
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of chiral-at-metal rhodium complexes from achiral tripodal tetradentate ligands: resolution and application to enantioselective Diels–Alder and 1,3-dipolar cycloadditions†
Alvaro G. Tejero,
María Carmona,
Ricardo Rodríguez *,
Fernando Viguri*,
Fernando J. Lahoz,
Pilar García-Orduña and
Daniel Carmona*
*,
Fernando Viguri*,
Fernando J. Lahoz,
Pilar García-Orduña and
Daniel Carmona*
Departamento de Catálisis y Procesos Catalíticos, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC – Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: dcarmona@unizar.es; riromar@unizar.es; fviguri@unizar.es
First published on 2nd December 2022
Abstract
An improved synthesis of the racemic rhodium compound [RhCl2(κ4C,N,N′,P-L1)] (1) containing an achiral tripodal tetradentate ligand is reported. Their derived solvate complexes [Rh(κ4C,N,N′,P-L1)(Solv)2][SbF6]2 (Solv = NCMe, 2; H2O, 3) are resolved into their two enantiomers. Complexes 2 and 3 catalyze the Diels–Alder (DA) reaction between methacrolein and cyclopentadiene and the 1,3-dipolar cycloaddition reaction between methacrolein and the nitrone N-benzylidenphenylamine-N-oxide. When enantiopure (ARh,RN)-2 was employed as the catalyst, enantiomeric ratios >99/1, in the R at C2 adduct, and up to 94/6, in the 3,5-endo isomer, were achieved in the DA reaction and in the 1,3-dipolar cycloaddition reaction, respectively. A plausible catalytic cycle that accounts for the origin of the observed enantioselectivity is proposed.
Introduction
Typically, a homogeneous chiral metal catalyst consists of a metal surrounded by a chiral ligand and accompanied by a range of achiral ligands. The source of chirality is the chiral ligand which configures a “chiral pocket” in the vicinity of the metal where the reaction between the catalytic substrates takes place.1As early as 1979, Brunner argued: “It therefore seemed promising to move the inducing chirality to the metal atom where the catalysis is occurring”.2 However, in 2000, 21 years later, Muñiz and Bolm recognized that asymmetric catalysis with enantiomeric chiral-at-metal complexes was still unknown and that the only approach to this type of catalysts was diastereomeric complexes in which the configuration of the metal centre is pre-determined by the coordination of a chiral ligand.3
It is interesting to point out that enantiomerically pure coordination compounds have been known since 1911 when Werner reported the resolution of the complex [CoX(en)2(NH3)]2+ (en = ethylenediamine, X = Cl or Br) into the individual mirror-imaged Δ- and Λ-enantiomers.4 However, it should be noted that the design and synthesis of efficient enantiopure catalysts in which the only stereogenic centre is the metal is not exempt from difficulties. On the one hand, given that different structural isomers are usually possible, the most suitable isomer must be selectively prepared or separated from the obtained mixture. In addition, this isomer will be obtained as a racemate that must be resolved following protocols that generally require a great deal of time with no guarantee of success.5 On the other, for these catalysts to achieve high efficiency, it is very convenient that they meet two requirements that seem contradictory a priori: configurational stability and labile coordination positions. Most likely for these reasons, the systematic development of chiral-only-at-metal catalysts has occurred only very recently. Indeed, although in 2003, Fontecave et al. demonstrated for the first time that chiral-only-at-metal bis-diimine Ru(II) complexes catalyze the oxidation of sulfides to sulfoxides by hydrogen peroxide with up to 59/41 er,6 only since 2013 the synthesis, resolution and catalytic applications of stereogenic-only-at-metal chiral catalysts has been systematically developed. From this date, Meggers group and others have been developing a family of bis-cyclometalated octahedral rhodium(III), iridium(III), ruthenium(II) and iron(II) complexes that efficiently catalyze a variety of organic transformations following Lewis acid,7 ligand sphere mediated7b–e,8 and photoredox7b–e,9 catalytic protocols.
Also in this line, Gladysz's group reported that octahedral enantiopure salts of the tris-chelate ethylenediamine complexes [M(en)3][Barf]n (M = Co, Cr, Rh, Ir, n = 3; M = Pt, n = 4; Barf = B(3,5-C6H3(CF3)2)4) catalyze the addition of malonates to cyclopentenone or trans-β-nitrostyrene and that of the methyl 2-oxocyclopentanecarboxylate to di-tert-butylazodicarboxylate following a second coordination sphere mechanism involving substrate activation by hydrogen bonding to the coordinate NH2 units. Modest enantioselectivities were achieved in all cases.10 Also, pseudo-octahedral geometry presents an enantiopure chiral-at-ruthenium complex containing a cyclometalated chelating NHC ligand that efficiently catalyzes the asymmetric cross metathesis, asymmetric ring closing metathesis and asymmetric ring opening cross metathesis of olefins.11
On the other hand, tetrahedral complexes with stereogenic metal centres are rarely used as chiral catalysts mainly because they are too labile to ensure the absolute configuration of the metal centre. However, Shionoya et al. have reported the preparation and resolution of the tetrahedral chiral zinc complex I with high configurational stability using the achiral tridentate ligand H2L depicted in Scheme 1. When enantiopure S-I was applied to the asymmetric oxa-Diels–Alder reaction of Danishefsky's diene with 1-naphthaldehyde the Diels–Alder R-adduct was obtained in 98% yield with 93.5/6.5 er.12
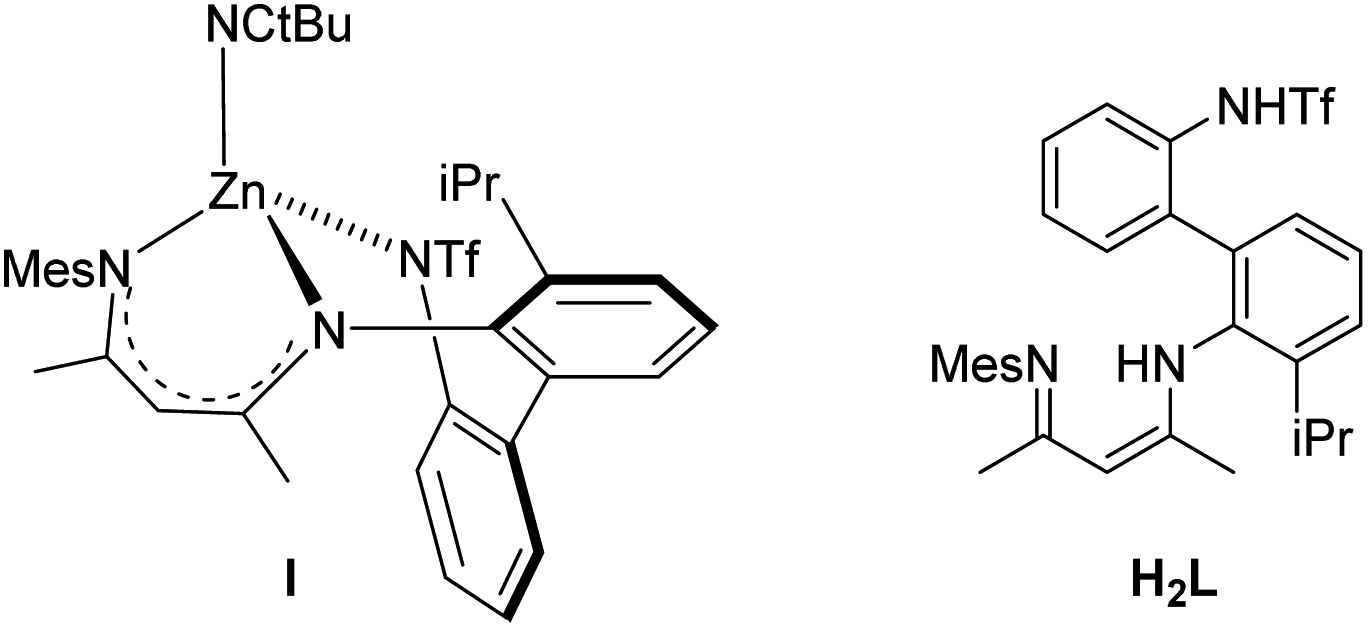 | ||
| Scheme 1 Chiral at zinc tetrahedral complex I and the achiral ligand H2L. | ||
We have recently reported the application of achiral tripodal tetradentate ligands to the synthesis of octahedral chiral-at-metal rhodium, iridium and ruthenium complexes.13 In particular, enantiopure rhodium complexes efficiently catalyze the enantioselectively Diels–Alder reaction between methacrolein and cyclopentadiene with enantiomeric ratios of up to >99/1 (ref. 13c) and, on the basis of experimental NMR studies and DFT calculations, a detailed mechanistic pathway for the Friedel–Crafts reaction of trans-β-nitrostyrene and N-methyl-2-methylindole reaction is proposed, including an explanation of the origin of the obtained enantioselectivity.13a
Herein, we report on the synthesis and stereochemical resolution of the rhodium complex 2 containing the tripodal tetradentate L1 (Scheme 2) and its application to enantioselective Diels–Alder and 1,3-dipolar cycloadditions.
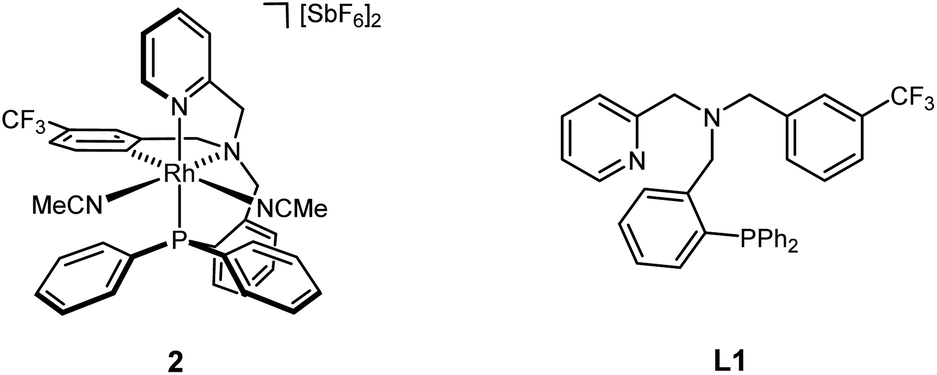 | ||
| Scheme 2 Chiral rhodium complex 2 bearing the achiral ligand L1. | ||
Results and discussion
Synthesis of neutral dichloride complex [RhCl2(κ4C,N,N′,P-L1)] (1)
The achiral tripodal tetradentate ligand L1 has been prepared as reported elsewhere.13e The synthesis of the dichlorido complex [RhCl2(κ4C,N,N′,P-L1)] (1) has been previously accomplished following the three steps protocol A depicted in Scheme 3.13e Reaction of RhCl3 with the ligand L1 in refluxing ethanol affords a mixture of fac-OC-6-43 (ref. 14) and mer-OC-6-41 trichlorido isomers that can be completely isomerized to the fac diastereomer by refluxing the mixture overnight in the same solvent. The cyclometalated isomer OC-6-54 of the dichlorido complex 1 could be obtained by refluxing in methanol pure trichlorido fac-OC-6-43 in the presence of NaOAc.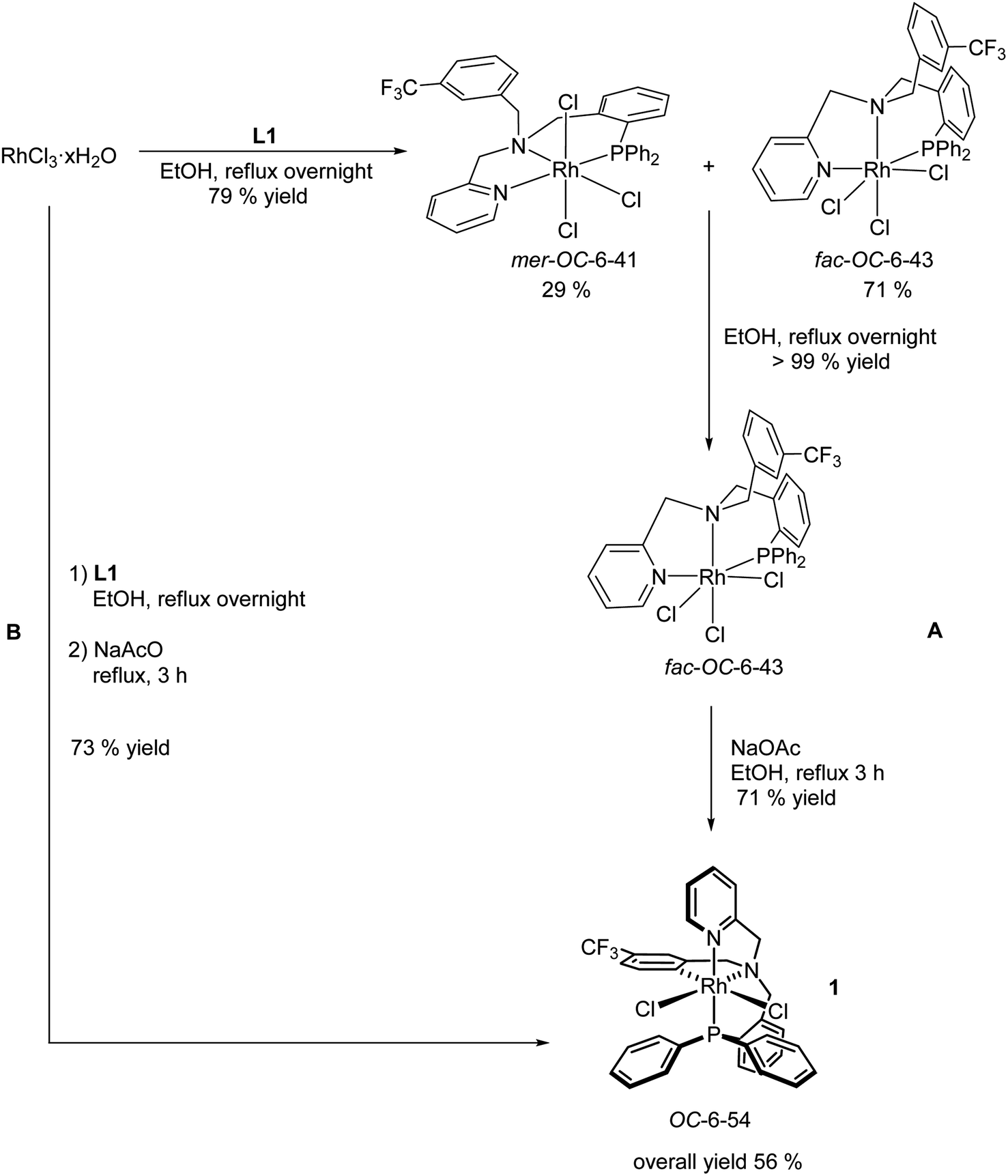 | ||
| Scheme 3 Synthesis of the dichlorido complex [RhCl2(κ4C,N,N′,P-L1)] as described in the bibliography.13e (A) and as reported in this work (B). | ||
We have developed an improved synthesis of the dichlorido complex 1 following a one-pot, two steps reaction. First, RhCl3 reacts with L1 in refluxing EtOH; then, the resulting mixture is heated in the same solvent in the presence of NaOAc for 3 h (protocol B, Scheme 3). Only metalation at C6 carbon of the trifluoro aryl substituent was observed in both preparative routes.13e
Synthesis of the solvate complexes [Rh(κ4C,N,N′,P-L1)(NCMe)2][SbF6]2 (2) and [Rh(κ4C,N,N′,P-L1)(OH2)2][SbF6]2 (3)
Treatment of the dichlorido complex 1 with AgSbF6 in acetonitrile or in wet dichloromethane affords the solvate complexes [Rh(κ4C,N,N′,P-L1)(NCMe)2][SbF6]2 (2) or [Rh(κ4C,N,N′,P-L1)(OH2)2][SbF6]2 (3), respectively (eqn (1)).13d
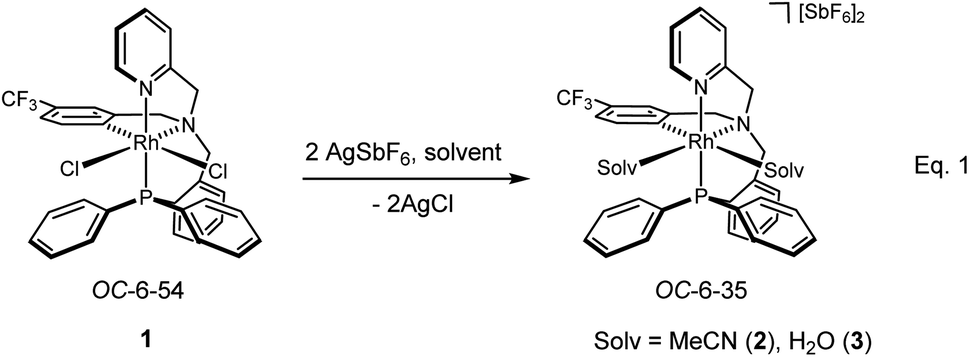 | (1) |
The compounds were characterized by analytical and spectroscopic methods (see Experimental) and by the X-ray determination of the crystal structure of the acetonitrile compound 2. The metal and the sp3 nitrogen atoms of the ligand are stereogenic centres. However, the 1H, 31P{1H} and 13C{1H} NMR spectra of complexes 2 and 3 consists of only one set of sharp resonance signals indicating that only one of the three possible isomers13e has been isolated. NMR measurements allow us to ascertain the geometry of the isolated isomer. As the spectroscopy features of both compounds are comparable, we will focus our discussion on the acetonitrile compound 2 as the model. The value of the J(PH) coupling constants together with COSY, HSQC and HMBC experiments permit the assignment of the three pairs of diastereotopic protons which have been labelled as CH2(Py), CH2(Ph) and CH2(P) (see Scheme 4A). Scheme 4B shows the NOE pattern measured for these six methylene protons. Additionally, the methyl protons of the acetonitrile ligand trans to the sp3 nitrogen atom exhibit NOE relationship with the 6-CH(Py) and 6-CH(Ph) protons of the tetradentate ligand. Accordingly, a J(PH) coupling constant of 4.0 Hz was measured for the proton-6 of the pyridine moiety (6-CH(Py)). At this point, it should be noted that when the ligand L1 coordinates with a metal in an octahedral geometry with a κ4C,N,N′,P coordination mode, the relative configuration of the sp3 nitrogen atom becomes pre-determined by the configuration at the metal. In particular, in the case of complex 2, the A (C) configuration14d at the rhodium atom forces an R (S) configuration at the sp3 nitrogen atom. Hence, we propose that compound 2 has been obtained as a racemic mixture of the isomers CRh,SN and ARh,RN.14,15 as, indeed, it has been corroborated by the X-ray determination of the crystal structure of compound 2.
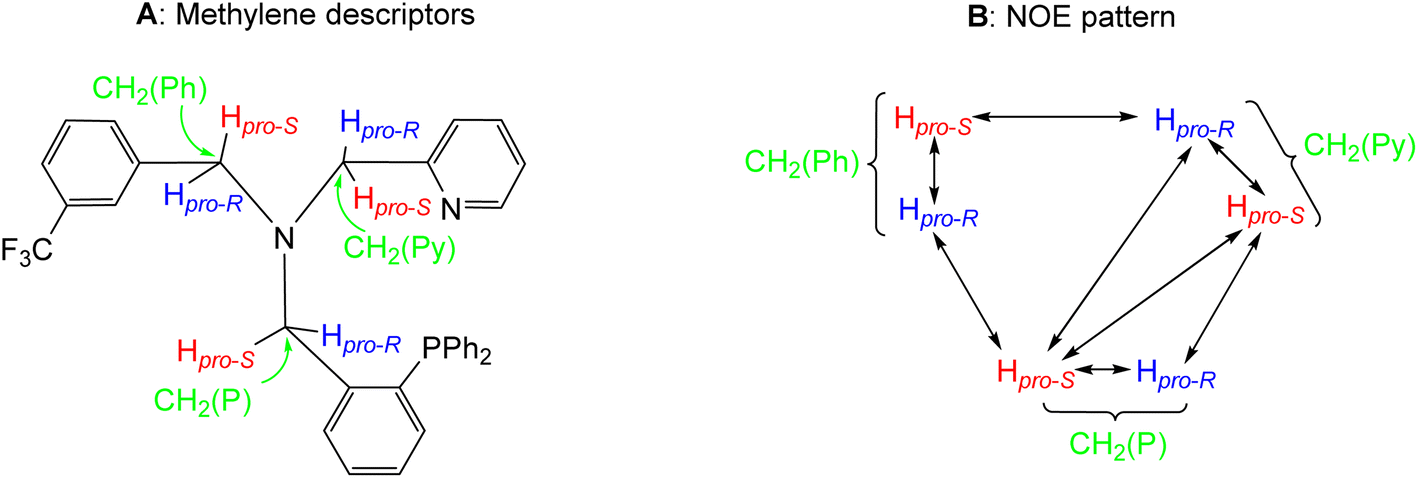 | ||
| Scheme 4 Assignment of the methylene descriptors (A) and NOE pattern (B) on OC-6-35 isomers of complex 2. | ||
A view of the structure of the CRh,SN isomer of the cation of the complex 2 (ref. 16) is depicted in Fig. 1 and relevant characteristics of the metal coordination sphere are summarized in Table 1. The cation presents an octahedral geometry in which four ligating atoms of L1 are coordinated in a tripodal fashion and two acetonitrile ligands occupy the two remaining coordination sites arranged cis to each other. The phosphorus atom is trans to the pyridinic nitrogen N(2), while the nitrile nitrogen atoms N(3) and N(4) are found to be trans to the sp3 nitrogen N(1) and to the aromatic C(28) carbon atoms, respectively. As a reflect of the different trans influence the Rh-N(4) bond distance (2.170(4) Å, trans to carbon atom) is about 0.142 Å greater than the Rh-N(3) bond distance (2.028(4) Å, trans to the sp3 nitrogen atom). The complex crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group of the triclinic crystallographic system and, therefore, the unit cell contains two enantiomers of the chiral complex OC-6-35 that, taking into account the arrangement of the coordination atoms around the metal, are the CRh,SN and ARh,RN diastereomers. It is important to remark that the relative configuration of the metal is retained when the two chloride ligands are released and exchanged by two solvent molecules. Changes in the notation of the descriptors are due to the application of the nomenclature rules for octahedral complexes.14d
space group of the triclinic crystallographic system and, therefore, the unit cell contains two enantiomers of the chiral complex OC-6-35 that, taking into account the arrangement of the coordination atoms around the metal, are the CRh,SN and ARh,RN diastereomers. It is important to remark that the relative configuration of the metal is retained when the two chloride ligands are released and exchanged by two solvent molecules. Changes in the notation of the descriptors are due to the application of the nomenclature rules for octahedral complexes.14d
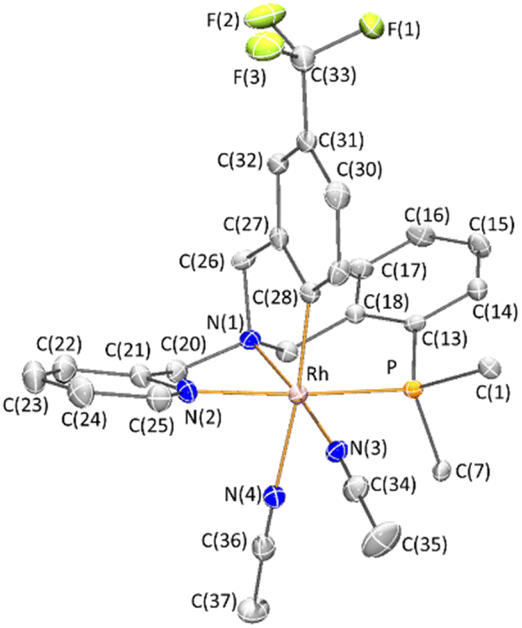 | ||
| Fig. 1 The crystal structure of racemic cation of 2. The (SN)-OC-6-35-C enantiomer of the racemate is depicted. For clarity, hydrogen atoms have been omitted, and only ipso-carbon atoms of phenyl groups are depicted. | ||
| Rh-P | 2.2968(12) | N(1)-Rh-N(2) | 80.10(15) |
| Rh-N(1) | 2.069(4) | N(1)-Rh-N(3) | 174.12(15) |
| Rh-N(2) | 2.102(4) | N(1)-Rh-N(4) | 90.59(14) |
| Rh-N(3) | 2.028(4) | N(1)-Rh-C(28) | 84.30(16) |
| Rh-N(4) | 2.170(4) | N(2)-Rh-N(3) | 94.02(15) |
| Rh-C(28) | 2.007(4) | N(2)-Rh-N(4) | 88.84(14) |
| P-Rh-N(1) | 95.17(11) | N(2)-Rh-C(28) | 86.84(16) |
| P-Rh-N(2) | 174.91(11) | N(3)-Rh-N(4) | 89.56(14) |
| P-Rh-N(3) | 90.69(11) | N(3)-Rh-C(28) | 95.16(16) |
| P-Rh-N(4) | 93.15(11) | N(4)-Rh-C(28) | 173.81(16) |
| P-Rh-C(28) | 90.81(13) |
From the point of view of the catalytic applications, an important feature of the chiral complex 2 is the relative disposition of the phenyl groups of the PPh2 fragment and the two coordinated acetonitrile molecules. As it can be seen in Fig. 2, whereas one face of the plane defined by the two linear acetonitrile molecules is shielded by the phenyl substituents of the diphosphino group, the other face is unencumbered. Therefore, when, in complex 2, prochiral substrates coordinate the metal displacing the labile acetonitrile ligands, the attack of a putative reagent would take preferably through the less hindered enantioface of the substrate originating enantioselectivity in the involved process.
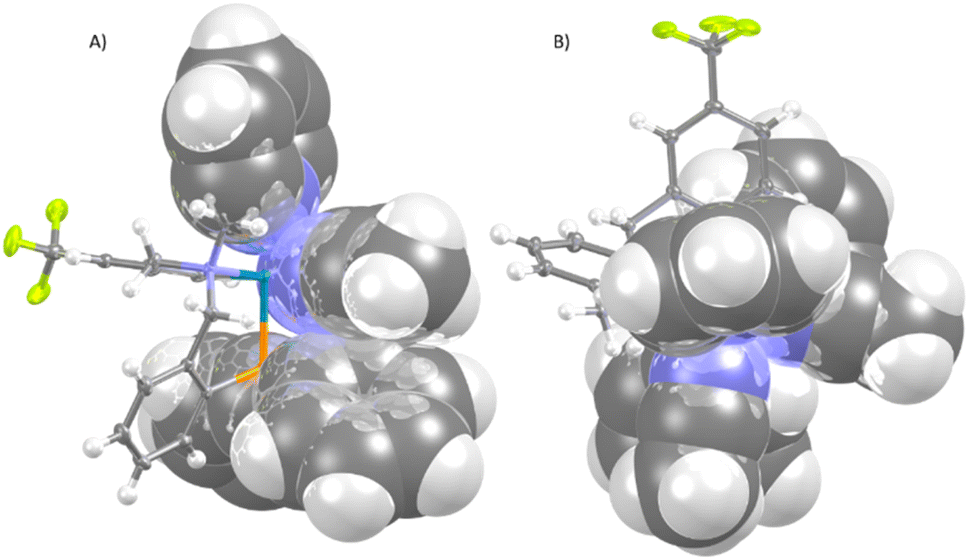 | ||
| Fig. 2 View of the cation of the complex (SN)-OC-6-35-C-2 along (A) and perpendicular to (B) the acetonitriles mean plane, showing the phenyl rings shielding. | ||
Resolution of the ARh,RN/CRh,SN racemate of the solvate complex 2
As above mentioned, complex 2 was obtained as an equimolar mixture of the CRh,SN and ARh,RN diastereomers. In order to apply this complex as an asymmetric catalyst, it is necessary to resolve the obtained racemate. To this end, we developed a diastereomerization protocol of resolution using (S)-phenylglycine as a chiral auxiliary.The determination of the absolute configuration of each of the complexes has been carried out by NMR from a mixed solution of the two diastereomers (see ESI†). In particular, the 6-CH(Py) proton (δ = 8.32 ppm) of one diastereomer exhibits a nuclear Overhauser effect (NOE) with the asymmetric C*H proton (δ = 3.98 ppm) determining the configuration for this diastereomer as (ARh,SN,SC)-4. In the other diastereomer, determined as (CRh,RN,SC)-4, the C*H proton (δ = 3.10 ppm) correlates by NOE with the phenyl protons of the PPh2 fragment (δ = 7.74 ppm). The 31P{1H} NMR spectrum of the mixture of diastereomers consists of two doublets centred at 29.67 ppm (J(RhP) = 130.8 Hz), assigned to the (ARh,SN,SC)-4 isomer, and at 28.83 ppm (J(RhP) = 130.1 Hz), attributed to the (CRh,RN,SC)-4 isomer (see ESI†). Crystallization of the 1/1 mixture of isomers from MeOH/Et2O affords a 57/43 molar ratio mixture from which individual diastereomers were separated by chromatography.
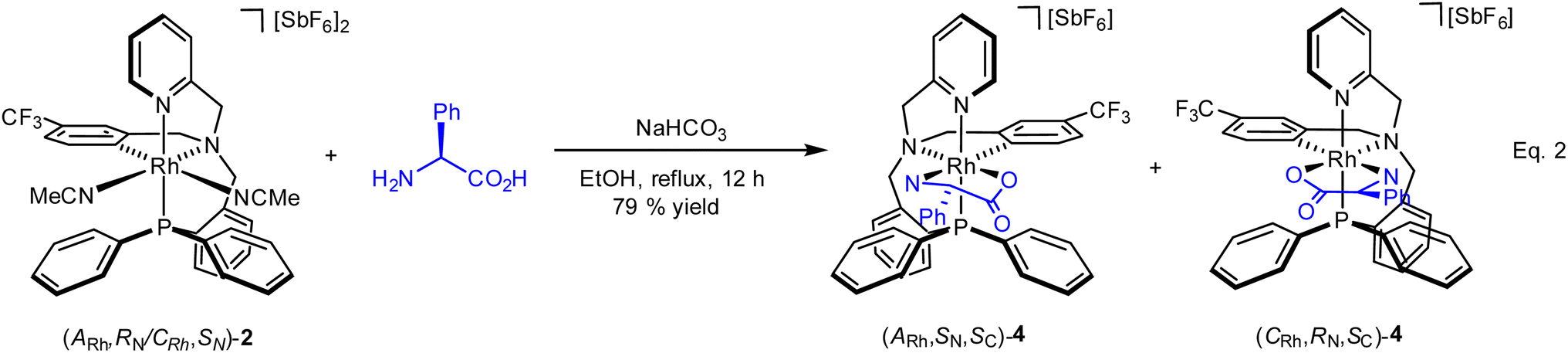 | (2) |
The 31P{1H} NMR spectrum of the starting mixture and those of each diastereomer, once separated by chromatography, are shown in Fig. 3.
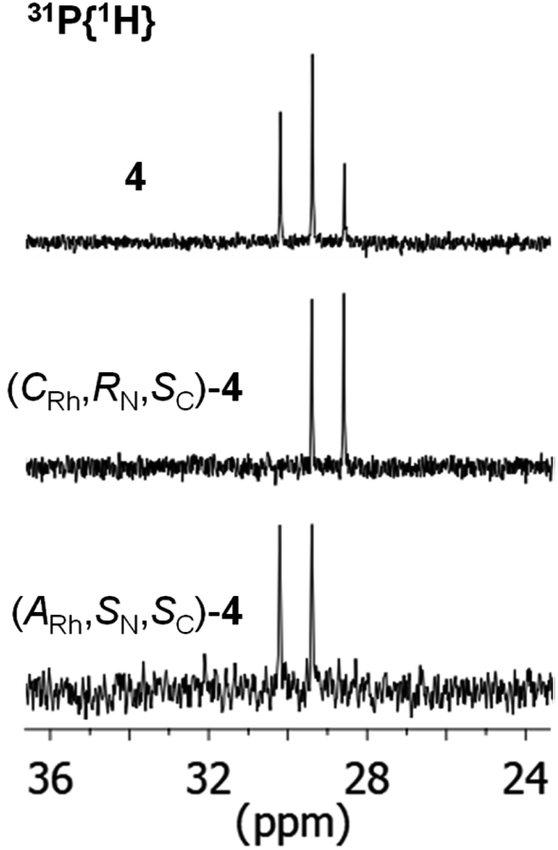 | ||
| Fig. 3 31P{1H} NMR of the starting mixture of complexes 4 (above) and the individual diastereomers (CRh,RN,SC)-4 (middle) and (ARh,SN,SC)-4 (below). | ||
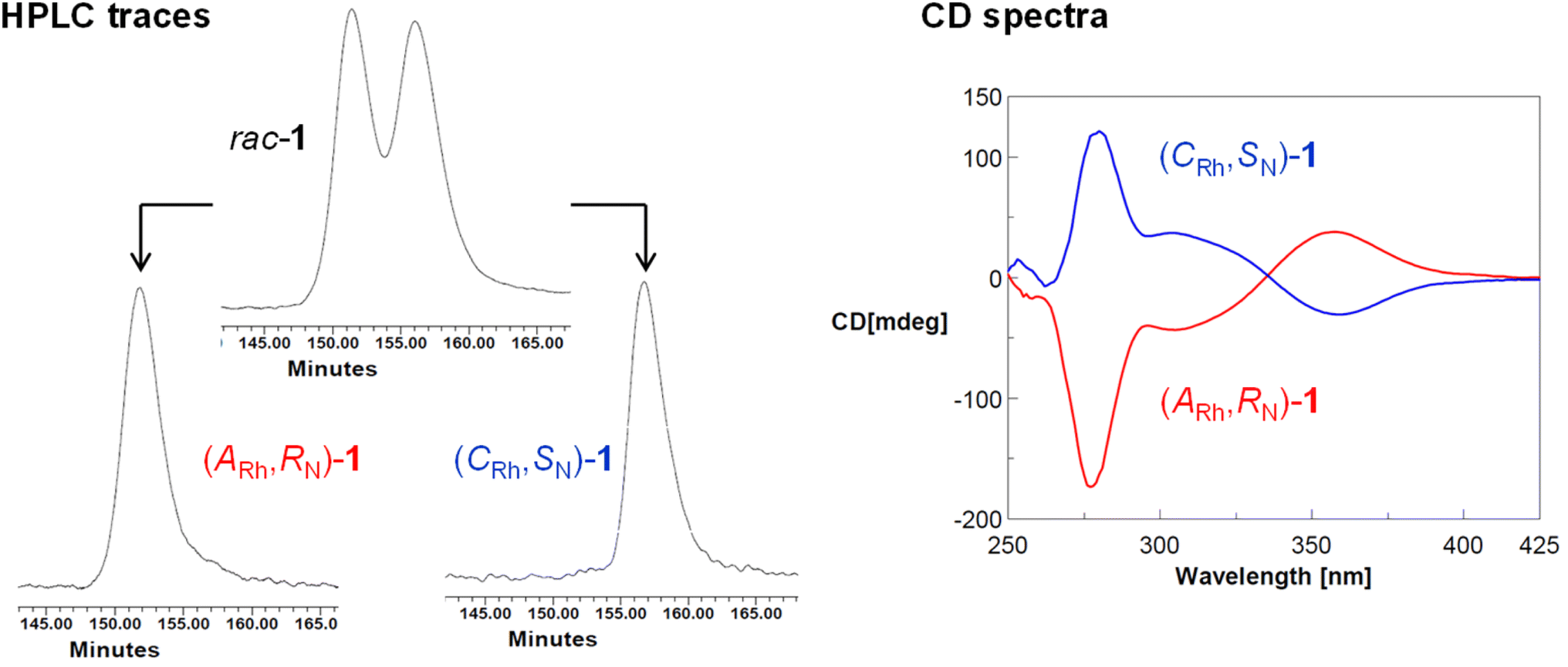 | ||
| Fig. 4 HPCL traces of rac-1, (ARh,RN)-1 and (CRh,SN)-1 and CD spectra of (ARh,RN)-1 (red) and (CRh,SN)-1 (blue). | ||
From the enantiopure chlorido complex (ARh,RN)-1, enantiopure solvate complex (ARh,RN)-2 was obtained following the treatment above reported for the preparation of racemic solvate complexes from racemic chlorido complex 1 (eqn (1)).
Catalytic Diels–Alder reaction between methacrolein and cyclopentadiene
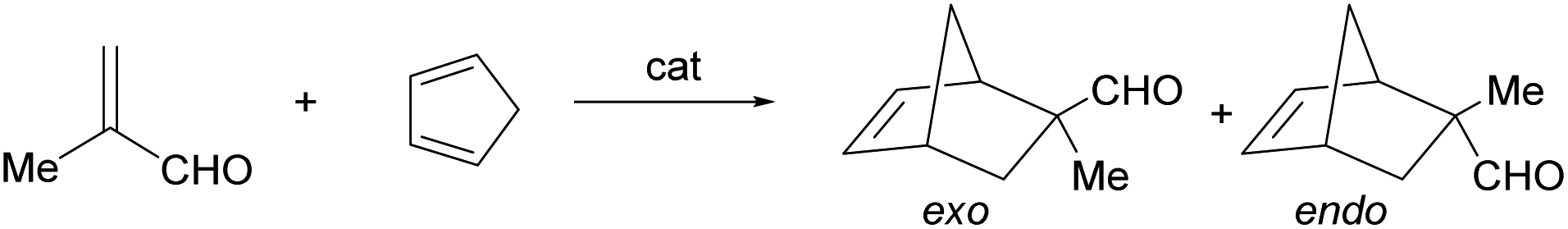
Complexes 2 and 3 catalyze the Diels–Alder reaction between methacrolein (MA) and cyclopentadiene (HCp) in dichloromethane. To determine the optimal conditions, we first tested the activity of rac-2 and rac-3. As shown in Table 2, at 298 K, to get good conversions with a catalyst loading of 5 or 10 mol%, several hours are needed (entries 1, 2, 5 and 6). Decreasing temperature increases the reaction time and diminishes the obtained exo/endo ratio of the adduct (entries 3 and 4). Complex 2 is more active than complex 3 (compare entries 1 and 2 with entries 5 and 6, respectively). At 5 mol% of catalyst loading, better exo/endo ratios than at 10 mol% catalyst loading were achieved but, to reach similar conversions, it is necessary to increase importantly the reaction time (entries 1, 2 and 5, 6). Taking into account all these data, we carried out the reaction between methacrolein and cyclopentadiene using 10 mol% of enantiopure (ARh,RN)-2, at 298 K. After 8 h of reaction, 97% of conversion was achieved with an exo/endo ratio of 93/7. Notably, enantioselectivity in the R at C2 isomer almost perfect was obtained (er > 99/1, entry 7). Comparing the catalytic results using the CF3-substituted (ARh,RN)-2 complex with those of the unsubstituted one13c (after 24 h at 298 K, conv. 93%; exo/endo ratio: 88/12; er: 91/9), the CF3-substituted complex has about 3 times higher catalytic activity, better diastereoselectivity and a remarkable increase in enantioselectivity, from 91/9 to >99/1 of er. These catalytic differences are understood by a large difference in the trans effect favourable to the CF3 group which enhances the catalytic activity by favouring the de-and coordination of the substrates. On the other hand, enantioselectivity increases because the non-catalytic route to products becomes less competitive versus the catalytic route.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Cat. loading (mol%) | T (K) | t (h) | Conv.b (%) | exo/endo | erc,d (R/S) |
| a Reaction conditions: catalyst, 1.16 × 10−3 or 2.32 × 10−3 mmol (5 or 10 mol%); MA, 23.0 × 10−3 mmol; HCp, 81.0 × 10−3 mmol and 10 mg of 4 Å MS, in 0.5 mL of CD2Cl2.b Based on MA. Determined by 1H NMR.c Determined for the exo isomer by integration of the 1H NMR signals in presence of the chiral shift reagent (+)-Eu(hfc)3.d Absolute configuration of the major exo adduct (R at C2) was established by comparison of the aldehyde proton splitting in presence of (+)-Eu(hfc)3 with previous work indicating the relative splitting of the (R)-exo and (S)-exo adducts.18 | |||||||
| 1 | (rac)-2 | 10 | 298 | 6 | 92 | 95/5 | — |
| 2 | (rac)-2 | 5 | 298 | 15 | 86 | 98/2 | — |
| 3 | (rac)-2 | 10 | 273 | 24 | 82 | 83/17 | — |
| 4 | (rac)-2 | 10 | 263 | 140 | 61 | 88/12 | — |
| 5 | (rac)-3 | 10 | 298 | 12 | 90 | 94/6 | — |
| 6 | (rac)-3 | 5 | 298 | 24 | 87 | 96/4 | — |
| 7 | (ARh,RN)-2 | 10 | 298 | 8 | 97 | 93/7 | >99/1 |
Catalytic 1,3-dipolar cycloaddition reaction between methacrolein and N-benzylidenphenylamine-N-oxide
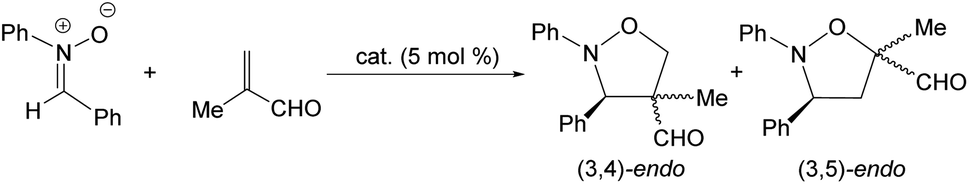
Complex 2 also catalyzes the 1,3-dipolar cycloaddition reaction between methacrolein and the nitrone N-benzylidenphenylamine-N-oxide. Table 3 lists a selection of the results obtained together with the reaction conditions employed. First, we tested rac-2 as the catalyst (entries 1–4). To achieve moderate to good conversions, 24 h of reaction are necessary even operating at 298 K (entry 1). Only endo isomers were detected and good regioselectivity in favour of the 3,5-endo isomer was obtained under all conditions explored. At 298 K, using (ARh,RN)-2 as the catalyst, an enantiomeric ratio of 94/6 was measured for the major adduct of the 3,5-endo diastereomer (entry 5). Lowering temperature to 273 K diminishes the er from 94/6 to 84/16 (entry 6). As long reaction times are necessary to reach acceptable conversions, a greater contribution of the thermal reaction is, probably, at the origin of the observed lowering of the er.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | T (K) | t (h) | Conv.b (%) | (3,4)/(3,5)c | erd |
| a Reaction conditions: catalyst 1.16 × 10−3 mmol (5.0 mol%); MA 70.0 × 10−3 mmol, 10 mg of 4 Å MS, and nitrone 35.0 × 10−3 mmol in 0.5 mL of CD2Cl2.b Based on nitrone. Determined by 1H NMR.c Only endo isomers were detected.d Determined for the 3,5-endo isomer by integration of the 1H NMR signals of the diastereomeric (S)-methylbenzylimine derivatives.19 | ||||||
| 1 | (rac)-2 | 298 | 24 | 64 | 5/95 | — |
| 2 | (rac)-2 | 281 | 24 | 17 | 2/98 | — |
| 3 | (rac)-2 | 273 | 24 | 8 | 6/94 | — |
| 4 | (rac)-2 | 263 | 24 | 0 | — | — |
| 5 | (ARh,RN)-2 | 298 | 24 | 78 | 4/96 | 94/6 |
| 6 | (ARh,RN)-2 | 273 | 164 | 68 | 12/88 | 84/16 |
Proposed mechanism and origin of the enantioselectivity
The proposed catalytic cycle is depicted in Fig. 5A. One molecule of methacrolein (MA) replaces the more labile molecule of acetonitrile trans to the aromatic carbon atom of the enantiopure catalyst (ARh,RN)-2. Assuming that the methacrolein coordinates as a planar molecule with an s-trans conformation for the double carbon–oxygen and carbon–carbon bonds and with an E-configuration around the carbon–oxygen double bond,20 its Si-face becomes much more accessible to the cyclopentadiene or to the nitrone than its Re-face, because, in the chiral pocket generated in the diastereopure catalyst, the Re-face is shielded by the phenyl rings of the PPh2 group (Fig. 5B). An attack of the cyclopentadiene substrate through the Si-face of the methacrolein renders the R at C2 enantiomer of the DA adduct as it was experimentally observed. On the other hand, an endo approach of the nitrone through the Si-face of the coordinated methacrolein would render the 3R,5R isomer of the corresponding isoxazolidine.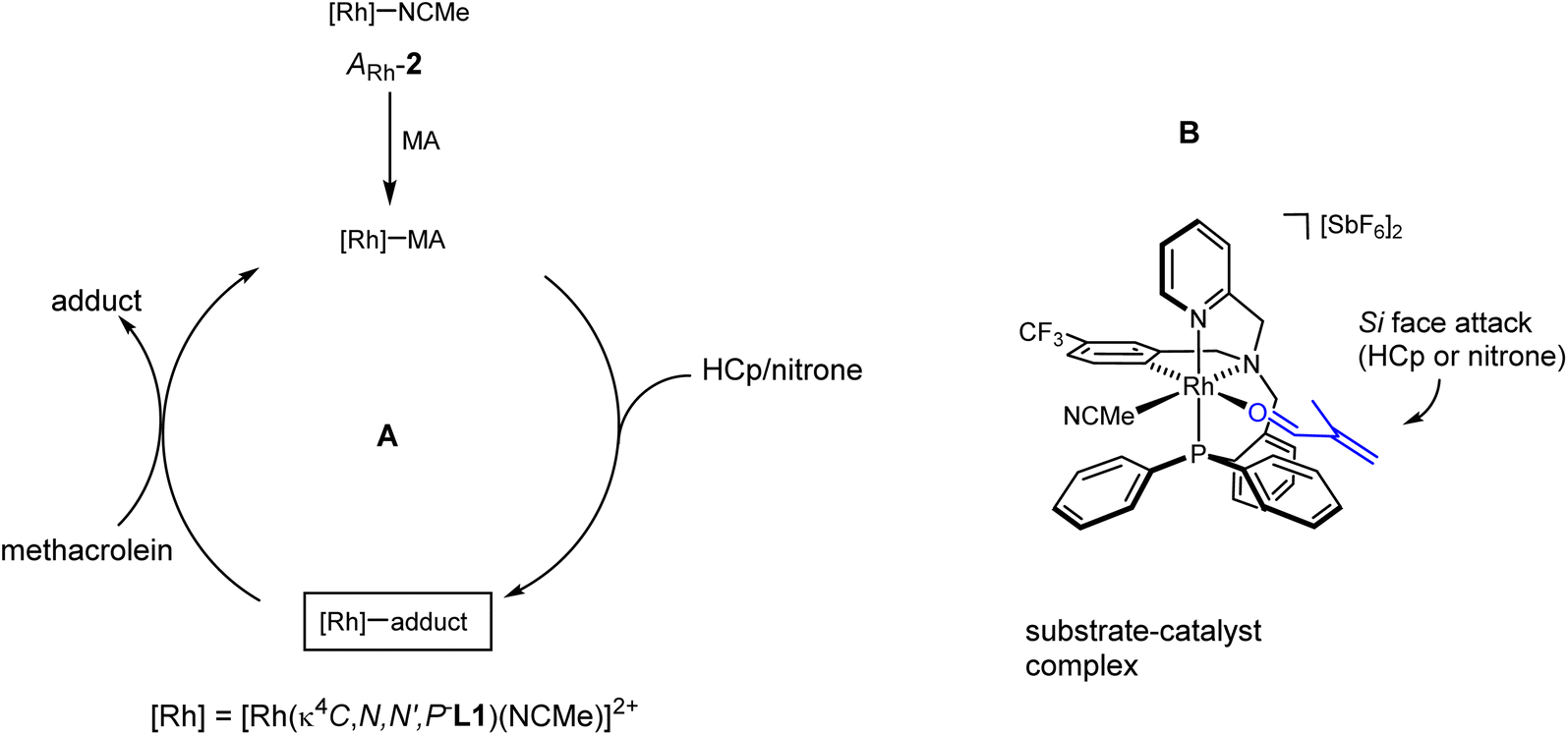 | ||
| Fig. 5 Proposed catalytic cycle (A) and the suggested model for the asymmetric induction (B). | ||
Conclusions
Judiciously designed achiral tripodal tetradentate ligands are able to generate octahedral rhodium complexes in which the metal is a stereogenic centre. This is the case of the dichlorido complex 1. The frame generated by the polydentate ligand confers configurational stability to the octahedral molecule even when the two remaining positions are occupied by labile solvent molecules as in complexes 2 and 3. Notably, the compounds 2 and 3 meet two important requirements that make them good candidates for asymmetric catalysts: configurational stability and labile coordination positions. Indeed, after stereochemical resolution the chiral complex (ARh,RN)-2 catalyzes the Diels–Alder reaction between methacrolein and cyclopentadiene with a >99/1 enantiomeric ratio. Furthermore, the same catalyst mediates the 1,3-dipolar cycloaddition reaction between methacrolein and the nitrone N-benzylidenphenylamine-N-oxide affording only endo-adducts with enantiomeric ratios of up to 94/6 for the 3,5-endo adduct. The molecular structure of complex 2, determined by X-ray diffraction analysis, reveals the presence of a chiral pocket around the labile solvent molecules coordination sites. When a prochiral substrate coordinates the metal inside this chiral pocket, the two phenyl groups of the tetradentate ligand shield one of its enantiofaces. Consequently, an incoming reagent preferentially approaches the coordinated substrate through its clearer enantioface resulting in enantioselection.Finally, the findings reported in this work open the door to the design and use of new polydentate ligands capable of generating stereogenic metal centres both in octahedral metal geometries and in metal complexes with other coordination numbers. Likewise, the studies can be extended to other metals and, in particular, to cheap, non-toxic and earth-abundant metals of the first transition series, with inherent economic and environmental benefits. Further work in this area is in progress in our laboratory and will be reported in due course.
Experimental
All preparations have been carried out under argon, unless otherwise stated. All solvents were treated in a PS-400-6 Innovative Technologies Solvent Purification System (SPS). Infrared spectra were recorded on PerkinElmer Spectrum-100 (ATR mode) FT-IR spectrometer. CD spectra were determined in MeOH (ca. 4 × 10−4 mol L−1 solutions) in a 1 cm path length cell by using a Jasco-810 apparatus. Carbon, hydrogen and nitrogen analyses were performed using a PerkinElmer 240 B microanalyzer. 1H, 13C and 31P spectra were recorded on a Bruker AV-300 (300.13 MHz), a Bruker AV-400 (400.16 MHz) or a Bruker AV500 (500.13 MHz) spectrometers. Chemical shifts are expressed in ppm upfield from SiMe4, 85% H3PO4 (31P) or CFCl3 (19F). COSY, NOESY, HSQC, HMQC, and HMBC 1H–X (X = 1H, 13C, 31P) correlation spectra were obtained using standard procedures. Flash chromatography was performed on an Interchim (PuriFlash 450) instrument using a PF-50SIHP column (25 g, 30 μm).Improved synthesis of the complex 1
At RT, to a suspension of RhCl3 × H2O (1.00 g, 3.85 mmol) in 20 mL of ethanol, 2.07 g of L1 were added. The resulting suspension was stirred under reflux overnight. During this time, the colour of the suspension gradually changes from pink-red to yellow. After the reaction time, the suspension was cooled to room temperature and 2.21 g (8.31 mmol) of NaAcO was added. The resulting suspension was stirred for 3 h under reflux. During this time, a yellow precipitate was formed. The solid was filtered off and extracted with CH2Cl2 (3 × 15 mL). The resulting yellow solution was vacuum-dried to give analytically pure compound 1. Yield: 2.01 g (73%). The spectroscopic data for complex 1 were in agreement with the previously reported in the literature.13ePreparation and characterization of the complex 2
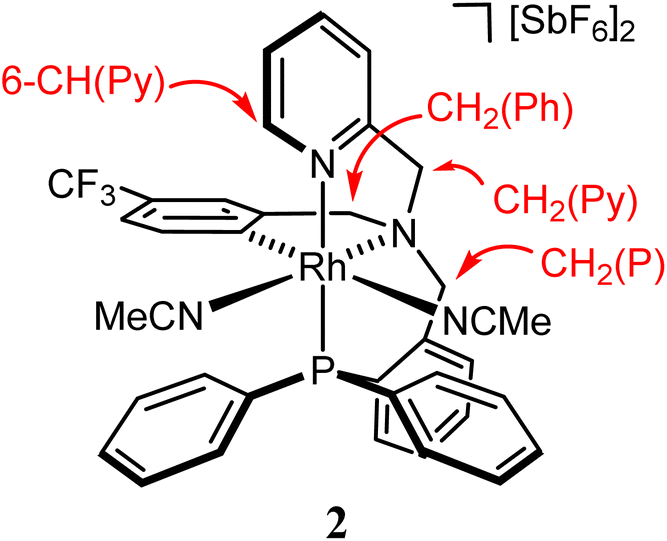 To a solution of [RhCl2(κ4C,N,N′,P-L1)] (1) (1.00 g, 1.40 mmol) in 40 mL of acetonitrile, 963.4 mg (2.81 mmol) of AgSbF6 were added. The resulting suspension was placed under reflux for 40 h and then filtered to remove any insoluble material. The resulting light yellow solution was evaporated to ca. 1 mL. The slow addition of Et2O (25 mL) led to the precipitation of a pale yellow solid that was filtered off, washed with Et2O (3 × 5 mL) and vacuum-dried. Yield: 1.54 g (92%) Anal. calcd C37H33F15N4PRhSb2·2CH2Cl2: C, 34.30; H, 2.73; N, 4.10. Found: C, 34.13; H, 2.67; N, 3.97. IR (solid, cm−1): ν(CN) 2327 (br), ν(CN) 2298 (br), ν(SbF6) 654 (s). 1H NMR (500.13 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 8.50 (broad pseudo-triplet, J = 4.6 Hz, 1H, 6-CH(Py)), 8.01 (ptd, J = 7.8, 1.4 Hz, 1H, CH(Ar)), 7.80–7.42 (m, 14H, H(Ar)), 6.97 (d, J = 9.6 Hz, 1H, 5-CH(Ph)), 6.84–6.78 (m, 4H, 2× CH(Ar), 6-CH(Ph), 3-CH(Ph)), 5.22 (d, J = 15.8 Hz, 1H, pro-S-CH2(Py)), 4.75 (d, J = 16.0 Hz, 1H, pro-R-CH2(P)), 4.74 (d, J = 14.0 Hz, 1H, pro-R-CH2(Py)), 4.64 (d, J = 17.9 Hz, 1H, pro-R-CH2(Ph)), 4.52 (ddd, J = 14.1, 5.0, 1.8 Hz, 1H, pro-S-CH2(P)), 4.07 (d, J = 17.9 Hz, 1H, pro-S-CH2(Ph)), 2.69 (s, 3H, NCMe cis to C), and 2.42 (s, 3H, NCMe trans to C). 13C{1H} NMR (125.77 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 155.79 (d, J = 1.9 Hz, 2-C(Py)), 149.30 (brdd, JRh-C = 29.1 Hz, JP-C = 8.7 Hz, CRh), 148.30 (s, 6-CH(Py)), 148.16 (s, 2-C(Ph)), 141.05–128.45 (m, 18C(H)(Ar)), 127.30 (q, JRh-C = 32.4 Hz, 4-C(Ph)), 127.08 (d, JRh-C = 5.8 Hz, NCMe cis to C), 123.52 (q, JF-C = 271.40 Hz, CF3), 125.98–120.26 (m, 6C(H)(Ar)), 124.13 (brs, NCMe trans to C), 74.31 (s, CH2(Py)), 66.95 (d, J = 7.9 Hz, CH2(P)), 66.24 (s, CH2(Ph)), 3.95 (s, NCMe cis to C), 1.08 (s, NCMe trans to C). 19F{1H} NMR (282.33 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = −65.52 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 28.58 (d, J = 114.1 Hz).
To a solution of [RhCl2(κ4C,N,N′,P-L1)] (1) (1.00 g, 1.40 mmol) in 40 mL of acetonitrile, 963.4 mg (2.81 mmol) of AgSbF6 were added. The resulting suspension was placed under reflux for 40 h and then filtered to remove any insoluble material. The resulting light yellow solution was evaporated to ca. 1 mL. The slow addition of Et2O (25 mL) led to the precipitation of a pale yellow solid that was filtered off, washed with Et2O (3 × 5 mL) and vacuum-dried. Yield: 1.54 g (92%) Anal. calcd C37H33F15N4PRhSb2·2CH2Cl2: C, 34.30; H, 2.73; N, 4.10. Found: C, 34.13; H, 2.67; N, 3.97. IR (solid, cm−1): ν(CN) 2327 (br), ν(CN) 2298 (br), ν(SbF6) 654 (s). 1H NMR (500.13 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 8.50 (broad pseudo-triplet, J = 4.6 Hz, 1H, 6-CH(Py)), 8.01 (ptd, J = 7.8, 1.4 Hz, 1H, CH(Ar)), 7.80–7.42 (m, 14H, H(Ar)), 6.97 (d, J = 9.6 Hz, 1H, 5-CH(Ph)), 6.84–6.78 (m, 4H, 2× CH(Ar), 6-CH(Ph), 3-CH(Ph)), 5.22 (d, J = 15.8 Hz, 1H, pro-S-CH2(Py)), 4.75 (d, J = 16.0 Hz, 1H, pro-R-CH2(P)), 4.74 (d, J = 14.0 Hz, 1H, pro-R-CH2(Py)), 4.64 (d, J = 17.9 Hz, 1H, pro-R-CH2(Ph)), 4.52 (ddd, J = 14.1, 5.0, 1.8 Hz, 1H, pro-S-CH2(P)), 4.07 (d, J = 17.9 Hz, 1H, pro-S-CH2(Ph)), 2.69 (s, 3H, NCMe cis to C), and 2.42 (s, 3H, NCMe trans to C). 13C{1H} NMR (125.77 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 155.79 (d, J = 1.9 Hz, 2-C(Py)), 149.30 (brdd, JRh-C = 29.1 Hz, JP-C = 8.7 Hz, CRh), 148.30 (s, 6-CH(Py)), 148.16 (s, 2-C(Ph)), 141.05–128.45 (m, 18C(H)(Ar)), 127.30 (q, JRh-C = 32.4 Hz, 4-C(Ph)), 127.08 (d, JRh-C = 5.8 Hz, NCMe cis to C), 123.52 (q, JF-C = 271.40 Hz, CF3), 125.98–120.26 (m, 6C(H)(Ar)), 124.13 (brs, NCMe trans to C), 74.31 (s, CH2(Py)), 66.95 (d, J = 7.9 Hz, CH2(P)), 66.24 (s, CH2(Ph)), 3.95 (s, NCMe cis to C), 1.08 (s, NCMe trans to C). 19F{1H} NMR (282.33 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = −65.52 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2/acetone-d6, RT, ppm): δ = 28.58 (d, J = 114.1 Hz).
Preparation and characterization of the complex 3
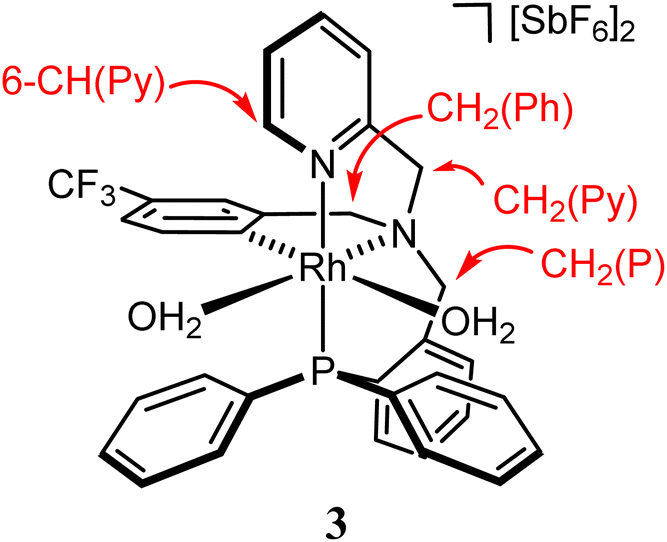 At room temperature, to a solution of [RhCl2(κ4C,N,N′,P-L1)] (1) (500.0 mg, 0.701 mmol) in 30 mL of wet dichloromethane, 1.55 mmol of AgSbF6 were added. The pale yellow suspension was stirred for 72 h and then was filtered to remove the AgCl formed. The solution was evaporated to ca. 5 mL. Addition of 10 mL Et2O and 10 mL of n-pentane led to the precipitation of a light yellow solid that was filtered off and vacuum-dried to afford complex 3 as a microcrystalline solid. Yield: 460.0 mg (57%). Anal. calcd C33H31F15N2O2PRhSb2: C, 34.47; H, 2.72; N, 2.44. Found: C, 34.16; H, 2.57; N, 2.51. IR (solid, cm−1): ν(OH) 3500 (br), ν(SbF6) 653 (s). 1H NMR (500.13 MHz, CD2Cl2, RT, ppm): δ = 8.32 (pt, J = 4.2 Hz, 1H, 6-CH(Py)), 7.96–6.66 (m, 20H, H(Ar)), 4.98 (d, J = 16 Hz, 1H, pro-S-CH2(Py)), 4.74–4.67 (overlapped, 4H, 2× OH2), 4.59 (d, J = 14.4 Hz, 1H, pro-R-CH2(P)), 4.53 (d, J = 15.6 Hz, 1H, pro-R-CH2(Py)), 4.49 (d, J = 18.5 Hz, 1H, pro-R-CH2(Ph)), 4.30 (dd, J = 14.8, 3.2 Hz, 1H, pro-S-CH2(P)), 3.94 (d, J = 18.2 Hz, 1H, pro-S-CH2(Ph)). 13C{1H} NMR (125.77 MHz, CD2Cl2, RT, ppm): δ = 155.87 (s, 2-C(Py)), 147.97 (s, 2-C(Ph)), 147.55 (s, 6-CH(Py)), 143.20 (dd, JRh-C = 32.2 Hz, JP-C = 12.5 Hz, 1H, CRh), 141.74–116.54 (m, 21C(H)(Ar)), 111.63 (q, JF-C = 300.7 Hz, CF3), 73.81 (s, CH2(Py)), 66.78 (d, J = 7.4 Hz, CH2(P)), 66.30 (s, CH2(Ph)). 19F{1H} NMR (282.33 MHz, CD2Cl2, RT, ppm): δ = −62.79 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2, RT, ppm): δ = 27.74 (d, J = 125.6 Hz).
At room temperature, to a solution of [RhCl2(κ4C,N,N′,P-L1)] (1) (500.0 mg, 0.701 mmol) in 30 mL of wet dichloromethane, 1.55 mmol of AgSbF6 were added. The pale yellow suspension was stirred for 72 h and then was filtered to remove the AgCl formed. The solution was evaporated to ca. 5 mL. Addition of 10 mL Et2O and 10 mL of n-pentane led to the precipitation of a light yellow solid that was filtered off and vacuum-dried to afford complex 3 as a microcrystalline solid. Yield: 460.0 mg (57%). Anal. calcd C33H31F15N2O2PRhSb2: C, 34.47; H, 2.72; N, 2.44. Found: C, 34.16; H, 2.57; N, 2.51. IR (solid, cm−1): ν(OH) 3500 (br), ν(SbF6) 653 (s). 1H NMR (500.13 MHz, CD2Cl2, RT, ppm): δ = 8.32 (pt, J = 4.2 Hz, 1H, 6-CH(Py)), 7.96–6.66 (m, 20H, H(Ar)), 4.98 (d, J = 16 Hz, 1H, pro-S-CH2(Py)), 4.74–4.67 (overlapped, 4H, 2× OH2), 4.59 (d, J = 14.4 Hz, 1H, pro-R-CH2(P)), 4.53 (d, J = 15.6 Hz, 1H, pro-R-CH2(Py)), 4.49 (d, J = 18.5 Hz, 1H, pro-R-CH2(Ph)), 4.30 (dd, J = 14.8, 3.2 Hz, 1H, pro-S-CH2(P)), 3.94 (d, J = 18.2 Hz, 1H, pro-S-CH2(Ph)). 13C{1H} NMR (125.77 MHz, CD2Cl2, RT, ppm): δ = 155.87 (s, 2-C(Py)), 147.97 (s, 2-C(Ph)), 147.55 (s, 6-CH(Py)), 143.20 (dd, JRh-C = 32.2 Hz, JP-C = 12.5 Hz, 1H, CRh), 141.74–116.54 (m, 21C(H)(Ar)), 111.63 (q, JF-C = 300.7 Hz, CF3), 73.81 (s, CH2(Py)), 66.78 (d, J = 7.4 Hz, CH2(P)), 66.30 (s, CH2(Ph)). 19F{1H} NMR (282.33 MHz, CD2Cl2, RT, ppm): δ = −62.79 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2, RT, ppm): δ = 27.74 (d, J = 125.6 Hz).
Preparation y characterization of the diastereomers (CRh,RN,SC/ARh,SN,SC)-4
To a suspension of 72.1 mg (0.477 mmol) of (S)-phenylglycine in 40 mL of EtOH, 60.1 mg (0.715 mmol) of NaHCO3 were added. After 15 min of vigorous stirring, 600.0 mg (0.502 mmol) of rac-[Rh(κ4C,N,N′,P-L1)(NCMe)2][SbF6]2 (2) were added and the resulting suspension was refluxed for 12 h. After cooling the reaction mixture to RT, the solvent was vacuum-evaporated until dryness and the residue was extracted with CH2Cl2 (4 × 15 mL). The resulting solution was vacuum-concentrated until ca. 3 mL. The addition of Et2O afforded a pale yellow solid which was filtered off, washed with Et2O (2 × 3 mL) and dried under vacuum. Yield: 406.2 mg (79%). Molar ratio (CRh,RN,SC)-4:(ARh,SN,SC)-4, 1/1. Crystallization of this mixture from MeOH/Et2O affords a (CRh,RN,SC)-4/(ARh,SN,SC)-4, 57/43 molar ratio mixture of diastereomers. Anal. calcd C41H35F9N3O2PRhSb: C, 49.77; H, 3.56; N, 4.24. Found: C, 49.38; H, 3.47; N, 4.00. IR (solid, cm−1): ν(NH2) 2959 (br), ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 1640 (s), ν(SbF6) 656 (s).
O) 1640 (s), ν(SbF6) 656 (s).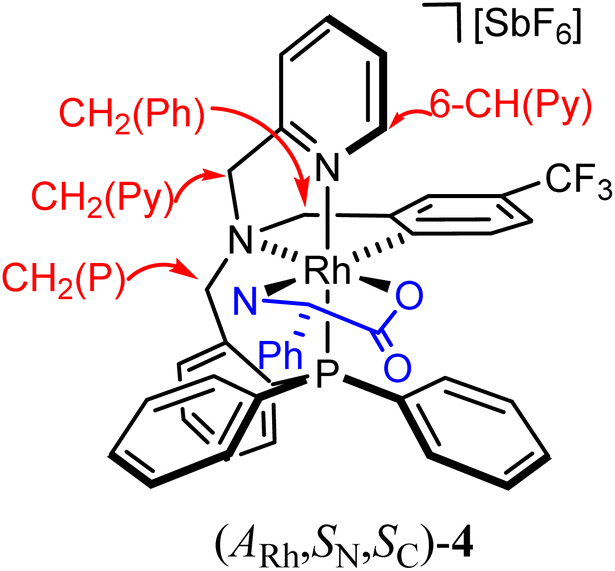
(ARh,SN,SC)-4 diastereomer. 1H NMR (500.13 MHz, CD2Cl2, RT, ppm): δ = 8.32 (broad pseudo-triplet, J = 4.0 Hz, 1H, 6-CH(Py)), 7.96–6.60 (m, 25H, CH(Ar)), 5.28 (d, J = 15.6 Hz, 1H, pro-S-CH2(Py)), 5.02 (dd, J = 11.0, 6.3 Hz, 1H, pro-S-NH2), 4.94 (d, J = 14.6 Hz, 1H, pro-S-CH2(P)), 4.67 (d, J = 15.9 Hz, 1H, pro-R-CH2(Py)), 4.60–4.54 (m, 2H, pro-S-CH2(P), pro-R-CH2(Ph)), 3.87 (d, J = 17.4 Hz, 1H, pro-R-CH2(Ph)), 3.98 (dd, J = 12.1, 6.3 Hz, 1H, C*H), 2.79 (pt, J = 11.2 Hz, 1H, pro-R-NH2). 13C{1H} NMR (125.77 MHz, CD2Cl2, RT, ppm): δ = 179.45 (s, COO), 160.93 (dd, JRh-C = 25.80 Hz, JP-C = 9.90 Hz, 1H, CRh), 156.11 (d, J = 1.7 Hz, 2 C(Py)), 147.38 (s, 6-CH(Py)), 145.45–115.98 (m, 34C(H)(Ar)), 74.72 (s, CH2(Py)), 68.65 (d, J = 5.6 Hz, CH2(P)), 67.26 (s, CH2(Ph)), 60.05 (s, C*). 19F{1H} NMR (282.33 MHz, CD2Cl2, RT, ppm): δ = −62.48 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2, RT, ppm): δ = 29.67 (d, J = 130.8 Hz).
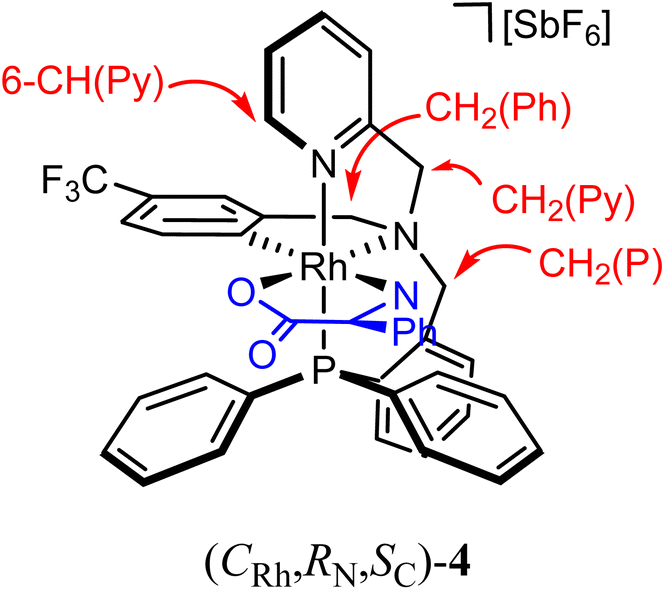
(CRh,RN,SC)-4 diastereomer. 1H NMR (500.13 MHz, CD2Cl2, RT, ppm): δ = 8.38 (broad pseudo-triplet, J = 3.9 Hz, 1H, 6-CH(Py)), 7.88–6.61 (m, 25H, CH(Ar)), 5.11 (d, J = 14.8 Hz, 1H, pro-R-CH2(P)), 5.07 (d, J = 15.9 Hz, 1H, pro-S-CH2(Py)), 4.49 (d, J = 16.0 Hz, 1H, pro-R-CH2(Py)), 4.64–4.53 (m, 3H, pro-S-CH2(Py), pro-S-CH2(P), pro-R-CH2(Ph)), 4.20 (dd, J = 11.5, 9.3 Hz, 1H, pro-R-NH2), 3.87 (dd, J = 17.4 Hz, 1H, pro-S-CH2(Ph)), 3.71 (dd, J = 12.0, 7.3 Hz, 1H, pro-S-NH2), 3.10 (pt, J = 8.0 Hz, 1H, C*H). 13C{1H} NMR (125.77 MHz, CD2Cl2, RT, ppm): δ = 180.06 (s, COO), 160.68 (dd, JRh-C = 24.29 Hz, JP-C = 10.56 Hz, 1H, CRh), 155.88 (d, J = 2.0 Hz, 2-C(Py)), 147.42 (s, 6-CH(Py)), 145.10–115.88 (m, 34C(H)(Ar)), 74.84 (s, CH2(Py)), 68.52 (d, J = 5.6 Hz, CH2(P)), 67.69 (s, CH2(Ph)), 58.61 (s, C*). 19F{1H} NMR (282.33 MHz, CD2Cl2, RT, ppm): δ = −62.54 (s). 31P{1H} NMR (202.46 MHz, CD2Cl2, RT, ppm): δ = 28.83 (d, J = 130.1 Hz).
Chromatographic separation of the diastereomers (CRh,RN,SC/ARh,SN,SC)-4
An equimolar mixture of diastereomers (CRh,RN,SC/ARh,SN,SC)-[Rh(κ4C,N,N′,P-L1)((S)-phenylglycinato)][SbF6] (200.0 mg, 0.194 mmol) was eluted with a CH2Cl2/MeOH (98.75/1.25, v/v, 30 mL min−1) mixture on a silica column (SI-HP, 25 g, 30 μm). Three fractions A, B and C, were consecutively collected. Fraction A (43.0 mg, 0.042 mmol, 21%) was diastereopure (ARh,SN,SC)-4. Fraction B (96.0 mg, 0.093 mmol, 48%) was an equimolar mixture of the diastereomers (CRh,RN,SC/ARh,SN,SC)-4. Fraction C (36.0 mg, 0.035 mmol, 18%) was diastereopure (CRh,RN,SC)-4.Elimination of the auxiliary. Crystallization of enantiopure (ARh,RN)-1 and (CRh,SN)-1
To a suspension of diastereopure (ARh,SN,SC)-4 (20.0 mg, 0.019 mmol) in 4 mL of EtOH, 0.20 mL of 12 M HCl(aq) were added. After 48 h at RT, a pale yellow microcrystalline solid precipitated from the bright yellow solution. It was collected by filtration and successively washed with EtOH (3 × 3 mL) and Et2O (3 × 3 mL). The second fraction of the product was obtained from the filtrate (12 mg). Total yield: 89%. The isolated solid was characterized as (ARh,RN)-1. The diastereomer (CRh,SN)-1 was similarly obtained starting from (CRh,RN,SC)-4 (yield: 87%). The optical purity of the samples was determined by HPLC with a Chiralpak IB column (0.46 × 25 cm) with IB guard (0.46 cm × 5 cm) (65/35, CH3CN/H2O, 0.1% CF3COOH; 0.4 mL min−1; tR 151.8 and 156.7 min for (ARh,RN)-1 and (CRh,SN)-1, respectively). Circular dichroism spectra: (ARh,RN)-1 (MeOH, 5.8 × 10−4 M, RT, λ(nm) (Δε)): 282 (−90.96), 205 (−25.28), 360 (+17.68). (CRh,SN)-1 (MeOH, 4.0 × 10−4 M, RT, λ(nm) (Δε)): 282 (+106.12), 205 (+26.53), 360 (−11.37).Preparation of the diastereopure solvate complex 2
Diastereopure (ARh,RN)-2 was prepared starting from the corresponding diastereopure chlorido complex (ARh,RN)-1, as reported above for rac-2.Catalytic procedure for the DA reaction between methacrolein and cyclopentadiene
At RT, in an NMR tube, to a solution of 1.16 × 10−3 or 2.32 × 10−3 mmol (5 or 10 mol%) of the corresponding catalyst in CD2Cl2 (0.5 mL), 2.0 μL (0.023 mmol) of methacrolein and 10 mg of 4 Å MS were added. After 5 min, 7.0 μL (0.081 mmol) of freshly distilled cyclopentadiene was added. After the indicated reaction times, the conversion and exo/endo ratio were determined by 1H NMR. The enantiomeric ratio for the exo isomer was determined by integration of the 1H NMR signals in presence of the chiral shift reagent (+)-Eu(hfc)3. Major enantiomer (R), δ = 17.10 ppm; minor enantiomer (S), δ = 19.09 ppm.18Catalytic procedure for the 1,3-dipolar cycloaddition reaction between methacrolein and N-benzylidenphenylamine-N-oxide
In an NMR tube, complex 2 (1.16 × 10−3 mmol (2.70 mg), 5 mol%) was dissolved in CD2Cl2 (0.2 mL) at −25 °C. Freshly distilled methacrolein (5 μL, 0.07 mmol) and 10.0 mg of activated 4 Å molecular sieves were added. After seeking the resulting suspension, a solution of N-benzylidenphenylamine-N-oxide (6.70 mg, 0.035 mmol) in CD2Cl2 (0.3 mL) was added. Regioselectivity was determined by the integration of the 1H NMR signal of the C3 proton on the crude mixture in CD2Cl2. Enantioselectivity was determined by the integration of the 1H NMR signals of the diastereomeric (S)-methylbenzylimine derivatives.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank the Ministerio de Ciencia, Innovación y Universidades (MCIU) of Spain, Agencia Estatal de Investigación (AEI) of Spain, Fondo Europeo de Desarrollo Regional (FEDER) (CTQ2018-095561-BI00 and PID2021-122406NB-I00) and Gobierno de Aragón (Grupo de Referencia: Catálisis Homogénea Enantioselectiva E05-20R) for financial support.References
- (a) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, vol. I–III Search PubMed; (b) Catalytic Asymmetric Synthesis, ed. I. Ojima, VCH, Weinheim, 2000 Search PubMed; (c) R. Noyori, Asymmetric Catalysis in Organic Synthesis, John Willey and Sons, New York, 1994 Search PubMed; (d) P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, 2009 Search PubMed.
- H. Brunner, Acc. Chem. Res., 1979, 12, 250–257 CrossRef CAS.
- K. Muñiz and C. Bolm, Chem.–Eur. J., 2000, 6, 2309–2316 CrossRef.
- A. Werner, Ber. Dtsch. Chem. Ges., 1911, 44, 1887–1898 CrossRef CAS.
- (a) L. Gong, M. Wenzel and E. Meggers, Acc. Chem. Res., 2013, 46, 2635–2644 CrossRef CAS PubMed; (b) E. Meggers, Eur. J. Inorg. Chem., 2011, 2911–2926 CrossRef CAS; (c) E. Meggers, Chem.–Eur. J., 2010, 16, 752–758 CrossRef CAS PubMed; (d) M. Fontecabe, O. Hamelin and S. Ménage, Top. Organomet. Chem., 2005, 15, 271–288 CrossRef.
- M. Chavarot, S. Ménage, O. Hamelin, F. Charnay, J. Pécaut and M. Fontecave, Inorg. Chem., 2003, 42, 4810–4816 CrossRef CAS PubMed.
- (a) G. M. Fusi, S. Gazzola and U. Piarulli, Adv. Synth. Catal., 2022, 364, 696–714 CrossRef CAS; (b) P. Dey, P. Rai and B. Maji, ACS Org. Inorg. Au, 2022, 2, 99–125 CrossRef CAS; (c) L. Zhang and E. Meggers, Chem.–Asian J., 2017, 12, 2335–2342 CrossRef CAS PubMed; (d) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed; (e) Z.-Y. Cao, W. D. G. Brittain, J. S. Fossey and F. Zhou, Catal. Sci. Technol., 2015, 5, 3441–3451 RSC.
- (a) V. A. Larionov, B. L. Feringa and Y. N. Belokon, Chem. Soc. Rev., 2021, 50, 9715–9740 RSC; (b) A. Fanourakis, A. Philip, J. Docherty, P. Chuentragool and R. J. Phipps, ACS Catal., 2020, 10, 10672–10714 CrossRef CAS PubMed; (c) S. K. Ghosh, A. Ehnbom, K. G. Lewis and J. A. Gladysz, Coord. Chem. Rev., 2017, 350, 30–48 CrossRef CAS; (d) A. Ehnbom, S. K. Ghosh, K. G. Lewis and J. A. Gladysz, Chem. Soc. Rev., 2016, 45, 6799–6811 RSC; (e) L. Gong, L.-A. Chen and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10868–10874 CrossRef CAS PubMed.
- (a) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (b) H. Huo and E. Meggers, Chimia, 2016, 70, 186–191 CrossRef CAS PubMed; (c) E. Meggers, Chem. Commun., 2015, 51, 3290–3301 RSC.
- (a) C. Ganzmann and J. A. Gladysz, Chem.–Eur. J., 2008, 14, 5397–5400 CrossRef CAS PubMed; (b) W. J. Maximuck, C. Ganzmann, S. Alvi, K. R. Hooda and J. A. Gladysz, Dalton Trans., 2020, 49, 3680–3691 RSC.
- (a) J. Hartung and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10183–10185 CrossRef CAS PubMed; (b) J. Hartung, P. K. Dornan and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 13029–13037 CrossRef CAS PubMed.
- K. Endo, Y. Liu, H. Ube, K. Nagata and M. Shionoya, Nat. Commun., 2020, 11, 6263 CrossRef CAS PubMed.
- (a) M. Carmona, R. Rodríguez, V. Passarelli and D. Carmona, Organometallics, 2019, 38, 988–995 CrossRef CAS; (b) J. Téllez, I. Méndez, F. Viguri, R. Rodríguez, F. J. Lahoz, P. García-Orduña and D. Carmona, Organometallics, 2018, 37, 3450–3464 CrossRef; (c) M. Carmona, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, J. Am. Chem. Soc., 2018, 140, 912–915 CrossRef CAS PubMed; (d) M. Carmona, L. Tejedor, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Chem.–Eur. J., 2017, 23, 14532–14546 CrossRef CAS PubMed; (e) M. Carmona, R. Rodríguez, I. Méndez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Dalton Trans., 2017, 46, 7332–7350 RSC.
- (a) R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., Int. Ed. Engl., 1966, 5, 385–415 CrossRef CAS; (b) V. Prelog and G. Helmchen, Angew. Chem., Int. Ed. Engl., 1982, 21, 567–583 CrossRef; (c) C. Lecomte, Y. Dusausoy, J. Protas, J. Tirouflet and A. Dormond, J. Organomet. Chem., 1974, 73, 67–76 CrossRef CAS . For the C/A convention for octahedral centres see ; (d) N. G. Connelly, T. Damhus, R. H. Hartshorn and A. T. Hutton, Nomenclature of Inorganic Chemistry, IUPAC Recommendations, RSC Publishing, Cambridge, UK, 2005, ch. IR-9.3.4.8, p. 189 Search PubMed.
- To simplify the notation of the stereochemical descriptors, we will label these diastereomers as CRh,RN and ARh,SN. R and S refer to the configuration at the sp3 nitrogen and C and A to the clockwise or anticlockwise arrangement of the donor atoms at the equatorial plane of the octahedral metal complex..
- CCDC-2206804.†.
- Due to the nomenclature rules, in this transformation, retention of the configuration implies the change of the metal descriptor from A to C, and vice versa..
- (a) K. Furuta, S. Shimizu, Y. Miwa and H. Yamamoto, J. Org. Chem., 1989, 54, 1481–1483 CrossRef CAS; (b) J. W. Faller, X. Liu and J. Parr, Chirality, 2000, 12, 325–337 CrossRef CAS PubMed; (c) D. Carmona, M. P. Lamata, F. Viguri, R. Rodríguez, C. Barba, F. J. Lahoz, M. L. Martín, L. A. Oro and L. Salvatella, Organometallics, 2007, 26, 6493–6496 CrossRef CAS.
- D. Carmona, M. P. Lamata, F. Viguri, R. Rodríguez, L. A. Oro, F. J. Lahoz, A. I. Balana, T. Tejero and P. Merino, J. Am. Chem. Soc., 2005, 127, 13386–13398 CrossRef CAS PubMed.
- (a) E. P. Kündig, C. M. Saudan and G. Bernardinelli, Angew. Chem., Int. Ed., 1999, 38, 1220–1223 CrossRef; (b) D. Carmona, C. Cativiela, S. Elipe, F. J. Lahoz, M. P. Lamata, M. P. López-Ram de Víu, L. A. Oro, C. Vega and F. Viguri, Chem. Commun., 1997, 2351–2352 RSC; (c) A. J. Davenport, D. L. Davies, J. Fawcett, S. A. Garrat and D. R. J. Russell, J. Chem. Soc., Dalton Trans., 2000, 4432–4441 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of complexes 2–4, HPLC chromatograms, and spectroscopic and structural characterization of complex 2. CCDC 2206804. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra06982b |
| This journal is © The Royal Society of Chemistry 2022 |