
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An environmentally benign and high-rate Appel type reaction†
Nicolas Mattias Del Rio
Fuenzalida
 ,
Eirin
Alme
,
Frida Johanne
Lundevall
and
Hans-René
Bjørsvik
*
,
Eirin
Alme
,
Frida Johanne
Lundevall
and
Hans-René
Bjørsvik
*
Department of Chemistry, University of Bergen, Allégaten 41, 5007 Bergen, Norway. E-mail: hans.bjorsvik@kj.uib.no
First published on 22nd April 2022
Abstract
A high rate, selective, and productive Appel type reaction was developed. The method allows for ipso-substitution of the alcoholic hydroxy group with halogens ∈ [Cl, Br, I]. The method demands mild reaction conditions that include a very short reaction time, <15 min only, versus reaction times of several hours or days using the classical Appel reaction conditions (PPh3 + CCl4 + R–OH). The method was demonstrated to operate with the cheap and easily available 1,3-dihalo-5,5-dimethylhydantoins and N-halo succinimides (halo ∈ [Cl, Br, I]) as the reagent that performs the halogenation of PPh3. The reaction protocol operated with several acceptable solvents rather than DCM that was used in the classical Appel reaction. Furthermore, the batch protocol was also translated and successfully implemented on a flow reactor platform (t < 5 min, y = 95%).
Introduction
Contemporary organic synthesis involves various Pd-catalysed cross-coupling reactions that demand halogenated reagents. Therefore, there is a continuous need for new and more benign and environmentally friendly halogenation methods.The classical Appel reaction1 is an organic reaction that converts alcohols into their corresponding alkyl chlorides. The reaction implicates a reaction between tetrachloromethane and triphenyl-phosphine that produces the “Appel salt”, Ph3P+–Cl−CCl3. In a subsequent reaction step, the Appel salt was initially used to convert alcohols to the corresponding chloride, Scheme 1(a), but the Appel salt has also been used as a dehydrating reagent2 and to convert carboxylic acids to oxazolidines,3Scheme 1 path (b).
 | ||
| Scheme 1 (a) The classical Appel reaction using PPh3 and CCl4 as reagents and an alcohol R–OH, where R ∈ [alkyl allyl propargyl]. (b) The Appel salt reacts with carboxylic acid to give the oxazolidine scaffold. | ||
The classical Appel reaction methods afford in general a high yield of the halogenated product under reasonably mild reaction conditions. However, the method needs stoichiometric quantities of tetrachloromethane as a chlorinating reagent and a lengthy reaction time. The reaction produces the target chlorinated product concomitant with the formation of stoichiometric quantities of two different by-products, namely chloroform and triphenyl-phosphine oxide.
Because of the prevailing disadvantages associated with the classical method, several attempts have been carried out with the goal of achieving an environmentally benign and efficient method. In this context, several attempts have been made to transform the method into a catalytic process with respect to triphenylphosphine.2,4a–f The new catalytic processes have led to methods that didn't demand stoichiometric quantities of triphenylphosphine, which per-se imply a significantly improved atom economy, however the said catalytic processes still comprise hazardous and environmentally demanding reagents (e.g. oxalyl chloride,4a,c,e,f benzo-trichloride4d) and produce stoichiometric quantities of halogen containing side-products (e.g. in the cases where CX4, X ∈ [Cl, Br, I] are still used as a halogen source). Furthermore, the new catalytic Appel reaction methods need reaction times in the range of 6–24 h. A few continuous flow chemistry processes have been disclosed, although these were also based on the classical Appel reagents, except that the phosphine was immobilized as a monolithic triphenylphosphine reagent that simplified the separation and purification of the post reaction mixture.5
Results and discussion
Previously, we have attained great results using 1,3-dihalo-5,5-dimethylhydantoins for both halogenation and oxidation reactions.6 The 1,3-dihalo-5,5-dimethylhydantoins are commercially available at low cost,7 except for 1,3-diiodo-5,5-dimehyl-hydantoin (DIH), but DIH can be synthesised at low cost by reacting I–Cl with 5,5-dimethylhydantoin.8
DCH, DBH, and DIH reagents held two halogens, which can both be utilised. This is of course an advantage from an environmental point of view as there are no left-over halogens in the consumed reagent.
Our idea for an improved Appel reaction method was to replace tetrahalomethane with DCH, DBH, and DIH. Preliminary experiments revealed that 1,3-dibromo-5,5-dimethylhydantoin reacted vigorously (exothermically) with triphenylphosphine. We believe that the Appel salt was formed with 3-bromo-5,5-dimethyl-2,4-dioxoimidazolidin-1-ide as a counter anion to the bromotriphenylphosphonium cation. This variant of the Appel salt turned out to be highly reactive towards the alcohol substrate. The reaction produces mono-bromo-5,5-dimethyl hydantoin and triphenyl-phosphine oxide as by-products. A proposal for a reaction mechanism is outlined in Scheme 2.
 | ||
| Scheme 2 Proposed reaction mechanism for the new Appel reaction procedure, where X ∈ [Cl, Br, I]. | ||
Reaction improvement by statistical experimental design
The initial exploratory experiments afforded varied and unpredictable results, which immediately propelled us to conduct a screening and optimization study involving statistical experimental design,9 multiple regression,10 and response surface methodology.11 The model approximation and graphical projections in terms of iso-contour maps of the response surfaces were carried out by means of the computer software MATLAB.12In the experimental design study, the experimental variables: z1 quantity of DBH [equiv.], z2 reaction temperature [°C], z3 solvent volume [mL], and z4 reaction time [h] were explored by means of a full factorial design with centre experiments, D (24 + 3), as displayed in Table 1. The variables z1–z4 were explored at two experimental levels, while all other experimental variables were kept at a fixed level throughout the experimentation. The model matrix M was created according to eqn (1) by means of the design matrix D (Table 1) that was scaled13 according to eqn (2) to facilitate the modelling and subsequent model interpretation.
| M = [1x1x2x3x4x1x2x1x3x1x4x2x3x2x4x3x4x1x2x3x1x2x4x1x3x4x2x3x4x1x2x3x4] | (1) |
 | (2) |
| y = Mβ ⇒ β = (MTM)−1MTy | (3) |
|
|
|||||
|---|---|---|---|---|---|
| Experimental levels | |||||
| # | Experimental variables | −1 | 0 | +1 | |
| z 1 | Quantity of DBH [equiv.] | 0.50 | 0.75 | 1.00 | |
| z 2 | Reaction temperature [°C] | 20 | 30 | 40 | |
| z 3 | Solvent volume [mL] | 5.0 | 7.5 | 10.0 | |
| z 4 | Reaction time [h] | 1 | 3 | 5 | |

| #a | Experimental variables | Response | |||
|---|---|---|---|---|---|
| z 1 | z 2 | z 3 | z 4 | y | |
| a The design table is given in standard order. b The experiments #1 (31.7), 16 (94.9), and 17 (65.9) were performed first to assess whether the experimental levels were properly selected. The actual variation was (94.9–31.7) ≈ 63% points, which reveals a good spread in the experimental space. The remaining experiments were then conducted in random order. | |||||
| 1b | 0.50 | 20 | 5.0 | 1 | 31.7 |
| 2 | 1.00 | 20 | 5.0 | 1 | 86.9 |
| 3 | 0.50 | 40 | 5.0 | 1 | 56.3 |
| 4 | 1.00 | 40 | 5.0 | 1 | 87.1 |
| 5 | 0.50 | 20 | 10.0 | 1 | 40.5 |
| 6 | 1.00 | 20 | 10.0 | 1 | 86.9 |
| 7 | 0.50 | 40 | 10.0 | 1 | 47.2 |
| 8 | 1.00 | 40 | 10.0 | 1 | 91.4 |
| 9 | 0.50 | 20 | 5.0 | 5 | 44.4 |
| 10 | 1.00 | 20 | 5.0 | 5 | 94.0 |
| 11 | 0.50 | 40 | 5.0 | 5 | 49.2 |
| 12 | 1.00 | 40 | 5.0 | 5 | 90.3 |
| 13 | 0.50 | 20 | 10.0 | 5 | 49.9 |
| 14 | 1.00 | 20 | 10.0 | 5 | 92.8 |
| 15 | 0.50 | 40 | 10.0 | 5 | 45.4 |
| 16b | 1.00 | 40 | 10.0 | 5 | 94.9 |
| 17b | 0.75 | 30 | 7.5 | 3 | 65.9 |
| 18 | 0.75 | 30 | 7.5 | 3 | 61.1 |
| 19 | 0.75 | 30 | 7.5 | 3 | 68.6 |
| a Product statistics: R2 = 0.9942, RAdj2 = 0.9654, RMSEP = 1.6136, RSD = 1.7059. | |||||
|---|---|---|---|---|---|
| β 0 | 67.605 | β 12 | −1.781 | β 123 | 2.331 |
| β 1 | 22.481 | β 13 | 0.394 | β 124 | 1.544 |
| β 2 | 2.169 | β 14 | 0.406 | β 134 | −0.181 |
| β 3 | 0.569 | β 23 | −1.712 | β 234 | 0.631 |
| β 4 | 2.056 | β 24 | −2.331 | β 1234 | −0.444 |
| β 34 | 0.069 | ||||
 | ||
| Fig. 1 Left: Cumulative normal probability (CND) plot of estimated model parameters (the β's) of the predicting model for yield% = f(quantity of DBH, reaction temperature, solvent volume, reaction time). Right: Stem plot showing the estimated coefficient spectrum (except β0). | ||
Furthermore, the model reveals a rather small numerical value for the coefficient related to the experimental variable “reaction time” (z4), which implies that both short and long reaction time can be used without a substantial impact on the reaction outcome.
For example, if a reaction time of 1 h is used, the model term predicts a contribution of 2% in the yield, while if a reaction time of 5 h is used, a contribution of 10% yield can be anticipated, which gives a difference in the yield of only 8%.
The experimental variable “reaction time” also takes part in an interaction term (x2 × x4) with “reaction temperature”, which advises the use of a short reaction time and high reaction temperature. Hence, the knowledge gained from the model suggests a very short reaction time compared to the demanded reaction time for the classical Appel reaction.1
However, interpreting each individual model term is cumbersome, especially when the absolute numerical values of the model terms are numerically similar and small. A painless approach is to simulate an iso-contour map of the response surface as depicted in Fig. 2 (here with non-scaled variables z1–z4). For this, an empirical model that possessed only the significant (revealed by the CND plot of Fig. 1) model terms was used, eqn (4).
| y = f(x1, x2, x3, x4) = 67.605 + 22.48 × x1 + 2.17 × x2 + 2.06x4 − 1.78 × x1 × x2 − 2.33 × x2 × x4 + 2.33 × x1 × x2 × x3 | (4) |
| R2 = 0.9853, RAdj2 = 0.9779, RMSEP = 2.5783, RSD = 2.7257 |
 | ||
| Fig. 2 Iso-contour projections of the response surfaces that display the yield of compound 3a. The plots above display the variations of the response “yield of 3a” (the iso-contour lines) when the four experimental variables z1 (quantity of DBH [equiv.]), z2 (reaction temperature [°C]), z3 (fixed solvent volume of 10 mL), and z4 (reaction time [h]) are varied. How to read the multi-dimensional iso-contour projection plot: the variation (abscissa) is the five discrete levels of reaction time (z4). Along with the reaction time axis is given five subplots displaying the iso-contour projections of the response surface when the abscissa, which is the quantity of DBH (z1), and the ordinate, which is the reaction temperature (z2), were varied. | ||
| # | Experimental variablesa | Responseb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| x 1 | z 1 | x 2 | z 2 | x 3 | z 3 | x 4 | z 4 | ŷ pred | y | |
| a z 1 quantity of DBH [equiv.], z2 reaction temperature [°C], z3 solvent volume [mL], and z4 reaction time [h]. x1–x4 scaled values [eqn (4)] of z1–z4. b Yield%. | ||||||||||
| 20 | +1.72 | 1.18 | +1 | 40 | +1 | 10 | −1.25 | 0.5 | 109 | 90 |
| 21 | +1.72 | 1.18 | +1 | 40 | +1 | 10 | +1.00 | 5 | 109 | 98 |
| 22 | +1.00 | 1.00 | +1 | 40 | +3 | 15 | +1.00 | 5 | 97 | 92 |
The reaction was completed within t = 10–15 min. It is important to use an anhydrous solvent and an inert (dry) atmosphere. The presence of water might lead to a lowered reaction yield of the halogenated target molecule as the counter anion of the Appel salt, 3-bromo-5,5-dimethyl-2,4-dioxo-imidazolidin-1-ide, can react with water in a similar mode to R–OH, see Scheme 2.
Scope and limitation
|
|
||||
|---|---|---|---|---|
| # | Alcohol | Product | GC | Isolated |
| n.i.: not isolated, difficult to purify and isolate. n.d.: not detected. | ||||
| 1 |
|

|
91% | 78% |
| 2 |

|

|
Quant. | 73% |
| 3 |

|

|
68% | n.i. |
| 4 |
|

|
84% | 73% |
| 5 |

|

|
91% | n.i. |
| 6 |

|
|
90% | 69% |
| 7 |

|

|
Quant. | 75% |
| 8 |

|

|
73% | 63% |
| 9 |

|

|
Quant. | 84% |
| 10 |

|

|
91% | 65% |
| 11 |

|

|
n.d. | 20% |
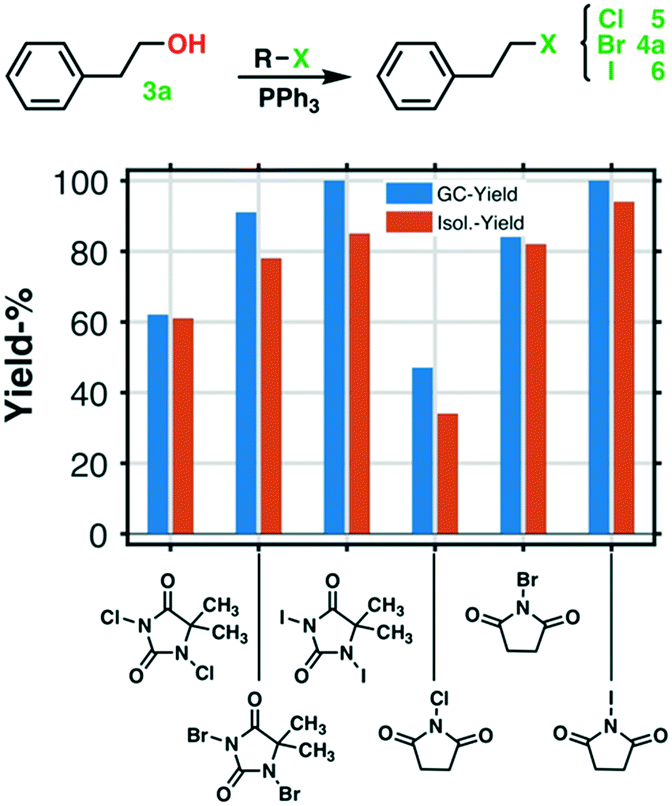 | ||
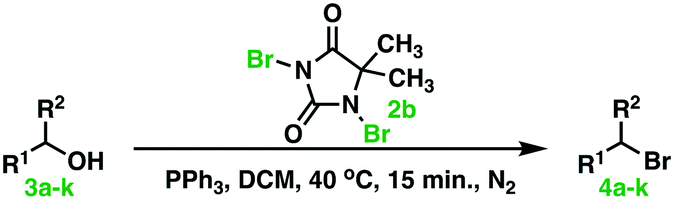
| Fig. 3 Various halogenation reagents used in the first step halogenation (of PPh3) in the novel Appel type reaction. | ||
Screening of solvents to replace the halogenated solvent DCM
In our attempt to transform the new Appel type reaction into an even more environmentally friendly version, we performed a solvent screening with the aim to replace DCM. DCM was the solvent originally used in the Appel reaction that we so far also used in our method development.The outcome of target molecules in two of the experiments in the solvent screening, diethyl ether and methyl tert-butyl ether, was modest only. However, the trials using THF, MeCN, 1,4-dioxane, and EtOAc resulted in high to excellent yields, namely in the range of 88–91%. All the said solvents can be used on a large scale, in particular ethyl acetate (afforded a yield of 89%) which is a cheap and green solvent that might be used on the industrial scale. It is also worth noting that none of the solvents used were subjected to extensive pre-treatment (only treatment with sodium sulphate used as obtained from the supplier) to remove water before being used in the reaction experiments.
Continuous flow synthesis
The excellent results achieved for the batch protocol of the new Appel type reaction prompted us to attempt to transform the batch method into a continuous flow reactor process. For this endeavour, a Vapourtec R-series system with a standard PFA coil (Vnet = 10 mL) tubular reactor was used. The set-up for the flow experiment is shown in Fig. 5.The reagent solubility became immediately a challenge in our attempt to transform the batch protocol into a continuous flow process. In fact, neither of the reagents PPh3 and DBH possessed good solubility in the potential and acceptable solvents that we tried as reaction media, Table 5.
| Entry | Solvent | Soluble | ||
|---|---|---|---|---|
| DBH | PPh3 | Appel salt | ||
| a Mixture A: DBH (1.26 g) was added to a flask with solvent (7.5 mL). Mixture B: PPh3 (2.46 g) was added to a flask with solvent (7.5 mL). Mixture A and B were combined (total 15 mL solvent) and mixed in a flask cooled on an ice-bath. DBH and PPh3 reacted and produced an Appel salt. | ||||
| 1 | DCM | No | Yes | Yes |
| 2 | THF | Yes | Yes | No |
| 3 | Acetonitrile | No | No | Yes |
| 4 | Ethyl acetate | No | Yes | No |
Unfortunately, due to the solubility problems, we therefore had to add an extra semi continuous step in the form of a batch reactor (B1) where the Appel salt could be produced in small portions and transferred to the reservoir tank R3.
The said batch process step was performed by filling B1 with DCM whereupon PPh3 was added and stirred until it was dissolved. The reaction with DBH was then performed by adding small portions of DBH to B1 over a period of 30 min. The produced Appel salt was afterwards transferred to reservoir R3 from which it was fed to the continuous flow reactor F1 by means of a pump (P3) concomitantly as a solution of the substrate (the alcohol 3a) in DCM (75 mL) placed in reservoir R2 was pumped using pump P2 to the flow reactor (F1) inlet. The pump rates were fine-tuned (Table 6) to deliver accurate mole equivalents. The right-hand column of Table 6 reveals the excellent outcomes (GC-yields) obtained from two different experiments.
| # | P2 rate | P3 rate | Tot. rate | Resid. t | Yield | |
|---|---|---|---|---|---|---|
| [mL × min−1] | [mL × min−1] | [mL × min−1] | [min] | [s] | [GC%] | |
| R2/P2: the substrate (alcohol 3a). R3/P3: the Appel salt. | ||||||
| 1 | 0.4 | 0.8 | 1.2 | 8 | 20 | 95% |
| 2 | 0.8 | 1.6 | 2.4 | 4 | 10 | 96% |
With enabling technology appropriate for feeding of slurries, a substantially improved and simplified flow platform could have been assembled. A reagent slurry would demand a much lower solvent volume and the batch step B1 could have been removed, overall, a safer process would have been the result. However, equipment for feeding of slurries is currently not commercially available.
Conclusions
This study demonstrates how an old classic named reaction, the Appel reaction, can be re-designed and optimized to fit into today's demands for green reaction solutions. Statistical experimental design with empirical modelling is a barely exploited methodology in explorative organic synthesis, but has become an every-day tool in organic process development. In the present study, however, such methodology was successfully used to develop and realize a novel Appel type reaction. In the first reaction step, the halogenation of triphenyl-phosphine was carried out using 1,3-dihalo-5,5-dimethyl-hydantoins or N-halo-succinimides (halo ∈ [Cl, Br, I]), as a substitute for tetrachloromethane that was used in the classical Appel reaction.The halogenation of PPh3via the novel Appel reaction was instant and exothermic. We believe that the Appel salt was formed with 3-bromo-5,5-dimethyl-2,4-dioxo-imidazolidin-1-ide as a counter anion to the bromotriphenyl-phosphonium cation. The high rate of this reaction allowed for a one-pot protocol where all the reagents are added in the beginning (not added sequentially). Such a set-up requires only 5–15 min to complete. The exchange of the halogenation reagent also resulted in a more environmentally friendly process as stoichiometric waste did not contain a halogen (in the case when N-halo-succinimide was used as a reagent) or one halogen (in the case when 1,3-dihalo-5,5-dimethyl hydantoin was used as a reagent). In contrast, the classic Appel reaction method produces stoichiometric amounts of a by-product that contains three halogen atoms (CHX3). The outcome of target halogenated molecules was high to excellent for the new method. Solvent screening for exchange of DCM that was the solvent used in the classical Appel reaction revealed several less toxic and non-halogen containing solvents that afforded the target product of the test reaction in yield >90%.
Experimental
Post-reaction mixtures were monitored by means of TLC (Merck Kieselgel 60 F254), GC-MS and/or HPLC. The TLC plates were observed under UV-light at l = 254 nm and l = 366 nm. Purification with Autoflash was conducted on a PuriFlash XS420 with Interchim soft V5. 1c.09 as software using a silica column (FSUD-0043-0050) from Biotage. GC analyses were performed with a capillary gas chromatograph equipped with a fused silica column (L 25 m, i.d. 0.20 mm, film thickness of 0.33 mm) at a helium pressure of 200 kPa, a split less/split injector and a flame ionization detector. Mass spectra were obtained with a GC-MS instrument, with a gas chromatograph equipped with a fused silica column (L 30 m, i.d. 0.25 mm i.d., film thickness of 0.25 mm) and helium as the carrier gas. HPLC analyses were performed with an Agilent Technologies 1260 HPLC with a multidiode array detector, an autoinjector and a 4.6 × 100 mm, 4 μm Agilent Technologies Poroshell 120 EC-C18 column. Solvents used were (MilliQ purified water–HCOOH 99.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1; v/v) and (acetonitrile–HCOOH 99.9:0.1; v/v). The ratio was determined by running 25–100% over 15 minutes with a flow rate of 1 mL min−1 and UV detection at 210 nm. 1H and 13C NMR spectra were recorded with a Bruker AV 500 MHz. 1H and 13C-NMR spectra were recorded at ambient temperature at a frequency of 500 and 125 MHz, respectively. The chemical shifts are reported in ppm relative to residual CHCl3 for proton (d = 7.26 ppm) and CDCl3 for carbon (d = 77.16 ppm) with tetramethylsilane as an external reference.
:0.1; v/v) and (acetonitrile–HCOOH 99.9:0.1; v/v). The ratio was determined by running 25–100% over 15 minutes with a flow rate of 1 mL min−1 and UV detection at 210 nm. 1H and 13C NMR spectra were recorded with a Bruker AV 500 MHz. 1H and 13C-NMR spectra were recorded at ambient temperature at a frequency of 500 and 125 MHz, respectively. The chemical shifts are reported in ppm relative to residual CHCl3 for proton (d = 7.26 ppm) and CDCl3 for carbon (d = 77.16 ppm) with tetramethylsilane as an external reference.
Procedure leading to (2-bromoethyl)benzene (4a) [103-63-9] explored as the model reaction with statistical experimental design, Table 1
Triphenylphosphine (0.656–1.377 g, 2.50–5.25 mmol, 1.0–2.1 equiv.) and 1,3-dibromo-5,5-dimethyl-imidazolidine-2,4-dione (DBH) (0.357–0.715 g, 1.25–2.50 mmol, 0.5–1.0 equiv.) were transferred to a pre-dried μW-reactor tube (10–20 mL) furnished with a magnetic stirrer bar. The reactor tube was sealed, flushed with N2, and immersed in an ice bath. Anhydrous dichloromethane (5–10 mL) was carefully added by means of a syringe. The ice bath was then removed. The mixture was continuously stirred for 30 min at 20–40 °C. The substrate 2-phenylethan-1-ol (0.300 mL, 2.50 mmol) was then, under stirring, added by means of a syringe to the mixture of the Appel salt and stirred for another 0.5–6.0 h at 20–40 °C. 1H NMR (500 MHz, CDCl3) δ 7.27–7.23 (m, 2H), 7.21–7.17 (m, 1H), 7.16–7.12 (m, 2H), 3.50 (t, J = 7.7 Hz, 2H), 3.09 (t, J = 7.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 138.92, 128.68, 128.64, 126.95, 39.45, 32.94.Scope: varying the alcohol substrate, see Table 4
:1 → 8:2) to provide the title compound as a transparent oil (43 mg, 20%). 1H-NMR (500 MHz, CDCl3): δ (ppm) = 7.54–7.49 (m, 3H), 7.30–7.27 (m, 2H), 7.04 (s, 1H), 4.81 (p, J = 1.4 Hz, 1H), 4.52 (t, J = 1.0 Hz, 1H), 1.86 (dd, J = 1.4, 0.8 Hz, 3H); 13C-NMR (126 MHz, CDCl3): δ (ppm) = 136.5, 135.8, 134.4, 132.0, 129.7, 129.5, 128.0, 127.2, 114.3, 22.1.
NCS, NBS, and NIS in the first halogenation step, see Fig. 3
Procedure for the solvent screening, Fig. 4
Triphenylphosphine (1.640 g, 6.25 mmol, 2.5 equiv.) and DBH (839 mg, 2.93 mmol, 1.17 equiv.) were transferred to a pre-dried μW-reactor tube (10–20 mL) that was sealed, flushed with N2 and equipped with a balloon filled with N2. The reactor tube was then cooled on an ice-bath whereupon anhydrous solvent (7 mL) was added. 2-Phenyletanol (0.3 mL, 2.50 mmol, 1 equiv.) in anhydrous solvent (3 mL) was added in one portion and then stirred at 40 °C for 15 min. Samples for GC-MS analyses were withdrawn after 15 min reaction time. | ||
| Fig. 4 Screening of a series of non-halogenated solvents EtOEt (diethyl ether), THF (tetrahydrofuran), MeCN (acetonitrile), 1,4-dioxane, EtOAc (ethyl acetate), and MTBE (methyl tert-butyl ether), which were compared with DCM (dichloromethane) that was used as a reaction medium through the method developed herein, but also for the classical Appel reaction. | ||
 | ||
| Fig. 5 Process flow diagram for the continuous flow process version of the new Appel type reaction that was implemented on a Vapourtec R-series continuous flow system using a standard PFA coil (Vnet = 10 mL) flow reactor (F1) that was heated at a temperature of 40 °C. Reservoir R2 was charged with the alcohol (4.5 mL, d = 1.017 g cm−1, m = 4.58 g) that was diluted in DCM (75 mL). Reservoir R3 was charged with a solution of modified Appel salt in DCM (150 mL), which was produced in the batch reactor. The production of the Appel salt was conducted in batch reactor B1 that was charged with DCM (150 mL) whereupon triphenylphosphine was added in portions (in total 24.6 g, 37.5 mmol) and DBH (in total 12.6 g, 44 mmol). The post-reaction mixture (120 mL) from the outlet of the flow reactor F1 was collected in reservoir R4. | ||
Author contributions
Hans-René Bjørsvik: conceptualization, methodology, design of the experiment, flow chemistry, computations, graphics, editing, and writing original draft. Equal contribution by Nicolas Mattias Del Rio Fuenzalida, Eirin Alme, and Frida Johanne Lundevall: experimental synthetic work, flow chemistry, chemical analyses, design of the experiment, writing, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. A. and F. J. L. are grateful to the Department of Chemistry at University of Bergen, for funding their research fellowships.Notes and references
- R. Appel, Angew. Chem., Int. Ed. Engl., 1975, 14, 801–811 CrossRef.
- Y. Nishida, Y. Shingu, H. Dohi and K. Kobayashi, Org. Lett., 2003, 5, 2377–2380 CrossRef CAS PubMed.
- H. Vorbrüggen and K. Krolikiewicz, Tetrahedron, 1993, 49, 9353–9372 CrossRef.
- (a) R. M. Denton, J. An and B. Adeniran, Chem. Commun., 2010, 46, 3025–3027 RSC; (b) H. A. van Kalkeren, S. H. A. M. Leenders, C. R. A. Hommersom, F. P. J. T. Rutjes and F. L. van Delft, Chem. – Eur. J., 2011, 17, 11290–11295 CrossRef CAS PubMed; (c) R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis and A. M. Poulton, J. Org. Chem., 2011, 76, 6749–6767 CrossRef CAS PubMed; (d) L. Longwitz, S. Jopp and T. Werner, J. Org. Chem., 2019, 84, 7863–7870 CrossRef CAS PubMed; (e) I. Pongener, K. Nikitin and E. M. McGarrigle, Org. Biomol. Chem., 2019, 17, 7531–7535 RSC; (f) A. Jordan, R. M. Denton and H. F. Sneddon, ACS Sustainable Chem. Eng., 2020, 8, 2300–2309 CrossRef CAS.
- (a) K. A. Roper, H. Lange, A. Polyzos, M. B. Berry, I. R. Baxendale and S. V. Ley, Beilstein J. Org. Chem., 2011, 7, 1648–1655 CrossRef CAS PubMed; (b) R. Morodo, P. Bianchi and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2020, 5236–5277 CrossRef CAS.
- (a) A. Drageset and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 436–444 RSC; (b) A. Drageset and H.-R. Bjørsvik, Eur. J. Org. Chem., 2018, 4436–4445 CrossRef CAS; (c) A. Drageset, N. Å. Frøystein, K. W. Törnroos and H.-R. Bjørsvik, React. Chem. Eng., 2019, 4, 41–51 RSC.
- Commercial prices retrieved from https://www.sigmaaldrich.com/NO/en/ (July 8, 2021): 1,3-Dichloro-5,5-dimethylhydantoin (DCH “available chlorine 68%”) - NOK 278 (€ 27)/250G, 1,3-dibromo-5,5-dimethylhydantoin (DBH “for synthesis”) - NOK 395 (€ 39)/250G, and 1,3-Diiodo-5,5-dimethylhydantoin(DIH ≥96%) - NOK 2790 (€ 279)/25G.
- M. Ferreri, A. Drageset, C. Gambarotti and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 379–386 RSC.
- (a) G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for Experimenters: An Introduction to Design, Data Analysis, and Model Building, Wiley, New York, 1978 Search PubMed; (b) D. C. Montgomery, Design and Analysis of Experiments, Wiley, New York, 3rd edn, 1991 Search PubMed.
- (a) N. R. Draper and H. Smith, Applied Regression Analysis, Wiley, New York, 3rd edn, 1998 CrossRef; (b) D. C. Montgomery and E. A. Peck, Introduction to linear regression analysis, Wiley, New York, 1982 Search PubMed.
- (a) G. E. P. Box and N. R. Draper, Empirical Model-Building and Response Surfaces, Wiley, New York, 1987 Search PubMed; (b) R. H. Myers and D. C. Montgomery, Response Surface Methodology, Process and Product Optimization Using Designed Experiments, Wiley, New York, 1995 Search PubMed.
- (a) The MATLAB program was utilized for the graphical representation of the results and models, see Using MATLAB Version 6 (Nov. 2000), The MathWorks, Inc., Natick, MA, USA; (b) Using MATLAB Graphics Version 6 (Nov. 2000), The MathWorks, Inc., Natick, MA, USA.
- Scaling of the experimental variables z1–z4 was performed according to eqn (2). The scaled low value is set at a value of −1, and the scaled high value was set at a value of +1.
- (a) C. Daniel, Technometrics, 1959, 1, 311–341 CrossRef; (b) C. Daniel, Application of Statistics to Industrial Experimentation, Wiley, New York, 1976 CrossRef; (c) G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for experimenters. Design, Innovation, and Discovery, Wiley, New York, 2nd edn, 2005, pp. 33–34 Search PubMed; (d) G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for experimenters. Design, Innovation, and Discovery, Wiley, New York, 2nd edn, 2005, pp. 203–205 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, and MS spectra. See DOI: https://doi.org/10.1039/d2re00071g |
| This journal is © The Royal Society of Chemistry 2022 |