
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous synthesis of Cu/ZnO/Al2O3 nanoparticles in a co-precipitation reaction using a silicon based microfluidic reactor†
Ghazal
Tofighi
 a,
Henning
Lichtenberg
ab,
Abhijeet
Gaur
ab,
Wu
Wang
c,
Stefan
Wild
b,
Karla
Herrera Delgado
b,
Stephan
Pitter
b,
Roland
Dittmeyer
d,
Jan-Dierk
Grunwaldt
ab and
Dmitry E.
Doronkin
*ab
a,
Henning
Lichtenberg
ab,
Abhijeet
Gaur
ab,
Wu
Wang
c,
Stefan
Wild
b,
Karla
Herrera Delgado
b,
Stephan
Pitter
b,
Roland
Dittmeyer
d,
Jan-Dierk
Grunwaldt
ab and
Dmitry E.
Doronkin
*ab
aInstitute for Chemical Technology and Polymer Chemistry (ITCP), Karlsruhe Institute of Technology (KIT), D-76131 Karlsruhe, Germany. E-mail: dmitry.doronkin@kit.edu; Fax: +49 721 608 44820; Tel: +49 721 608 48090
bInstitute of Catalysis Research and Technology (IKFT), Karlsruhe Institute of Technology (KIT), D-76344 Eggenstein-Leopoldshafen, Germany
cInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), D-76344 Eggenstein-Leopoldshafen, Germany
dInstitute for Micro Process Engineering (IMVT), Karlsruhe Institute of Technology (KIT), D-76344 Eggenstein-Leopoldshafen, Germany
First published on 17th December 2021
Abstract
CuO/ZnO/Al2O3 catalysts were continuously synthesized in a microfluidic reactor, analyzed by X-ray diffraction (XRD), physisorption (BET), chemisorption, electron microscopy and X-ray absorption spectroscopy (XAS), and tested for methanol synthesis from CO-rich synthesis gas. The results were compared to those obtained from CuO/ZnO and CuO/ZnO/Al2O3 produced by conventional co-precipitation in a batch reactor. The predominant phase of the aged precursor from microfluidic co-precipitation was identified as zincian malachite. After calcination the microfluidically synthesized catalyst exhibited smaller CuO crystallites, a larger BET surface area, a rather uniform morphology and a homogeneous distribution of Cu and Zn compared to catalysts prepared by batch co-precipitation. H2-Temperature programmed reduction (TPR) showed that Cu species in CuO/ZnO/Al2O3 from microfluidic co-precipitation were more easily reducible. In situ Cu and Zn K-edge XAS during the TPR indicated reduction of Cu2+ to Cu0 between 150 °C and 240 °C, without detectable reduction of Zn. N2O pulse chemisorption evidenced an enlarged Cu surface area of the nanoparticles from the microfluidic synthesis. Based on activity tests in methanol synthesis, at 250 °C the microfluidically synthesized Cu/ZnO/Al2O3 catalysts showed better performance than the catalyst from batch preparation when 1 mol% CO2 was present in the synthesis gas. Dimethyl ether formed as a side product. As the microreactor is specially designed for high X-ray transmission with a thin Si/glass observation window, this study opens interesting perspectives for investigating the formation of catalyst precursors at the early stage of precipitation in future.
Introduction
Methanol (MeOH) is not only an essential industrial bulk chemical but can also be used as a fuel additive or precursor for clean fuels.1–4 Due to its high energy density methanol plays an important role in chemical energy and hydrogen storage including fuel-cell applications.5–8 The most prominent catalytic system for industrial methanol synthesis since decades ago is based on Cu/ZnO/(Al2O3) and enormous efforts have been dedicated to the enhancement of its catalytic activity based on rational design via different preparation methods.3,5,9–19 One widely applied method for the synthesis of Cu/ZnO/(Al2O3) is co-precipitation of soluble copper, zinc and aluminum precursors using e.g. sodium carbonate.9,20–22Specific synthesis parameters such as pH, temperature, ageing time and mixing conditions play key roles in the resulting catalytic performance.5,9,21,23 Fundamental studies in this field e.g. by Behrens et al.20 show that co-precipitation temperature between 60 °C and 70 °C and pH values between 6–7 as well as the ageing time and temperature are crucial to achieve optimum material properties. Sufficient ageing time is required for meso-structuring of the precipitate.9,24 These parameters directly influence the formation of the – in this context important – zincian malachite (CuxZny)2(OH)2CO3 phase. Calcination, typically in the range of 320 °C to 430 °C, promotes further nano-structuring during the phase transition to the mixed metal oxide.20,25 Most preparation methods focus on batch procedures. However, for larger quantities and industrial applications a semi-batch or continuous process would be advantageous.26–29
Catalytic performance of Cu/ZnO/(Al2O3) is particularly influenced by the surface area of copper (SACu) and the area of reactive interface with ZnO in a nanoparticulate and porous structure.30 Metallic Cu clusters/particles are considered to be the most active sites for methanol synthesis, strongly improved by ZnO,2,25 and Al2O3 as a structural promoter25,29 reducing NP sintering. Fast and homogeneous mixing of the soluble metal precursors and the precipitating agent are essential for the most uniform precipitate.27 This is difficult to obtain in conventional stirred batch reactors where initial precipitation may already start before the reactants have been efficiently mixed, resulting in spatial and temporal material gradients. Every single added drop of reactants induces concentration changes during the entire process time. Redissolution, reprecipitation and ion exchange during ageing, washing and drying of the catalyst precursor may additionally influence the catalytic properties.20,25,29 In contrast, during co-precipitation in a continuous microflow29 the precipitates are formed in a small volume.27,31 This results in defined and stable preparation conditions, once they are established. In the last decade, micromixing techniques have been increasingly applied for precipitation reactions.26,27,29,32 Advanced micromixing devices such as microfluidic reactors, T-mixers and confined impinging jet reactors are designed for fast and homogeneous mixing based on high mass transfer and short residence time.27–29
In earlier studies, we used a novel microfluidic reactor especially designed for X-ray spectroscopic in situ studies to investigate colloidal noble metal NP formation during fast reduction reactions, specifically focusing on the critical early stages of the process.33–36 The experiments described here aimed at extending the application range of this type of microfluidic device towards co-precipitation reactions, focusing on Cu/ZnO/Al2O3 catalysts for methanol synthesis as a case study. For this purpose, two streams of the reactants were injected separately into the micromixers integrated in the microfluidic chip. For comparison, Cu/ZnO and Cu/ZnO/Al2O3 catalysts from conventional batch synthesis under conditions adopted from the literature were also characterized and tested in order to illustrate potential advantages of microfluidic mixing during co-precipitation.
Materials and methods
Materials
Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, Sigma-Aldrich, >99% purity), zinc nitrate hexahydrate (Zn(NO3)·6H2O, Alfa Aesar, 99.99% purity), zinc oxide (ZnO, Fluka, >99% purity), aluminum nitrate nonahydrate (Al(NO3)3·9H2O, Sigma-Aldrich, >98% purity), sodium carbonate (Na2CO3, Sigma-Aldrich, >99.95% purity) and nitric acid (HNO3, Merck, 65% solution) were used without further purification.Batch synthesis of Cu/ZnO and Cu/ZnO/Al2O3
Throughout the manuscript we denote all catalysts “Cu/ZnO/(Al2O3)-BR/MF”, irrespective of the copper phases present, where BR stands for batch reactor and MF for microfluidic synthesis.Nitrate-derived Cu/ZnO and Cu/ZnO/Al2O3 reference catalysts were synthesized via co-precipitation in a batch reactor. For this purpose, a procedure adopted from Behrens et al.20 was applied. 21.6 mmol Cu(NO3)2·3H2O, 9.6 mmol ZnO and 4.4 mmol Al(NO3)3 were dissolved in 38 mL distilled water with 2.5 mL of concentrated nitric acid. 23 mL of 1.6 M aqueous solution of Na2CO3 was used as a precipitating agent. The two solutions were injected dropwise into the batch reactor using individual syringe pumps (neMESYS, Cetoni GmbH, with 50 mL glass syringes). The flow rates for injecting metal precursor and precipitating agent solutions were 1 and 0.56 mL min−1, respectively. The batch reactor used was a round-bottom flask containing 100 mL distilled water stirred at ∼65 °C. The pH of the precipitate was kept constant at 6–7. Subsequently, the precipitate was aged for 3 h, filtered and washed several times. This process was followed by drying overnight at 70 °C and calcination at 330 °C for 3 h. This procedure resulted in 2 g of catalyst. For synthesizing Cu/ZnO the same procedure was applied, cf.Table 1.
| Catalyst | Synthesis technique | Metal precursor (mmol) | Na2CO3 (mmol) | ||
|---|---|---|---|---|---|
| Cu2+ | Zn2+ | Al3+ | |||
| The amount of distilled water for dissolving the metal precursors and precipitating agent for batch reactor synthesis was 38 and 23 mL, and for microfluidic synthesis 40 and 40 mL, respectively. | |||||
| Cu/ZnO | Batch reactor | 21.6 | 9.6 | — | 36.8 |
| Cu/ZnO/Al2O3 | Batch reactor | 21.6 | 9.6 | 4.4 | 36.8 |
| Cu/ZnO/Al2O3 | Microfluidic reactor | 7.2 | 3.6 | 1.2 | 14.4 |
Microfluidic synthesis of Cu/ZnO/Al2O3
A nitrate-derived ternary Cu/ZnO/Al2O3 catalyst was prepared by co-precipitation of aqueous metal nitrate solution with Na2CO3 in a microfluidic chip (Fig. 1) fabricated in collaboration with GeSiM GmbH and the Institute of Semiconductors and Microsystems at Technische Universität Dresden (IHM TUD). It is made of Si-bonded glass and was specifically designed for in situ characterization using X-ray spectroscopic and scattering techniques.36 A metal nitrate solution with a molar Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:Al ratio of 60:30:10 was prepared by dissolving the precursors in 40 ml distilled water (total metal ion concentration 0.3 M). 1 ml concentrated nitric acid was added to the solution resulting in pH 1. The Na2CO3 concentration was 0.36 M. To avoid heavy deposition of precipitates in the channel and clogging, the precursors were diluted. However, the metal ratio was kept at the same level as for the batch synthesis. Each of the two reactant solutions (40 mL) was loaded in a 50 mL glass syringe and injected into the microfluidic chip at a flow rate of 22 mL min−1 (neMESYS syringe pump, Cetoni GmbH), and the precipitate was transferred into a flask containing 100 mL distilled water under stirring at 65 °C. After ∼15 min a color change from blue to turquoise was observed. The pH of the precipitate was constant at 6–7, and the same ageing, washing, drying and calcination conditions as in the previous section were applied. This procedure provided 0.8 g catalyst.
:Zn:Al ratio of 60:30:10 was prepared by dissolving the precursors in 40 ml distilled water (total metal ion concentration 0.3 M). 1 ml concentrated nitric acid was added to the solution resulting in pH 1. The Na2CO3 concentration was 0.36 M. To avoid heavy deposition of precipitates in the channel and clogging, the precursors were diluted. However, the metal ratio was kept at the same level as for the batch synthesis. Each of the two reactant solutions (40 mL) was loaded in a 50 mL glass syringe and injected into the microfluidic chip at a flow rate of 22 mL min−1 (neMESYS syringe pump, Cetoni GmbH), and the precipitate was transferred into a flask containing 100 mL distilled water under stirring at 65 °C. After ∼15 min a color change from blue to turquoise was observed. The pH of the precipitate was constant at 6–7, and the same ageing, washing, drying and calcination conditions as in the previous section were applied. This procedure provided 0.8 g catalyst.
 | ||
| Fig. 1 (a) Microfluidic synthesis of Cu/ZnO/Al2O3via co-precipitation at constant pH and a total flow rate of 44 mL min−1, (b–e) color change during precipitation from 10 to 81 min. | ||
Catalytic testing: methanol synthesis
MeOH synthesis was performed using the CuO/ZnO and CuO/ZnO/Al2O3 pre-catalysts. The pre-catalysts were pressed and sieved into fractions of 250–500 μm particle size before use. To minimize hot spot formation and potential sintering, the pre-catalyst was mixed with SiC (Mineraliengrosshandel Hausen GmbH, Telfs, Austria) in a mass ratio of 1:5. The reaction was performed in a continuous-flow, stainless-steel fixed bed reactor (460 mm length, 12 mm inner diameter) which was filled with 5 g of the pre-catalyst/SiC mixture. The temperature was measured at the outside of the reactor wall, details about the equipment are reported elsewhere.37 Prior to the experiments, pre-catalysts were heated to 100 °C in Ar for 1 h. Catalyst reduction was performed by heating to 200 °C in 5% H2 in Ar with a heating rate of 0.33 °C min−1, followed first by increasing the H2 content to 50% and then the temperature to 240 °C with a heating rate of 0.2 °C min−1. These conditions were kept constant for 5 h. Every catalyst was tested at 50 bar, 230 °C and 250 °C, with two feed gas compositions (H2/N2/CO/CO2/Ar: 34:15:15:0:36 and 34:15:14:1:36, respectively) and gas hourly space velocity (GHSV) of 24000 mL h−1 g−1 to ensure conversion in the kinetic region. Further details on feed compositions and data recording during time on stream (ToS) are provided in Table S1.† Reaction products were analyzed using gas chromatography on an Agilent G1530A equipped with RT®-Msieve 5A and RT®-U-BOND columns, a thermal conductivity detector (TCD) and a flame ionization detector (FID). The performance indicators productivity and selectivity were calculated using the equations provided in the ESI.†
Characterization methods
Afterwards, the sample was cooled down to 50 °C and temperature-programmed reduction (TPR) at ambient pressure in a flow of 5% H2 in Ar (30 mL min−1) through the sample was started. The temperature was increased at a rate of 1 °C min−1 up to 250 °C and kept at 250 °C for 30 min. The effluent gas passed through a cold trap at 0 °C to condense any water formed. The dry gas was then analyzed by the TCD and the MS in series. The H2 signal of the MS (m/z = 2) was calibrated with 5% H2 in Ar before and after each measurement and used for quantification of the overall H2 consumption during TPR.
Subsequently, N2O pulse chemisorption was performed. The sample was cooled to room temperature and flushed with 30 mL min−1 He for 30 min. Afterwards, 40 pulses with 0.32 mL of 30% N2O in He were dosed onto the sample. The effluent gas from the sample passed through a cooling trap at −196 °C to separate any N2O not converted to N2. N2 formed by partial Cu oxidation was then analyzed with the TCD and the MS in series. The TCD data were used to determine the metallic Cu surface area and Cu dispersion (Cu atomic area 0.068 nm2, stoichiometry factor 2.0).
EXAFS data analysis was performed using the software packages Athena and Artemis.39Athena was first used to process the raw data (absorption coefficient μ(E) vs. E) by background subtraction, normalization, converting μ(E) data to χ(k) data and Fourier transforming the resulting spectrum from k-space to R-space. For the EXAFS background extraction, the input parameter Rbkg, was set to 1.0 Å. EXAFS fitting was performed using Artemis by generating theoretical models from available crystallographic data of reference compounds, i.e. CuO and Cu. The models were fitted to the experimental data in k-space and R-space to determine energy shifts (ΔE0) and structural parameters, including changes in the scattering path length (ΔR), passive electron reduction factor (S02), coordination number (N) and relative mean-square displacement of the atoms (Debye–Waller factor, σ2).
In situ XANES spectra were analyzed by linear combination fitting (LCF) using the spectra measured at 20 °C and 260 °C as internal references.
Results and discussion
Comparison of Cu/ZnO/(Al2O3) produced in the microfluidic and batch reactor
The ternary Cu, Zn, (Al) hydroxycarbonate precursor materials were co-precipitated at constant pH in the magnetically stirred-batch reactor and the microfluidic reactor and then, after ageing, washing and drying, analyzed by XRD. In both cases the acquired XRD patterns clearly show reflections of zincian malachite [(Cu1−xZnx)2(OH)2CO3] with x < 0.3 (Fig. 2a). The XRD data of the precipitates from the batch reactor show additional reflections attributed to gerhardtite [Cu2(OH)3NO3],20 whereas the diffraction patterns of the precipitates from the microfluidic reactor indicate presence of rosasite phase with poor crystallinity.29 The formation of different phases is attributed to the efficient micromixing effect (homogeneous mixing in short time) and the spatially controlled nucleation of the primary precipitates in small volumes flowing in the microchannel. | ||
| Fig. 2 XRD patterns of the (a) uncalcined ternary CuZnAl precipitates prepared by different methods indicating formation of malachite/rosasite and malachite/gerhardtite phases in precipitates synthesized in (I) microfluidic and (II) batch reactor, respectively; (b) calcined (III) Cu/ZnO/Al2O3-MF, (IV) Cu/ZnO/Al2O3-BR and (V) Cu/ZnO-BR. | ||
According to literature,20,25,29 the formation of different crystalline phases during co-precipitation is a dynamic process, during or at the end of which other processes such as partial dissolution and reprecipitation can occur. It was reported earlier that such exchange reactions between malachite [Cu2(OH)2CO3] and hydrozincite [Zn5(OH)6(CO3)2] lead to rosasite [(Cu, Zn)2(OH)2CO3] and aurichalcite [(Cu, Zn)5(OH)6(CO3)2].21,25,40,41 During the precipitation process the following main reactions occur:
| 2Cu2+ + CO32− + 2OH− → Cu2(CO3)(OH)2 | (1) |
| 5Zn2+ + 2CO32− + 6OH− → Zn5(CO3)2(OH)6 | (2) |
| 2Cu2+ + NO3− + 3OH− → Cu2(NO3)(OH)3 | (3) |
| Cu2(NO3)(OH)3 + CO32− → Cu2(CO3)(OH)2 + NO3− + OH− | (4) |
| Cu2(CO3)(OH)2 + xZn2+ → (Cu2−x,Znx)(CO3)(OH)2 + xCu2+ | (5) |
| Zn5(CO3)2(OH)6 + xCu2+ → (Zn5−x,Cux)(CO3)2(OH)6 + xZn2+ | (6) |
The pre-catalysts, including the Cu/ZnO-BR, calcined at 330 °C were also investigated by XRD. Reflections of Al2O3 were not observed for the Al containing materials due to its amorphous phase structure.24,42 The results in Fig. 2b show the dominant CuO and ZnO phases in all three samples. In the case of Cu/ZnO/Al2O3-MF catalyst, the reflections are broader and less intense. The CuO reflection at 2θ = 38.8° in the XRD patterns of Cu/ZnO/Al2O3-MF was weaker compared to the Cu/ZnO/Al2O3-BR indicating smaller CuO crystallites. In the literature, it was repeatedly reported that small particle sizes and well-distributed copper clusters are important for the catalytic performance (activity and selectivity) in methanol synthesis via hydrogenation of CO/CO2.25,29 Thus, better catalytic performance is expected from Cu/ZnO/Al2O3-MF.
Table 2 summarizes the main textural characteristics of the Cu/ZnO/(Al2O3) samples prepared by different methods. Based on the ICP-OES results, the molar Cu:Zn:Al ratios in Cu/ZnO/Al2O3 catalysts produced in the microfluidic and batch reactors are similar (77:6:17 and 76:8:16, respectively). This enables sufficient comparability of their catalytic performance. In the case of Cu/ZnO-BR catalyst a molar Cu:Zn ratio of 77:23 was obtained.
| Catalyst | Synthesis technique | ICP-OES wt% | BET m2 g−1 | N2O chemisorption | ||||
|---|---|---|---|---|---|---|---|---|
| Cu | Zn | Al | Metal dispersion% | SACua m2 g−1 | Active particle diameter nm | |||
| a Per gram metal. | ||||||||
| Cu/ZnO | Batch reactor | 58.9 | 18.6 | — | 29 | 1.6 | 10.3 | 65 |
| Cu/ZnO/Al2O3 | Batch reactor | 59.0 | 6.5 | 5.2 | 36 | 1.5 | 9.5 | 71 |
| Cu/ZnO/Al2O3 | Microreactor | 59.2 | 4.5 | 5.2 | 49 | 2.6 | 17.0 | 39.5 |
BET surface areas of Cu/ZnO/Al2O3 synthesized in the batch reactor and the microreactor were 36 m2 g−1 and 49 m2 g−1, respectively. Cu/ZnO-BR had the lowest BET surface area (29 m2 g−1), which could be due to the absence of alumina.
H2-TPR was applied to study the reduction of copper entities in the three calcined catalysts (Fig. 3). The Cu/ZnO/Al2O3-BR reduction peak is slightly broader with its maximum at higher temperature compared to the Cu/ZnO/Al2O3-MF TPR profile. This indicates a narrower size distribution29 of CuO particles synthesized in the microreactor. Moreover, in the TPR data of the batch synthesized Cu/ZnO-BR and Cu/ZnO/Al2O3-BR a pronounced shoulder at 136 °C and 164 °C, respectively, suggests various CuO entities. N2O pulse chemisorption (Table 2) shows a smaller Cu particle diameter, higher metallic surface area and higher metal dispersion for the Cu/ZnO/Al2O3-MF, which is beneficial for improved catalytic activity.
 | ||
| Fig. 3 TPR profiles of the calcined catalysts prepared in the batch and microfluidic reactor. | ||
Finally, the samples were studied by transmission electron microscopy combined with EDX mapping. Fig. 4 shows TEM images of calcined Cu/ZnO produced in the batch reactor. STEM measurements combined with EDX mapping (Fig. 5a, S1 and S4a and Table S2†) on selected nanoparticles indicate variations in the Cu:Zn ratio and a non-uniform distribution of CuO and ZnO in the sample. The morphology and the EDX mapping of different regions of Cu/ZnO/Al2O3 nanoparticles produced in the batch reactor are shown in Fig. 5b, S2 and S4b and Table S3.† They reveal uniform distributions of O, Al, Cu and Zn as well as relatively uniform Cu:Zn ratio in the nanoparticles of this catalyst. This more uniform distribution compared to the Cu/ZnO-BR sample is attributed to the presence of alumina which prevents random sintering of nanoparticles.25 Interestingly, metal distribution in the calcined Cu/ZnO/Al2O3-MF is somewhat more uniform (lower variance in Cu:Zn ratios at different positions) and the morphology looks more finely structured compared to nanoparticles produced in the batch reactor (Fig. 5c, S3 and S4c and Table S4†).
 | ||
| Fig. 4 TEM images of calcined Cu/ZnO nanoparticles produced in the batch reactor. | ||
 | ||
| Fig. 5 STEM images and the corresponding elemental maps obtained from STEM-EDX spectrum imaging in the areas marked by the orange box of calcined (a) Cu/ZnO, (b) Cu/ZnO/Al2O3 nanoparticles produced in the batch reactor and (c) Cu/ZnO/Al2O3 nanoparticles produced in the microfluidic reactor. Scale bars for the elemental maps are 10 nm (parts a and b) and 5 nm (part c). | ||
X-ray absorption spectroscopy on Cu/ZnO/Al2O3 nanoparticles
Fig. 6a shows normalized XANES spectra at Cu K-edge at room temperature (RT) for the two Cu/ZnO/Al2O3 catalysts produced in microfluidic and batch reactors and Fig. 6b shows their corresponding Fourier transforms (FT). A very weak pre-edge feature at 8976 eV is present indicating Cu in +2 oxidation state. Another feature is the shoulder S attributed to square planar geometry43 that has been found to be weaker in the spectrum of the microfluidically synthesized sample (MF) indicating higher geometrical distortion. The EXAFS analysis (Table 3) shows that Cu is coordinated to 2 O atoms with a Cu–O bond distance of 1.95 Å, similar to the typical Cu–O bond distance of 1.95 Å in bulk CuO. We note that XAS spectra measured in transmission geometry show considerable damping effect probably attributable to higher harmonics in the beam in combination with strongly absorbing samples. This results in decreased coordination numbers (the true CN of oxygen in the first coordination shell should be 4 for square planar geometry typical for Cu(II)) but does not influence positions of features in EXAFS and XANES. Hence, the EXAFS-derived CN values can serve only as a trend for each specific catalyst rather than to identify particle sizes. The second shell consisting of Cu/Zn atoms at 2.94 Å slightly differs from the typical Cu–Cu bond distances of 2.90 or 3.083 Å in tenorite. Thus, the sample prepared from microfluidic synthesis probably has almost the same Cu structure as the catalyst obtained from the batch reactor. Fig. 6c shows normalized XANES spectra at the Zn K-edge for the two catalysts at RT. The shape of the edge for both Cu/ZnO/Al2O3 samples is quite different from that of the ZnO reference. These XANES features correspond to a mixed aluminate phase in which Zn possesses octahedral geometry.44 | ||
| Fig. 6 (a) XANES spectra at Cu K-edge, (b) corresponding magnitude of the Fourier transformed k3-weighted EXAFS data at Cu K-edge, and (c) XANES spectra at Zn K-edge of Cu/ZnO/Al2O3 produced in microfluidic (MF) and batch reactor (BR). | ||
| As prepared | Synthesis technique | Cu–O shell | Cu–(Cu/Zn) shell | ||||
|---|---|---|---|---|---|---|---|
| R (Å) | CNa | DW factor | R (Å) | CNa | DW factor | ||
| a CN = coordination number (affected by spectral damping), R = distance (Å), σ2 = Debye–Waller factor (DW) (Å−2). b Fixed during fitting. | |||||||
| Cu/ZnO/Al2O3 | Microreactor | 1.96 ± 0.01 | 2.1 ± 0.4 | 0.0036 ± 0.0020 | 2.94 ± 0.043.11 ± 0.04 | 3.6 ± 0.82b | 0.0150 ± 0.0030b |
| Cu/ZnO/Al2O3 | Batch reactor | 1.96 ± 0.01 | 1.9 ± 0.4 | 0.0042 ± 0.0022 | 2.93 ± 0.033.11 ± 0.03 | 3.4 ± 0.72b | 0.0150 ± 0.0030b |
Fig. 7a and b shows background subtracted and normalized XANES spectra at Cu K and Zn K-edges, respectively, measured in situ during the reduction of the catalyst produced in the microreactor. The reduction of Cu(II) to Cu(0) is reflected by the decrease in the intensity of white line and shift of the edge to lower energies starting at a temperature close to 150 °C and is almost completed at 240 °C. The XANES features of the reduced catalyst at 260 °C (Fig. S5†) closely resemble the profile of the copper foil (spectral damping taken into account), indicating that the reduction has proceeded to the apparent full reduction of Cu2+ into Cu0 metal. This is confirmed by a Cu–Cu distance corresponding to metallic Cu (Table 4). In this case, no contribution from Cu(I) was observed during the TPR which is similar to the results recently reported by Frei et al.,45 however there are examples in literature where relatively low (<10%) contribution from Cu(I) were also reported.23
 | ||
| Fig. 7 In situ XANES spectra at (a) Cu K and (b) Zn K-edges of Cu/ZnO/Al2O3 produced in the microreactor measured during the TPR (from RT up to 260 °C), along with (c) first derivative XANES spectra at Zn K-edge and (d) comparison of the LCF results at Cu K-edge during TPR for catalysts produced in microfluidic and batch reactor. | ||
| Synthesis technique | T (°C) | Cu–Cu shell | Cu–Cu shell (2nd shell) | ||||
|---|---|---|---|---|---|---|---|
| R (Å) | CN | DW factor | R (Å) | CN | DW factor | ||
| a Fixed during fitting. | |||||||
| Microreactor | At 260 | 2.53 ± 0.02 | 5.4 ± 0.6 | 0.0134 ± 0.0012 | 3.56 ± 0.05 | 2a | 0.0163 ± 0.0042 |
| After cooling down to RT | 2.54 ± 0.01 | 5.7 ± 0.5 | 0.0082 ± 0.0007 | 3.58 ± 0.03 | 3a | 0.0118 ± 0.0015 | |
| Batch reactor | At 260 | 2.54 ± 0.01 | 5.0 ± 0.4 | 0.0135 ± 0.0009 | 3.57 ± 0.03 | 2a | 0.0152 ± 0.0016 |
| After cooling down to RT | 2.54 ± 0.01 | 5.2 ± 0.4 | 0.0085 ± 0.0007 | 3.59 ± 0.02 | 4a | 0.0156 ± 0.0016 | |
Fig. 7c shows the derivative XANES spectra at Zn K-edge during the same TPR to 260 °C. The changes observed in the Zn K-edge in Fig. 7b are weak, and to highlight these the spectra are shown as derivative. The reduction of the copper oxide phase that occurs between 150 and 240 °C does not strongly affect the Zn K-edge XANES profile, showing that the segregation and reduction of the Cu2+ to Cu0 metal particles is an independent phenomenon. The decrease in the white line intensity accompanied by a shift of the edge to lower energies have been interpreted as indicators of the transformation of Zn(II) from octahedral to tetrahedral coordination.46,47 These effects are more evident in the corresponding first derivatives that display a regular shift in energy and decrease in the intensity of the Zn2+ 1s → 4p peak from ∼9662.5 eV to ∼9662.0 eV.48 As the reduction temperature increases, the Zn containing phase is undergoing a regular transformation from the mixed aluminate phase in which Zn possesses an octahedral geometry to a highly dispersed and amorphous ZnO phase with Zn in tetrahedral coordination. Also, the appearance of the ∼9658 eV feature, typical of metallic Zn,45 is a clear indication that Zn(0) formation is just initiated at the end of the TRP experiment. By comparing the results of LCF of Cu K XANES spectra of TPR obtained for the two catalysts (Fig. 7d) it can be observed that the reduction of Cu species from the batch reactor starts at lower temperature than of those from the microreactor.
Methanol synthesis
After activation, catalysts prepared by the batch and continuous microfluidic precipitation were compared in order to assess their catalytic performance during methanol synthesis from CO-rich syngas. Reaction conditions are summarized in Table S1.† The three main reactions are as follows:CO hydrogenation to methanol
 | (7) |
 | (8) |
 | (9) |
000 mL g−1 h−1. In addition to the CO syngas feed, a second feed was used with a CO/CO2 ratio of 14:1, as it is known that small quantities of CO2 increase methanol productivity for Cu/ZnO-based catalysts.49–52 As expected, in pure CO synthesis gas feed the methanol productivity of all catalysts increased by a factor of between 2.1 and 3.4 at 250 °C compared to 230 °C. The methanol productivity for the two feed compositions increased by a factor of 6.9–14.8. Note that this factor is the highest for the catalyst prepared by microfluidic synthesis. The results also show that Cu/ZnO/Al2O3-MF in pure CO synthesis gas feed yields dimethyl ether (DME) with selectivity of 14.1% at 230 °C and 16.9% at 250 °C. This may be attributed to higher surface area of the Cu/ZnO/Al2O3-MF catalyst which results in more available acidic sites catalyzing dehydration of methanol (reaction (10)). For technical methanol synthesis DME formation would require a purification step and should be avoided by e.g. further optimizing the microfluidic synthesis procedure to decrease the amount of acidic sites.
000 mLN h−1 g−1
| Reaction conditions | Catalysts | S DME mol% | S MeOH mol% | P DME mmol h−1 gcat−1 | P MeOH mmol h−1 gcat−1 |
|---|---|---|---|---|---|
| 230 °C | Cu/ZnO/Al2O3-MF | 14.1 | 41.5 | 0.2 | 0.6 |
| 15 mol% CO | Cu/ZnO/Al2O3-BR | 0.0 | 47.7 | 0.0 | 1.3 |
| Cu/ZnO-BR | 0.0 | 34.7 | 0.0 | 0.7 | |
| 230 °C | Cu/ZnO/Al2O3-MF | 1.3 | 98.7 | 0.1 | 8.9 |
| 14 mol% CO | Cu/ZnO/Al2O3-BR | 0.0 | 100 | 0.0 | 13.4 |
| 1 mol% CO2 | Cu/ZnO-BR | 0.0 | 100 | 0.0 | 8.2 |
| 250 °C | Cu/ZnO/Al2O3-MF | 16.9 | 65.9 | 0.4 | 1.5 |
| 15 mol% CO | Cu/ZnO/Al2O3-BR | 0.4 | 60.2 | 0.0 | 2.7 |
| Cu/ZnO-BR | 0.0 | 58.3 | 0.0 | 2.4 | |
| 250 °C | Cu/ZnO/Al2O3-MF | 1.8 | 98.1 | 0.4 | 21.9 |
| 14 mol% CO | Cu/ZnO/Al2O3-BR | 0.0 | 99.9 | 0.0 | 19.6 |
| 1 mol% CO2 | Cu/ZnO-BR | 0.1 | 99.8 | 0.0 | 16.6 |
MeOH dehydration
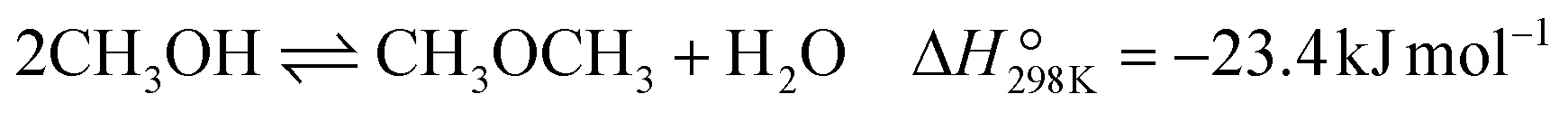 | (10) |
:1) CO2 formation via WGSR can be suppressed in the investigated operation range, leading to nearly 100% MeOH selectivity (see Table 5), also the water produced through reaction (8) inhibits the formation of DME. At 230 °C and in presence of 1 mol% CO2, methanol productivity over the Cu/ZnO/Al2O3-BR is by a factor of 1.5 higher than over the Cu/ZnO/Al2O3-MF, whereas at 250 °C in the same gas mixture the catalyst from the microfluidic reactor produces >10% more methanol compared to its counterpart from the batch reactor.
Overall, the Cu/ZnO/Al2O3-MF catalyst also shows the highest observed methanol productivity (21.9 mmol h−1 gcat−1 at 250 °C, CO/CO2 = 14) in all experiments, suggesting that the microfluidic co-precipitation method has a promoting effect on catalytic activity under these conditions, while further optimization of the MF synthesis is needed to control the DME selectivity.
Conclusion
A continuously operated microfluidic reactor was used to synthesize a nitrate-derived Cu/ZnO/(Al2O3) catalyst for CO2 dehydrogenation to methanol and DME. The obtained catalyst was thoroughly characterized by multiple physicochemical techniques and benchmarked against materials made via co-precipitation in a conventional stirred batch reactor using the same precursors and the same metal ratios. XRD measurements revealed that in both materials zincian malachite was the main phase of the precipitated precursor. Additionally, some gerhardtite and rosasite phases were observed in the Cu/ZnO/Al2O3 synthesized in the batch and microfluidic reactors, respectively. After calcination, the sample made via microfluidic synthesis had smaller CuO crystallites leading to higher dispersion and active surface area of Cu in the final catalyst.The catalyst synthesized in the batch reactor demonstrates higher MeOH productivity at 230 °C, whereas at 250 °C the microfluidically synthesized catalyst provides higher MeOH and DME yields. Hence, the study shows that a continuous synthesis using a micromixer is an attractive process for preparation of highly active methanol synthesis catalyst, as it is well-controllable and scalable. The used microfluidic reactor with optically and X-ray transmissible windows, furthermore, offers a possibility to investigate the early stages of methanol catalyst formation during co-precipitation with a multitude of X-ray spectroscopic and scattering techniques.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Virtual Institute VI-403 “In situ Nano Imaging of Biological and Chemical Processes”, the BMBF (projects 05K10VK1, 05K13VK2) and KIT (Helmholtz-programs “Storage and Crosslinked Infrastructures” (SCI) and “Materials and Technologies for the Energy Transition” (MTET)) are gratefully acknowledged for financial support. We would like to thank the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz research infrastructure at KIT for providing STEM-EDX and ICP-OES measurements and Angela Deutsch (ITCP) for BET measurements. Special thanks to Simon Wodarz and Sabrina Polierer (IKFT) for the N2O chemisorption experiments. Finally, we thank the KIT synchrotron (operated by KIT-IBPT) for providing beamtime at the CAT-ACT beamline, and Dr. Tim Prüßmann and Dr. Anna Zimina (IKFT) for their help and technical support during XAS experiments.References
-
G. A. Olah, A. Goeppert and G. S. Prakash, Beyond Oil and Gas: The Methanol Economy, John Wiley & Sons, 2018 Search PubMed
.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr and B.-L. Kniep, Science, 2012, 336, 893–897 CrossRef CAS PubMed
- T. Lunkenbein, F. Girgsdies, T. Kandemir, N. Thomas, M. Behrens, R. Schlögl and E. Frei, Angew. Chem., Int. Ed., 2016, 55, 12708–12712 CrossRef CAS PubMed
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS
-
J. B. Hansen and P. E. H. Nielsen, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, 2nd edn, Wiley-VCH, Weinheim, 2008 Search PubMed
- J.-K. Lee, I.-B. Lee and J. Han, J. Ind. Eng. Chem., 2019, 75, 77–85 CrossRef CAS
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed
- K. F. Kalz, I. R. Kraehnert, P. M. Dvoyashkin, I. R. Dittmeyer, R. Gläser, I. U. Krewer, K. Reuter and J.-D. Grunwaldt, ChemCatChem, 2017, 9, 17 CrossRef CAS PubMed
- C. Kiener, M. Kurtz, H. Wilmer, C. Hoffmann, H.-W. Schmidt, J.-D. Grunwaldt, M. Muhler and F. Schüth, J. Catal., 2003, 216, 110–119 CrossRef CAS
- G. Chinchen, K. Waugh and D. Whan, Appl. Catal., 1986, 25, 101–107 CrossRef CAS
- G. Chinchen, P. Denny, J. Jennings, M. Spencer and K. Waugh, Appl. Catal., 1988, 36, 1–65 CrossRef CAS
- S. Vukojević, O. Trapp, J.-D. Grunwaldt, C. Kiener and F. Schüth, Angew. Chem., Int. Ed., 2005, 44, 7978–7981 CrossRef PubMed
- Q.-C. Zhang, Z.-W. Liu, X.-H. Zhu, L.-X. Wen, Q.-F. Zhu, K. Guo and J.-F. Chen, Ind. Eng. Chem. Res., 2015, 54, 8874–8882 CrossRef CAS
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef CAS
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed
- B. L. Kniep, T. Ressler, A. Rabis, F. Girgsdies, M. Baenitz, F. Steglich and R. Schlögl, Angew. Chem., Int. Ed., 2004, 43, 112–115 CrossRef PubMed
- Y. Zhang, Q. Sun, J. Deng, D. Wu and S. Chen, Appl. Catal., A, 1997, 158, 105–120 CrossRef CAS
- Z. Chu, H. Chen, Y. Yu, Q. Wang and D. Fang, J. Mol. Catal. A: Chem., 2013, 366, 48–53 CrossRef CAS
- R. Van Den Berg, G. Prieto, G. Korpershoek, L. I. Van Der Wal, A. J. Van Bunningen, S. Lægsgaard-Jørgensen, P. E. De Jongh and K. P. De Jong, Nat. Commun., 2016, 7, 1–7 Search PubMed
- M. Behrens, D. Brennecke, F. Girgsdies, S. Kißner, A. Trunschke, N. Nasrudin, S. Zakaria, N. F. Idris, S. B. A. Hamid and B. Kniep, Appl. Catal., A, 2011, 392, 93–102 CrossRef CAS
- B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem. – Eur. J., 2003, 9, 2039–2052 CrossRef CAS PubMed
- M. Behrens, Catal. Today, 2015, 246, 46–54 CrossRef CAS
- J.-D. Grunwaldt, C. Kiener, F. Schüth and A. Baiker, Phys. Scr., 2005, 2005, 819 CrossRef
- M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef CAS
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS
- S. Polierer, D. Guse, S. Wild, K. Herrera Delgado, T. N. Otto, T. A. Zevaco, M. Kind, J. Sauer, F. Studt and S. Pitter, Catalysts, 2020, 10, 816 CrossRef CAS
- M. Schur, B. Bems, A. Dassenoy, I. Kassatkine, J. Urban, H. Wilmes, O. Hinrichsen, M. Muhler and R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 3815–3817 CrossRef CAS PubMed
- T. Kirner, J. Albert, M. Günther, G. Mayer, K. Reinhäckel and J. Köhler, Chem. Eng. J., 2004, 101, 65–74 CrossRef CAS
- Q.-C. Zhang, K.-P. Cheng, L.-X. Wen, K. Guo and J.-F. Chen, RSC Adv., 2016, 6, 33611–33621 RSC
- M. M. Günter, B. Bems, R. Schlögl and T. Ressler, J. Synchrotron Radiat., 2001, 8, 619–621 CrossRef PubMed
- X. Jiang, X. Qin, C. Ling, Z. Wang and J. Lu, AIChE J., 2018, 64, 2647–2654 CrossRef CAS
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183–1202 RSC
- G. Tofighi, A. Gaur, D. E. Doronkin, H. Lichtenberg, W. Wang, D. Wang, G. N. Rinke, A. Ewinger, R. Dittmeyer and J.-D. Grunwaldt, J. Phys. Chem. C, 2018, 122, 1721–1731 CrossRef CAS
- G. Tofighi, X. Yu, H. Lichtenberg, D. E. Doronkin, W. Wang, C. Wöll, Y. Wang and J.-D. Grunwaldt, ACS Catal., 2019, 9(6), 5462–5473 CrossRef CAS
- G. Tofighi, D. Degler, B. Junker, S. Müller, H. Lichtenberg, W. Wang, U. Weimar, N. Barsan and J.-D. Grunwaldt, Sens. Actuators, B, 2019, 292, 48–56 CrossRef CAS
- G. Tofighi, H. Lichtenberg, J. Pesek, T. L. Sheppard, W. Wang, L. Schöttner, G. Rinke, R. Dittmeyer and J.-D. Grunwaldt, React. Chem. Eng., 2017, 2, 876–884 RSC
- S. Wild, S. Polierer, T. A. Zevaco, D. Guse, M. Kind, S. Pitter, K. Herrera Delgado and J. Sauer, RSC Adv., 2021, 11, 2556–2564 RSC
- A. Zimina, K. Dardenne, M. Denecke, D. Doronkin, E. Huttel, H. Lichtenberg, S. Mangold, T. Pruessmann, J. Rothe and T. Spangenberg, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS PubMed
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed
- B. Zhang and N. Yan, Catalysts, 2013, 3, 543–562 CrossRef CAS
- Z. Li, S. Yan and H. Fan, Fuel, 2013, 106, 178–186 CrossRef CAS
- P. Gao, F. Li, F. Xiao, N. Zhao, W. Wei, L. Zhong and Y. Sun, Catal. Today, 2012, 194, 9–15 CrossRef CAS
- A. Gaur, W. Klysubun, N. N. Nair, B. Shrivastava, J. Prasad and K. Srivastava, J. Mol. Struct., 2016, 1118, 212–218 CrossRef CAS
- R. Revel, D. Bazin and A.-M. Flank, J. Synchrotron Radiat., 1999, 6, 717–718 CrossRef CAS PubMed
- E. Frei, A. Gaur, H. Lichternberg, L. Zwiener, M. Scherzer, F. Girgsdies, T. Lunkenbein and R. Schlögl, ChemCatChem, 2020, 12, 4029–4033 CrossRef CAS
- L. Galoisy, L. Cormier, G. Calas and V. Briois, J. Non-Cryst. Solids, 2001, 293, 105–111 CrossRef
- G. Waychunas, C. Fuller and J. Davis, Geochim. Cosmochim. Acta, 2002, 66, 1119–1137 CrossRef CAS
- D. Grandjean, V. Pelipenko, E. D. Batyrev, J. C. van den Heuvel, A. A. Khassin, T. M. Yurieva and B. M. Weckhuysen, J. Phys. Chem. C, 2011, 115, 20175–20191 CrossRef CAS
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley and F. Abild-Pedersen, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS
- K. Klier, V. Chatikavanij, R. Herman and G. Simmons, J. Catal., 1982, 74, 343–360 CrossRef CAS
- X.-M. Liu, G. Lu, Z.-F. Yan and J. Beltramini, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef CAS
- M. Bowker, ChemCatChem, 2019, 11, 4238 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1re00499a |
| This journal is © The Royal Society of Chemistry 2022 |