
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe design of experiments (DoE) in optimization of an aerobic flow Pd-catalyzed oxidation of alcohol towards an important aldehyde precursor in the synthesis of phosphatidylinositide 3-kinase inhibitor (CPL302415)†
Stanisław Michałek ab,
Lidia Gurba-Bryśkiewicza,
Wioleta Maruszaka,
Marcin Zagozdaa,
Anna M. Maj*a,
Zbigniew Ochalb,
Krzysztof Dubiela and
Maciej Wieczoreka
ab,
Lidia Gurba-Bryśkiewicza,
Wioleta Maruszaka,
Marcin Zagozdaa,
Anna M. Maj*a,
Zbigniew Ochalb,
Krzysztof Dubiela and
Maciej Wieczoreka
aCelon Pharma S.A., Ul. Marymoncka 15, 05-152 Kazuń Nowy, Poland. E-mail: anna.maj@celonpharma.com
bFaculty of Chemistry, Warsaw University of Technology, Ul. Noakowskiego 3, 00-664 Warsaw, Poland
First published on 23rd November 2022
Abstract
Herein, we describe the development of a green, scalable flow Pd-catalyzed aerobic oxidation for the key step in the synthesis of CPL302415, which is a new PI3Kδ inhibitor. Applying this environmental-friendly, sustainable catalytic oxidation we significantly increased product yield (up to 84%) and by eliminating of workup step, we improved the waste index and E factor (up to 0.13) in comparison with the stoichiometric synthesis. The process was optimized by using the DoE approach.
The first class of PI3K (phosphoinositide 3-kinase) inhibitors including four heterodimeric proteins (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ) is considered to be very attractive for the treatment of many diseases like SLE (Systemic Lupus Erythematosus), MS (Multiple Sclerosis), asthma and another inflammatory, autoimmune and respiratory diseases.1 Recently we published the synthesis, biological activity, and toxicology of a new PI3Kδ inhibitor based on the pyrazolo[1,5-a]pyrimidine core the CPL302415 (Fig. 1) which is now under evaluation for the treatment of Systemic Lupus Erythematosus.2 A critical step in its synthesis is oxidation of primary alcohol the {5-[2-(difluoromethyl)-2,3-dihydro-1H-1,3-benzodiazol-1-yl]-7-(morpholin-4- yl)pyrazolo[1,5-a]pyrimidin-2-yl}methanol (1) to an aldehyde 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (3) in penultimate step. The selective oxidation of pharmaceutical precursors, which are often complex organic molecules bearing multiple functional groups, is generally very demanding.3 In our case, not only catalyst must selectively carry out the oxidation of the alcohol group to the aldehyde with high efficiency but also recover one double bond in the benzimidazole ring, which is very important for CPL302415 stability. Also, the whole oxidation step should be economically and environmentally sustainable and easily integrate with large-scale production of active pharmaceutical ingredient (API) (CPL302415). An additional problem in this reaction is the very low solubility of 1.
 | ||
| Fig. 1 Structure of CPL302415. | ||
For this transformation we have already explored 15 oxidation procedures therein: MnO2,4 Dess–Martin Periodinane (DMP),5 ABNO/CuI/NMI,6 TEMPO/NaBr/NaOCl,7 TEMPO/tBuONO/HCl,7 TEMPO/tBuNBr/NaOCl,7 TEMPO/BuNBr/OXONE®,8 IBX,9 IBX/Bu4NHSO4/OXONE®,10 NaOCl/Bu4NBr,11 Pt–Bi/C/KOH/air,12 Pt–Bi/C/O2,13 Pt–Bi/C/H2O2,13 Ru/Al2O3/Air14 or O2,15 Aurolite®/O2,16 and only two methods resulted in the formation of the desired product. Although the oxidations with activated MnO2 and DMP were sufficient to produce 250 g of 3, due to unsatisfactory yield on a large-scale, purification problems, and a huge quantity of waste generated in those reactions as well as commercial goals the alternative oxidation procedure was necessary. The Dess–Martin procedure is too expensive in large-scale production while oxidation with MnO2 requires 10-fold excess of MnO2. Besides it, the reaction is strongly dependent on the quality of MnO2 lot and sometimes a longer reaction time is required to obtain a high yield. Moreover, we also confronted the problem with the adsorption of the desired product on the surface of MnO2. Thus we turned towards an aerobic oxidation with molecular oxygen as a stoichiometric oxidant, which is preferred on large scale because of its low cost, and insignificant environmental impact, as well into flow techniques which are the safe and scalable technology leading at intensified conditions to maximizing yield and throughput.17–19 The aim of this work was to develop flow aerobic oxidation of {5-[2-(difluoromethyl)-2,3-dihydro-1H-1,3-benzodiazol-1-yl]-7-(morpholin-4- yl)pyrazolo[1,5-a]pyrimidin-2-yl}methanol (1) to 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4-yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (3) the precursor of CPL302415 in the presence of cheap and environmental-friendly catalyst, in order to achieve the commercial goal. In this objective, we adopted the already known Pd(OAc)2/pyridine catalytic system,20 but to our knowledge, it has not yet been used in the oxidation of such a complicated molecule with the pharmaceutical interest. For this purpose, we also applied DoE approach,21 a structured, cost-effective statistical method to organize, limit the number of experiments, determine critical process parameters and their interactions as well, and set the optimal reaction conditions for high yield and low levels of impurities.22
Flow experiments were performed using a combined two Vapourtec easy-Medchem systems comprising peristaltic pumps and together with four PFA tubular reactors (10 mL, id = 1 mm) (Fig. 2). The second Vapourtec easy-Medchem system was used only for heating additional two reactors, the temperature was set manually. The two liquid feeds were introduced with peristaltic pumps and oxygen gas was introduced through a mass flow controller (Vapourtec SF-10 pump; input pressure 5 bar). The System Solvent Bottle was filled with toluene. The substrate feed and gas feed were mixed using a Y-shaped mixer, then run through a 28 cm (id = 1 mm) tube to enable substrate solution to saturate it with oxygen and later combined with catalyst solution. The reaction was performed first within two heated PFA tubular reactors (10 mL, id = 1 mm). Next, in order to extend the reaction time, the reaction mixture feed was supplemented with oxygen and transferred into two additional heated PFA tubular reactors (10 mL, id = 1 mm). The pressure was applied by using an adjustable back pressure regulator (BPR – Vapourtec SF-10 pump, set up pressure = 5 bar). The oxygen feeds were always set at the same value for each mass flow controller. For all flow experiments, fractions were collected at the end of the reaction and analyzed offline with UHPLC. The different reactions were controlled with FlowWizard™ software which calculated reaction time, and operated the easy-Medchem system and the collection/waste valve. Previously,20 the catalyst has been reported to slowly decompose in absence of oxygen and it was oxygenated before adding the substrate but surprisingly, in our case, we observed better results when the substrate was oxygenated first and then the solution of the catalyst was added. The DoE study and statistical analysis were performed by using the design of experiment tools of STATISTICA software (v.13.3).23,24
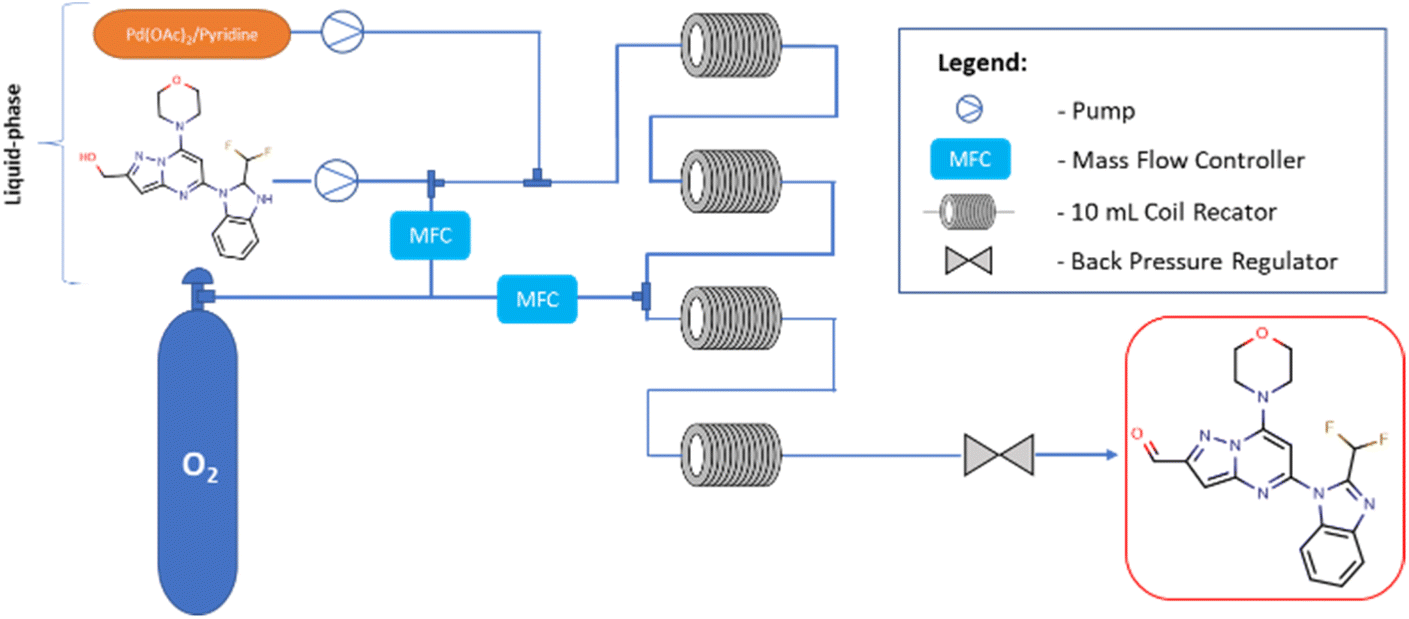 | ||
| Fig. 2 Continuous flow setup for the Pd-catalyzed oxidation of alcohol 1 to aldehyde 3. | ||
For the first screening, we have implemented a six-parameter two-level fractional factorial experimental design plan (2^(6–3)) (Table 1) containing ten experiments and including two repeats at a center point for the reproducibility study.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst loading (mol%) | Pyridine eq. per catalyst | T (°C) | PO2 (bar) | V of O2 (mL min−1) | V of reagents (mL min−1) | Conv. of 1b (%) | Yield of 2b (%) | Yield of 3b (%) | Yield of 4b (%) |
a Standard reaction conditions: 20 mg (0.05 mmol) dissolved in 2 mL toluene/caprolactone = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.b % determined by UHPLC for details see supplementary material. :1.b % determined by UHPLC for details see supplementary material. |
||||||||||
| 1 | 5 | 1.3 | 80 | 5 | 1.0 | 1.0 | 9.7 | 7.4 | 2.3 | 0.0 |
| 2 | 5 | 1.3 | 120 | 5 | 0.1 | 0.1 | 12.3 | 0.0 | 12.2 | 0.0 |
| 3 | 5 | 4 | 80 | 2 | 0.1 | 1.0 | 39.0 | 38.9 | 0.0 | 0.0 |
| 4 | 5 | 4 | 120 | 2 | 1.0 | 0.1 | 4.8 | 4.8 | 0.0 | 0.0 |
| 5 | 22.5 | 2.65 | 100 | 3.5 | 0.55 | 0.55 | 51.6 | 0.0 | 51.6 | 0.0 |
| 6 | 22.5 | 2.65 | 100 | 3.5 | 0.55 | 0.55 | 51.1 | 0.0 | 51.1 | 0.0 |
| 7 | 40 | 1.3 | 80 | 2 | 1.0 | 0.1 | 21.7 | 0.0 | 21.6 | 0.0 |
| 8 | 40 | 1.3 | 120 | 2 | 0.1 | 1.0 | 80.2 | 0.0 | 80.2 | 0.0 |
| 9 | 40 | 4 | 80 | 5 | 0.1 | 0.1 | 44.2 | 0.0 | 44.2 | 0.0 |
| 10 | 40 | 4 | 120 | 5 | 1.00 | 1.0 | 60.6 | 0.0 | 60.6 | 0.0 |
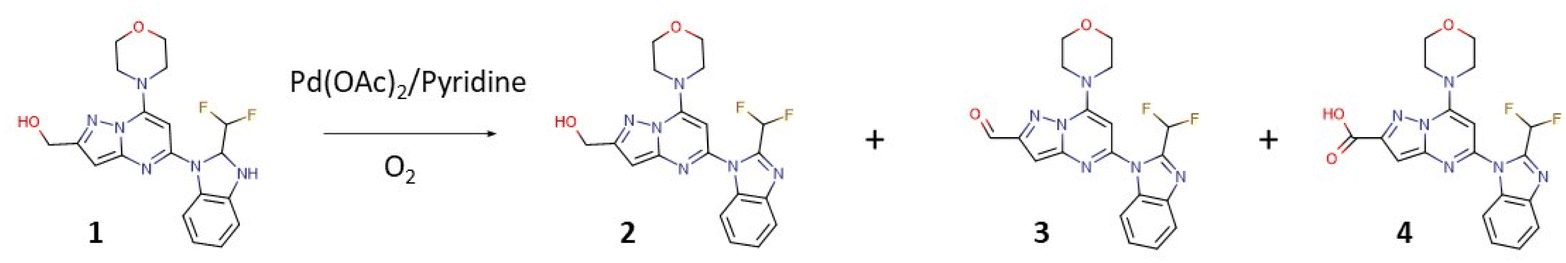
The following parameters were considered for the multivariate optimization: catalyst loading, equivalents of pyridine per catalyst, temperature, oxygen pressure, the flow of oxygen, and finally, the flow of the catalyst and substrate solutions. In order to facilitate the adequate mixing of the streams, and secure the appropriate substrate/catalyst ratio, flows of the catalyst and substrate solutions were identical. To ensure the solubility of all the reaction components, substrate 1 was dissolved in a mixture of toluene and caprolactone, and in addition, we carried out our flow experiments at a very low concentration (0.0125 M). The temperature varied between 80-120 °C, the oxygen pressure between 2–5 bar, the rate of reagents and oxygen between 0.1–1.0 mL min−1, the catalyst loading from 5–40%, and base equivalents from 1.3 to 4.0 equivalents per catalyst. The aerobic flow oxidation of 1 (the CPL302415 precursor) may lead to multiple products: the alcohol with recovered double bond in benzimidazole ring (2), the desired aldehyde with recovered double bond (3), and overoxidized acidic product 4. In our case, we observed mainly products 2 and 3, and the acidic product was formed in a very limited quantity under specific conditions (see ESI†).
Based on the DoE screening study results gathered in Table 1 the mathematical model with the main linear effects for the product 3 yield was generated with a good fit to the experimental data, R2 = 0.86. ANOVA analysis shows that catalyst loading, temperature, reagents flow rate, and O2 pressure have a statistically significant (p < 0.05) influence on the product 3 yields. The quantity of catalyst has the most important positive effect (p = 0.0032) on the aldehyde yield. Next, the temperature (p = 0.0072) and reagents flow speed (p = 0.0094) have also a beneficial effect on the yield of 3 however, the flow speed of O2 has a negative influence (p = 0.0116), diminished the yield of the desired product. O2 pressure has a small positive effect (p = 0.0347) whereas the effect of equivalents of pyridine per catalyst is near the significance level limit (p = 0.052) (Fig. S1†).
Because the screening study with fractional factorial design examines only main linear effects further optimization study was performed by using central composite design (CCD) and response surface methodology (RSM). It has been performed with three variable parameters (with the most significance from screening DoE) i.e.: catalyst loading, temperature, and reagents flow rate (Table 2). Values of the other parameters were selected to maximize the aldehyde product yield based on the results of the analysis screening step. Oxygen pressure was set up at 5 bar (the higher, the higher efficiency), the flow of oxygen at 0.1 mL min−1 (the lower, the greater efficiency), and equivalents of pyridine per catalyst on 1.3 eq. (no significant effect on efficiency).
| Entry | Catalyst loading (mol%) | T (°C) | V of reagents (mL min−1) | Conv. of 1b (%) | Yield of 2b (%) | Yield of 3b (%) |
|---|---|---|---|---|---|---|
| a Standard reaction conditions: substrate 1 = 20 mg (0.05 mmol) dissolved in 2 mL toluene/caprolactone = 1:1; PO2 = 5 bar; catalyst/pyridine = 1/1.3; VO2 = 0.1 mL min−1.b % determined by UHPLC for details see ESI. |
||||||
| 1 | 5 | 80 | 0.1 | 0.4 | 0.4 | 0.0 |
| 2 | 5 | 80 | 1.0 | 17.0 | 17.0 | 0.0 |
| 3 | 5 | 120 | 0.1 | 12.3 | 0.0 | 12.2 |
| 4 | 5 | 120 | 1.0 | 6.4 | 0.4 | 6.0 |
| 5 | 40 | 80 | 0.1 | 20.8 | 0.0 | 20.8 |
| 6 | 40 | 80 | 1.0 | 41.2 | 0.0 | 41.2 |
| 7 | 40 | 120 | 0.1 | 36.3 | 0.0 | 36.3 |
| 8 | 40 | 120 | 1.0 | 70.4 | 0.0 | 70.4 |
| 9 | 5 | 100 | 0.55 | 6.0 | 4.4 | 1.6 |
| 10 | 40 | 100 | 0.55 | 27.4 | 0.0 | 27.4 |
| 11 | 22.5 | 80 | 0.55 | 6.5 | 0.0 | 6.5 |
| 12 | 22.5 | 120 | 0.55 | 39.1 | 0.0 | 39.1 |
| 13 | 22.5 | 100 | 0.1 | 22.3 | 0.0 | 22.3 |
| 14 | 22.5 | 100 | 1.0 | 15.0 | 0.0 | 15.0 |
| 15 | 22.5 | 100 | 0.55 | 24.1 | 0.0 | 24.1 |
| 16 | 22.5 | 100 | 0.55 | 22.6 | 0.0 | 22.6 |
| 17 | 22.5 | 100 | 0.55 | 22.6 | 0.0 | 22.6 |
CCD model has good fit, R2 = 0.92. The main statistically significant effects of tested parameters on the aldehyde yield are linear, similarly to those obtained from fractional factorial design. Only an additional interaction effect of the catalyst loading with the flow of reagents as a statistically significant positive effect (p = 0.0157) was identified. Quadratic effects are not statistically significant (Fig. S2†). The catalyst loading has the most positive influence (p = 0.0024) on the aldehyde product yield based on this CCD (RSM) model. Then, the temperature (p = 0.0080) and the flow of the reactants (p = 0.0414) have a positive effect as well.
As it results from the CCD model of maximum predicted aldehyde product yields in the tested range are not greater than 80% at the maximum value of the catalyst loading (40%), temperature (120 °C), and reactants flow (1.0 mL min−1) (Fig. 3).
 | ||
| Fig. 3 Surface response for aldehyde product yield, central composite design model 2^(3), temperature = 120 °C, PO2 = 5 bar, VO2 = 0.1 mL min−1. | ||
Based on the literature data25 and the experience with catalytic reactions, our doubts were raised by the lack of dependence of the reaction efficiency on pyridine eq. per catalyst. We conducted additional DoE experiments according to the D-optimal plan with three variable parameters, catalyst loading, equivalents of pyridine per catalyst, and reagents flow (Table 3). Oxygen pressure was set up at 5 bar, the flow of oxygen at 0.1 mL min−1, and the temperature at 120 °C. The obtained model confirms that pyridine eq. per catalyst has little effect on the reaction yield (Fig. S3†).
| Entry | Catalyst loading (mol%) | Pyridine eq. per catalyst | V of reagents (mL min−1) | Conv. of 1b (%) | Yield of 2b (%) | Yield of 3b (%) |
|---|---|---|---|---|---|---|
| a Standard reaction conditions: substrate 1 = 20 mg (0.05 mmol) dissolved in 2 mL toluene/caprolactone = 1:1; PO2 = 5 bar; T = 120 °C; VO2 = 0.1 mL min−1.b % determined by UHPLC for. |
||||||
| 1 | 5 | 1.3 | 0.1 | 51.3 | 0.0 | 44.2 |
| 2 | 5 | 1.3 | 1.0 | 22.1 | 1.1 | 19.5 |
| 3 | 5 | 2.65 | 0.55 | 19.7 | 0.0 | 18.4 |
| 4 | 5 | 4 | 1.0 | 44.3 | 1.7 | 36.5 |
| 5 | 22.5 | 1.3 | 0.55 | 63.6 | 0.0 | 57.7 |
| 6 | 22.5 | 2.65 | 1.0 | 70.4 | 0.0 | 70.4 |
| 7 | 22.5 | 4 | 0.1 | 83.0 | 0.0 | 70.1 |
| 8 | 40 | 1.3 | 0.1 | 97.5 | 3.3 | 52.1 |
| 9 | 40 | 1.3 | 1.0 | 92.4 | 0.0 | 81.2 |
| 10 | 40 | 4 | 1.0 | 88.5 | 1.6 | 79.4 |
Moreover, the obtained results confirmed that in the investigated range, the maximum predicted performance is 85% with the maximum value of catalyst loading equaling 40%, the flow of each liquid reagent equals 1.0 ml min−1 and pyridine eq. per catalyst equals 4, (84% for pyridine eq. per catalyst equals 1.3)
Initially, we carried out the oxidation of 1 in pure toluene. At 120 °C under 5 bars of oxygen, and in reagents flow rate = 0.4 mL min−1 we observed 89% yield of 3, while the original stoichiometric procedures with MnO2 or DMP gave us only 68% or 78% yield respectively. Yet in some cases using pure toluene the substrate precipitated in the inlet feeding tubes before the pump even when the substrate solution was preheated and with additional isolation of inlet tubes, this made the procedure uncertain and unrepetitive. The addition of caprolactone to toluene let us solubilize alcohol 1 and carry out the reaction in flow conditions without precipitation of 1, thus we performed the optimization in the toluene/caprolactone mixture. However, we observed a similar or higher yield in comparison to stoichiometric methods (78% of 3), the caprolactone, due to its high boiling temperature (241 °C) and good miscibility with water and organic solvents, generated problems in the purification of the final product (CPL302415) in large scale and it was almost impossible to separate it without using column chromatography. Thus, we also tried oxidation of 1 in a mixture of toluene and ethyl acetate (1:1) and we observed similar tendencies as in the case of reactions performed in the toluene/caprolactone mixture (see ESI† Table S3). The toluene/ethyl acetate mixture turned out to be the most promising option for us, because of better solubility of our substrate and product in this mixture than in pure even preheated toluene. The substrate did not precipitate any more in inlet tubes and the procedure becomes repetitive. The 1 g test carried out under the best conditions i.e. catalyst = 20 mol%; catalyst/pyridine = 1/1.3; T = 120 °C; PO2 = 5 bar; VO2 = 0.1 mL min; Vreagents = 1 mL min−1) and operated during 100 min resulted in productivity 0.589 g h−1 of 3. What's more, we obtained very good results towards selective oxidation of alcohol to an aldehyde group, 84% yield of product 3 having a double bond in the benzimidazole ring we also got less alcohol 2 (Fig. 4). While the counterpart experiment carried out in a batch autoclave gave only 45% yield (see ESI†). That solvent mixture had also two additional and pivotal advantages, especially regarding large scale production, can be use as the reaction mixture without any workup and proceed with the next and simultaneously final step in the synthesis of the 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4- yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde our CPL302415 and beside the EtOAc is four times cheaper than caprolactone.
 | ||
| Fig. 4 Other solvents screened for the Pd-catalyzed oxidation of 1. Standard reaction conditions: substrate 1 = 20 mg (0.05 mmol) dissolved in 2 mL; catalyst = 20 mol%; catalyst/pyridine = 1/1.3; T = 120 °C. | ||
The comparison of the qualitative and quantitative green metrics26 between Pd(OAc)2/pyridine in toluene/EtOAc mixture, MnO2, and Dess–Martin procedures (Table 4) clearly shows the benefits of the flow Pd-catalyzed process over the stoichiometric methods. Using Pd(OAc)2/pyridine we got a higher yield of more than 6 or 17% respectively, moreover, the catalytic process is also characterized by a higher atom economy (AE) and higher reaction mass efficiency (RME).




Contrary to the Dess–Martin process, which necessities the undesirable DMF, the synthesis with Pd(OAc)2/Pyridine and MnO2 use the mixture of toluene and EtOAc or BuOAc admitted as acceptable or green solvents.26,27 Furthermore from the environmental point of view as manganese is considered a critical element,28 the supply of which may run out in the next 5–50 years, thus the tenfold excess of MnO2 required to achieve high efficiency is one of the major drawbacks of the MnO2 protocol. While the palladium reserve is expected to be 100–500 years. In addition, when the reaction is carried out on large scale, we observed adsorption of our desired product on the surface of MnO2 and we faced problems with the recovery of 3 from the reaction mixture.28 Another drawback of the reaction with MnO2 is its lower energy efficiency in comparison with the procedure applied Pd(OAc)2/pyridine catalyst. The oxidation with MnO2 is carried out under atmospheric pressure at reflux and thus is less energy efficient than the reaction in the presence of Pd(OAc)2/pyridine which is carried out also at 120 °C but under 5 bars of O2 and the energy input required to run the reaction may be smaller especially when heating flow reactors with small diameter tubes. It is well known that running a reaction at reflux results in a 6-fold increase in energy consumption as opposed to doing so at 5 °C below,28 thus we consider the flow process as less energy-consuming. Moreover, the complicated workup, additionally requiring washing with non-green solvent (DCM) in the MnO2 oxidation process does not allow easy and cost-effective recycling of the solvents. While in the case of aerobic oxidation with Pd(OAc)2/pyridine, no workup is necessary to proceed the following step of the synthesis. This and the large excess of MnO2 necessary for high yield results in more than 1600-fold higher waste index for this process compared to the Pd(OAc)2/pyridine oxidation. No workup and solvent recycling causes also much better E factor for the Pd-catalyzed flow process in comparison with MnO2 or DMP oxidations (Table 4). Over and above all these arguments the flow process provides higher spacetime yield in comparison to batch oxidation with MnO2, thus is more favorable for industrial application.
Conclusion
The development and DoE optimization of this study was realized in order to improve the synthesis of the 5-[2-(difluoromethyl)-1H-benzimidazol-1-yl]-7-(morpholin-4- yl)pyrazolo[1,5-a]pyrimidine-2-carbaldehyde (3) an important precursor in the production of our new PI3Kδ inhibitor (CPL302415). The difficulty of this work relies on the oxidation of relatively complicated molecule which may potentially lead to at least two undesired by-products. Besides it, our substrate is low soluble and tends to adsorb on the MnO2 surface. The catalytic flow gas–liquid aerobic oxidation of 1 in the presence of Pd(OAc)2/pyridine is characterized by higher yield, better atom economy, lower environmental impact and consume less energy. That allows us to refine our process of the CPL302415 production compared to two already existing stoichiometric methods of {5-[2-(difluoromethyl)-2,3-dihydro-1H-1,3-benzodiazol-1-yl]-7-(morpholin-4- yl)pyrazolo[1,5-a]pyrimidin-2-yl}methanol oxidation. Moreover, the flow synthesis let us skip the complicated workup and reduce about 1600-fold the waste intensity factor thus we consider that procedure a green synthesis.The use of various tools of the DoE approach made it possible to find important factors influencing the efficiency of the process and determine the operational range that gives the maximum product yield.
Abbreviations
Acronyms| AE | Atom economy |
| DCM | Dichloromethane |
| DoE | Design of experiment |
| DMF | Dimethylfuran |
| DMP | Dess–Martin Periodinane |
| E factor | Environmental factor |
| OE | Optimum Efficiency |
| PFA | Perfluoroalkoxy alkanes |
| PMI | Process Mass Intensity |
| RME | Reaction Mass Efficiency |
| WI | Waste Index |
Conflicts of interest
The authors declare the following financial interest/personal relationships which may be considered as potential competing interests. All contributors to this work (except Z. Ochal) at the time of their direct involvement in the project were full-time employees of Celon Pharma SA. M. Wieczorek is the CEO of Celon Pharma S.A. Some of the authors are the shareholders of Celon Pharma S.A. This work was financially supported by The National Centre for Research and Development (POIR.01.02.00-00-0085/18-00).Acknowledgements
This research was co-financed by the National Centre for Research and Development “Narodowe Centrum Badan i Rozwoju” and Celon Pharma. SA., project “KICHAI – Preclinical and clinical development of innovative lipid kinases inhibitor as a candidate for the treatment of steroid-resistant and severe inflammatory lung diseases”, grant number POIR.01.02.00-00-0085/18-00.Notes and references
- (a) T. Saurat, F. Buron, N. Rodrigues, M. L. de Tauzia, L. Colliandre, S. Bourg, P. Bonnet, G. Guillaumet, M. Akssira, A. Corlu, C. Guillouzo, P. Berthier, P. Rio, M. L. Jourdan, H. Bénédetti and S. Routier, J. Med. Chem., 2014, 57(3), 613 CrossRef CAS PubMed; (b) J. A. Engelman, J. Luo and L. C. Cantley, Nat. Rev. Genet., 2006, 7(8), 606 CrossRef CAS PubMed; (c) P. J. Parker, Biochem. Soc. Trans., 2004, 32(6), 893 CrossRef CAS PubMed; (d) J. G. Foster, M. D. Blunt, E. Carter and E. S. G. Ward, Pharmacol. Rev., 2012, 64(4), 1027 CrossRef CAS PubMed.
- M. Stypik, S. Michałek, N. Orłowska, M. Zagozda, M. Dziachan, M. Banach, P. Turowski, P. Gunerka, D. Zdżalik-Bielecka, A. Stańczak, U. Kędzierska, K. Mulewski, D. Smuga, W. Maruszak, L. Gurba-Bryśkiewicz, A. Leniak, W. Pietruś, Z. Ochal, M. Mach, B. Zygmunt, J. Pieczykolan, K. Dubiel and M. Wieczorek, Pharmaceuticals, 2022, 15, 927 CrossRef CAS PubMed.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Organomet. Chem., 2015, 67, 91 CrossRef.
- (a) R. J. Gritter and T. J. Wallace, J. Org. Chem., 1959, 24(8), 1051 CrossRef CAS; (b) G. Tojo and M. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones, Springer, 2006, pp. 289.
- J. S. Yadav, B. V. S. Reddy, A. K. Basak and A. V. Narsaiah, Tetrahedron, 2004, 60(9), 2131 CrossRef CAS.
- J. E. Stevens, Y. Preger, J. R. Martinelli, C. j. Welch, T. W. Root, J. M. Hawkins and S. S. Sthal, Org. Process Res. Dev., 2015, 19(11), 1548 CrossRef.
- (a) R. A. Sheldon, I. W. C. E. Arends, G. J. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774 CrossRef CAS PubMed; (b) P. D. Hampton, M. D. Whealon, L. M. Roberts, A. A. Yaeger and R. Boydson, Org. Process Res. Dev., 2008, 12, 946 CrossRef CAS; (c) A. Hinzmann, M. Stricker, J. Busch, S. Glinski, K. Oike and H. Gröger, Eur. J. Org. Chem., 2020, 16, 2399 CrossRef.
- C. Bolm, A. S. Magnus and J. P. Hildebrand, Org. Lett., 2000, 2(8), 1173 CrossRef CAS PubMed.
- (a) A. Schulze and A. Giannis, Synthesis, 2006, 2, 257 Search PubMed; (b) J. D. More and N. S. Finney, Org. Lett., 2002, 4(17), 3001 CrossRef CAS PubMed.
- Y. X. Yanga, X. Q. Ana, M. Kanga, W. Zenga, Z. W. Yanga and H. C. Ma, Russ. J. Org. Chem., 2020, 56(3), 521 CrossRef.
- A. B. Leduc and T. F. Jamison, Org. Process Res. Dev., 2012, 16(5), 1082 CrossRef CAS.
- R. K. Bowman, A. D. Brown, J. H. Cobb, J. F. Eaddy, M. A. Hatcher, M. R. Leivers, J. F. Miller, M. B. Mitchell, D. E. Patterson, M. A. Toczko and S. Xie, J. Org. Chem., 2013, 78(23), 11680 CrossRef CAS PubMed.
- R. Anderson, K. Griffin, P. Johnston and P. L. Alsters, Adv. Synth. Catal., 2003, 345(4), 517 CrossRef CAS.
- (a) Y. Bessard and J. Heveling, WO 99/25696, 1999; (b) Y. Bessard and J. Heveling, US Pat., 6258958B1, 1999 Search PubMed; (c) Y. Kon, H. Yazawa, Y. Usui and K. Sato, Chem.–Asian J., 2008, 3, 1642 CrossRef CAS PubMed; (d) Y. Kon, T. Nakashima, A. Yada, T. Fujitani, S. Onozawa, S. Kobayashi and K. Sato, Org. Biomol. Chem., 2021, 19, 111 RSC.
- (a) N. Zotova, K. Hellgardt, G. H. Kesall, A. S. Jessiman and K. K. (M. ). Hii, Green Chem., 2010, 12, 2157 RSC; (b) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41(23), 4538 CrossRef CAS.
- C. Marsden, E. Taarning, D. Hansen, L. Johansen, S. K. Klitgaard, K. Egeblad and C. H. Christensen, Green Chem., 2008, 10, 168 RSC.
- (a) A. Gavrilidis, A. Constantinou, K. Hellgardt, K. K. (M. ). Hii, G. J. Hutchings, G. L. Brett, S. Kuhn and S. P. Marsden, React. Chem. Eng., 2016, 1, 595 RSC; (b) S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456 RSC; (c) H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noël, Chem. Soc. Rev., 2016, 45, 83 RSC.
- A. I. Alfano, M. Brindisi and H. Lange, Green Chem., 2021, 23, 2233 RSC.
- C. A. Hone and C. O. Kappe, Chem. Methods, 2021, 1, 454 CrossRef CAS.
- (a) T. Nishimura, T. Onoue, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 6750 CrossRef CAS PubMed; (b) J. Muzart, Tetrahedron, 2003, 59(31), 5789 CrossRef CAS; (c) X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180 RSC.
- (a) B. Durakovic, Period. Eng. Nat. Sci., 2017, 3, 421 Search PubMed; (b) S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605 CrossRef CAS.
- J. C. Taylor, A. Baker, M. R. Chapman, W. R. Reynolds, K. E. Jolley, G. Clemens, G. E. Smith, A. J. Blacker, T. W. Chamberlain, S. D. R. Christie, B. A. Taylor and R. A. Bourne, J. Flow Chem., 2021, 11, 75 CrossRef.
- TIBCO Software Inc., Data Science Textbook, 2020, https://docs.tibco.com/data-science/textbook, accessed: 19th September 2022 Search PubMed.
- StatSoft's Electronic Statistics Textbook, StatSoft Inc., 2006, http://www.statsoft.pl/textbook/stathome.html, accessed 19th September 2022 Search PubMed.
- B. A. Steinhoff, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2004, 126, 11268 CrossRef CAS PubMed.
- (a) R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32 CrossRef CAS; (b) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehadad and P. J. Dunn, Green Chem., 2016, 18, 288 RSC.
- D. Prat, J. Haylerb and A. Wells, Green Chem., 2014, 16, 4546 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra07003k |
| This journal is © The Royal Society of Chemistry 2022 |