate, and a combination of amines with formic acid), revealed Pd/C and ammonium formate as the most effective (Table 4, entry 1). Notably, the yield decreased with extended reaction time (entries 1 and 2), suggesting that the product is unstable under these conditions.
| Entry | Catalyst | H-Surrogate | Time/h | Yieldb/% |
|---|---|---|---|---|
|
a Reactions conditions: imine 13 (0.2 mmol), DME/MeOH (2:1, 0.05 M), 5 mol% catalyst and hydrogen surrogate (5 equiv.), 50 °C.
b Yield of lapatinib determined by HPLC using 1,3,5-trimethoxybenzene as internal standard.
|
||||
| 1 | Pd/C | HCO2NH4 | 1 | 70.2 |
| 2 | Pd/C | HCO2NH4 | 2 | 60.8 |
| 3 | Pd/C | NEt3 + HCO2H | 1 | 16.2 |
| 4 | Pd/C | DIPEA + HCO2H | 1 | 14.4 |
| 5 | Pd(OAc)2 | HCO2NH4 | — | n.d. |
| 6 | Pd(OAc)2 | NEt3 + HCO2H | — | n.d. |
| 7 | Pd(OAc)2 | DIPEA + HCO2H | — | n.d. |
In contrast, no product was detected when Pd(OAc)2 was employed under homogeneous conditions (entries 5–7), implying that the reduction requires supported Pd(0) species. Consequently, the catalytic reaction was subjected to further optimization using Pd/C as the catalyst (Table 5). At 50 °C and 5 mol% catalyst loading, the reaction is practically complete within 30 min (entry 1); further increases in the amount of Pd led only to deleterious product decomposition (entries 2 and 3). As may be expected, the reaction rate is dependent upon the amount of ammonium formate, with seven equivalents being optimal (entry 5).
Given that both the SM reaction and the reductive amination utilise Pd catalysts and proceeded well in the DME–MeOH mixture, we attempted to telescope the two reactions into a ‘one-pot procedure’ to improve the mass intensity. Our first attempt utilised Pd/C for the SM reaction, followed by the addition of the amino-sulfone reactant and ammonium formate as the H-surrogate. Unfortunately, the reaction sequence terminated with the formation of the imine intermediate 13 (Scheme 6, eqn (1)). A likely explanation is that the presence of excess triethylamine (leftover from the SM cross coupling) may be inhibiting the transfer hydrogenation (supported by earlier observations: Table 4, entries 3–4, 6–7); for example, by scavenging Pd–H species.
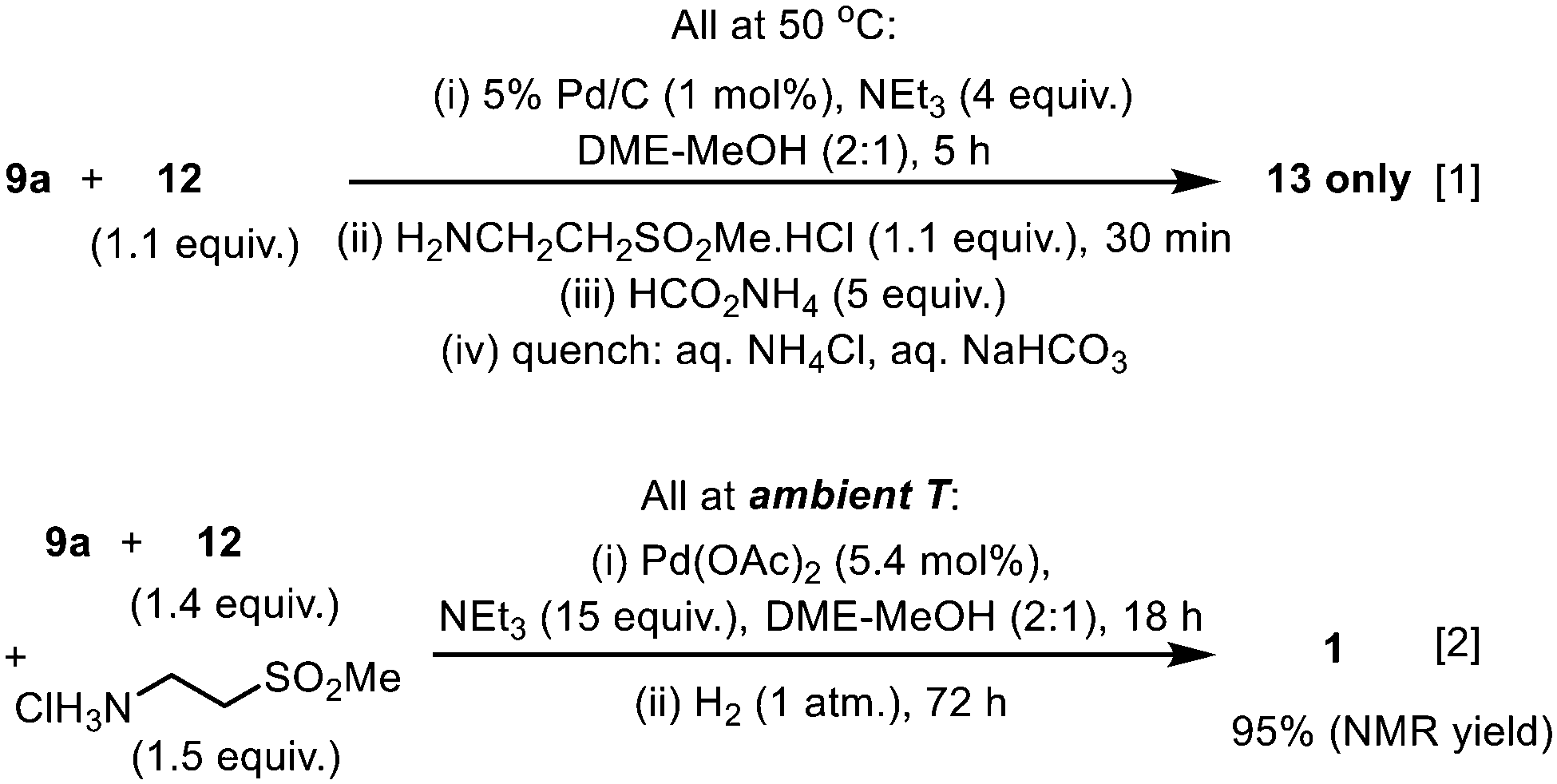 | ||
| Scheme 6 Attempts to achieve sequential Suzuki Miyaura-reductive amination in ‘one-pot’, with transfer hydrogenation (eqn (1)) and catalytic hydrogenation (eqn (2)) as terminating steps. | ||
Subsequently, the second step was replaced by a catalytic hydrogenation protocol. In this attempt, the reaction vessel was charged initially with all the reactants, reagent, and Pd(OAc)2 under a N2 atmosphere. Once the C–C coupling was complete, the reaction mixture was exposed to H2, whereupon catalytic reduction of the imine 13 was effected by the residual Pd(0) in situ (Scheme 6, eqn (2)), affording lapatinib 1 with 95% conversion (ESI†). Critically, in contrast to the transfer hydrogenation protocol, the presence of triethylamine did not inhibit the catalytic hydrogenation of the imine, thus allowing the C–C coupling and CN reduction to be telescoped, using a single charge of Pd catalyst. In principle, the overall process is extremely atom-efficient, whilst also reducing the amounts of solvent and catalyst.
However, it should be noted that the protocol will be difficult to duplicate at scale, due to safety concerns in deploying flammable H2 for a prolonged period in a batch reactor. These problems can be mitigated by performing the catalytic hydrogenation in flow. In this part of the work, the imine 13 was pre-formed in situ by mixing 11 and the 2-aminoethylmethylsulfone hydrochloride in the presence of triethylamine (to release the free base), and the reaction mixture was directly subjected to catalytic hydrogenation by passing it through a catalytic packed bed reactor (H-Cube Pro). The preliminary study, performed on a laboratory scale (Table 6), showed that good single-pass conversion of 13 to 1 can be attained at a reaction temperature of 50 °C@0.5 mL min−1 (entry 3) or, for a higher productivity, 60 °C@1 mL min−1 (entry 6). The single-pass conversion may be further improved by elevating the H2 pressure (entries 3–5). By precise control of residence time, the competitive product decomposition at higher temperature can be suppressed.
| Entry | Flow rate/mL min−1 | P(H2)/bar | T/°C | Yieldb/% |
|---|---|---|---|---|
|
a The imine was generated in situ (ESI†), 5% Pd/C, DME–MeOH (2:1 v/v, 4 mL).
b HPLC yield using 1,3,5-trimethoxybenzene as internal standard, recorded at steady state conversion (aliquots collected after 3 reactor volumes).
|
||||
| 1 | 1 | 1 | 50 | 70 |
| 2 | 2 | 1 | 50 | 61 |
| 3 | 0.5 | 1 | 50 | 87 |
| 4 | 1 | 2 | 50 | 81 |
| 5 | 1 | 5 | 50 | 84 |
| 6 | 1 | 1 | 60 | 85 |
| 7 | 1 | 1 | 40 | 23 |
Subsequently, the catalytic reductive amination reaction was employed on a Gram-scale to produce lapatinib in 71% isolated yield (ESI†). The lower yield (compared to Table 6, entry 3) is attributed to possible Pd deactivation/leaching. This will be investigated in our further work, involving time-on-line studies and modification of the reactor, which will be best performed on a pilot scale. The information gathered up to this point is, nevertheless, sufficient to support the techno-economic assessment of a proposed API production process in South Africa.
Conclusions
The commercial route for the synthesis of lapatinib has been revisited and substantial improvements to the sustainability of the process have been achieved (Scheme 7). The synthetic route was used to produce 10 g of the final product lapatinib and the key intermediates. In summary, the overall synthesis route comprises of 6 reactions, performed in 5 steps, with an overall yield of 53.3%, compared to 8 steps (including a deprotection) in the original report (Scheme 1).33 While the use of toxic DME is undesirable from a health and safety perspective and cannot be eliminated from the SM reaction, the environmental impact can be alleviated by telescoping reaction steps and reducing solvent switches, which also enables a much lower amount of the Pd catalyst (a critical material) to be used. Similarly, we have shown that the SM cross-coupling could potentially be telescoped with the catalytic reductive amination performed in continuous flow, eliminating the use of a stoichiometric hydride reagent. Finally, all the intermediates and final product can be obtained in high purity after a simple filtration or crystallization, and no column chromatography was required.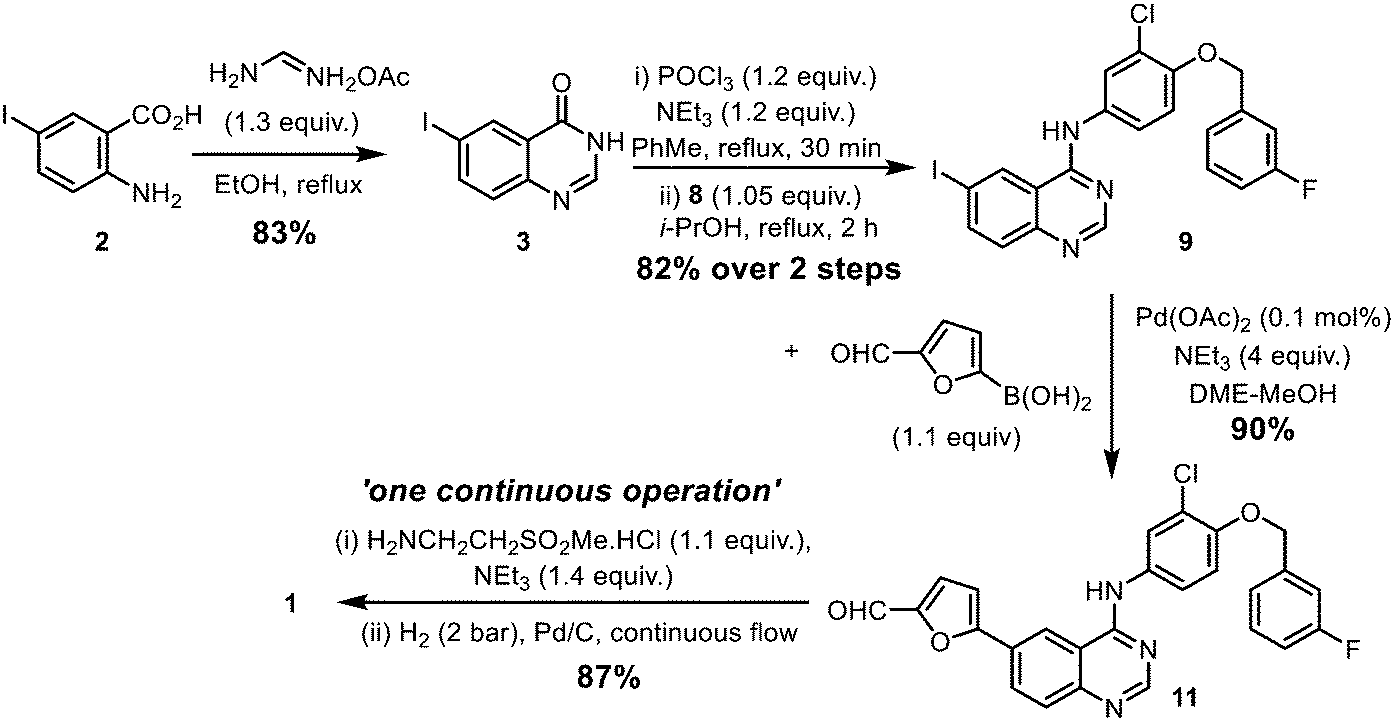 | ||
| Scheme 7 Overall synthetic scheme. | ||
Perhaps one of the most surprising results from this work is the comparisons of the sustainability metrics between the two-step and one-pot procedures (Scheme 2), as well as the Suzuki–Miyaura cross coupling and the direct C–H arylation reactions (Scheme 3vs.Scheme 4), which revealed that the latter processes are not necessarily ‘greener’, as may be expected intuitively. This highlights the importance and value of these quantification tools to accompany the 12 Principles of Green Chemistry, in the evaluation and demonstration of the sustainability of chemical processes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Imperial College London-Research England GCRF Institutional Awards (2018 and 2019). OJN was supported by a doctoral training award with additional contributions by Eli Lilly (as part of the Pharmacat Consortium). We also acknowledge funding of the ROAR facility by EPSRC (Strategic Equipment Grant, EP/R008825/1).Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 1–41 Search PubMed.
- C. Turnbull and N. Rahman, Annu. Rev. Genomics Hum. Genet., 2008, 9, 321–345 CrossRef CAS PubMed.
- R. Sharma, Breast Cancer Res. Treat., 2021, 187, 557–567 CrossRef PubMed.
- W. Y. Joko-Fru, E. Jedy-Agba, A. Korir, O. Ogunbiyi, C. P. Dzamalala, E. Chokunonga, H. Wabinga, S. Manraj, A. Finesse, N. Somdyala, B. Liu, P. McGale, A. Jemal, F. Bray and D. M. Parkin, Int. J. Cancer, 2020, 147, 2131–2141 CrossRef CAS PubMed.
- K. Togawa, B. O. Anderson, M. Foerster, M. Galukande, A. Zietsman, J. Pontac, A. Anele, C. Adisa, G. Parham, L. F. Pinder, F. McKenzie, J. Schuz, I. dos Santos-Silva and V. McCormack, Int. J. Cancer, 2021, 148, 2212–2226 CrossRef CAS PubMed.
- C. H. Barrios, T. Reinert and G. Werutsky, Ecancer, 2019, 13, 898 Search PubMed.
- M. C. Carter, G. S. Cockerill, S. B. Guntrip, K. E. Lackey and K. J. Smith, WP99/35146, 1999.
- W. Xia, R. J. Mullin, B. R. Keith, L. H. Liu, H. Ma, D. W. Rusnak, G. Owens, K. J. Alligood and N. L. Spector, Oncogene, 2002, 21, 6255–6263 CrossRef CAS PubMed.
- Y. Zhang, H. Gao, R. Liu, J. Liu, L. Chen, X. Li, L. Zhao, W. Wang and B. Li, Bioorg. Med. Chem. Lett., 2017, 27, 4309–4313 CrossRef CAS PubMed.
- M. Pereira, C. S. Verma and G. Fuentes, PLoS One, 2013, 8, e77054 CrossRef CAS PubMed.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K. J. Alligood, D. W. Rusnak, T. M. Gilmer and L. Shewchuk, Cancer Res., 2004, 64, 6652–6659 CrossRef CAS PubMed.
- K. G. Petrov, Y.-M. Zhang, M. Carter, G. S. Cockerill, S. Dickerson, C. A. Gauthier, Y. Guo, R. A. Mook, D. W. Rusnak, A. L. Walker, E. R. Wood and K. E. Lackey, Bioorg. Med. Chem. Lett., 2006, 16, 4686–4691 CrossRef CAS PubMed.
- N. H. Al-Ziftawi, A. A. Shafie and M. I. M. Ibrahim, Expert Rev. Pharmacoeconomics Outcomes Res., 2021, 21(4), 655–666 CrossRef PubMed.
- K. E. Lackey, N. L. Spector, E. R. Wood III and W. Xia, WO02/056912, 2002.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- G. Erickson, J. S. Guo, M. McClure, M. Mitchell, M. C. Salaun and A. Whitehead, Tetrahedron Lett., 2014, 55, 6007–6010 CrossRef CAS.
- S. Mahboobi, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2010, 53, 8546–8555 CrossRef CAS PubMed.
- E. C. Taylor, W. A. Ehrhart and M. Kawanisi, Org. Synth., 1966, 46, 39 CrossRef CAS.
- J. C. Kath, N. J. Tom, Z. Liu, E. D. Cox, S. K. Bhattacharya and J. Morris, WO00/44728, 2000.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, D. J. S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- G. Patel, C. E. Karver, R. Behera, P. J. Guyett, C. Sullenberger, P. Edwards, N. E. Roncal, K. Mensa-Wilmot and M. P. Pollastri, J. Med. Chem., 2013, 56, 3820–3832 CrossRef CAS PubMed.
- E. A. Arnott, L. C. Chan, B. G. Cox, B. Meyrick and A. Phillips, J. Org. Chem., 2011, 76, 1653–1661 CrossRef CAS PubMed.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- L. Metsger, S. Yurkovski, S. Gorohovsky-Rosenberg, N. Kipinis and D. Lavy, WO2010/017387, 2010.
- R. J. Prasad, B. R. A. K. Satya and N. V. Chowdary, WO2011/039759 A1, 2011.
- R. Tung, PCT/US2007/018655, 2008.
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 28, 1–18 Search PubMed.
- https://echa.europa.eu/substance-information/-/substanceinfo/100.003.451, accessed 3 February 2021.
- C. Jimenez-Gonzalez, Curr. Opin. Green Sustainable Chem., 2019, 18, 66–71 CrossRef.
- M. A. Raheem, G. Weeratunga, C. Zetina-Rocha and E. G. Cammisa, US8952154, 2011.
- https://echa.europa.eu/documents/10162/0ea58491-bb76-4a47-b1d2-36faa1e0f290, accessed 3 February 2021.
- Further comparisons using green metrics are not possible as not all the yields of the original synthesis were reported in the patent.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, addition optimization studies, characterization data and copies of NMR spectra of isolated intermediates and final product. See DOI: https://doi.org/10.1039/d2re00267a |
| This journal is © The Royal Society of Chemistry 2022 |