
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature CO oxidation over Rh/Al2O3 in a stagnation-flow reactor†
Nawaf M.
Alghamdi
 *a,
Ribhu
Gautam
a,
Jorge
Gascon
b,
Dionisios G.
Vlachos
cd and
S. Mani
Sarathy
a
*a,
Ribhu
Gautam
a,
Jorge
Gascon
b,
Dionisios G.
Vlachos
cd and
S. Mani
Sarathy
a
aClean Combustion Research Center, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955 Saudi Arabia. E-mail: nawaf.alghamdi@kaust.edu.sa
bKAUST Catalysis Center, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955 Saudi Arabia
cDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE, 19716 USA
dCatalysis Center for Energy Innovation, RAPID Manufacturing Institute, Delaware Energy Institute (DEI), 221 Academy Street, Newark, DE, 19716 USA
First published on 12th October 2022
Abstract
Anthropogenic activities are responsible for nearly half of the total CO emissions in the US. A significant amount of CO is emitted by the transportation sector. Three-way catalytic converters are widely employed to treat CO emissions from gasoline engines; however, current kinetic mechanisms for CO oxidation and the water-gas shift (WGS) reaction on Rh are limited and were built based on data collected over a narrow range of conditions. To fill in this gap, we conducted low-temperature CO oxidation and WGS experiments on 5 wt% Rh/Al2O3 in a stagnation-flow reactor, which allows for reducing the problem to one dimension and simplifies the development of accurate kinetic models. We characterized the catalyst via N2-physisorption, ICP, XRD, H2-chemisorption, H2-TPR, STEM and EELS. We studied the effects of pressure, temperature, flowrate, and the presence of H2O on the conversion of CO to CO2 and on the WGS reaction over the temperature range relevant to aftertreatment systems. The total operating pressure affected the resolution of the experimental measurements. Higher temperatures resulted in higher CO2 production due to faster kinetics. Investigating the reaction order with respect to O2 showed three distinct kinetic regimes, where the order is positive below the stoichiometric ratio, beyond which a negative order was observed which decreased with increasing O2 content. With respect to CO, the order was positive below the stoichiometric ratio, beyond which the order was negative. When increasing and reducing the O2 content, we observed bistability manifested as a hysteresis behavior, which is attributed to the oxidation (by O2) and reduction (by CO) of the metal. This thorough experimental study aids in developing accurate and versatile CO oxidation on Rh kinetic mechanisms that predict reactivity over a wide range of conditions.
Introduction
According to the United Nations, air pollution causes the premature death of nearly 7 million people, including 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 children, every year.1 One of the six criteria pollutants responsible for air pollution is carbon monoxide (CO). In the US, the transport sector alone produces 44% of CO emissions.2 Moreover, up to 95% of all CO emissions in US cities are due to motor vehicle exhaust.3 Even though electrification of passenger vehicles seems to be promising from an economic point of view, it faces many non-economic barriers especially in emerging markets and developing countries, where there is a heavy reliance on second-hand markets, an absence of governmental environmental pledges, and a severe lack of cheap and fast charging infrastructure.4 Therefore, a pragmatic and more immediate way of reducing the current emissions resulting from the 1 billion passenger vehicles on the road today is to improve the current aftertreatment systems.
000 children, every year.1 One of the six criteria pollutants responsible for air pollution is carbon monoxide (CO). In the US, the transport sector alone produces 44% of CO emissions.2 Moreover, up to 95% of all CO emissions in US cities are due to motor vehicle exhaust.3 Even though electrification of passenger vehicles seems to be promising from an economic point of view, it faces many non-economic barriers especially in emerging markets and developing countries, where there is a heavy reliance on second-hand markets, an absence of governmental environmental pledges, and a severe lack of cheap and fast charging infrastructure.4 Therefore, a pragmatic and more immediate way of reducing the current emissions resulting from the 1 billion passenger vehicles on the road today is to improve the current aftertreatment systems.
Noble metals have been used as catalysts for the oxidation of CO, as they show high stability and excellent performance with various supports and dopants.5 Feng et al.5 comprehensively reviewed CO oxidation performance and reaction mechanisms on platinum (Pt), gold (Au), palladium (Pd), silver (Ag), ruthenium (Ru), rhodium (Rh), and iridium (Ir), and highlighted the need for developing noble metal catalysts that are active for CO oxidation over the range of 100–250 °C. In a review of CO oxidation on Rh and Ru, Dey et al.6 stated that Rh and Ru-based catalysts are the most effective for CO oxidation at low temperatures.
In the context of passenger vehicle aftertreatment, three-way catalysts are implemented in gasoline-engine vehicles to simultaneously remove carbon monoxide (CO), nitric oxide (NO), and unburned hydrocarbons (UHC). Of particular interest are the CO emissions because they constitute up to 1 wt% of the exhaust emissions.7 The three-way catalyst technology relies on a combination of platinum (Pt) and rhodium (Rh) noble metals as the active sites, supported on alumina (Al2O3) for high surface area and on cerium oxides for oxygen storage.7,8 On the surface of the three-way catalyst, CO is oxidized by O2 to CO2 (eqn 1). This reaction has been extensively studied, especially on Pt, which is an excellent oxidation catalyst (see ref. 9–14). In the presence of H2O, such as in aftertreatment systems, the water-gas shift (WGS) reaction becomes important (eqn 2).
| 2CO + O2 → 2CO2 | (1) |
| CO + H2O → H2 + CO2 | (2) |
| Karakaya et al.19,24 | Shumokuri et al.18 | Maestri et al.22 | |
|---|---|---|---|
| Reactor configuration | Stagnation-flow | Honeycomb + FTIR | Annular reactor |
| Temperature (°C) | 250, 400, and 600 | 150 to 325 | 350 to 920 |
| Pressure (bar) | 0.5 | — | 1 |
| Rh metal loading | 5 wt% Rh | 0.7 g L−1 | 4 wt% |
| Dispersion (%) | 1.2 | 97 | 70 |
| Inlet gases | CO, O2, Ar | CO, O2, N2 | N2, O2, CH4, CO, H2O, CO2, H2 |
| Inlet composition | Fixed at each temperature | Stoichiometric | Multiple conditions |
Accurate kinetic data can be obtained via a variety of reactors, such as the annular reactor used by Maestri et al.22 and the honeycomb monolith used by Shumokuri et al.18 However, for a large number of species, assumptions are often made to simplify reactor modeling and reduce the computational time, which can result in uncertainties. An attractive alternative is to utilize reactors whose flow-fields are established and easy to model, such as channel and stagnation flows.25 These reactors allow for testing kinetic models under well-defined near-surface convective-diffusive transport conditions, which may be difficult to establish in configurations where access to the catalyst surface, such as in a plug-flow reactor, is not possible.26 The stagnation flow reactor concept is particularly useful in kinetic studies because it can be modeled as a boundary layer problem described by a set of ordinary differential equations.27 The boundary layer is adjacent to the catalyst surface where the concentration is uniform in the radial direction and only changes in the axial/vertical direction away from the surface.27 This simplifies the numerical simulations of the reactor, whose flow field is 2D, to a 1D problem.25 These attributes of the stagnation-flow reactor have led many groups to utilize it.28–30
There are no studies on CO oxidation and the WGS reaction on Rh in a stagnation-flow reactor in the low temperature regime typical of vehicle aftertreatment systems. As the existing literature experimental data are in a narrow range from either a temperature, flowrate or inlet composition perspectives, the mechanisms generated tend to be applicable to a narrow range of conditions. Therefore, the aim of this study is to obtain a better understanding of the direct oxidation of CO and the WGS reaction on 5 wt% Rh/Al2O3 using a stagnation-flow reactor at temperatures relevant to aftertreatment systems (175 to 275 °C). The reason behind choosing 5 wt% was to mimic the catalyst of Karakaya et al.;19,24 however, the results may be applicable to other metal loadings since CO oxidation on Rh/Al2O3 is independent of metal loading.31 This thorough, low-temperature study includes the effects of temperature, flowrate, pressure, inlet composition, and the addition of H2O. The combination of the various experimental conditions as well as the nature of the one-dimensional reactor paves the way for accurate kinetic modeling of low-temperature CO oxidation and the WGS reaction in the future.
Methodology
Catalyst preparation
The catalyst is 5 wt% Rh/Al2O3, purchased from Sigma-Aldrich (article number 212857). 200 mg of the catalyst powder was dispersed in 12 mL of DI water. The mixture was stirred at 1000 rpm and 40 °C for 12 hours. The resulting slurry was coated on an α-Al2O3 ceramic crucible. The coating was done using a spin coater (specialty coating systems G3P-8) with a heating element (120 °C, 1000 RPM), which has the advantage of creating homogeneous and thin coating layers on surfaces, beneficial for minimizing diffusion effects. Before coating, we treated the crucible surface with plasma to increase its hydrophilicity, which in turn increased the wettability (and therefore the coating quality) of the surface. After coating the catalyst, the crucible was calcined in air at 700 °C for 2 hours (as implemented in the literature19,24) then finally loaded into the reactor.Characterization of the catalyst powder
We characterized the catalyst powder via N2-physisorption, inductively coupled plasma optical emission spectroscopy (ICP-OES), X-ray diffraction (XRD), temperature-programmed reduction (TPR), H2-chemisorption, scanning transmission electron microscopy (STEM), and electron energy loss spectroscopy (EELS).The N2-physisorption test was conducted at 77 K on a Micromeritics ASAP 2420 with around 50 mg of the catalyst powder after calcination in air at 700 °C for 2 hours and applying vacuum overnight. The surface area was estimated by the Brunauer–Emmett–Teller (BET) method in a relative pressure range of 0.05–0.25. The pore volume was determined by the Barrett–Joyner–Halenda (BJH) method (desorption branch).
To determine the exact wt% of the Rh metal and confirm the specifications of the catalyst, we conducted experiments in 5110 ICP-OES (Agilent Technologies). First, the catalyst was digested in aqua regia (a mixture of nitric acid and hydrochloric acid with a 1:3 molar ratio). Around 10 mg of the catalyst and 8 mL of aqua regia was loaded into Polytetrafluoroethylene (PTFE) vessels. The sample solution was digested at 220 °C for 20 minutes in an ETHOS 1 closed vessel microwave-assisted digestion instrument. After cooling to room temperature, the sample was diluted in deionized water and then tested via ICP-OES.
To determine the crystallinity of the catalyst support, we conducted XRD experiments on the catalyst powder before and after calcination (in air at 700 °C for 2 hours) using a Bruker XRD D8 ADVANCE instrument in the Bragg–Brentano configuration using CuKα radiation and an EIGER2 R detector. The step size was 0.02° and the 2θ range was 10–90°. The Joint Committee on Powder Diffraction Standards (JCPDS) cards were used to identify the crystalline phase of the catalyst support.
To determine the reducibility of the catalysts, H2-temperature programmed reduction (H2-TPR) experiments were performed in a Micromeretics AutoChem 2950 HP instrument. About 200 mg of the calcined catalyst was packed into a quartz tube. To degas the powder, the catalyst was purged by high purity helium at 50 mL min−1 and 120 °C for one hour then cooled to room temperature. The sample was heated to 800 °C at a rate of 10 °C min−1 under 10% H2/Ar flow of 50 mL min−1. A thermal conductivity detector (TCD) was used to monitor the consumption of hydrogen. An H2/Rh ratio of 2:1 was used (i.e. H2 adsorbs dissociatively, where one H atom adsorbs to one Rh site).32,33
Static H2-chemisorption measurements were performed at 34 °C using a Micromeritics ASAP 2020 instrument. Around 100 mg of the catalyst (calcined in air at 700 °C for 2 hours) was reduced in situ under H2 flow for one hour at 300 °C (the same reduction temperature used in the experiment), evacuated at that temperature for 3 hours, and then cooled to 34 °C. A total chemisorption profile was obtained from 100 to 450 mmHg in increments of 50 mmHg. The sample was degassed for a few hours to remove the physisorbed amount of H2, then another chemisorption profile was obtained (over the same pressure range) to capture the amount of physisorbed H2. The difference between the two isotherms corresponded to the chemisorbed H2. By extrapolating the chemisorbed H2 isotherm to zero pressure, we determined the hydrogen uptake and therefore the catalyst dispersion and average particle size.
STEM images of the reduced catalyst were obtained using Titan Themis Z (Thermo-Fisher Scientific) to examine the catalyst morphology and quantify the particle size distribution. The analysis was performed by operating the microscope at an acceleration voltage of 300 kV. Prior to STEM imaging, the powder catalyst was loaded in a packed bed reactor and reduced by heating the catalyst to 300 °C (the same reduction temperature used in the experiment) at a rate of 10 °C min−1 and flowing H2 (8 mL min−1) for 1 hour. After reduction, the reactor was cooled with helium, then sealed and moved into a glovebox. In the glovebox, where the O2 and H2O concentrations were lower than 1 ppm, the reduced catalyst was mixed with isopropanol, and a small amount of the solution was drop-casted onto a carbon-coated copper TEM grid. The grid was placed in a TEM vacuum transfer holder to avoid exposure to air outside the glovebox. Bright-field (BF) and high-angle annular dark field (HAADF) images were obtained while simultaneously generating elemental maps using EELS. In addition, EELS line scan analysis was performed to ensure the absence of RhOx and therefore confirm the success of the catalyst reduction and accuracy of the particle size distribution.
Stagnation-flow reactor design
The experiments were conducted in a stagnation-flow reactor (depicted in Fig. 1), which allows to numerically model the system as a one-dimensional reacting flow, enabling accurate kinetic modeling and a coupling of gas and surface phase chemistry effects.27 Reactant species were flowed from gas cylinders using MKS mass-flow controllers towards the bottom of the reactor chamber (shown as “A” in Fig. 1). The chamber was oriented vertically for zero buoyancy effects and the chamber pressure was measured by an MKS baratron transducer. Reactant species were then perfectly mixed inside the chamber via a mixing device (shown as “B” in Fig. 1), whose design is based on that of a McKenna burner.34 Reactant species flowed upwards towards the catalyst coated onto a heated ceramic stagnation plate made of α-Al2O3 (“C” in Fig. 1) and held in place via a pedestal (“D” in Fig. 1).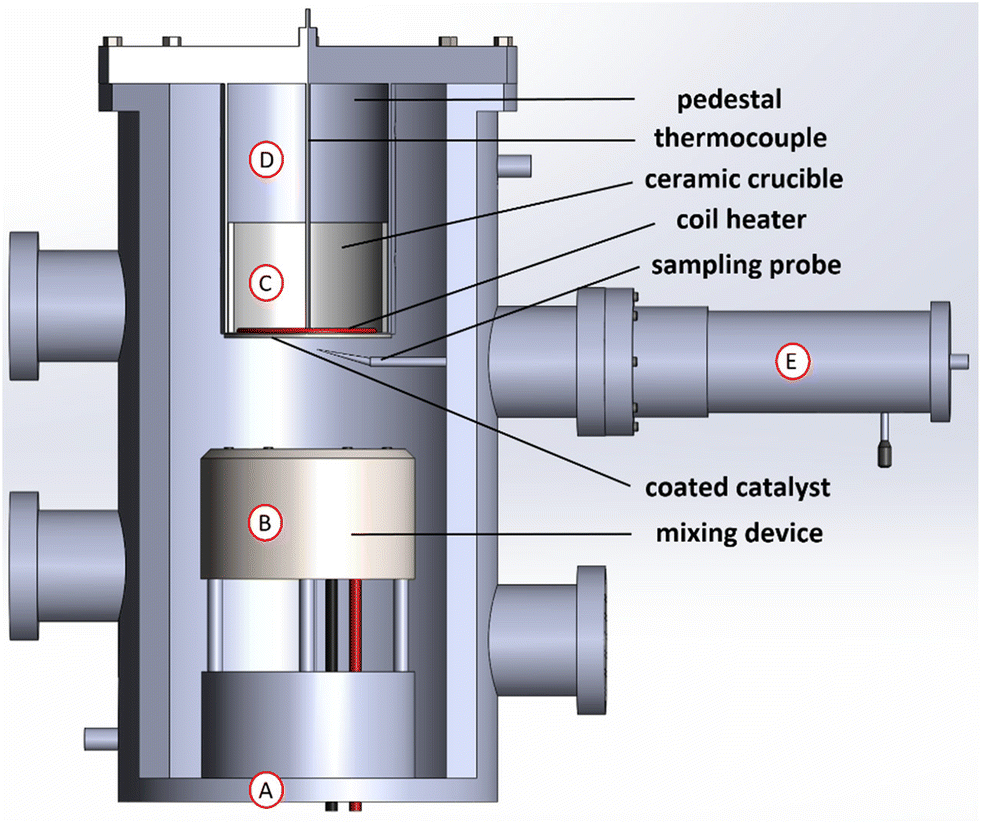 | ||
| Fig. 1 Schematic depiction of the stagnation-flow reactor: “A” is the bottom of the reactor where the inlet gases were flowed upwards, “B” is for mixing the inlet gases, “C” is the ceramic crucible on which Rh/Al2O3 was coated, “D” is the pedestal holding the crucible in place, and “E” is the sampling probe manual step manipulator. | ||
The plate was heated by a coil heater (purchased from Micropyretics Heaters International), controlled by a feedback loop based on measurements from a type-K thermocouple placed right above the coil. The temperature of the catalyst surface was measured using another type-K thermocouple placed directly on the catalyst surface (i.e. the bottom of the stagnation plate), whereby the thermocouple was subjected to the same flow conditions to which the catalyst surface was subjected, especially the cooling effect due to the high flowrates involved. The thermocouple was ensured to be placed immediately on the surface by visual inspection through an observation window mounted on the chamber.
Probe-based sampling was used to measure gaseous reactant and stable product species concentrations as a function of distance from the catalyst surface. A probe with an outer diameter of 250 μm was used and placed slightly off-center, to minimize the probe disturbance of the flow. The probe was moved up and down via a step manipulator (shown as “E” in Fig. 1, purchased from CHI-VAC), which was mounted on the chamber from the outside and operated manually. The probe was ensured to be placed at the catalyst surface for distance-zero measurements by visual inspection. The steady-state conversion of CO to CO2 (and production of H2 in the case of the water-gas shift reaction experiments) was measured directly using gas chromatography (GC). The GC used was the Agilent Refinery Gas Analysis system (7890 A), following the ASTM D1945 and D1946 methods. One TCD was used to detect O2, N2, CO, and CO2 (for CO oxidation), and another TCD to detect H2 (for WGS). The GC was calibrated using a standard calibration gas. In addition to the sampling line, another output of the chamber was towards the exhaust, where a suction pump (Varian, model DS 302) was installed.
To add H2O to the feed, a nebulizer (purchased from Precision Glassblowing) was used to vaporize the H2O as part of a vaporization chamber (design taken from ref. 35). The water was fed to the nebulizer via a syringe pump (purchased from New Era Pump Systems). The vaporization chamber as well as the tubing which fed the vapor to the system were heated to 120 °C to prevent condensation. To ensure that the vapor would not condense upon mixing with the feed CO, O2, and N2 gases, the entire gaseous reactant feed needed to be heated. An in-line heater (purchased from Kanthal) was used to heat the gaseous reactant species to 165 °C before mixing with the vapor. All the feedlines were insulated to minimize heat losses before entering the reactor, at which point the temperature was measured using a type-K thermocouple. Additionally, an H2O adsorption column (clean gas moisture filter, purchased from Agilent) was installed downstream the reactor sampling port to prevent H2O from entering the GC. Lastly, given that the Varian suction pump has a limited temperature rating of 40 °C, the exhaust line of the reactor chamber was cooled to 20 °C using a chiller (purchased from Julabo) before entering the pump.
Testing procedure and reaction conditions
Before conducting CO oxidation experiments, a deactivation test was performed, where CO2 production at a fixed distance (zero), fixed surface temperature (275 °C), and fixed CO:O2 molar ratio (2:1) was measured as a function of time for over five hours. The point in time at which the production of CO2 started decreasing was deemed the deactivation time, which was the maximum operating time for the experiments. Henceforward, whenever the deactivation time was approached, the catalyst was regenerated in situ by oxidation at 300 °C for 30 minutes under 10% O2/N2 flow then reduction at 300 °C for 30 minutes under 10% H2/N2 flow. In addition to testing for deactivation, initial experiments were performed to quantify the relative error of the gas chromatograph.
Table 2 summarizes the experimental conditions. Three pressures (300, 500 and 700 mbar) were tested to investigate the effect of pressure on the resolution of the CO profile as a function of distance from the catalyst surface. This was done at 24 g min−1 and 275 °C under lean conditions. Two flowrates were tested to examine the effect of residence time: 24 g min−1 (corresponding to 0.6 bar and 20 SLM of dry gas total flow) and 35 g min−1 (corresponding to 0.8 bar and 30 SLM of dry gas total flow). At each flowrate, two inlet compositions were tested: stoichiometric (CO:O2 = 2:1), and lean (CO:O2 = 1:1), with N2 as the balance gas. At each inlet composition, reactivity profiles as a function of distance away from the catalyst surface were obtained from catalyst temperature of 175 to 275 °C in increments of 25 °C. From each reactivity profile, the conversion of CO was calculated as shown in eqn (3). After collecting reactivity profiles over the temperature range under one inlet composition, the catalyst was regenerated as stated above, before testing another inlet composition over the temperature range again.
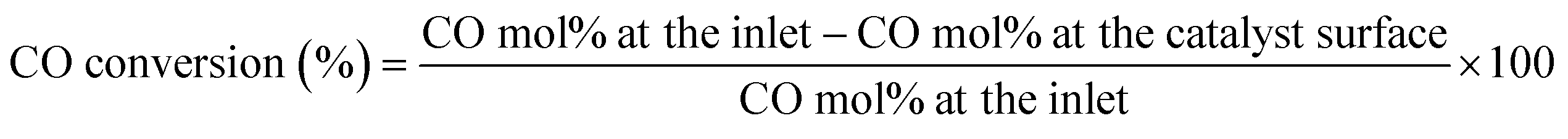 | (3) |
| Procedure | Flowrate (g min−1) | CO:O2 ratio |
N2 content (SLM) | Temperature (°C) | H2O content (mol%) | Total pressure (bar) |
|---|---|---|---|---|---|---|
| Deactivation | 24 | 2:1 |
18 | 275 | 0 | 0.6 |
| Effect of pressure | 24 | 1:1 |
18 | 275 | 0 | 0.3, 0.5, 0.7 |
| CO oxidation | 24 | 18 & 19 | 0 | 0.6 | ||
| 35 | 2:1 & 1:1 |
27 & 28 | 175, 200, 225, 250, 275 | 0 | 0.8 | |
| 35 | 27 & 28 | 1.0 | 0.8 | |||
| 35 | 27 & 28 | 1.7 | 0.8 | |||
| Reaction Order & Hysteresis | O2:CO = 0.4 to 4.0 |
14 to 18 | 300 | |||
| 24 | CO:O2 = 0.4 to 4.0 |
14 to 18 | 300 | 0 | 0.6 | |
| 1:1 |
17 | 200, 225, 250, 275 | ||||
| Water-gas shift | 35 | CO:H2O = 1:1 |
30 | 275 | 1.7 | 0.8 |
Additionally, reaction orders and hysteresis behavior were investigated by: (1) changing the O2 inlet composition at the expense of the inert (N2, see Table 2) from an O2:CO ratio of 0.4 to 4 and then back to 0.4 while the temperature was fixed at 300 °C, (2) changing the CO inlet composition at the expense of the inert (N2, see Table 2) from a CO:O2 ratio of 0.4 to 4 and then back to 0.4 while the temperature was fixed at 300 °C, and (3) testing a fixed, lean inlet composition at four temperatures from 200 °C to 300 °C and then back to 200 °C.
To test for the effect of H2O on CO oxidation at low temperature, reactivity profiles were obtained for stoichiometric and lean inlet compositions in the presence of 1 mol% and 1.7 mol% of H2O over the temperature range and at a total flow of 35 g min−1. Lastly, the water-gas shift reaction (eqn (2)) was tested by feeding 1.7 mol% of H2O and 1.7 mol% of CO at 275 °C and 35 g min−1 of total flow.
Results and discussion
Catalyst characterization results
The BET area of the Rh/Al2O3 catalyst is 116 m2 g−1 and the BJH pore volume is 0.50 cm3 g−1, which are typical values are for Al2O3-supported Rh.36–40 The N2-physisorption isotherm is type IV and is attached in the ESI† (Fig. S1) The ICP results confirm that the metal content is approximately 5 wt%. The XRD results shed some light on the effect of calcining the catalyst in air at 700 °C for 2 hours. More specifically, as the catalyst was heated, the alumina support partially transitioned from the γ phase to the δ phase, which is the expected next phase in the well-established γ → δ → Θ → α sequence.41 The XRD patterns of the as-purchased and calcined catalysts are shown in Fig. S2.† Given that the γ phase was still present after calcination, not much reduction in surface area was expected, which is confirmed by the fact that we attained similar physisorption results to those in the literature for Rh/Al2O3.36–40Fig. S3† shows the H2-TPR profile of the calcined Rh/Al2O3 catalyst. A broad peak with two shoulders can be observed: a first main shoulder near 160 °C along with a second one near 500 °C. This is attributed to the reduction of surface Rh (160 °C)42–45 and Rh strongly interacting with Al2O3. Indeed, Burch et al.46 studied the effect of calcination temperature on TPR of Rh/Al2O3. They show that the higher the calcination temperature, the later the TPR peak appeared and the broader the peak became. More specifically, when they calcined Rh/Al2O3 at 700 °C, they observed a broad peak in the range of 350–500 °C similar to the one shown in Fig. S3.† Additionally, the reduced catalyst H2-chemisorption results show that the dispersion is 20% and the active particle diameter is 5.4 nm. This corroborates the STEM particle diameter of 5.1 nm, averaged from diameter measurements of over 100 particles from a few STEM images (distribution shown in Fig. 2(c)). The H2-chemisorption profiles as a function of pressure are shown in Fig. S4.†
 | ||
| Fig. 2 Bright (a) and dark (b) field STEM images of the reduced Rh/Al2O3 along with (c) the particle size distribution attained from measuring over 100 particles, with a log-normal distribution fit and average diameter of 5.1 nm. | ||
Low magnification bright field and high magnification dark field STEM images are shown in Fig. 2(a and b). The EELS images of the reduced catalyst are shown in Fig. 3; (a) is the raw HAADF image; (b), (c), and (d) highlight Al, O, Rh, respectively, and (e) shows the metal and the support together. Fig. 4 shows the EELS line scan analysis (a) and the HAADF image on which the scan line analysis was performed (b and c). Fig. 4(a) shows that, prior to STEM imaging, the catalyst was successfully reduced. More specifically, the line scan analysis shows that Rh is present on the surface of AlOx; however, while there is an O signal, it is significantly reduced when the Rh signal increases, excluding the possibility of the presence of RhOx.
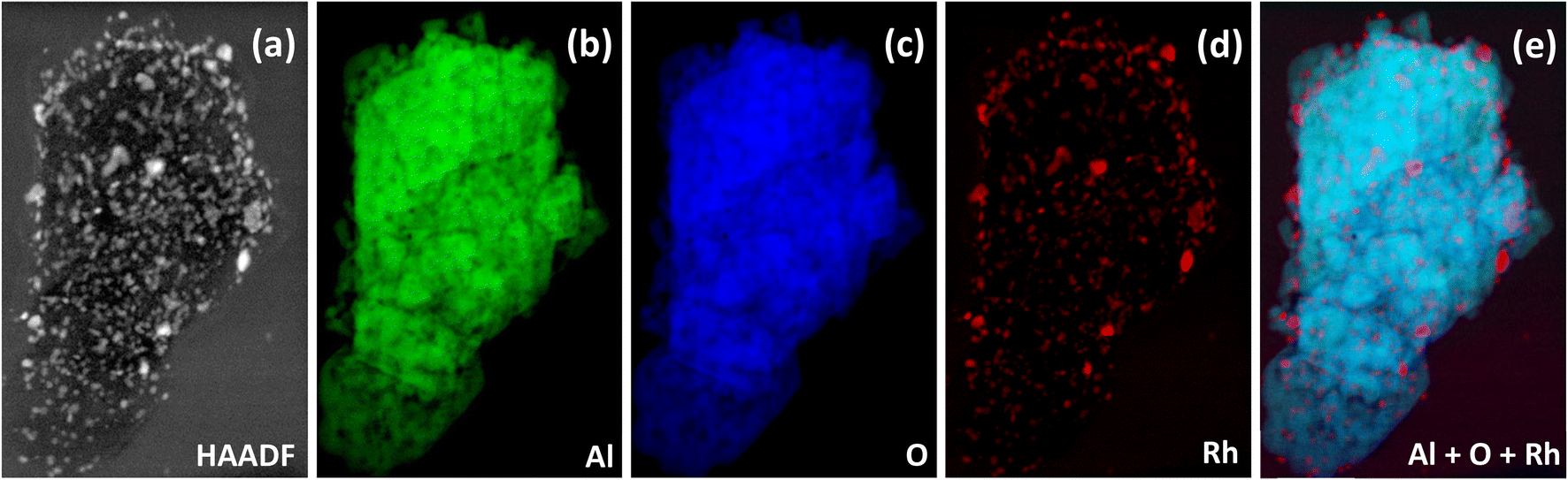 | ||
| Fig. 3 EELS images of the reduced catalyst: (a) raw HAADF image on which EELS was performed, (b) Al alone, (c) O alone, (d) Rh alone, and (e) the combination of Al, O and Rh. | ||
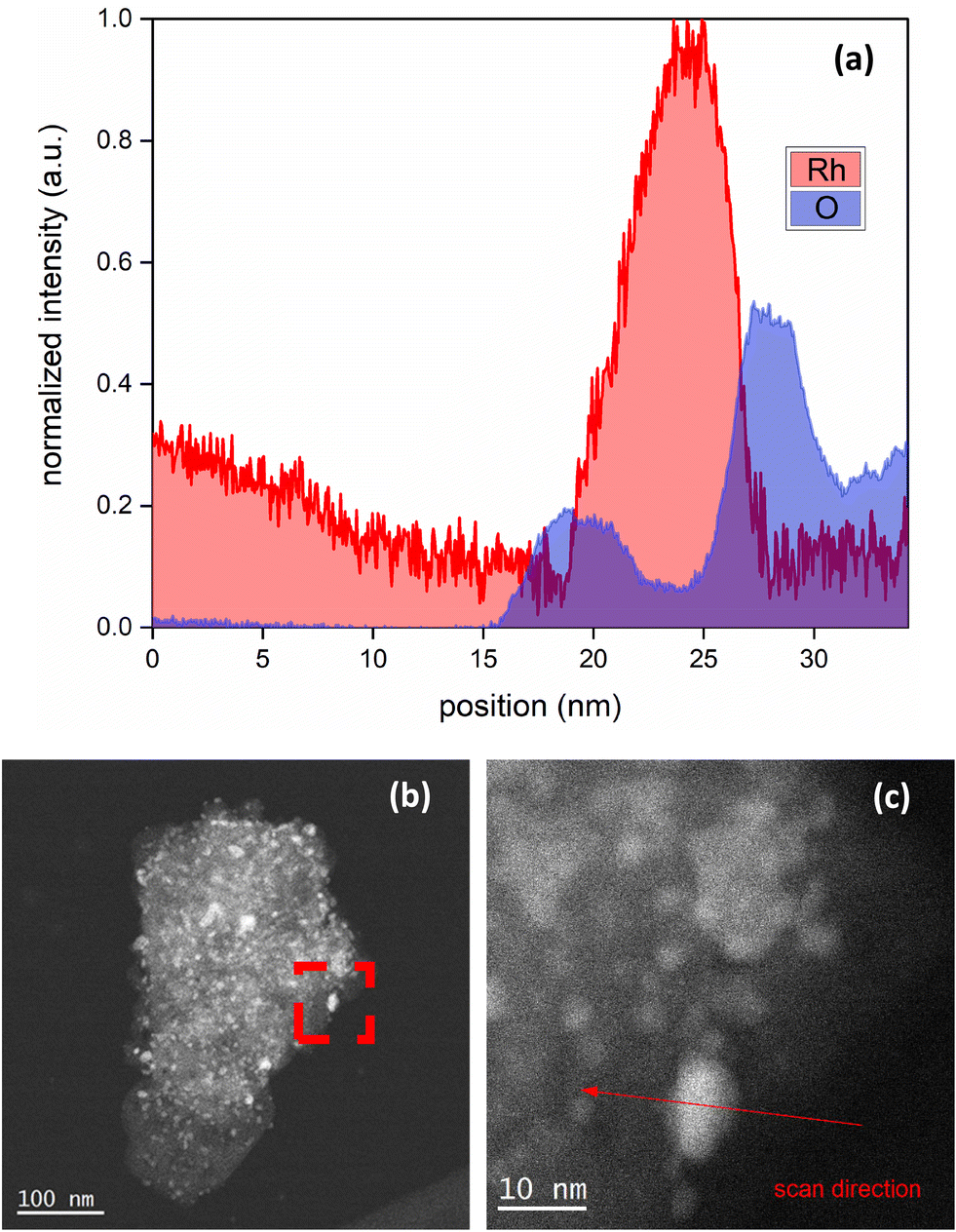 | ||
| Fig. 4 (a) EELS line scan analysis, with (b) the low-magnification raw HAADF image chosen for the analysis and (c) a zoomed-in version of (b) on which the analysis was performed and the scan direction is shown. | ||
Deactivation results
We collected CO2 production data as a function of time to determine the point at which the catalyst deactivated, which was set as the maximum for our experimental time window. For a conservative estimate, this was done at the highest experimental temperature (275 °C), the most reactive inlet composition (stoichiometric), and the highest residence time (total flow of 24 g min−1). The results are shown in Fig. S5.† After oxidizing and reducing the catalyst, the oxidation of CO to CO2 showed consistent results for nearly 3.5 hours. Therefore, whenever we performed experiments, we would regenerate the catalyst when we reached 3 hours of testing or when we finished a set of temperatures at the same inlet composition, whichever came first. Upon regenerating the catalyst, the same deactivation behavior was observed at roughly 3.5 hours as well. Lastly, measurements of the reactive system repeatedly showed an error of less than 0.5% in conversion values. This demonstrated the regenerability of the catalyst.Effects of pressure, residence time, temperature, and inlet composition on CO oxidation
The pressure played a role when optimizing the reactor to attain well-resolved species profiles of the reactive system. On the catalyst surface, CO2 is produced from the oxidation of CO by O2. CO2 diffuses back towards the inlet, making it possible to detect a decreasing CO2 mole fraction and increasing O2 and CO mole fractions as the sampling probe is moved away from the catalyst surface towards the inlet (Fig. 6 and 7 are examples of this). Fig. 5 shows the effect of pressure on the resolution of the CO profile as a function of distance from the catalyst surface. The CO conversion remained the same at all pressures tested, but at a pressure of 700 mbar, the CO mole percentage reached the inlet composition 16 mm away from the catalyst surface. At 500 mbar, the distance was reduced to 12 mm. At 300 mbar, the distance was further reduced to 8 mm. This is explained by the fact that reducing the pressure results in increasing the inlet velocity. However, the sampling gas flowrate at 300 mbar was significantly reduced such that it would take a prohibitively long time to collect samples. So, high pressures cause species profiles to be resolved at a further distance from the surface, while low pressures hinder species detection.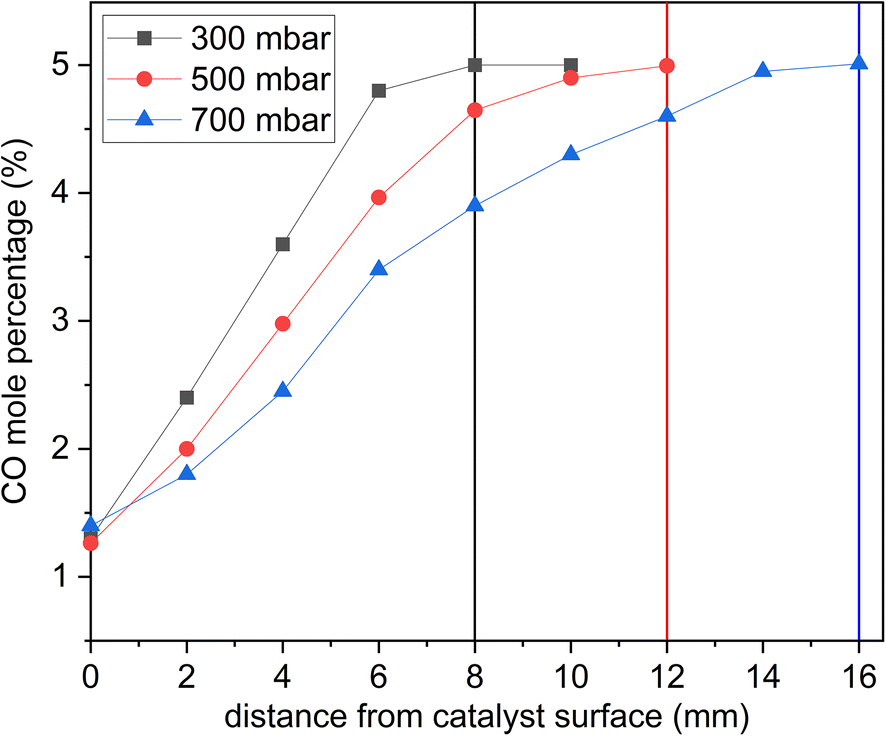 | ||
| Fig. 5 CO mol% as a function of pressure at 24 g min−1 of total flow, 275 °C, and 5 mol% inlet of CO. The vertical lines show where the CO mol% reaches inlet composition levels (5 mol%) away from the catalyst surface. The experimental data points are connected by lines for ease of presentation. | ||
To demonstrate the effect of residence time, Fig. 6 shows sample species profiles at 250 °C and a stoichiometric inlet composition under 24 and 35 g min−1 of total flow. The production of CO2 was higher at 24 g min−1, which led the CO2 diffusion distance back towards the feed to be longer. Specifically, the CO2 mole percentage at 24 g min−1 reached nearly 5.5%, but at 35 g min−1, it was only 2.5%. Additionally, the CO2 mole percentage reached near-zero levels 10 mm away from the catalyst surface at 24 g min−1, but it did so earlier, at 8 mm, under a total flow of 35 g min−1.
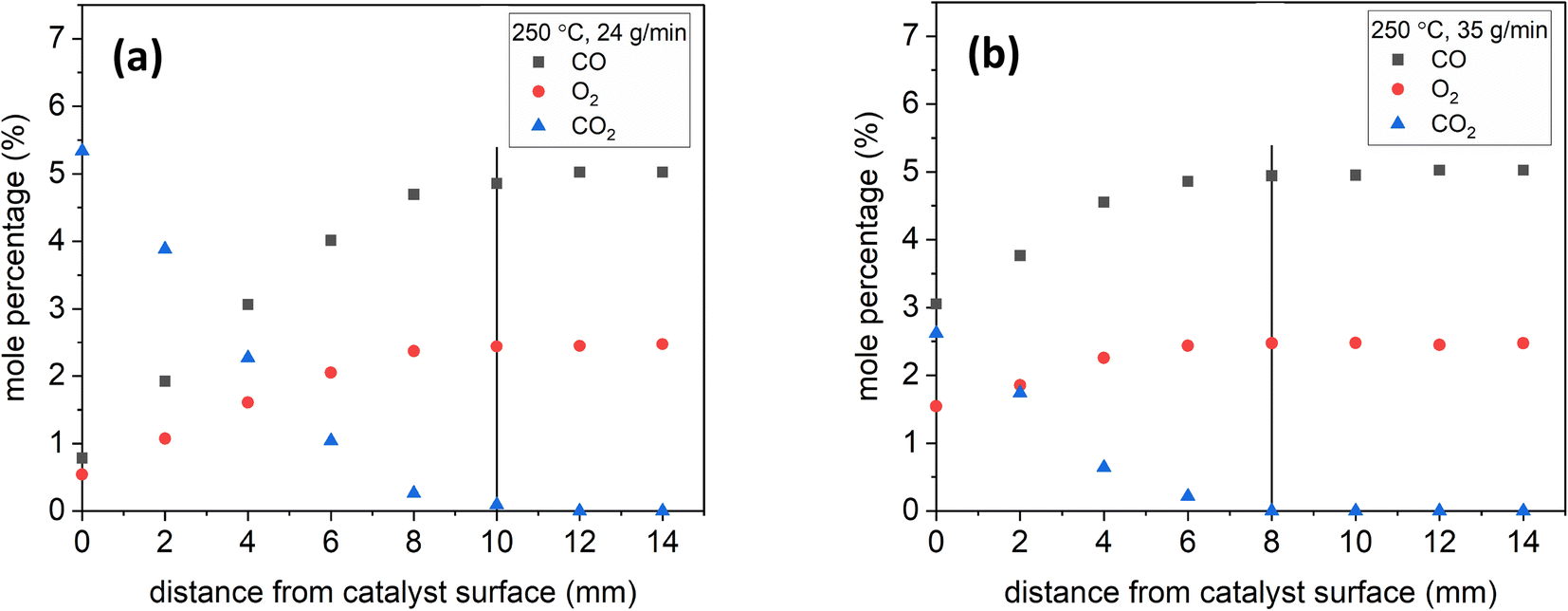 | ||
| Fig. 6 Species profiles as a function of distance at 250 °C, stoichiometric inlet composition, and (a) 24 & (b) 35 g min−1. The vertical lines show where the CO2 near-zero levels were reached away from the surface. | ||
To demonstrate the effect of temperature, Fig. 7 shows sample species profiles at a lean inlet composition under 35 g min−1 at 275 °C and 225 °C. The production of CO2 was higher at higher temperatures: nearly 4% at 275 °C as opposed to 1.5% at 225 °C. The higher CO2 production led the CO2 mole percentage to reach near-zero levels at 8 mm at 275 °C, whereas it did so at 6 mm at 225 °C.
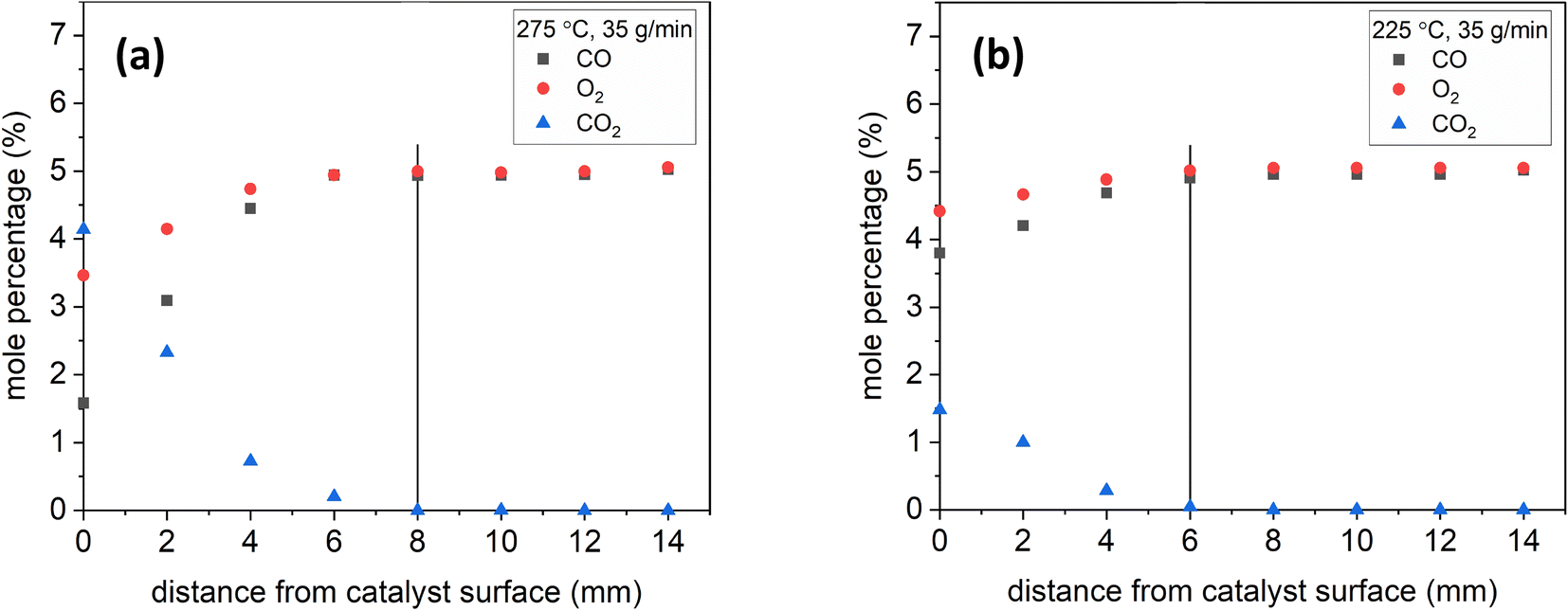 | ||
| Fig. 7 Species profiles as a function of distance at 35 g min−1, lean inlet composition, and 275 °C (a) & 225 °C (b). The vertical lines show where the CO2 near-zero levels were reached away from the surface. | ||
Table 3 shows the effects of residence time, inlet composition, and temperature on the CO conversion under all conditions tested. First, at the same inlet composition and temperature, the CO conversion levels were always higher at the lower flowrate. The higher reactivity is due to the higher residence time. Second, at the same flowrate and inlet composition, the reactivity increased as the temperature increased, which is due to higher reaction rates at higher temperatures.
| Temperature (°C) | 35 g min−1 | 24 g min−1 | ||
|---|---|---|---|---|
| Lean | Stoichiometric | Lean | Stoichiometric | |
| 275 | 68 | 68 | 75 | 85 |
| 250 | 60 | 38 | 72 | 84 |
| 225 | 23 | 9 | 55 | 70 |
| 200 | <5 | <5 | 13 | 19 |
| 175 | 0 | 0 | <5 | <5 |
As for the effect of inlet composition, the stoichiometric ratio was favored at the lower flowrate (24 g min−1), but the lean ratio was slightly favored at the higher flowrate (35 g min−1). Under the low flowrate and lean inlet composition, there was an abundance of O2 that adsorbed on the surface and occupied more active sites at the expense of CO, which reduced the reactivity. Under the low flowrate and stoichiometric inlet composition, there was little competition on active sites, which resulted in higher conversion levels at the stoichiometric composition. The fact that we observed higher reactivity at the stoichiometric conditions agrees with findings by Gopinath et al.,47 who reported maximum CO oxidation levels at the stoichiometric inlet ratio of CO and O2 near 230 °C.
At the higher flowrate (35 g min−1), the trend was reversed: lean conditions showed higher reactivity at temperatures below 250 °C. This agrees with findings by Bunluesin et al.,16 who studied CO oxidation on Rh/Al2O3 and Rh/CeO2 and observed that the adsorption of O2 was rate limiting on the Al2O3 support. Campbell and White48 reported that the adsorption and desorption reactions of O2 and CO on Rh can be highly temperature dependent. They showed that at low temperatures, the rate of CO adsorption is high and inhibits the adsorption of O2, leading to lower CO2 production. More specifically, they reported that CO starts desorbing at 257 °C, allowing for more O2 to adsorb. As the temperature increases, the inhibitive effect of CO coverage decreases, eventually becoming negligible near 275 °C.48 So, at 35 g min−1 and below 250 °C, where CO coverage is significant, more O2 in the inlet resulted in higher adsorption of O2 and therefore higher CO conversion levels compared to the stoichiometric inlet composition. Additionally, the likelihood of adsorption can be quantified in the form of a sticking coefficient, which represents the probability (ranging from 0 to 1) for a certain molecule to adsorb on the surface. In the mechanisms generated by Mhadeshwar and Vlachos,23 Maestri et al.22 and Karakaya et al.,19,24 the sticking coefficient for O2 ranges from 0.01 to 0.05, whereas that of CO is around 0.5. Given the much lower sticking coefficient for O2, more O2 may be needed to compensate for the low adsorption of O2, which is more pertinent at sufficiently high flowrates.
Reaction order, hysteresis behavior, and the water-gas shift reaction
To determine the reaction order with respect to CO, we changed the CO:O2 volume ratio from 0.4 to 4 by fixing the O2 content and increasing that of CO at the expense of the inert at 300 °C and 24 g min−1 of total flow. Fig. 8(a) shows the mol% of O2 consumed as a function of CO partial pressure. Fig. 8(b) shows that below the stoichiometric ratio of 2, the order is positive. Beyond the stoichiometric ratio, the order is negative, indicating an inhibition effect.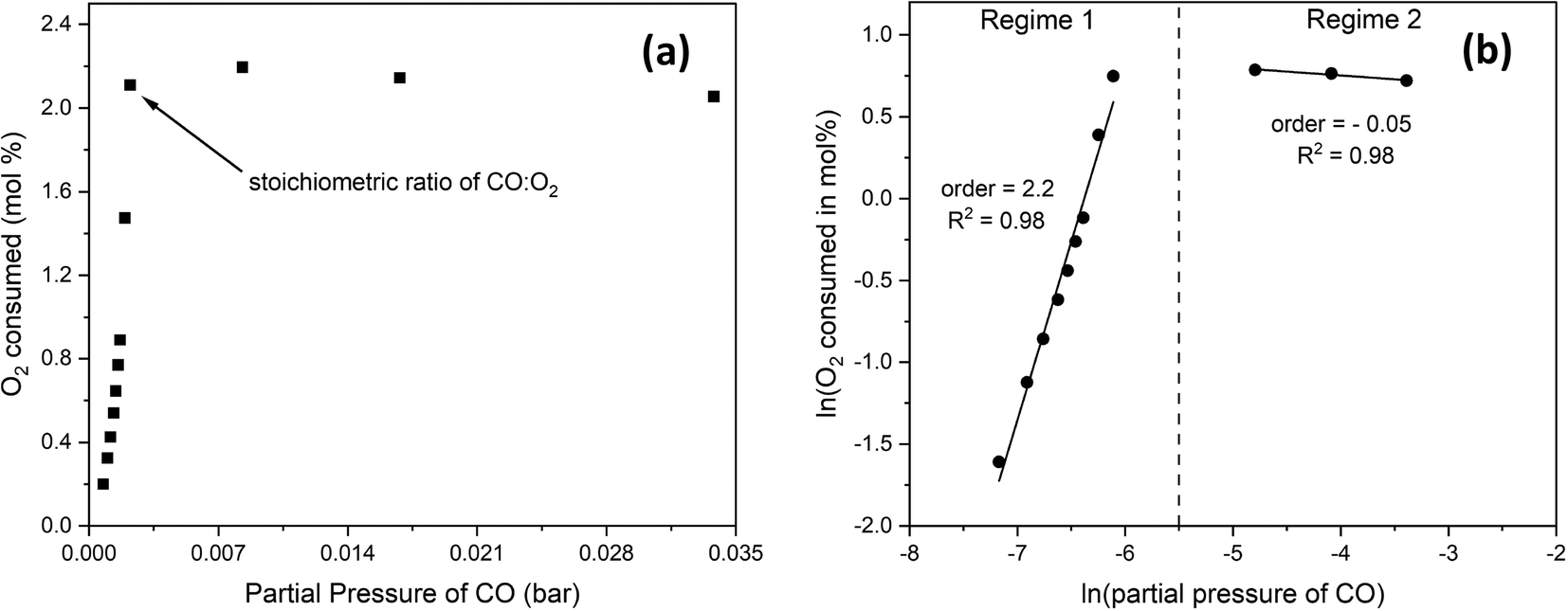 | ||
| Fig. 8 (a) the mol% of O2 consumed as a function of CO partial pressure at 300 °C and 24 g min−1, with (b) showing the natural logarithm values where reaction orders can be extracted. | ||
Similarly, to determine the reaction order with respect to O2, we changed the O2:CO volume ratio from 0.4 to 4.0 by fixing the CO content and increasing that of O2 at the expense of the inert at 300 °C and 24 g min−1 of total flow. Fig. 9(a) shows the mol% of CO consumed as a function of the partial pressure of O2. Three kinetic regimes are observed (Fig. 9(b)). Below the stoichiometric ratio of 0.5, the order is positive. Beyond the stoichiometric ratio, a second regime exists in which the order is negative, indicating that increasing the partial pressure of O2 beyond the stoichiometric ratio inhibits the CO oxidation. At higher partial pressures of O2, a lower, negative order is observed, where the effect of O2 is inhibiting but to a lesser extent.
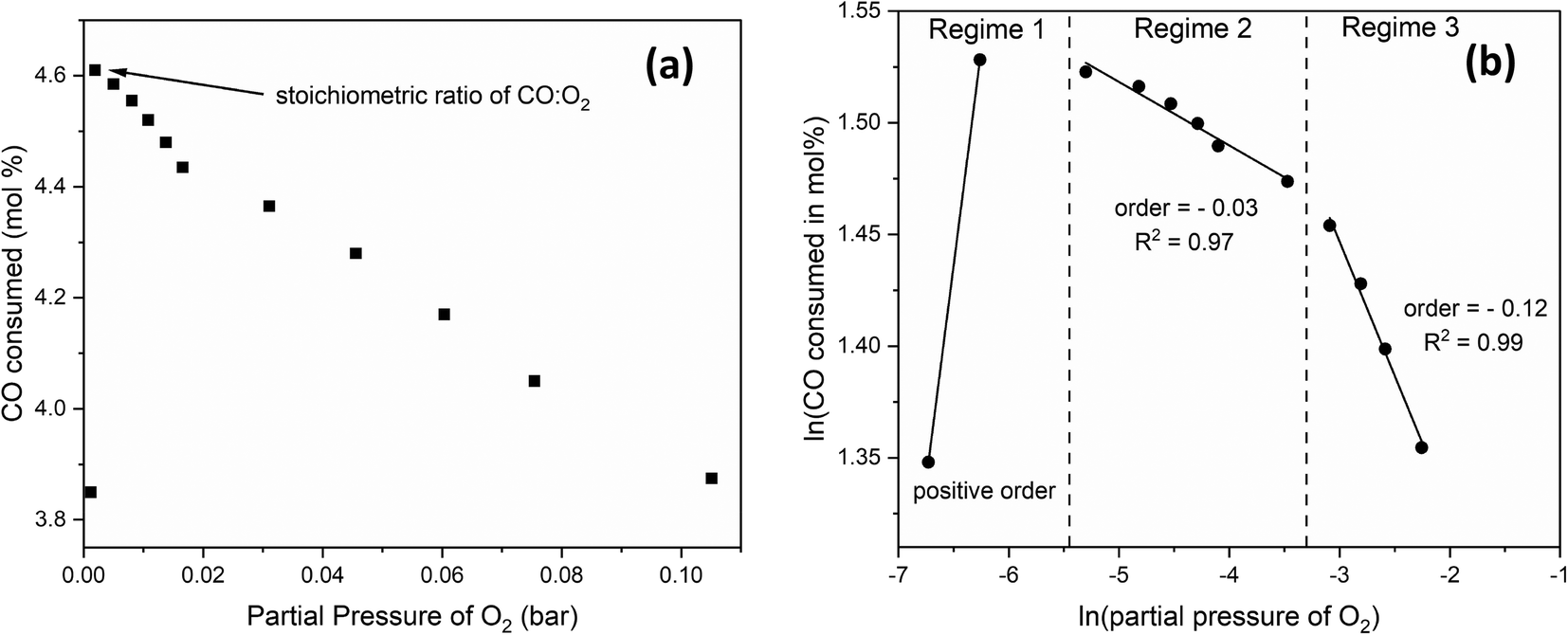 | ||
| Fig. 9 (a) the mol% of CO consumed as a function of O2 partial pressure at 300 °C and 24 g min−1, with (b) showing the natural logarithm values where reaction orders can be extracted. | ||
Lee et al.49 studied the low-temperature oxidation of methane on Pt, Pd and Ag–Pd catalysts, where they also observed three kinetic regimes: a positive order with respect to O2 below the stoichiometric ratio then decreasing orders as the partial pressure of O2 increased. Using in situ X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS), they attributed the reduction in catalyst activity at high O2 partial pressure to the oxidation of the metals. While their study was on methane oxidation on Pt, Pd, and Ag–Pd catalysts, we expect Rh to behave similarly under CO oxidation conditions. Therefore, we propose that as the O2 content increased, Rh was oxidized, resulting in lower activity. Additionally, Lee et al.49 observed hysteresis as they decreased the partial pressure of O2, and they attributed the observed bistability to the reduction of the metal by methane at decreasing partial pressures of O2.
To investigate whether hysteresis would occur on Rh under CO oxidation conditions, we performed a hysteresis study, where after increasing the partial pressure of O2 at the expense of the inert as shown in Fig. 9(a), we decreased the O2 content back to the starting point. Hysteresis was indeed observed, shown in Fig. 10 in terms of steady-state CO conversion as a function of O2:CO volume ratio. As the O2 content decreased, CO reduced RhOx, restoring the activity of the catalyst. Upon decreasing the O2 content, the catalyst showed lower activity at the same ratio compared to when increasing the O2 content. The difference in reactivity between the two routes was lower as the stoichiometric ratio was approached, at which point bistability was no longer observed.
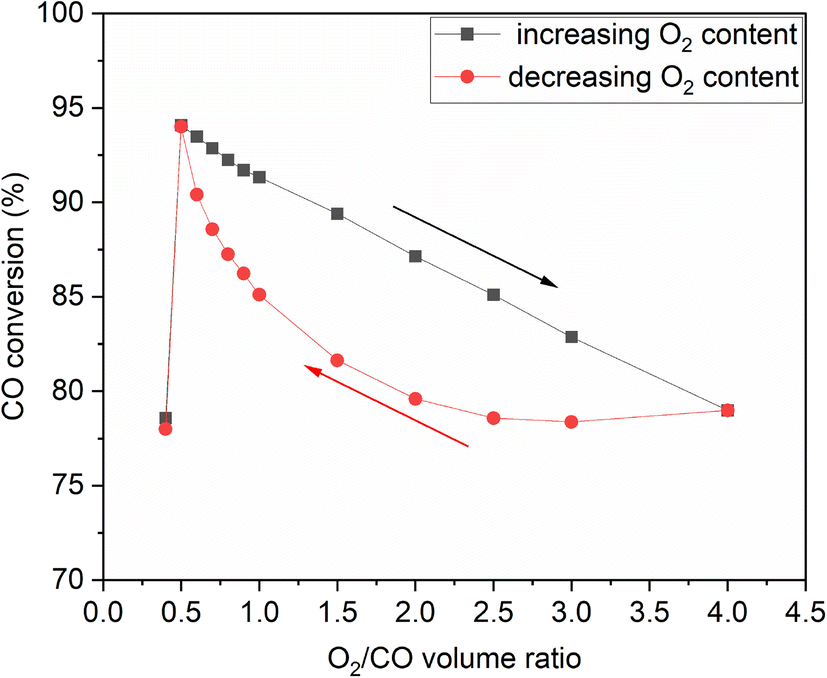 | ||
| Fig. 10 Hysteresis observed when changing the O2 content at the expense of the inert at 300 °C and 24 g min−1. | ||
A few theories have been developed to explain the irregularities of CO oxidation, such as bistability, on platinum group metals (PGM). The theories include effects of surface coverage, formation of metal oxides, formation of sub-surface oxygen during the reaction, and changes to the surface by the adsorbates.50–53 In particular, hysteresis during low-temperature oxidation reactions of hydrocarbons on PGM catalysts is a common phenomenon, the extent of which depends on the oxophilicity of the catalyst and the reduction ability of the hydrocarbon.54
No hysteresis was observed when changing the CO:O2 ratio by increasing and decreasing the CO content at the expense of the inert (Fig. S6†). This signifies no change in the oxidation state of the catalyst by the presence of CO. No hysteresis was observed when changing the temperature from 200 °C to 300 °C and back to 200 °C (Fig. S7†) either, which indicates no sintering of the catalyst upon short-time exposure to 300 °C. As for the water-gas shift reaction, we observed no reactivity in the temperature range studied, which is supported by previous literature reports.55–57 Similarly, the addition of H2O had no influence on the oxidation of CO in the temperature range studied, irrespective of inlet composition.
Conclusions
In this study we investigated Rh-catalyzed CO oxidation and WGS reaction in a stagnation-flow reactor. The catalyst characterization techniques show a change of the Al2O3 phase from γ to δ upon calcination in air at 700 °C for 2 hours, but the BET surface area and BJH pore volume remained similar to those typically found in the literature for Rh/Al2O3. Calcination did cause some Rh to be strongly bound to the surface, but H2-TPR shows that easily accessible Rh, reducible at 160 °C, was still present. STEM and EELS images show the morphology of the reduced catalyst, with an average particle size of 5 nm, which corroborates the H2-chemisorption results.The stagnation-flow reactor experimental results show how the pressure, flowrate, temperature, inlet composition, and presence of H2O affected reactivity. At 24 g min−1 of total flow, as we increased the temperature from 175 to 275 °C, the CO conversion increased from less than 5% to 75% and to 85% under lean and stoichiometric inlet compositions, respectively. The lower reactivity under lean conditions is attributed to O2 occupying active sites at the expense of CO. At 35 g min−1 of total flow, there was a preference for the lean inlet composition at temperatures below 250 °C, due to CO surface coverage inhibiting the adsorption of O2, which is compensated for by running lean. Investigating the reaction order with respect to O2 revealed three kinetic regimes where the reaction order with respect to O2 is positive below the stoichiometric ratio and negative with decreasing orders at increasing O2 partial pressures. The reaction order with respect to CO is positive below the stoichiometric ratio, at which point the reaction order with respect to CO becomes negative. Lastly, we observed catalyst bistability as we increased and decreased the O2 content at the expense of the inert, and we attribute that to the oxidation of the metal at sufficiently high O2 content and the reduction of the metal by CO at decreasing O2 content. We observed no hysteresis behavior while changing the CO content or the temperature, and we observed no water-gas shift reactivity over the temperature range tested. The current, thorough investigation aids in kinetic modeling of CO oxidation on Rh over a wide range of conditions, where accurate reaction rates can be obtained.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by King Abdullah University of Science and Technology Office of Sponsor Research with funds given to the Clean Combustion Research Center (CCRC) and KAUST Catalysis Center (KCC). We would like to thank Dr. Et-touhami Es-sebbar, Dr. Adamu Alfazazi, Dr. Can Shao and Yitong Zhai for their help with the experimental setup, and Dr. Abdulrahman El Labban for his help with coating the catalyst. In addition, our thanks go to the KAUST Core Labs and the KAUST Catalysis Center staff for their help with the characterization experiments.References
- Air pollution: The silent killer that claims 7 million lives each year, retrieved from https://www.ohchr.org, 2019 Search PubMed.
- National Emission Inventory, retrieved from https://www.epa.gov, 2021 Search PubMed.
- EPA's Report on the Environment (ROE), 2021 Search PubMed.
- World Energy Outlook, 2021 Search PubMed.
- C. L. Feng, X. L. Liu, T. Y. Zhu and M. K. Tian, Environ. Sci. Pollut. Res., 2021, 28, 24847–24871 CrossRef CAS PubMed.
- S. Dey and G. C. Dhal, Polytechnica, 2020, 3, 26–42 CrossRef.
- G. C. Koltsakis and A. M. Stamatelos, Prog. Energy Combust. Sci., 1997, 23, 1–39 CrossRef CAS.
- H. Shinjoh, Catal. Surv. Asia, 2009, 13, 184–190 CrossRef CAS.
- R. Kissel-Osterrieder, F. Behrendt, J. Warnatz, U. Metka, H. R. Volpp and J. Wolfrum, Proc. Combust. Inst., 2000, 28, 1341–1348 CrossRef CAS.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- P. Aghalayam, Y. K. Park and D. G. Vlachos, Proc. Combust. Inst., 2000, 28, 1331–1339 CrossRef CAS.
- A. B. Mhadeshwar and D. G. Vlachos, J. Phys. Chem. B, 2004, 108, 15246–15258 CrossRef CAS.
- A. B. Mhadeshwar and D. G. Vlachos, Combust. Flame, 2005, 142, 289–298 CrossRef CAS.
- S. Raimondeau and D. G. Vlachos, Comput. Chem. Eng., 2002, 26, 965–980 CrossRef CAS.
- A. V. Kalinkin, A. V. Pashis and V. I. Bukhtiyarov, Kinet. Catal., 2007, 48, 298–304 CrossRef CAS.
- T. Bunluesin, H. Cordatos and R. J. Gorte, J. Catal., 1995, 157, 222–226 CrossRef CAS.
- A. B. Mhadeshwar and D. G. Vlachos, J. Catal., 2005, 234, 48–63 CrossRef CAS.
- D. Shimokuri, H. Murakami, S. Hinokuma, Y. Matsumoto, S. Naing, K. Naito, H. Yokohata, H. Takebayashi and A. Miyoshi, Combust. Sci. Technol., 2021, 193, 2137–2157 CrossRef CAS.
- H. Karadeniz, C. Karakaya, S. Tischer and O. Deutschmann, Chem. Eng. Sci., 2013, 104, 899–907 CrossRef CAS.
- Y. P. Cai, H. G. Stenger and C. E. Lyman, J. Catal., 1996, 161, 123–131 CrossRef CAS.
- M. J. P. Hopstaken and J. W. Niemantsverdriet, J. Chem. Phys., 2000, 113, 5457–5465 CrossRef CAS.
- M. Maestri, D. G. Vlachos, A. Beretta, G. Groppi and E. Rronconi, AIChE J., 2009, 55, 993–1008 CrossRef CAS.
- A. B. Mhadeshwar and D. G. Vlachos, J. Phys. Chem. B, 2005, 109, 16819–16835 CrossRef CAS PubMed.
- C. Karakaya, PhD, Karlsruher Institut für Technologie (KIT), 2012 Search PubMed.
- T. Yuan, Y. H. Lai and C. K. Chang, Combust. Flame, 2008, 154, 557–568 CrossRef CAS.
- R. W. Sidwell, H. Y. Zhu, R. J. Kee, D. T. Wickham, C. Schell and G. S. Jackson, Proc. Combust. Inst., 2002, 29, 1013–1020 CrossRef CAS.
- R. J. Kee, M. E. Coltrin and P. Glarborg, Chemically reacting flow: theory and practice, Wiley-Interscience, Hoboken, NJ, 2003 Search PubMed.
- O. Deutschmann, L. I. Maier, U. Riedel, A. H. Stroemman and R. W. Dibble, Catal. Today, 2000, 59, 141–150 CrossRef CAS.
- Y. K. Park, N. E. Fernandes and D. G. Vlachos, Chem. Eng. Sci., 1999, 54, 3635–3642 CrossRef CAS.
- G. Veser, M. Ziauddin and L. D. Schmidt, Catal. Today, 1999, 47, 219–228 CrossRef CAS.
- S. H. Oh and C. C. Eickel, J. Catal., 1991, 128, 526–536 CrossRef CAS.
- J. M. Gatica, R. T. Baker, P. Fornasiero, S. Bernal, G. Blanco and J. Kaspar, J. Phys. Chem. B, 2000, 104, 4667–4672 CrossRef CAS.
- S. Fuentes and F. Figueras, J. Catal., 1980, 61, 443–453 CrossRef CAS.
- D. W. Senser, J. S. Morse and V. A. Cundy, Rev. Sci. Instrum., 1985, 56, 1279–1284 CrossRef CAS.
- A. D. Abid, J. Camacho, D. A. Sheen and H. Wang, Energy Fuels, 2009, 23, 4286–4294 CrossRef CAS.
- B. H. Zhao, R. Ran, Y. D. Cao, X. D. Wu, D. Weng, J. Fan and X. Y. Wu, Appl. Surf. Sci., 2014, 308, 230–236 CrossRef CAS.
- E. Diaz, J. A. Casas, A. F. Mohedano, L. Calvo, M. A. Gilarranz and J. J. Rodriguez, Ind. Eng. Chem. Res., 2008, 47, 3840–3846 CrossRef CAS.
- P. Osorio-Vargas, C. H. Campos, R. M. Navarro, J. L. G. Fierro and P. Reyes, Appl. Catal., A, 2015, 505, 159–172 CrossRef CAS.
- R. Buchel, A. Baiker and S. E. Pratsinis, Appl. Catal., A, 2014, 477, 93–101 CrossRef CAS.
- M. Bilal and S. D. Jackson, Appl. Catal., A, 2017, 529, 98–107 CrossRef CAS.
- A. Boumaza, L. Favaro, J. Ledion, G. Sattonnay, J. B. Brubach, P. Berthet, A. M. Huntz, P. Roy and R. Tetot, J. Solid State Chem., 2009, 182, 1171–1176 CrossRef CAS.
- J. C. Vis, H. F. J. Vantblik, T. Huizinga, J. Vangrondelle and R. Prins, J. Mol. Catal., 1984, 25, 367–378 CrossRef CAS.
- M. Ojeda, M. L. Granados, S. Rojas, P. Terreros, F. J. Garcia-Garcia and J. L. G. Fierro, Appl. Catal., A, 2004, 261, 47–55 CrossRef CAS.
- S. S. Kim, S. J. Lee and S. C. Hong, Chem. Eng. J., 2011, 169, 173–179 CrossRef CAS.
- J. M. Li, F. Y. Huang, W. Z. Weng, X. Q. Pei, C. R. Luo, H. Q. Lin, C. J. Huang and H. L. Wan, Catal. Today, 2008, 131, 179–187 CrossRef CAS.
- R. Burch, P. K. Loader and N. A. Cruise, Appl. Catal., A, 1996, 147, 375–394 CrossRef CAS.
- C. S. Gopinath and F. Zaera, J. Catal., 2001, 200, 270–287 CrossRef CAS.
- C. T. Campbell and J. M. White, J. Catal., 1978, 54, 289–302 CrossRef CAS.
- G. Lee, W. Q. Zheng, K. A. Goulas, I. C. Lee and D. G. Vlachos, Ind. Eng. Chem. Res., 2019, 58, 17718–17726 CrossRef CAS.
- E. A. Lashina, N. A. Chumakova, G. A. Chumakov and A. I. Boronin, Chem. Eng. J., 2009, 154, 82–87 CrossRef CAS.
- E. I. Latkin, V. I. Elokhin and V. V. Gorodetskii, J. Mol. Catal. A: Chem., 2001, 166, 23–30 CrossRef CAS.
- K. Krischer, M. Eiswirth and G. Ertl, J. Chem. Phys., 1992, 96, 9161–9172 CrossRef CAS.
- M. Eiswirth, K. Krischer and G. Ertl, Surf. Sci., 1988, 202, 565–591 CrossRef CAS.
- G. Lee, W. Q. Zheng, I. C. Lee and D. G. Vlachos, Appl. Catal., A, 2019, 587 Search PubMed.
- G. G. Olympiou, C. M. Kalamaras, C. D. Zeinalipour-Yazdi and A. M. Efstathiou, Catal. Today, 2007, 127, 304–318 CrossRef CAS.
- A. Donazzi, A. Beretta, G. Groppi and P. Forzatti, J. Catal., 2008, 255, 241–258 CrossRef CAS.
- C. Karakaya, R. Otterstatter, L. Maier and O. Deutschmann, Appl. Catal., A, 2014, 470, 31–44 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00235c |
| This journal is © The Royal Society of Chemistry 2022 |